
Abstract
For several decades, microglia were considered to be subordinated to neurons. However, growing evidence indicates that microglia play key roles in the normal functioning of the nervous system, as well as in age-dependent changes and neurodegenerative diseases. As the brain ages, microglia acquire a phenotype that can be increasingly inflammatory and cytotoxic (dysfunctional microglia), generating a hostile environment for neurons. There is mounting evidence that this process facilitates the development of neurodegenerative diseases, for which the greatest risk factor is age. In neurodegenerative diseases, the abnormal inflammatory response can depend on the impairment of the endogenous activation control of aging microglia that potentiate the release of potentially detrimental factors such as cytokines and oxidative stress mediators. This chapter will discuss key aging-dependent changes occurring in microglia, the inflammatory and oxidative environment they establish, their impaired regulation, and their interaction and effect on neurons. In addition, the role of complement in the neuron-microglia interaction and their modeling of neural circuits through microglia-mediated phagocytosis in development will be highlighted, as well as the growing evidence on its contribution in neurodegenerative processes.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
1 Introduction
Microglia are the resident macrophages and professional phagocytes of the central nervous system (CNS). They acquire a characteristic phenotype that distinguishes them from blood-derived monocytes. Microglia have a highly dynamic surveillance branched phenotype, which changes into various activated phenotypes in response to diverse stimuli (Li et al., 2019; Simon et al., 2019), being the main orchestrators of the neuroimmune response.
Microglia are in permanent surveillance of the brain environment. They undergo changes in gene expression that confer them a dynamic response to environmental changes (Subhramanyam et al., 2019). Through this monitoring, microglia detect the environmental signals and transduce, integrate, and respond to them to maintain brain homeostasis.
Microglia have an active role in both healthy and dysfunctional brains (Cherry et al., 2014). There is increasing evidence of the existence of various microglia types that show characteristic morphological and functional identities in diverse contexts, recognizing functional states that can be distinguished from those observed in neuroinflammation.
However, the role of microglia in a “healthy” context is not completely understood, as well as their function during adulthood and aging. Experimental limitations prevented to answer these questions until novel markers and in vivo techniques revealed that microglia are highly active in the non-injured (Nimmerjahn et al., 2005) and injured (Davalos et al., 2005) CNS.
2 The Microglia-Neuron Interaction
Neuron-microglia interactions are crucial for CNS development and homeostasis (Li et al., 2012). Research in murine models shows that microglia engulf and eliminate synapses during development, eliminating supernumerary neurons (Paolicelli et al., 2011). This is coherent with experimental findings of neuronal hyperconnectivity in the early stages of development, which progressively diminishes as the brain matures (Marín-Teva et al., 2004). Microglia within the juvenile visual cortex modify their association with dendritic spines in response to changes in visual sensory experience, and they appear to phagocytose dendritic spines (Tremblay et al., 2010). While the underlying molecular mechanisms are not clear, recent work has identified complement factors associated with the microglia that participate in this process (Schafer et al., 2012).
Being such versatile cells, microglia can fulfill multiple functional roles that, depending on environmental signals, allow them to adopt different phenotypes and interact with neurons in various ways. The microglia secrete several neurotrophic factors, such as nerve growth factor (NGF) and basic fibroblast growth factor (bFGF), which help maintain neuronal cell survival and circuit formation (Ueno et al., 2013). There is increasing evidence on the importance of neuron-microglia crosstalk, their impact on neuronal activity, on the phenotypic changes in response to neuronal injury, and their participation in various pathologies.
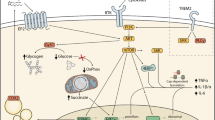
Neurons present membrane receptors and secrete neurotransmitters and neurotrophins to signal constantly their healthy or unhealthy state to the microglia. Thus, neurons are not simply passive targets for microglia, but rather regulate their activity through the expression of “on” and “off” signals (Simon et al., 2019) and, therefore, can influence their activity either by activating them or by promoting their inactivation (Biber et al., 2006), including multiple activated phenotypes (Fig. 1). These signals are of different nature. The “on” signals are inducible, initiate a defined program of microglial activation, and range from changes on the cell membrane, pathogens, aggregates of abnormal proteins (e.g., β-amyloid), and apoptotic cells (Biber et al., 2007). These ignition signals operate from endangered or damaged neurons, because of the appearance of structures associated with bacterial cell walls, viral envelopes or their DNA and RNA, identified as signs of infection, which can initiate an inflammatory state of the microglia. The molecules released after tissue damage are signals that induce particularly robust microglial responses, which adopt a phagocytic phenotype, with shorter and less branched processes.

Through the interactions of various cell interaction systems, neurons contribute to maintaining microglia in a homeostatic phenotype. A dysregulation of key pathways impairs the regulation and leads to a shift in the microglial phenotype to an activated inflammatory profile. C′ (complement system)
ATP is an important molecular chemoattractant mediating “find-me” signals for immune cells. Microglial processes extend rapidly toward local tissue damage and to locally applied ATP (Davalos et al., 2005; Elliott et al., 2009). The local increase of neuronal activity by glutamate or a global alteration of neuronal activity induces changes in the morphology and motility of microglia, which processes are directed toward highly active neurons and facilitate their contact. NMDA receptor activation in dendrites triggers the release of ATP in a pannexin-1-independent manner inducing the outgrowth of microglial processes in mouse hippocampal slices, suggesting a purine-mediated neuron-microglia communication (Dissing-Olesen et al., 2014). This process can also involve membrane depolarization-activated pannexin-1 hemichannels on neurons (Chekeni et al., 2010) and the activation of small Rho GTPase Rac in microglia via ATP/P2 purinergic receptors, involved in cytoskeleton dynamics (Haynes et al., 2006).
Signaling ensures adequate maintenance of the cleaning process until the resolution and repair of the initial damage, allowing the transition from a reactive inflammatory phenotype to a noninflammatory one to avoid neuronal damage in unaffected brain areas.
The “off” signals are the direct responsibility of neurons. The neuronal signals responsible for turning off microglia are, among others, chemokines. One of the most relevant chemokines is CX3CL1 (fractalkine), which is constitutively present in healthy neurons and can be membrane bound or soluble. It is not yet clear whether the membrane-bound or the soluble fractalkine is responsible for the inhibition of microglia (Ruqayya et al., 2020). Fractalkine receptor (CX3CR1) is expressed by microglia (Crews et al., 2021; Verge et al., 2004). In wild-type mice, neurotoxic microglial activity is suppressed by CX3CL1-CX3CR1 signaling. The genetic ablation of CX3CR1 in various inflammation models and in microglia-neuron cocultures treated with lipopolysaccharide (LPS) shows an increase in neuronal death in an inflammatory environment in the absence of fractalkine signaling (Bruttger et al., 2015; Huang et al., 2018; Vainchtein & Molofsky, 2020).
This delicate balance between surveillance and “inflamed” microglia undergoes various changes. Microglia change as a function of aging and those changes translate in a decreased capacity of microglia to carry out their homeostatic functions (Simon et al., 2019). Given the cooperative interaction between neurons and microglia, aging has a significant impact on neuronal integrity and function, as well as on their protein expression (von Bernhardi et al., 2015; Beltrán-Castillo et al., 2018).
Dysfunctional or perturbed microglial homeostasis could have direct consequences on the onset of neurodegenerative or neuropsychiatric disorders across the whole lifespan (Ruqayya et al., 2020; Zhan et al., 2014). Impaired microglial remodeling of neuronal circuits can impair learning and memory (Maggi et al., 2011; Rogers et al., 2011; Nguyen et al., 2020). Fractalkine signaling deficiency (Maggi et al., 2011; Rogers et al., 2011), microglial BDNF deletion (Parkhurst et al., 2013), and microglial depletion (Zhan et al., 2014) have similar effects resulting in cognitive impairment, motor behavior, and fear conditioning. In addition, CX3CR1 knockout mice display social interaction deficits that have been associated with autism spectrum disorders in humans, both early in life and during adulthood (Zhan et al., 2014).
Other mutations, like those in TREM2, a gene exclusively expressed by microglia in the intact CNS, have been also associated with an autosomal recessive form of early-onset dementia (Guerreiro et al., 2013) and to confer an increased risk of Alzheimer’s disease (AD) and neurodegeneration (Jonsson et al., 2013). Thus, in disease conditions, glial cells can induce changes leading to neuronal dysfunction, indicating the importance of neuron-glia interaction in the pathophysiology of neurological disorders (Ruqayya et al., 2020).
3 Microglia and the Complement
Complement proteins are “eat me” signals that mark apoptotic cells and pathogens for removal by macrophages that express C3 receptors (CR3; CD11b/CD18). They are part of the innate immune recognition system and participate in the trafficking and elimination of unwanted endogenous and exogenous material (Moriyama et al., 2011) and are synthesized by neurons, microglia, astrocytes, and oligodendrocytes. Complement activation leads to the cleavage of several cascade proteins, including key proteins such as C3b, which binds to immune complexes, and C5b, which initiates the assembly of the membrane attack complex C5b-9 (MAC), involved in death on the one hand and cellular activation on the other (Peterson et al., 2017). Furthermore, the release of the anaphylactic peptides C3a and C5a recruits inflammatory cells and induces inflammation (Rahpeymai et al., 2006; Stevens et al., 2007) and participates also in neurogenesis (Peterson et al., 2017) and synaptic plasticity (Stevens et al., 2007; Schafer et al., 2012) through functions analogous to those in the systemic immune system, the clearance of cellular material “labeled” for its elimination by phagocytosis (Bialas & Stevens, 2013).
The formation of mature neuronal circuits in the CNS requires the pruning of inappropriate synapses dependent on neuronal activity during development, and the complement system intervenes by eliminating weak, immature, or unused synapses through microglia-mediated phagocytosis (Stevens et al., 2007; Schafer et al., 2012). In the developing visual system, this process involves microglia-mediated internalization of synaptic structures through complement receptor 3. However, just as the complement is actively involved in the modeling of a functional neuronal circuit, it appears to be also involved in chronic inflammatory responses that contribute to neurodegeneration (Benoit & Tenner, 2011), as will be discussed.
4 The Aging Microglia
A hallmark of brain aging is the increased oxidative stress and lipid peroxidation. One hypothesis is that the accumulation of free radicals leads to increased neuroinflammation along with a reduction in growth and antioxidant capacity in the brain of aged rodents (Godbout et al., 2005). Thus, the increase in inflammatory responses during aging is, at least partly, the result of alterations in the activation and function of the microglia.
Aged microglia have a reduced proliferation capacity (senescence) that progresses toward characteristic morphological changes: a larger soma with numerous short and less complex ramifications and, eventually, cytoplasmic fragmentation resulting in dystrophic microglia. When exposed to injury, cell migration and phagocytosis of aged microglia as well as the dynamics of their response are decreased (Hefendehl et al., 2014). These changes on aged microglia suggest that, rather than becoming overactive, aged microglia become dysregulated and lose the balance between their protective activity and their cytotoxicity (von Bernhardi et al. 2007, 2015; Cornejo et al., 2018), which often results in maladaptive responses and chronic inflammation.
Microglial metabolism changes from oxidative phosphorylation to glycolysis, which results in increased levels of lactate production (Ruqayya et al., 2020). Additionally, aged mice show the transcriptional profile of activated microglia (Godbout et al., 2005) with an increased expression of mRNA for pro-inflammatory cytokines, tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), IL-6, and interferon γ (IFNγ); decreased anti-inflammatory cytokines, IL-10 and transforming growth factor β (TGFβ) (Sierra et al., 2007; Frank et al., 2007); and increased pro-inflammatory receptors, MHC II (Henry et al., 2009) and CD86 (Frank et al., 2006), suggesting a change into a persistent inflammatory phenotype. Neurons also produce inflammatory mediators, including eicosanoids, C-reactive protein, amyloid protein, and complement factors, capable of stimulating the inflammatory response of microglia.
During persistent injury, aged microglia respond with an inflammatory profile, with high and prolonged production of pro-inflammatory cytokines, reactive oxygen species (ROS) and reactive nitrogen species (RNS), and lipid mediators (Simon et al., 2019), driven by the aforementioned metabolic changes, and reduction of mitochondrