
Abstract
Selenium (Se) is an essential element, and severe deficiency of Se is incompatible with human life. Se is part of a restricted group of 25 proteins, the selenoproteins. The selenoproteins have the rare amino acid selenocysteine (Sec), which is an analog of cysteine (Cys). The -SeH (selenol) group of Sec is an active redox center in selenoproteins, which are part of the endogenous antioxidant system, for instance, the oxidoreductases glutathione peroxidase (GPX) and thioredoxin reductase (TXNRD). The selenoproteins iodothyronine deiodinases (DIO1 and DIO2) are involved in the metabolism of triiodothyronine (T3) and thyroxine (T4) thyroid hormones; and methionine sulfoxide reductase B1 (MSRB1) reduces the oxidized methionine sulfoxide to methionine. Selenoprotein P (SELENOP) plays an important role in the body Se distribution and homeostasis. The brain is particularly dependent on Se supply via SELENOP and is spared from Se deficiency. Although Se deficiency can have neuropathological effects, the chronic exposure to Se levels above the nutritional requirement can be associated with neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD). Paradoxically to Se role as antioxidant in the -SeH group of selenoproteins, the metabolites of Se in the body (particularly the selenide intermediates and methylselenol) are highly reactive and can generate cytotoxic prooxidant metabolites (e.g., methylselenyl radical). This chapter will explore the role of Se (both excess and deficiency) as physiological neuroprotective or neurotoxic agent potentially involved in chronic neurodegenerative pathologies, such as ALS, AD, and Parkinson’s disease (PD).
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
- Selenoproteins
- Alzheimer’s disease
- Amyotrophic lateral sclerosis
- Parkinson’s disease
- Organoselenium compounds
1 Introduction
Selenium (Se) is a chemical element discovered by Jöns Jacob Berzelius in 1817 and belongs to the chalcogen family and, along with oxygen (O), sulfur (S), tellurium (Te), and polonium (Po), forms the group 16 of the periodic table (Barbosa et al. 2017). It is an essential trace element and has fundamental importance for human health, and nutritionally it has a narrow range between deficiency and toxicity (Fig. 1) (Nogueira et al. 2021). In mammals, Se has critical functions in the central nervous system (CNS), and the brain is one of the organs of the body that has the highest demand for Se, particularly upon dietary deficiency (Kühbacher et al. 2009).

Inverted U relationship between blood Se levels and human pathologies. The ideal levels of Se for adequate selenoprotein biosynthesis were arbitrarily based on the quantity of Se in blood that was associated with maximal glutathione peroxidase activity in adult humans from New Zealand (for original citations, see Nogueira et al. 2021). The adult normal range of Se is between 70 and 130 μg/L (or 0.9–1.6 μmol/L)
The main biochemical task of Se is dictated by the functional group selenol (-SeH), which is found in 25 selenoproteins in humans. The well-established biochemistry of selenoproteins is related to their catalytic role as antioxidant enzymes. For instance, selenoenzymes such as glutathione peroxidases (GPXs), thioredoxin reductases (TXNRDs), and methionine sulfoxide reductase (MSRB) are central in the regulation of cellular redox state. In addition to these enzymes, mammalian cells express a dozen other selenoproteins, which are not well characterized in terms of their exact biochemical role in cell chemistry. For instance, iodothyronine deiodinase 1 (DIO1), iodothyronine deiodinase 2 (DIO2), thioredoxin reductase 3 (TXNRD3), selenoprotein F (SELENOF), selenoprotein K (SELENOK), selenoprotein M (SELENOM), selenoprotein N (SELENON), selenoprotein S (SELENOS), and selenoprotein T (SELENOT) are found in the endoplasmic reticulum and modulate several complexes process (Table 1).
The potential role of Se as a part of antioxidant enzymes (selenoproteins) in Parkinson’s disease (PD) and Alzheimer’s disease (AD) has been suggested by different authors (Adani et al. 2020; Cardoso et al. 2017). Adequate intake of Se for brain homeostasis is not surprising, given the critical role of selenoproteins as antioxidants. Furthermore, several selenoproteins participate in the modulation of inflammatory responses (Hariharan and Dharmaraj 2020). Moreover, there are studies indicating a decrease in Se levels in the plasma, blood, and several brain regions in AD patients (Reddy et al. 2017). However, other studies have provided conflicting results, and no association or even a negative correlation between Se levels in the brain and AD was observed (Vinceti et al. 2017, 2019a). There are several suggestions indicating that inflammation may cause a decrease in the body burden of Se. For instance, a decrease in blood Se levels has been reported in sepsis and severe SARS-CoV-2 infection (Nogueira et al. 2021). In short, it is still unclear if the low levels of Se found in some studies were a consequence or the cause of exaggerated inflammatory response associated with AD and other acute and chronic pathologies. Another critical point regarding Se and AD is the role of its speciation as a predicting factor of disease (Vinceti et al. 2017). In short, Se has essential roles in brain biochemistry, but its intake and blood and body levels must be finely adjusted to be within the ideal range needed to support the adequate selenoprotein biosynthesis (Fig. 1 and Sect. 2.1).
It has also been posited that the overexposure to inorganic forms of Se is involved in the etiology of amyotrophic lateral sclerosis (ALS) (Vinceti et al. 2013). The neural toxicity of Se is played by its reactive metabolites (for instance, methylselenol, methylselenyl radical, and HSe-) (Nogueira et al. 2021).
2 Sources of Se
Se can be found in diverse organic (selenomethionine, methylselenocysteine, and selenocysteine – Sec) and inorganic chemical forms (selenate and selenite). The comparison of bioavailability of organic (particularly, selenomethionine) and inorganic Se compounds is not fully consistent, though the organic forms seem to be more effectively absorbed in the gastrointestinal tract (Vendeland et al. 1992) and are less prone to induce toxic effects (Kiełczykowska et al. 2018).
Se is found in soils and is taken up by plants. The bulk of Se in the food system resides in the soil (0.1–2 mg Se/kg), primarily because of the weathering of Se-containing rocks, as well as volcanic activity and dust (Hariharan and Dharmaraj 2020; Oliveira et al. 2017). Other factors that can influence the levels of Se in foods are the ability of certain plants to accumulate it, cultivation and breeding methods, climatic conditions, and methods of preparing food products (Kieliszek and Błazejak 2016). In water, Se is encountered in low quantities, especially as selenate and selenite. The acceptable limit of Se in drinking water is 10 μg/L (Filippini et al. 2018).
Diet is the main source of Se to humans. The selenomethionine synthesized by plants is incorporated non-specifically in proteins in the place of methionine and can be utilized by consumers at all levels of the food web. The main sources of Se in European populations are cereals, meat, fish, and dairy products (Filippini et al. 2018). The content of Se in some typical food products is presented in Table 2.
Brazil nuts and mushrooms can exhibit high levels of organic Se, mainly selenomethionine, and the Se concentration varies depending on the Se-soil content (British Nutrition Foundation 2001). High concentrations of Se have also been found in plants of the Brassica genus (broccoli, cabbage, cauliflower, and kohlrabi) (Kieliszek and Błazejak 2016). Meat, egg yolk, and fish are also good sources of Se, but the Se concentration will heavily depend on Se-soil and on the Se-feed supplements content (British Nutrition Foundation 2001).
The effect of milling, wash, cooking, and general preparation of foods can change the Se bioavailability. However, studies have suggested that loss of Se during usual cooking procedures is likely to be insignificant for most foods (British Nutrition Foundation 2001).
2.1 Dietary Recommendation of Se Intake
The Food and Nutrition Board at the Institute of Medicine of the National Academies, USA, established that the Se recommended dietary allowance (RDA) for individuals higher than 14 years old is 55 μg Se/day (Institute of Medicine 2000) (Table 3). It is important to stressed that the Se RDA to children is lower than the RDA to adolescents and adult individuals (Table 3) once the acceptable levels of blood Se in children is lower than adults (Table 4). Moreover, an ingestion of an extra 15 μg Se/day is recommended during lactation period, based on the level of Se in human milk of 10–20 ng/mL (British Nutrition Foundation 2001; Institute of Medicine 2000).
According to the US National Academy of Sciences (NAS), the tolerable upper intake level (UL) of Se, i.e., the threshold that should not be exceeded, is 400 μg Se/day (Table 3). However, the toxic dose was established as greater than 700 μg Se/day for adults (Kiełczykowska et al. 2018). However, a chronic intake above 100 μg Se/day may cause toxicity after long-term of exposure. Accordingly, the Denmark PRECISE study has demonstrated that the intake of 300 μg/day of Se-enriched-yeast (mainly, selenomethionine) for 5 years increased the mortality rate 10 years later (Rayman et al. 2018).
In the United Kingdom, the Se recommended reference nutrient intake (RNI) for adults is 60 μg/day for adult women and 75 μg/day for lactating women and adult men (British Nutrition Foundation 2001). The RNI is based on an intake where blood Se levels are near to the concentration of 100 μg/L in the blood. At this concentration, the blood activity of GPX reaches a plateau (Nogueira et al. 2021).
The blood Se levels are influenced by pathologies that are associated with deficiency or excess of the element. For instance, inflammatory processes can be associated with low levels of Se in the blood (Nogueira et al. 2021). The ideal level of Se in the blood is in the range of 70–110 μg/L, but the acceptable limits vary according to the developmental stage (Table 4) (Nogueira et al. 2021; Institute of Medicine 2000). Of nutritional importance, body Se burden will vary depending on the Se concentration in the ingested food and on the chemical Se form (British Nutrition Foundation 2001; Filippini et al. 2018; Fox et al. 2004).
3 Chemistry of Se: From the Diet to the Selenoproteins
After entering the body, the Se fate will depend on its chemical form. In rats, selenite and selenate are readily taken up by the small intestine (particularly in the ileum) and the absorption of selenate is much faster than that of selenite. The large intestine (colon and cecum) can also absorb Se, but to much less extent than jejunum and ileum (Vendeland et al. 1992). Selenomethionine is moved by the transporters involved in methionine uptake, and the uptake of Se from selenomethionine is faster than that of selenite or selenate (Vendeland et al. 1992). In adult humans (men and women), the absorption of Se is high (near 90% from fish and selenate, but about 50% from yeast) (Fox et al. 2004).
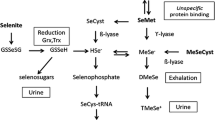
The metabolism of eletrophilic Se forms (selenite and selenate) involves their reduction to selenide anion (HSe−) either chemically or enzymatically (Tobe and Mihara 2018). HSe− is central in the metabolism of Se once it is incorporated into selenoproteins (Sect. 3.1). Indeed, both inorganic and organic forms of Se must be metabolized to HSe− as an obligatory step for incorporation into Sec. Possibly selenopersulfide (formed by a direct interaction of reduced glutathione (GSH) with electrophilic Se forms) and some Se delivery proteins seem to play a role in the regulation of HSe− in living cells (Tobe and Mihara 2018).
Selenite and selenate can be directly reduced by low molecular mass molecules (e.g., GSH) or by NADPH-dependent thioredoxin reductase or glutathione reductase (Tobe and Mihara 2018). The metabolism of organic forms of Se is more complicated and depends on the type of compound. Selenomethionine, which is the most common form of organic Se found in human diet, can be metabolized by different enzymes to form either Sec (which is expected to be oxidized to selenocystine) or methylselenol. The Sec is transformed to methylselenol that is then demethylated to form HSe− (Tobe and Mihara 2018). The Sec derived from the catabolism of selenoproteins can enter the same pathway described for that derived from selenomethionine by the transelenation pathway (Tobe and Mihara 2018) and can be re-incorporated in newly synthesized selenoproteins (Fig. 2).

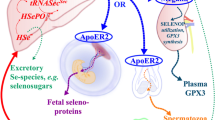
Representation of the Se metabolic pathway. SeO4−2 (selenate), SeO3−2 (selenite), Sec (selenocysteine), SeM (selenomethionine), MeSec (methylselenocysteine), and proteins can be obtained from diets and metabolized to HSe−, which posteriorly will be directed to selenoprotein synthesis via the co-translational incorporation in the tRNA[Ser][Sec] (Sec-tRNA). The selenoprotein P (SELENOP) has 10 Sec residues and is involved in the transport of Se to several organs (for instance, the brain). SELENOP provides Se for brain selenoprotein biosynthesis, which is critical during Se deficiency states. The highly reactive toxic MeSe• is derived from methylselenol (MeSeH)
Of particular importance for the toxicity of Se, methylselenol is highly reactive and can originate the methylselenyl free radical (MeSe•), which can oxidize thiol groups and other biomolecules. As depicted in Fig. 1, the toxic effects of overexposure to Se might be associated with several pathologies, which can be due to high concentrations of HSe−, methylselenol, and MeSe•.
3.1 Sec Synthesis
All organisms that synthesize Sec for incorporation in selenoproteins do it in their tRNA (tRNA[Ser]Sec) (Hatfield et al. 2006). For Sec synthesis, Se must be available in the HSe− form, as noted above. Sec synthesis begins with the aminoacylation of serine (Ser) in tRNA[Ser]Sec by seryl-tRNA synthetase (SerS) with the break of one ATP molecule (Hatfield et al. 2006). Ser differs structurally from Sec only in the lateral functional group (-OH group), and Ser is the source of the carbon backbone for Sec synthesis (Oliveira et al. 2017). Subsequently, phosphoseryl-tRNA kinase (PSTK) phosphorylates the seryl moiety in a process that requires ATP consumption (Hatfield et al. 2006; Oliveira et al. 2017). Concomitantly, monoselenophosphate is synthesized by selenophosphate synthase 2 (SEPHS2) from HSe− and ATP as substrate (Hatfield et al. 2006; Oliveira et al. 2017). There are two selenophosphate synthase-coding genes in mammals SEPHS1 and SEPHS2, wherein SEPHS2 is a selenoprotein that participates in autoregulation of its biosynthesis and the SEPHS1 seems to have another cellular function other than Sec or selenoprotein synthesis (Turanov et al. 2011). The Se incorporation in the Ser is catalyzed by phosphoseryl-tRNA selenium transferase (SEPSecS) and results in the synthesis of the Sec residue and the release of pyrophosphate (Hatfield et al. 2006; Oliveira et al. 2017) (Fig. 3).

Synthesis of Sec and its incorporation into the nascent polypeptide. Sec synthesis occurs in its own tRNA[Ser]Sec. A Ser residue is aminoacylated in the tRNA[Ser]Sec by the enzyme SerS and is subsequently phosphorylated by the PSTK enzyme. At the same time, HSe− is converted to monoselenophosphate by SEPHS2. The synthesis of Sec itself is carried out by SEPSecS which incorporates Se into the phosphorylated Ser and releases a pyrophosphate. Virtually, all stages of Sec synthesis have ATP consumption. Once Sec is synthesized, it is incorporated into the nascent polypeptide chain by the UGA codon, canonically a stop codon. For Sec to be inserted, cis-elements are needed, such as selenocysteine insertion sequence (SECIS), and trans, highlighting SECIS binding protein (SBP2) and selenocysteine elongation factor (EFSec). SBP2, EFSec, SECIS, and the tRNA[Ser]Sec form a complex that interacts with the UGA codon allowing the insertion of Sec and preventing the completion of the translation
It is noteworthy that in rodents the presence of cysteine (Cys) in place of Sec in proteins that normally contain Se in a state of Se deficiency has been reported (Turanov et al. 2011). Cys and Sec differ in their molecular structure only in the side chain chalcogen atom (S or Se, respectively). Cys is encoded by the UGU and UGC codons (Fig. 4a), but in this specific case, it is inserted in the UGA codon (i.e., Sec codon) by an unknown mechanism. Turanov et al. (2011) speculated that SEPHS2 could use sulfide anion (HS−) instead of HSe− as a sulfur donor for the synthesis of Cys in the tRNA[Ser]Sec. Cys was found in TXNRD1 in a SECIS-dependent manner in NIH 3 T3 cells and in TXNRD1 and TXNRD3 of mice liver fed a Se-deficient diet (Turanov et al. 2011). In this sense, it is proposed that in a situation of Se deficiency, Cys can be synthesized in mammalian tRNA[Ser]Sec.

Genetic code with 21 proteinogenic amino acids. Initiation codon Met is indicated in orange, two stop codons (UAA and UAG) are indicated in green, and the UGA Sec or the third stop codon are in blue (a). Mammalian tRNA[Ser]Sec (b). The modified nucleotides are highlighted in orange and pink. Pink is the uracil in position 34 (U34), which can be facultatively methylated according to the Se status of the body. In a context of Se sufficiency, uracil is methylated, forming 5-5-methoxycarbonylmethyl-20-O-methyluridine (mcm5Um), and this tRNA[Ser]Sec is involved in the stress-related selenoproteins expression. On the contrary, in a context of Se deficiency, uracil is not methylated, remaining 5-methoxycarbonylmethyluridine (mcm5U), and this tRNA[Ser]Sec is involved in the housekeeping selenoproteins expression
3.2 tRNA[Ser]Sec
The tRNA[Ser]Sec is the longest tRNA in eukaryotes (90–93 nt) and has only five modified nucleotides (two possible modifications at position 34 and one at positions 37, 55, and 58) (Hatfield et al. 2006) (Fig. 4b). Modification of uracil from position 34 generates 5-methoxycarbonylmethyluridine (mcm5U) and subsequent methylation, forming 5-methoxycarbonylmethyl-20-O-methyluridine (mcm5Um). In fact, the synthesis of mcm5Um is the last nucleotide modification that occurs in the tRNA[Ser]Sec (Hatfield et al. 2006; Turanov et al. 2011). The methylation process is facultative and allows the existence of two major isoforms mcm5U e mcm5Um that present distinct secondary and tertiary structures (Hatfield et al. 2006). Se levels influence the methylation process in such a way that in the context of Se sufficiency, there is more mcm5Um isoform than mcm5U and the opposite occurs in Se deficiency, particularly, in the liver, kidney, heart, and muscles (Hatfield et al. 2006; Oliveira et al. 2017; Turanov et al. 2011). The presence of mcm5Um is correlated with an increase in the stress-related selenoprotein expression, and its absence (i.e., presence of mcm5U) is associated with a predominant expression of housekeeping selenoproteins (Oliveira et al. 2017). The methylation of mcm5U to mcm5Um is a key regulatory covalent modification in the tRNA[Ser]Sec that canalizes the scarce Se to the synthesis of hierarchically more important selenoproteins, for instance, GPX4 and TXNRDs.
3.3 Selenoproteins Translation
To recognize the UGA codon, a canonical stop codon, as Sec insertion, cis- and trans-elements are needed during the synthesis of selenoproteins. In animals, selenocysteine insertion sequence (SECIS) is a cis-element present in the 3′-untranslated region (3′-UTR) of selenoprotein mRNAs that forms a stem-loop-stem-loop structure with approximately 100 nt (Bulteau and Chavatte 2015). The SECIS of eukaryotes presents (I) two or three helixes separated by an internal loop; (II) two GA-AG pairs at the base of the second helix (SECIS core or quartet), responsible for a kink turn generating a peculiar SECIS 3D-conformation; and (III) an apical loop with two or three unpaired adenines (AAA/G) that are important for the recognition of SECIS binding protein (SBP2) (Fig. 5). The tandem GA-AG pairs in the SECIS core and unpaired AAA/G are conserved sequences and are necessary for the functioning of SECIS (Bulteau and Chavatte 2015). Eukaryotes have two classes of SECIS structures, i.e., type 1 (present in the 3′-UTR of DIO1, GPX1, GPX2, and the second element of SELENOP mRNAs) and type 2 (present in the 3′-UTR of DIO2, DIO3, GPX3, GPX4, the first element of SELENOP, SELENOW, SELENOF, and SEPHS2 mRNAs) which differ in the position of the adenines and in the presence of an additional helix in the apical loop (Fig. 5) (Bulteau and Chavatte 2015). It is important to highlight that the selenoprotein mRNAs present only one SECIS element, except for SELENOP which has two (Burk and Hill 2005). SELENOP has ten Sec residues in its structure, one residue in the N-terminal region, and nine residues in the C-terminal region (Table 1) (Burk and Hill 2005). It is proposed that the second SECIS element interacts with the first UGA codon, and the first SECIS element, more efficient, interacts with the other nine UGA codons to insert Sec residues (Bulteau and Chavatte 2015). In addition to SELENOP, DIO2 and SELENON have more than one Sec, i.e., two residues of this amino acid. SELENON has two variants (accession numbers in NCBI NM_206926.2 and NM_020451.3) derived from alternative splicing. Variant 2 has 590 amino acids and two Sec residues (Table 1), and variant 1 presents 556 amino acids and a Sec residue at position 428. Specifically, the exon removed by alternative splicing has the UGA codon.

Consensus structure of SECIS type 1 and type 2 of eukaryotes (B). Tandem GA-AG pairs and AAA/G conserved nucleotides were indicated in green. The nucleotide amount of the internal and apical loops is variable, so they have not been indicated. • means non-conventional base pairs
The SBP2 and the Sec elongation factor (EFSec) are the main trans-elements, but there are other proteins involved in the Sec insertion, for instance, rpL30, termination factor eRF1, and the eRF1- and ribosome-dependent GTPase eRF3 (Hatfield et al. 2006). Briefly, the C-terminal region of EFSec interacts with SBP2 forming a complex which is stimulated by the presence of selenocysteinyl-tRNA[Ser]Sec. SBP2 binds to the SECIS core and to the 28S rRNA for the co-translational insertion of the Sec into the nascent polypeptide chain in the UGA codon (Fig. 3) (Bulteau and Chavatte 2015). The translation stop codon in selenoproteins is predicted to be UAG or UAA. It is estimated that SECIS cannot be closer than 55–111 nt from the UGA codon to support Sec insertion, probably due to the lack of space for SECIS to interact with trans-elements and UGA (Bulteau and Chavatte 2015).
As briefly described in Sect. 3.2, the methylation of mcm5U in the tRNA[Ser]Sec to mcm5Um in the abundance of Se is an important level of control for selenoprotein biosynthesis. In addition, there are some additional levels of control of UGA recoding efficiency. For instance, the affinity of SECIS binding proteins for specific selenoprotein mRNA molecules can be higher for those of essential selenoproteins (e.g., GPX4 and TXNRD1) than for non-essential ones. Furthermore, the levels of specific mRNA molecules are also modulated by the Se availability (Sect. 3.4). However, the mechanism behind this process is not well understood. There is evidence that Sec insertion competes with normal translation termination and nonsense-mediated decay (NMD) (Zupanic et al. 2016). NMD consists of the degradation of mRNAs that have premature stop codons. Se deficiency is correlated with increased susceptibility of selenoprotein mRNAs to NMD, and increasing Se levels increases the stability of these mRNAs (Zupanic et al. 2016).
3.4 Hierarchy of Selenoprotein Expression
The bioavailability of Se from food or culture cell media influences the expression of several selenoproteins, forming the bases for why a hierarchy of selenoprotein expression concept was developed. In fact, in a Se deficiency state, the organism does not support the synthesis of all selenoproteins; therefore, it is stipulated that housekeeping selenoprotein expressions are maintained at the expense of stress-related selenoproteins (Oliveira et al. 2017).
Selenoproteins affected by Se deficiency differ in various cell types (Zupanic et al. 2016). Of particular importance for this chapter, the brain has special requirements of selenoproteins for its proper functioning (Sect. 3.5). Thus, even upon Se deficiency, levels of Se in the brain do not differ considerably, unlike other organs, such as the liver and kidneys (Kühbacher et al. 2009; Oliveira et al. 2017). The supply of Se in the brain is maintained by SELENOP (Oliveira et al. 2017).
The level of Se in different organs of rats varies considerably (the kidney has about 6 mg Se/kg of tissue, the liver 3 mg Se/kg, and the brain 0.5 mg/kg), which at first glance could give the impression that Se is not so important to the brain. However, after imposing a Se deficiency for some generations by feeding rats a diet poor in Se (maximum of 5 μg/kg of diet; the ideal levels of Se for rats are about 100–300 μg/kg), the kidney lost about 85% and the liver 99% of its total Se content. In contrast, the brain lost only about 30% of its total Se (Kühbacher et al. 2009). Thus, the hierarchy of selenoprotein synthesis seems to have different levels of control, which is possibly influenced by the preferential distribution of Se to organs such as the brain in detriment of others (for instance, the liver). Furthermore, inside the brain, Se levels also varied depending on the region. For instance, cerebellum had the highest levels (approximately 0.6 mg/kg of tissue), whereas the brain stem had the lowest levels (near 0.35 mg/kg). Hippocampus, hindbrain, and forebrain had about 0.5 mg of Se/kg of tissue (Kühbacher et al. 2009). In short, the preferential distribution of Se to the brain under Se deficiency indicates the presence of physiological mechanism(s) of selenoproteins control, which is(are) based on the tight regulation of cerebral Se levels under Se deficiency.
3.5 Selenoproteins and the Brain
Several selenoproteins have a wide range of functions such as antioxidant, anti-inflammatory, and thyroid hormones regulator, as well as uncharacterized functions (Hatfield et al. 2006; Oliveira et al. 2017). In the brain, Se as -SeH group of Sec play critical biochemical roles in GPXs (chiefly in GPX1 and GPX4) and TXNRDs (TXNRD1 and TXNRD2).
The exact role played by each of the 25 selenoproteins encoded in human genome in brain physiology is still elusive, but both antioxidant and anti-inflammatory roles played by selenoproteins and selenoenzymes are essential for adequate brain development and functioning. Zhang et al. (2007) demonstrated that the mouse brain selenoprotein-enriched regions are cerebral cortex, olfactory areas, hippocampus, and cerebellar cortex, as well as the highly expressed selenoproteins are GPX4, SELENOF, SELENO K, SELENOM, SELENOP, and SELENOW (Table 5).
As commented in Sect. 3.4, the SELENOP is the major Se transporter to all the brain regions (Oliveira et al. 2017) and with a minor contribution other Se-containing molecules, for instance, selenite or selenomethionine (Valentine et al. 2008) (Fig. 6). To demonstrate the importance of SELENOP to the brain and, consequently, individual development, several studies have been carried out with knockout of SELENOP in rodents (Byrns et al. 2014; Caito et al. 2011; Valentine et al. 2008). The knockout animals developed severe neurological dysfunction and neurodegeneration (Byrns et al. 2014; Caito et al. 2011; Valentine et al. 2008).

Se from the diet to the brain. Briefly, the dietary Se is taken up by the liver and incorporated into the SELENOP in the 8–10 residues of the amino acid selenocysteine. The hepatic SELENOP is secreted to the bloodstream and distributed to the other organs. Regarding the brain, hepatic SELENOP crosses the blood-brain barrier through the interaction with the ApoER2 receptor. In cases of a Se deficiency diet, the cerebral SELENOP serves as a form of Se “storage” and can supply the cerebral needs of Se to the synthesis of housekeeping selenoproteins (Oliveira et al. 2017). Moreover, there is evidence that small inorganic and organic molecules from the diet, for instance, selenite and selenomethionine, can cross the blood-brain barrier and supply the brain Se needs (Valentine et al. 2008). The selenomethionine non-specifically incorporated in the place of methionine in brain cell protein can also be used for Sec synthesis after selenomethionine-containing protein degradation. Thus, the ingestion of selenomethionine can serve either as direct or indirect source of selenide (HSe−)
Interestingly, few case reports have demonstrated that humans with mutations in the Sec machinery synthesis have progressive pontocerebellar and optic nerve atrophy and microcephaly (Pavlidou et al. 2016; Van Dijk et al. 2018).
4 Neurodegenerative Diseases
Neurodegenerative diseases are a group of complex pathologies that affect the CNS. The incidence of neurodegenerative diseases has increased in the last several decades because population longevity has been increasing due to the success of the prevention and treatment of other chronic degenerative diseases. Neurodegenerative diseases can be classified into two main forms: (I) familial forms (early-onset) which are associated with a genetic mutation and (II) sporadic forms (late-onset) which have unknown etiologies. Despite this distinction, there is also mounting evidence that sporadic neurodegeneration is a combination of genetic predisposition and environmental factors with overlapping disease mechanisms as seen in the familial forms (Jellinger 2009).
Neurodegenerative diseases can therefore be considered as distinct entities; however, there is increasing evidence of clinical, pathological, and genetic overlapping of the spectrum of etiologies culminating in final common pathway(s) of neuronal death. The pathogenic mechanisms underlying neurodegeneration are complex, but the universal clinical factors are aging, protein aggregation, neuroinflammation, and mitochondrial dysfunction (Fig. 7) (Sheikh et al. 2013). The combined feature of neurodegenerative diseases is progressive degenerations and loss of neuronal cells. However, despite this commonality neurodegeneration appears to be cell-specific, for instance, predominantly affecting entorhinal and neocortical glutamatergic and nucleus basalis cholinergic neurons in AD, nigrostriatal neurons in PD, and spinal motor neurons in ALS (Fig. 7) (Sheikh et al. 2013).

The brain regions’ representative where the most prevalent neurodegenerative diseases start: Alzheimer’s disease (in the temporal lobe in cortical neurons), Parkinson’s disease (in substantia nigra pars in dopaminergic neurons located in the basal ganglia), and amyotrophic lateral sclerosis (in anterior spinal cord in motor neurons). Illustrative hypothesis of common final mechanisms of neuronal death and loss in neurodegenerative diseases, including inflammation and oxidative stress, mitochondrial dysfunction, defect in protein aggregation, degradation of defective proteins, and a putative role for neurotoxic metals. Antioxidant selenoproteins block or modulate negatively the inflammatory and oxidative stress, protect mitochondrial from redox unbalance, and indirectly can decrease protein aggregation
Neurodegenerative diseases can be triggered by a combination of exogenous and endogenous factors that will be associated with the death of specific and predominant types of neurons in definite brain areas. Neuronal damage leads to the loss of specific brain function and culminate in diseases such as PD, ALS, AD, and others, such as Huntington’s disease or multisystemic atrophy. The relationship between epigenetic variables and genetic susceptibility plays a critical role in mediating the increase or decrease in risks of diseases development. Allied with other risks, the aging is the most critical factor for the appearance of all neurodegenerative diseases, as discussed above. Next, the effects of Se as a risk factor or as preventive agent will be discussed (Sect. 5). In fact, Se has a dual and opposing role in neurodegenerative diseases (Fig. 1). At ideal levels, Se has antioxidant and anti-inflammatory roles, which can delay the development of chronic neurodegenerative diseases; however, at above nutritional and physiological adequate levels, Se will be a pro-oxidant and may be a contributing factor for neurodegeneration.
4.1 Alzheimer’s Disease (AD)
AD is the most prevalent cause of dementia, as well as the most common neurodegenerative disease, characterized by progressive loss of memory, presenting a gradual evolution with a progressive and sequential decline in cognitive, behavioral, and motor functions, interfering with individual daily functionality and quality of life (Scheltens et al. 2016).
In the early stages, AD-afflicted individuals may experience symptoms like depression. In the advanced stages, the triad aphasia, apraxia, and agnosia are observed, characterized by a significant loss of language and ability to perform tasks and name people and objects (Scheltens et al. 2016). In addition, the psychiatric and behavioral changes, such as agitation, aggression, delusions, and hallucinations, are present in up to 75% of cases. Moreover, AD individuals have a decline in motor function causing significant strain for caregivers and requiring specific pharmacological interventions (Scheltens et al. 2016).
AD is increasingly prevalent with advancing age, with a prevalence of 10–30% in those over 65 years old, and the incidence doubles every 10 years after 60 years old (Eratne et al. 2018). The risks to develop the AD are related to lower education, apolipoprotein E’s polymorphism, sex (mainly women), and age (particularly in those over 80 years old). The lifestyle and other modifiable factors such as smoking, hypertension, obesity, diabetes, physical and mental inactivity, depression, and diet play a complex role in dementia. It has been proposed that approximately one-third of AD worldwide might be due to potentially modifiable risk factors (Eratne et al. 2018).
The diabetes as factor related with AD has a strongly relation with immunological mechanisms in the brain and triggers an innate immune response, characterized by the release pro-inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor- α (TNF-α). Additionally, reactive glia is a common feature of the AD brain, with both microglia and astrocytes observed surrounding Aβ plaques, and the dominant source of innate immune mediator production in the brain (Heppner et al. 2015). Of particular importance, Se supplementation (above the ideal nutritional levels; see Fig. 1) can increase the risk of type 2 diabetes mellitus. Consequently, chronic and non-adequate over supplementation with Se can facilitate the progression of AD (Nogueira et al. 2021).
There is evidence that insulin deficiency and resistance are mediators of AD neurodegeneration. This is explained for the loss of insulin receptor-bearing neurons which may precede or accompany initial stage of AD. This state is referred to as “type 3 diabetes” and seems to progress with AD such that, in the terminal stages, it worsens and becomes global. Oxidative stress, impaired glucose and energy metabolism, tau hyperphosphorylation, and APP-Aβ deposition have been linked to perturbation in insulin signaling (Ahmed et al. 2015).
4.2 Parkinson’s Disease (PD)
PD is the second neurodegenerative disease more common after AD; its description dates to 1817 when James Parkinson described the cardinal symptoms of this disease later designated with his name (Poewe et al. 2017). PD clinical manifestations are bradykinesia and either resting tremor or rigidity. In addition to the motor manifestations, there is a long list of nonmotor symptoms, several of which occur before motor signs and are considered early PD signs (Poewe et al. 2017). The nonmotor symptoms are cognitive deterioration, depression, anxiety, psychosis, apathy, fatigue, gastrointestinal complaints (dysphagia and constipation autonomic as retention or urgency urge), sexual dysfunction, sialorrhea, hyperhidrosis, hypotension orthostatic, sensory manifestations (hyposmia and ache), visual manifestations (changes in perception of the contrast, illusions, and visual hallucinations disorders), sleep with vivid dreams and night hypermotor activity in the disturbance of rapid eye movement (REM) sleep behavior, daytime hypersomnolence, and syndrome of restless legs (Poewe et al. 2017).
PD generally arises between the 50 and 80 years old, with a peak in the seventh decade of life, being more prevalent in men. The PD prevalence and incidence increase exponentially with age and peak after 80 years old (Poewe et al. 2017). Recently, Chohan et al. (2021) demonstrated a causal effect of type 2 diabetes mellitus on PD risk and some evidence of an effect on motor deficit progression, but not on cognitive deficit progression. As commented for AD, excess of Se intake can increase the risk of type 2 diabetes mellitus and, indirectly, may increase the risk of incidence of some symptoms of PD.
4.3 Amyotrophic Lateral Sclerosis (ALS)
ALS typical form is sporadic; only 5–10% of ALS individuals have a familial form of the disease with an autosomal dominant, but X-linked and recessive inheritance patterns also occur. The mutations of the SOD1 gene have been shown to be associated with ALS in some families (Hardiman et al. 2017).
ALS patients can present with symptoms of predominantly upper motor neuron dysfunction (spasticity and weakness), with the involvement of lower motor neurons only becoming evident at later stages of disease (Hardiman et al. 2017). Conversely, patients can present symptoms of lower motor neuron dysfunction, which includes fasciculations, cramps, and muscle wasting. Approximately one-third of patients with ALS present with bulbar-onset disease, which is characterized by progressive dysarthria, followed by dysphagia and often with associated emotional lability (Chiò et al. 2013). Limb-onset disease accounts for 60% of cases, is usually asymmetrical in presentation, and can first develop in the upper or lower limbs. Up to 5% of patients present respiratory problems, and these patients are often observed in cardiology and pulmonology clinics before they are referred to neurology clinics (Chiò et al. 2013). ALS has an inflammatory component that contributes to progression of the disease and is associated with oxidative stress in specific regions of the motor neurons. Consequently, in addition to selenoproteins role as oxidoreductase antioxidant enzymes (e.g., GPX, TXNRD, and MSRB1), the regulation of inflammatory signaling by selenoproteins (for instance, TXNRD) is expected to delay the progression of the disease. However, the Se intake cannot exceed the ideal levels of the element because excess of Se can be associated with an increase in the incidence of ALS (Vinceti et al. 2014; Sect. 5.1).
In Europe, the incidence ranges from 2 to 3 cases per 100,000 individuals (Chiò et al. 2013). The incidence of ALS differs based on ancestral origin, for instance, studies in populations of European origin have shown a crude incidence of >3 cases per 100,000 individuals, but incidence is lower in East Asia (~0.8 cases per 100,000 individuals) and South Asia (~0.7 cases per 100,000 individuals) (Chiò et al. 2013). The main limitation of global ALS epidemiology is that ~80% of studies have been conducted in Europe and the United States and they comprise patient cohorts of northern European ancestry. The age of onset peaks at 65 years (Hardiman et al. 2017).
5 Selenium and Neurodegenerative Diseases
Several studies have been carried out to address the role of Se in the neurodegenerative diseases. Although the essentiality of Se to human life maintenance, and particularly for the proper functioning of the brain, there is a tenuous agreement about where the Se nutritional effects vanish and the Se harmful effects commence. To understand the effects of Se, we need to take into consideration the neurodegenerative disease to be studied, the Se chemical form, the amount of Se bioaccumulated into the body, as well as the individual ability to metabolize particular Se compounds.
5.1 Evidence that Overexposure to Se Can Trigger Neurodegenerative Diseases
Epidemiologic studies have been demonstrating that the environmental overexposure to Se can be involved in the etiology of some neurodegenerative diseases (Oliveira et al. 2017; Vinceti et al. 2013, 2017). Here, the most recent scientific literature concerning Se as a potential environmental factor involved in the neurodegenerative diseases’ development will be highlighted and summarized.
The toxic effects of Se exposure were first observed in farm animals, mainly in livestock. Animals chronically exposed to Se, usually from Se-accumulators’ plants, had lack of vitality, anemia, stiffness of joints, deformed and sloughed hooves, roughened hair coat, and lameness. Moreover, animals have a garlic-like breath odor, which is characteristic of Se intoxication due to Se metabolization to dimethylselenide. Interestingly, cases of neurotoxicity were not reported in animals environmentally exposed to Se, despite the fact that experimental animals exposed to Se presented several biochemical, behavioral, and physiological alterations in the nervous system, as elegantly reviewed by Vinceti et al.(2014).
In recent years, evidence has demonstrated that individuals living in seleniferous areas have hypertension (Vinceti et al. 2019b), nausea and vomiting, chest pain, hair and nail abnormalities and loss, garlic odor, edema, and spontaneous abortion (Chawla et al. 2020). Regarding neurodegenerative diseases, important epidemiological studies have raised the hypothesis that the chronic overexposure to Se, mainly the inorganic chemical form, can increase the risk of neurodegenerative diseases development, with a great emphasis on ALS (Vinceti et al. 2013, 2014).
The first evidence that the environmental overexposure to Se could be involved in the etiology of ALS was published in 1977 (for historical details, see Vinceti et al. 2014). Subsequently, in an Italian population exposed to drinking water with high Se content for approximately 15 years, Vinceti et al. (1996) observed an increase in ALS incidence, and, approximately 15 years after the drinking water high Se exposure was corrected and ended, the relative risk to ALS development in the same population decreased (Vinceti et al. 2019c). Vinceti et al. (2013) observed, in newly diagnosed ALS patients, an increase in the total content of Se, especially the inorganic form, selenite, in the cerebrospinal fluid (CSF) when compared with health patients.
Reinforcing the evidence that the CSF levels of Se seem to be a potential marker to ALS and perhaps to other neurodegenerative diseases, a recent systematic reviews and meta-analysis paper summarized that PD patients have the CSF Se levels increased when compared to health individuals, while the blood Se levels were unchanged (Adani et al. 2020). The study by Maass et al. (2020) was the first to evaluate the Se speciation in the CSF of PD patients, and interestingly the authors observed a slight increase in the levels of organic Se species. However, more studies are necessary with large sample sizes and in different populations to determine if the organic Se species are potential markers of PD.
On the other hand, regarding AD there is no consensus in the literature data. Cardoso et al. (2017) did not observe significant differences in the blood or CSF Se levels among AD patients and age-matched controls. On the contrary, in a systematic review and meta-analyses study, Reddy et al. (2017) observed a decrease of Se levels in the blood and CSF of AD patients in comparison with healthy controls; but the CSF meta-analyses included only two papers; thus the risk of bias is high, and meta-analyses including greater number of studies are necessary.
Interestingly, Vinceti et al. (2017) in a cohort study observed that mild cognitive impairment (MCI) patients with CSF inorganic Se (selenate) above the median levels are more likely to develop AD (hazard ratio of 3.1), though, when the same research group evaluate, in a case-control study, the CSF Se speciation in AD and MCI patients, the risk of AD development was inversely correlated with inorganic Se species (Vinceti et al. 2019a). Based on the contradictory results, the authors suggested that case-control studies are not a good design to understand the Se effects in the AD etiology.
5.2 Evidence that Se Can Protect from Neurodegenerative Diseases Development
As commented in the previous section, in the first decades after Se discovery, it was considered a toxic substance. However, after the studies of Schwarz and Foltz, where it was demonstrated that Se could be one of the factors preventing liver degeneration caused by a vitamin E-deficient diet (Nogueira et al. 2021), the interest in the nutritional Se effects increased. The biochemical essentiality of Se was proved almost two decades after the studies of Schwarz and Foltz, when Flohe and colleagues identified Se as component of an important antioxidant protein, GPX (Nogueira et al. 2021), and since the classical study of Dr. Flohe’s group, more 24 selenoproteins were identified in the human genome (Sect. 3).
In relation to the potential neuroprotective role of Se, an extensive number of studies have been carried out investigating the pharmacological effects of several Se-containing molecules in animal models of neurodegenerative diseases. The most used models of AD consist of genetically modified animals (Zhang et al. 2020), but some chemical agents (e.g., aluminum) have also been considered in some studies (Ji et al. 2020). The Se-containing molecules studied varied considerably, and inorganic Se species (selenate), natural organic selenium compounds (selenomethionine), and synthetic organoselenium compounds (Ebselen, 7-chloro-4-(phenylselanyl) quinoline, among others) have been shown to exhibit interesting pharmacological effects (Barbosa et al. 2017; Nogueira et al. 2021). The mechanisms of Se action seem to involve the modulation of the redox balance either directly or indirectly (as source of Se for the selenoproteins synthesis).
On the other hand, the studies with humans are inconclusive. The work of Cardoso et al. (2016) explored the effects of Brazil nut (a Se-rich nut; Sect. 2) supplementation for 6 months on adults with MCI. The authors observed improvements in verbal fluency and constructional praxis after the Brazil nut supplementation; but the long-term effects were not evaluated. Recently, Cardoso et al. (2019) observed that supranutritional sodium selenate increased the CSF Se and selenoprotein levels and caused a stabilization in the AD individual in the Mini-Mental Status Examination. The idea of a supranutritional Se supplementation must be carefully considered, once as discussed in the Sects. 2.1 and 5.1, an increase of blood Se levels above 110 μg/L can be toxic and the overaccumulation of Se in the CSF could be a trigger to some types of neurodegenerative disease. The critical point here is to determine the Se status in individual terms before considering any nutritional intervention with Se supplements.
Until now, no study has been conducted on Se supplementation in humans having PD and ALS. In the meantime, the Se effects have been explored on PD-like animal models, and Se have demonstrated promising results mainly related to increased antioxidant defense and increased synthesis of selenoproteins (Nogueira et al. 2021).
6 Loss of Se from Selenoproteins and Its Influence on Neurodegenerative Diseases
As previously mentioned, selenoproteins are essential to CNS due to their antioxidant activity (GPX, TXNRD, and MSRB1) and Se transport (SELENOP), contributing to keep the intercellular reducing microenvironments (Oliveira et al. 2017). For example, GPX, which presents the Sec residue in the active site, can reduce the harmful hydrogen peroxide (H2O2) to water (H2O) very efficiently (Hatfield et al. 2006). In this sense, the -SeH moiety plays an essential role due to its reactivity and sequential oxidation and reduction reactions, which is kinetically better when compared with thio-GPX, which have the Cys residue in the Sec position (Barbosa et al. 2017). In general, the Se from Sec has a larger atom size, lower pKa, and lower redox potential than the S from Cys, which makes –SeH more efficient than –SH group as redox and antioxidant center in proteins (Oliveira et al. 2017). Computational quantum-chemistry studies using the density functional theory (DFT) calculations also demonstrated that the energetic profile of the catalytic cycle for a Sec-GPX is lower than the Cys-GPX (Orian et al. 2015).
Of neurotoxicological significance, the inactivation of selenoproteins by environmental relevant electrophiles may contribute to the development of neurodegenerative diseases. For instance, it has been suggested that some toxic metals (e.g., Hg) could be causative factors in brain disorders (e.g., neurodegenerative disorders) (Oliveira et al. 2017; Nogara et al. 2019). Although it is still debatable whether toxic metals and other relevant electrophiles have a role in the development of brain diseases, the disruption of selenoproteins function are intuitively expected to disrupt normal brain cell physiology. Here, the hypothesis that interaction of electrophiles (mainly with methylmercury - MeHg) with selenoproteins facilitating Se loss as a potential mechanism that triggers neurodegeneration will be discussed.
According to the GPX catalytic cycle (Fig. 8a), the -SeH from Sec reacts with H2O2 (or organic peroxides, ROOH) leading to the water or ROH and selenoxide (-Se = O), which is isomerized to the selenenic acid (-Se-OH) (or also the -SeH can be oxidized directly to the -Se-OH) (Orian et al. 2015). The -Se-OH is reduced by one GSH molecule, forming the selenosulfide (-Se-S-) intermediate, which reacts with other GSH, leading the oxidized GSH (GSSG) and regenerating the -SeH group (Barbosa et al. 2017). However, under oxidative stress conditions (where the oxidants species levels are higher than the reductants and/or there is depletion of low molecular mass thiols (LMM-SH), such as GSH), the overoxidation of Sec residues could be irreversible (forming the selenonic acid (-SeO3H), analogous to sulfonic acids R-SO3H from Cys) leading to the protein inactivation and consequently cell damage. In fact, the oxidation of Sec and Cys residues could lead to the dehydroalanine (Dha) formation and the loss of Se/S from the protein. The Dha formation is a non-enzymatic process, which occurs via syn-β-elimination, where the Se undergo facile elimination in biological conditions (Ma et al. 2003).

Proposed mechanism of GPX catalytic activity (a), the overoxidation of Sec (b), the loss of Se by the Dha formation (c), and formation of selenenamide 8-membered ring (d). The loss of Se can occur by the overoxidation of Se atom (b) and by the selenoxide elimination from the alkyl/MeHg-adducts from Sec (c). In addition, the β-elimination reaction of the Sec-HgMe adduct could lead to mercury selenide (HgSe) (c), by an unknown mechanism. The mechanisms discussed for GPX enzymes can also occur in all the other selenoproteins, and hypothetically the simultaneous disruption of several selenoproteins may have catastrophic effects of cell redox balance
As demonstrated in Fig. 8b, the overoxidation of Se can lead to the -SeO3H and β-elimination, this last, via intramolecular abstraction of the Hα, leading to the Dha and selenious acid (H2SeO3, or selenite: SeO32− + 2H+). In addition, the conversion of -SeH to other functional groups, such as alkyl derivatives, may contribute to the Dha formation (Fig. 8c), as suggested for TXNRD, GPX, and SELENOP (Ma et al. 2003; Wang et al. 2011). The industrial pollutants acrolein, acrylonitrile, and methylvinyl ketone can act as alkylating agents and could react with the Sec residue, consequently, inhibiting selenoenzymes (Fig. 9a) (Marie et al. 2020). These type 2 alkenes can cause nerve terminal damage by forming Michael-type adducts with Cys/Sec residues and possibly mediate synaptotoxicity, which would increase the risk of developing neurodegenerative diseases (LoPachin et al. 2008).

Proposed mechanism for selenoenzymes inhibition by alkylating agents, such as acrolein (a), and alkyl metals, such as MeHg+ (b). In both cases occurs the nucleophilic attack from Se on the electrophilic site of the neurotoxicants
Metals may be associated with neurodegenerative diseases incidence, for example, in vitro and in vivo studies reported that Pb, Hg, and Mn increase Aβ peptides, phosphorylation of tau protein, aggregation of α-synuclein (α-syn), and oxidative stress, causing neuronal death (Cicero et al. 2017). However, their effects on selenoproteins have been little explored chemically. One possible mechanism can involve the binding of metals to the Se atom from Sec, which blocks the catalytic reaction via the adduct formation (-Se-M, M = metal) (Fig. 9b) (Nogara et al. 2019). The other hypothesis is that the Se could be removed from the proteins because after the adduct formation, the Se moiety could be oxidized by H2O2. The formed selenoxide bound to MeHg (alkyl metal-selenoxide complex-Se(O)-R, R = MeHg), can undergo the β-elimination reactions, as proposed in Fig. 8c. The products of the reaction (Dha and the respective R-SeOH or seleninic acid – R-SeO2H) are sequentially released.
Considering these observations, one possible chemical mechanism, that could be associated with neurodegenerative diseases and Se, is that the overoxidation of the Se atom from Sec residues (in its -SeH form or bonded to alkyl or metal groups) could lead to the loss of Se and irreversible selenoprotein denaturation, via β-elimination reaction (which could be prevented by the formation of an 8-membered ring between Sec and a vicinal residue; Fig. 8d) (Orian et al. 2015). A similar mechanism could occur with other selenoproteins besides GPX, such as TXNRD and SELENOP. However, more studies involving the β-cleavage reaction are necessary to confirm this hypothesis.
Experimental support to the hypothesis that MeHg+ binding to Sec residues can facilitate the release of Se increase Hg neurotoxicity has been provided in the literature. Accordingly, insoluble nanoparticles of the inorganic mercury selenide (HgSe) have been found in human and long-living mammals brains. Therefore, the Se protection against Hg has a significant cost and may be associated with brain Se depletion (Nogara et al. 2019; Oliveira et al. 2017).
Furthermore, in addition to facilitate the loss of Se and inactivation of selenoprotein function, MeHg+ can also downregulate the antioxidant selenoprotein synthesis at transcriptional level, which can facilitate the overproduction of reactive oxygen species (ROS) and disruption of seleno- and thiol-proteins function, notably TXNRD and GPX (Nogara et al. 2019; Oliveira et al. 2017).
6.1 Synthetic Organoselenium Compounds: Small Molecules with Weak Selenoprotein-Like Activity
Synthetic Se-organic compounds are characterized by the presence of a Se atom in an organic molecule. The first organoselenium, the diethylselenide, was synthetized by C. J. Löwig in 1836, and after that, many different organoselenium molecules have been reported (Barbosa et al. 2017). More recently, studies have indicated the neuroprotective effects of organoselenium compounds (Nogueira et al. 2021; Oliveira et al. 2017).
In relation to the use of organoselenium compounds as neuroprotective therapeutic agents, the Ebselen (2-phenyl-1,2-benzoselenazol-3-one) and diphenyl diselenide (DPDSe) are the most studied molecules (Barbosa et al. 2017; Nogueira et al. 2021). Ebselen is a benzoisoselenazole derivative, which presents a five-membered ring selenenamide group, while DPDSe is the simplest diaryl diselenide (Fig. 10a).

(a) Organoselenium molecules and their (b) hypothetical mechanism of action on neuroprotective disorders either as mimetic of selenoproteins or as activators of antioxidant transcription factor
In general, the pharmacology of organoselenium compounds is associated with their antioxidant activity, due to the ability to mimic the GPX (GPX-like activity) and to be a substrate of TXNRD. The conversion of organoselenium compounds to their selenol intermediates (R′-SeH) is critical for their neuroprotective effects. The conversion is performed indirectly by TXNRD or directly by GSH (Fig. 10b) (Barbosa et al. 2017). The neuroprotective effects of Se compounds in animal models of PD and AD are related to their antioxidant activities (Nogueira et al. 2021).
However, as reported by Barbosa et al. (2017), the organoselenium compounds can be only weak mimics of selenoproteins, and their properties of modifying thiol groups in proteins can be a more reasonable explanation to their pharmacological mechanisms of action. The oxidation of thiol-containing transcription factor proteins can activate antioxidant enzymes synthesis. Accordingly, organoselenium compounds have been shown to activate the KEAP1/NFE2L2 signaling pathway via the oxidation of critical cysteinyl residues in Kelch-like ECH-associated protein 1 (KEAP1) which is a transcription factor inhibitor of NFE2L2. KEAP1 oxidation release the nuclear factor, erythroid 2 like factor 2 (NFE2L2), a trans-regulatory factor, which bind to antioxidant response elements (AREs), a cis-element, leading to transcription of antioxidant enzymes and detoxification systems (Barbosa et al. 2017).
In addition, Ebselen, DPDSe, and its derivatives can inhibit critical enzymes involved in pathologies, for instance, the acetylcholinesterase (AChE) in AD and monoamine oxidases A and B (MAO-A/B) in PD (Barbosa et al. 2017; Nogueira et al. 2021). The inhibition of AChE increases the synaptic levels of the neurotransmitter acetylcholine, which is depleted in AD (Eratne et al. 2018). In turn, PD is characterized by the progressive dopamine depletion and degeneration of dopaminergic neurons, and MAO inhibitors (mainly MAO-B) are used to improve dopamine levels (Poewe et al. 2017).
As briefly discussed above, intoxication with Hg and Pb may be associated with neurodegenerative diseases. The neurotoxicity of Hg is caused via complex formation with critical thiol groups of thio(seleno)proteins and/or by their ability to activate the process of free radical oxidation (Cicero et al. 2017; Oliveira et al. 2017). Ebselen and DPDSe can form complexes with neurotoxic metals and facilitate their excretion or neutralize their reactivity (Barbosa et al. 2017; Oliveira et al. 2017). In short, in addition to enhance the activation of AREs, organoselenium compounds can protect selenoproteins from the neurotoxicant by forming stable complexes with the electrophilic metals (particularly Hg).
7 Conclusion
In summary, despite their relatively low abundance and low diversity, selenoproteins are essential to mammalian brain via modulation of cell redox balance. Selenoproteins are involved in the regulation of complex processes such as mitochondrial dysfunction, endoplasmic reticulum stress, and inflammatory and immune response, which can explain the neuroprotective effects of Se against chronic neurodegenerative diseases. However, it is still not known if the biochemical role of some of the selenoproteins is indirect (via modulation of oxidative and reductive stress) or direct via modulation of specific metabolic pathway. Despite this, the extreme reactivity and cytotoxicity of metabolites of Se (particularly HSe−, methylselenol, and methylselenyl radical) indicates that Se supplementation should be considered only when the blood levels are below the minimum needed to support proper selenoprotein synthesis. In short, overexposure to Se can be as neurotoxic as its deficiency. Consequently, to perform its neuroprotective functions, Se must be maintained within specific blood levels (Fig. 1).
Abbreviations
- 3′-UTR:
-
3′-untranslated region
- AChE:
-
Acetylcholinesterase
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- AREs:
-
Antioxidant response elements
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- Cys:
-
Cysteine
- DFT:
-
Density functional theory
- Dha:
-
Dehydroalanine
- DIO1:
-
Iodothyronine deiodinase 1
- DIO2:
-
Iodothyronine deiodinase 2
- DIO3:
-
Iodothyronine deiodinase 3
- DPDSe:
-
Diphenyl diselenide
- EFSec:
-
Selenocysteine elongation factor
- GPX:
-
Glutathione peroxidase
- GPX1:
-
Glutathione peroxidase 1
- GPX2:
-
Glutathione peroxidase 2
- GPX3:
-
Glutathione peroxidase 3
- GPX4:
-
Glutathione peroxidase 4
- GPX6:
-
Glutathione peroxidase 6
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidized GSH
- H2O:
-
Water
- H2O2:
-
Hydrogen peroxide
- H2SeO3:
-
Selenious acid
- HgSe:
-
Mercury selenide
- HS−:
-
Sulfide
- HSe−:
-
Selenide
- IL-1β:
-
Interleukin-1β
- KEAP1:
-
Kelch-like ECH-associated protein 1
- LMM-SH:
-
Low molecular mass thiol
- MAO-A/B:
-
Monoamine oxidases A and B
- MCI:
-
Mild cognitive impairment
- mcm5U:
-
5-methoxycarbonylmethyluridine
- mcm5Um:
-
5-5-methoxycarbonylmethyl-20-O-methyluridine
- MeHg:
-
Methylmercury
- MeSe•:
-
Methylselenyl
- MeSec:
-
Methylselenocysteine
- MeSeH:
-
Methylselenol
- MSRB1:
-
Methionine sulfoxide reductase B1
- NAS:
-
National Academy of Sciences
- NFE2L2:
-
Nuclear factor, erythroid 2 like factor 2
- NMD:
-
Nonsense-mediated decay
- O:
-
Oxygen
- PD:
-
Parkinson’s Disease
- Po:
-
Polonium
- PSTK:
-
Phosphoseryl-tRNA kinase
- R′-SeH:
-
Selenol intermediates
- RDA:
-
Recommended dietary allowance
- REM:
-
Rapid eye movement
- RNI:
-
Reference nutrient intake
- ROOH:
-
Organic peroxides
- ROS:
-
Reactive oxygen species
- R-SeO2H:
-
Seleninic acid
- S:
-
Sulfur
- SBP2:
-
SECIS binding protein
- Se:
-
Selenium
- -Se=O:
-
Selenoxide
- Sec:
-
Selenocysteine
- SECIS:
-
Selenocysteine insertion sequence
- -SeH:
-
Selenol
- SELENOF:
-
Selenoprotein F
- SELENOH:
-
Selenoprotein H
- SELENOI:
-
Selenoprotein I
- SELENOK:
-
Selenoprotein K
- SELENOM:
-
Selenoprotein M
- SELENON:
-
Selenoprotein N
- SELENOO:
-
Selenoprotein O
- SELENOP:
-
Selenoprotein P
- SELENOS:
-
Selenoprotein S
- SELENOT:
-
Selenoprotein T
- SELENOV:
-
Selenoprotein V
- SELENOW:
-
Selenoprotein W
- SeM:
-
Selenomethionine
- SeO3−2:
-
Selenite
- −SeO3H:
-
Selenonic acid
- SeO4−2:
-
Selenate
- -Se-OH:
-
Selenenic acid
- SEPHS2:
-
Selenophosphate synthase 2
- SEPHS2:
-
Selenophosphate synthetase 2
- SEPSecS:
-
Phosphoseryl-tRNA selenium transferase
- Ser:
-
Serine
- SerS:
-
Seryl-tRNA synthetase
- -Se-S-:
-
Selenosulfide
- T3:
-
Triiodothyronine
- T4:
-
Thyroxine
- Te:
-
Tellurium
- TNF-α:
-
Tumor necrosis factor- α
- TXNRD:
-
Thioredoxin reductase
- TXNRD1:
-
Thioredoxin reductase 1
- TXNRD2:
-
Thioredoxin reductase 2
- TXNRD3:
-
Thioredoxin reductase 3
- UL:
-
Upper intake level
- α-syn:
-
α-synuclein
References
Adani, G., Filippini, T., Michalke, B., & Vinceti, M. (2020). Selenium and other trace elements in the etiology of Parkinson’s disease: A systematic review and meta-analysis of case-control studies. Neuroepidemiology, 54(1), 1–23. https://doi.org/10.1159/000502357
Ahmed, S., Mahmood, Z., & Zahid, S. (2015). Linking insulin with Alzheimer’s disease: Emergence as type III diabetes. Neurological Sciences, 36(10), 1763–1769. https://doi.org/10.1007/s10072-015-2352-5
Barbosa, N. V., Nogueira, C. W., Nogara, P. A., De Bem, A. F., Aschner, M., & Rocha, J. B. T. (2017). Organoselenium compounds as mimics of selenoproteins and thiol modifier agents. Metallomics, 9(12), 1703–1734. https://doi.org/10.1039/c7mt00083a
British Nutrition Foundation. (2001). Selenium and health. In British Nutrition Foundation (pp. 1–38). https://www.nutrition.org.uk/attachments/145_Seleniumandhealth.pdf
Bulteau, A.-L., & Chavatte, L. (2015). Update on selenoprotein biosynthesis. Antioxidants & Redox Signaling, 23(10), 775–794. https://doi.org/10.1089/ars.2015.6391
Burk, R. F., & Hill, K. E. (2005). Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annual Review of Nutrition, 25, 215–250. https://doi.org/10.1146/annurev.nutr.24.012003.132120
Byrns, C. N., Pitts, M. W., Gilman, C. A., Hashimoto, A. C., & Berry, M. J. (2014). Mice lacking selenoprotein P and selenocysteine lyase exhibit severe neurological dysfunction, neurodegeneration, and audiogenic seizures. Journal of Biological Chemistry, 289(14), 9662–9674. https://doi.org/10.1074/jbc.M113.540682
Caito, S. W., Milatovic, D., Hill, K. E., Aschner, M., Burk, R. F., & Valentine, W. M. (2011). Progression of neurodegeneration and morphologic changes in the brains of juvenile mice with selenoprotein P deleted. Brain Research, 1398, 1–12. https://doi.org/10.1016/j.brainres.2011.04.046
Cardoso, B. R., Apolinário, D., Bandeira, V. S., Busse, A. L., Magaldi, R. M., Jacob-Filho, W., & Cozzolino, S. M. (2016). Effects of Brazil nut consumption on selenium status and cognitive performance in older adults with mild cognitive impairment: A randomized controlled pilot trial. European Journal of Nutrition, 55(1), 107–116. https://doi.org/10.1007/s00394-014-0829-2
Cardoso, B. R., Hare, D. J., Bush, A. I., Li, Q. X., Fowler, C. J., Masters, C. L., Martins, R. N., Ganio, K., Lothian, A., Mukherjee, S., Kapp, E. A., & Roberts, B. R. (2017). Selenium levels in serum, red blood cells, and cerebrospinal fluid of Alzheimer’s disease patients: A report from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL). Journal of Alzheimer’s Disease, 57(1), 183–193. https://doi.org/10.3233/JAD-160622
Cardoso, B. R., Roberts, B. R., Malpas, C. B., Vivash, L., Genc, S., Saling, M. M., Desmond, P., Steward, C., Hicks, R. J., Callahan, J., Brodtmann, A., Collins, S., Macfarlane, S., Corcoran, N. M., Hovens, C. M., Velakoulis, D., O’Brien, T. J., Hare, D. J., & Bush, A. I. (2019). Supranutritional sodium selenate supplementation delivers selenium to the central nervous system: Results from a randomized controlled pilot trial in Alzheimer’s disease. Neurotherapeutics, 16(1), 192–202. https://doi.org/10.1007/s13311-018-0662-z
Chawla, R., Filippini, T., Loomba, R., Cilloni, S., Dhillon, K. S., & Vinceti, M. (2020). Exposure to a high selenium environment in Punjab, India: Biomarkers and health conditions. Science of the Total Environment, 719, 134541. https://doi.org/10.1016/j.scitotenv.2019.134541
Chiò, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., & White, L. A. (2013). Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology, 41(2), 118–130. https://doi.org/10.1159/000351153
Chohan, H., Senkevich, K., Patel, R. K., Bestwick, J. P., Jacobs, B. M., Bandres Ciga, S., Gan-Or, Z., & Noyce, A. J. (2021). Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Movement Disorders. https://doi.org/10.1002/mds.28551
Cicero, C. E., Mostile, G., Vasta, R., Rapisarda, V., Signorelli, S. S., Ferrante, M., Zappia, M., & Nicoletti, A. (2017). Metals and neurodegenerative diseases. A systematic review. Environmental Research, 159, 82–94. https://doi.org/10.1016/j.envres.2017.07.048
Eratne, D., Loi, S. M., Farrand, S., Kelso, W., Velakoulis, D., & Looi, J. C. (2018). Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australasian Psychiatry, 26(4), 347–357. https://doi.org/10.1177/1039856218762308
Filippini, T., Michalke, B., Wise, L. A., Malagoli, C., Malavolti, M., Vescovi, L., Salvia, C., Bargellini, A., Sieri, S., Krogh, V., Ferrante, M., & Vinceti, M. (2018). Diet composition and serum levels of selenium species: A cross-sectional study. Food and Chemical Toxicology, 115, 482–490. https://doi.org/10.1016/j.fct.2018.03.048
Fox, T. E., Van den Heuvel, E. G. H. M., Atherton, C. A., Dainty, J. R., Lewis, D. J., Langford, N. J., Crews, H. M., Luten, J. B., Lorentzen, M., Sieling, F. W., van Aken-Schneyder, P., Hoek, M., Kotterman, M. J. J., van Dael, P., & Fairweather-Tait, S. J. (2004). Bioavailability of selenium from fish, yeast and selenate: A comparative study in humans using stable isotopes. European Journal of Clinical Nutrition, 58(2), 343–349. https://doi.org/10.1038/sj.ejcn.1601787
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., Shaw, P. J., Simmons, Z., & van den Berg, L. H. (2017). Amyotrophic lateral sclerosis. Nature Reviews Disease Primers, 3, 17071. https://doi.org/10.1038/nrdp.2017.71
Hariharan, S., & Dharmaraj, S. (2020). Selenium and selenoproteins: it’s role in regulation of inflammation. Inflammopharmacology, 28, 667–695. https://doi.org/10.1007/s10787-020-00690-x
Hatfield, D. L., Carlson, B. A., Xu, X.-M., Mix, H., & Gladyshev, V. N. (2006). Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Progress in Nucleic Acid Research and Molecular Biology, 81, 97–142. https://doi.org/10.1016/S0079-6603(06)81003-2
Heppner, F. L., Ransohoff, R. M., & Becher, B. (2015). Immune attack: The role of inflammation in Alzheimer disease. Nature Reviews Neuroscience, 16(6), 358–372. https://doi.org/10.1038/nrn3880
Institute of Medicine (US). (2000). Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. The National Academies Press (US).
Jellinger, K. A. (2009). Recent advances in our understanding of neurodegeneration. Journal of Neural Transmission, 116(9), 1111–1162. https://doi.org/10.1007/s00702-009-0240-y
Ji, D., Wu, X., Li, D., Liu, P., Zhang, S., Gao, D., Gao, F., Zhang, M., & Xiao, Y. (2020). Protective effects of chondroitin sulphate nano-selenium on a mouse model of Alzheimer’s disease. International Journal of Biological Macromolecules, 154, 233–245. https://doi.org/10.1016/j.ijbiomac.2020.03.079
Kasaikina, M. V., Fomenko, D. E., Labunskyy, V. M., Lachke, S. A., Qiu, W., & Moncaster, J. A. (2011). Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. Journal of Biological Chemistry, 286, 33203–33212. https://doi.org/10.1074/jbc.M111.259218
Kiełczykowska, M., Kocot, J., Paździor, M., & Musik, I. (2018). Selenium – A fascinating antioxidant of protective properties. Advances in Clinical and Experimental Medicine, 27(2), 245–255. https://doi.org/10.17219/acem/67222
Kieliszek, M., & Błazejak, S. (2016). Current knowledge on the importance of selenium in food for living organisms: A review. Molecules, 10(5), 609. https://doi.org/10.3390/molecules21050609
Kühbacher, M., Bartel, J., Hoppe, B., Alber, D., Bukalis, G., Bräuer, A. U., Behne, D., & Kyriakopoulos, A. (2009). The brain selenoproteome: Priorities in the hierarchy and different levels of selenium homeostasis in the brain of selenium-deficient rats. Journal of Neurochemistry, 110(1), 133–142. https://doi.org/10.1111/j.1471-4159.2009.06109.x
LoPachin, R. M., Gavin, T., & Barber, D. S. (2008). Type-2 alkenes mediate synaptotoxicity in neurodegenerative diseases. Neurotoxicology, 29, 871–882. https://doi.org/10.1016/j.neuro.2008.04.016
Ma, S., Caprioli, R. M., Hill, K. E., & Burk, R. F. (2003). Loss of selenium from selenoproteins: Conversion of selenocysteine to dehydroalanine in vitro. Journal of the American Society for Mass Spectrometry, 14, 593–600. https://doi.org/10.1016/S1044-0305(03)00141-7
Maass, F., Michalke, B., Willkommen, D., Schulte, C., Tönges, L., Boerger, M., Zerr, I., Bähr, M., & Lingor, P. (2020). Selenium speciation analysis in the cerebrospinal fluid of patients with Parkinson’s disease. Journal of Trace Elements in Medicine and Biology, 57, 126412. https://doi.org/10.1016/j.jtemb.2019.126412
Marie, E. J., Wehrle, R. J., Haupt, D. J., Wood, N. B., Van Der Vliet, A., Previs, M. J., Masterson, D. S., & Hondal, R. J. (2020). Can selenoenzymes resist electrophilic modification? Evidence from thioredoxin reductase and a mutant containing α-methylselenocysteine. Biochemistry, 59, 3300–3315. https://doi.org/10.1021/acs.biochem.0c00608
Nogara, P. A., Oliveira, C. S., Schmitz, G. L., Piquini, P. C., Farina, M., Aschner, M., & Rocha, J. B. T. (2019). Methylmercury’s chemistry: From the environment to the mammalian brain. Biochimica et Biophysica Acta – General Subjects, 1863(12), 129284. https://doi.org/10.1016/j.bbagen.2019.01.006
Nogueira, C. W., Barbosa, N. V., & Rocha, J. B. T. (2021). Toxicology and pharmacology of synthetic organoselenium compounds: An update. Archives of Toxicology, 95, 1179–1226. https://doi.org/10.1007/s00204-021-03003-5
Oliveira, C. S., Piccoli, B. C., Aschner, M., & Rocha, J. B. T. (2017). Chemical speciation of selenium and mercury as determinant of their neurotoxicity. In M. Aschner & L. Costa (Eds.), Neurotoxicity of metals (Advances in neurobiology) (Vol. 18, pp. 53–83). Springer. https://doi.org/10.1007/978-3-319-60189-2_4
Orian, L., Mauri, P., Roveri, A., Toppo, S., Benazzi, L., Bosello-Travain, V., De Palma, A., Maiorino, M., Miotto, G., Zaccarin, M., Polimeno, A., Flohé, L., & Ursini, F. (2015). Selenocysteine oxidation in glutathione peroxidase catalysis: An MS-supported quantum mechanics study. Free Radical Biology and Medicine, 87, 1–14. https://doi.org/10.1016/j.freeradbiomed.2015.06.011
Pavlidou, E., Salpietro, V., Phadke, R., Hargreaves, I. P., Batten, L., McElreavy, K., Pitt, M., Mankad, K., Wilson, C., Cutrupi, M. C., Ruggieri, M., McCormick, D., Saggar, A., & Kinali, M. (2016). Pontocerebellar hypoplasia type 2D and optic nerve atrophy further expand the spectrum associated with selenoprotein biosynthesis deficiency. European Journal of Paediatric Neurology, 20(3), 483–488. https://doi.org/10.1016/j.ejpn.2015.12.016
Pitts, M. W., Reeves, M. A., Hashimoto, A. C., Ogawa, A., Kremer, P., Seale, L. A., & Berry, M. J. (2013). Deletion of selenoprotein M leads to obesity without cognitive deficits. Journal of Biological Chemistry, 288(36), 26121–26134. https://doi.org/10.1074/jbc.M113.471235
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., Schrag, A. E., & Lang, A. E. (2017). Parkinson disease. Nature Reviews Disease Primers, 3, 17013. https://doi.org/10.1038/nrdp.2017.13
Raman, A. V., Pitts, M. W., Seyedali, A., Hashimoto, A. C., Bellinger, F. P., & Berry, M. J. (2013). Selenoprotein W expression and regulation in mouse brain and neurons. Brain and Behavior, 3(5), 562–574. https://doi.org/10.1002/brb3.159
Rayman, M. P., Winther, K. H., Pastor-Barriuso, R., Cold, F., Thvilum, M., Stranges, S., Guallar, E., & Cold, S. (2018). Effect of long-term selenium supplementation on mortality: Results from a multiple-dose, randomised controlled trial. Free Radical Biology and Medicine, 127, 46–54. https://doi.org/10.1016/j.clnesp.2019.07.002
Reddy, V. S., Bukke, S., Dutt, N., Rana, P., & Pandey, A. K. (2017). A systematic review and meta-analysis of the circulatory, erythrocellular and CSF selenium levels in Alzheimer’s disease: A metal meta-analysis (AMMA study-I). Journal of Trace Elements in Medicine and Biology, 42, 68–75. https://doi.org/10.1016/j.jtemb.2017.04.005
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., & Van der Flier, W. M. (2016). Alzheimer’s disease. Lancet, 388(10043), 505–517. https://doi.org/10.1016/S0140-6736(15)01124-1
Sheikh, S., Safia, Haque, E., & Mir, S. S. (2013). Neurodegenerative diseases: Multifactorial conformational diseases and their therapeutic interventions. Journal of Neurodegenerative Diseases, 2013, 563481. https://doi.org/10.1155/2013/563481
Tobe, R., & Mihara, H. (2018). Delivery of selenium to selenophosphate synthetase for selenoprotein biosynthesis. Biochimica et Biophysica Acta (BBA)-General Subjects, 1862(11), 2433–2440. https://doi.org/10.1016/j.bbagen.2018.05.023
Turanov, A. A., Xu, X.-M., Carlson, B. A., Yoo, M.-H., Gladyshev, V. N., & Hatfield, D. L. (2011). Biosynthesis of selenocysteine, the 21st amino acid in the genetic code, and a novel pathway for cysteine biosynthesis. Advances in Nutrition, 2, 122–128. https://doi.org/10.3945/an.110.000265
Valentine, W. M., Abel, T. W., Hill, K. E., Austin, L. M., & Burk, R. F. (2008). Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. Journal of Neuropathology and Experimental Neurology, 67(1), 68–77. https://doi.org/10.1097/NEN.0b013e318160f347
Van Dijk, T., Vermeij, J. D., van Koningsbruggen, S., Lakeman, P., Baas, F., & Poll-The, B. T. (2018). A SEPSECS mutation in a 23-year-old woman with microcephaly and progressive cerebellar ataxia. Journal of Inherited Metabolic Disease, 41(5), 897–898. https://doi.org/10.1007/s10545-018-0151-x
Vendeland, S. C., Butler, J. A., & Whanger, P. D. (1992). Intestinal absorption of selenite, selenate, and selenomethionine in the rat. The Journal of Nutritional Biochemistry, 3(7), 359–365. https://doi.org/10.1016/0955-2863(92)90028-H
Verma, S., Hoffmann, F. W., Kumar, M., Huang, Z., Roe, K., Nguyen-Wu, E., Hashimoto, A. S., & Hoffmann, P. R. (2011). Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. Journal of Immunology, 186(4), 2127–2137. https://doi.org/10.4049/jimmunol.1002878
Vinceti, M., Guidetti, D., Pinotti, M., Rovesti, S., Merlin, M., Vescovi, L., Bergomi, M., & Vivoli, G. (1996). Amyotrophic lateral sclerosis after long-term exposure to drinking water with high selenium content. Epidemiology, 7(5), 529–532.
Vinceti, M., Solovyev, N., Mandrioli, J., Crespi, C. M., Bonvicini, F., Arcolin, E., Georgoulopoulou, E., & Michalke, B. (2013). Cerebrospinal fluid of newly diagnosed amyotrophic lateral sclerosis patients exhibits abnormal levels of selenium species including elevated selenite. Neurotoxicology, 38, 25–32. https://doi.org/10.1016/j.neuro.2013.05.016
Vinceti, M., Mandrioli, J., Borella, P., Michalke, B., Tsatsakis, A., & Finkelstein, Y. (2014). Selenium neurotoxicity in humans: Bridging laboratory and epidemiologic studies. Toxicology Letters, 230(2), 295–303.
Vinceti, M., Chiari, A., Eichmüller, M., Rothman, K. J., Filippini, T., Malagoli, C., Weuve, J., Tondelli, M., Zamboni, G., Nichelli, P. F., & Michalke, B. (2017). A selenium species in cerebrospinal fluid predicts conversion to Alzheimer’s dementia in persons with mild cognitive impairment. Alzheimer’s Research & Therapy, 9(1), 100. https://doi.org/10.1186/s13195-017-0323-1
Vinceti, M., Michalke, B., Malagoli, C., Eichmüller, M., Filippini, T., Tondelli, M., Bargellini, A., Vinceti, G., Zamboni, G., & Chiari, A. (2019a). Selenium and selenium species in the etiology of Alzheimer’s dementia: The potential for bias of the case-control study design. Journal of Trace Elements in Medicine and Biology, 53, 154–162. https://doi.org/10.1016/j.jtemb.2019.03.002
Vinceti, M., Chawla, R., Filippini, T., Dutt, C., Cilloni, S., Loomba, R., Bargellini, A., Orsini, N., Dhillon, K. S., & Whelton, P. (2019b). Blood pressure levels and hypertension prevalence in a high selenium environment: Results from a cross-sectional study. Nutrition, Metabolism and Cardiovascular Diseases, 29(4), 398–408. https://doi.org/10.1016/j.numecd.2019.01.004
Vinceti, M., Filippini, T., Malagoli, C., Violi, F., Mandrioli, J., Consonni, D., Rothman, K. J., & Wise, L. A. (2019c). Amyotrophic lateral sclerosis incidence following exposure to inorganic selenium in drinking water: A long-term follow-up. Environmental Research, 179, 1–6. https://doi.org/10.1016/j.envres.2019.108742
Wang, S. K., Weaver, J. D., Zhang, S., & Lei, X. G. (2011). Knockout of SOD1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic GPX1 protein. Free Radical Biology and Medicine, 51(1), 197–204. https://doi.org/10.1016/j.freeradbiomed.2011.03.018
Wirth, E. K., Conrad, M., Winterer, J., Wozny, C., Carlson, B. A., Roth, S., Schmitz, D., Bornkamm, G. W., Coppola, V., Tessarollo, L., Schombure, L., Köhrle, J., Hateld, D. L., & Schweizer, U. (2010). Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. The FASEB Journal, 24, 844–852. https://doi.org/10.1096/fj.09-143974
Zhang, Y., Zhou, Y., Schweizer, U., Savaskan, N. E., Hua, D., Kipnis, J., Hatfield, D. L., & Gladyshev, V. N. (2007). Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. Journal of Biological Chemistry, 283(4), 2427–2438. https://doi.org/10.1074/jbc.M707951200
Zhang, Z. H., Chen, C., Jia, S. Z., Cao, X. C., Liu, M., Tian, J., Hoffmann, P. R., Xu, H. X., Ni, J. Z., & Song, G. L. (2020). Selenium restores synaptic deficits by modulating NMDA receptors and selenoprotein K in an Alzheimer’s disease model. Antioxidants and Redox Signaling. https://doi.org/10.1089/ars.2019.7990
Zupanic, A., Meplan, C., Huguenin, G. V. B., Hesketh, J. E., & Shanley, D. P. (2016). Modeling and gene knockdown to assess the contribution of nonsense-mediated decay, premature termination, and selenocysteine insertion to the selenoprotein hierarchy. RNA, 22, 1076–1084. https://doi.org/10.1261/rna.055749.115
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this entry

Cite this entry
Oliveira, C.S. et al. (2022). Selenium Neuroprotection in Neurodegenerative Disorders. In: Kostrzewa, R.M. (eds) Handbook of Neurotoxicity. Springer, Cham. https://doi.org/10.1007/978-3-031-15080-7_238
Download citation
DOI: https://doi.org/10.1007/978-3-031-15080-7_238
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-15079-1
Online ISBN: 978-3-031-15080-7
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences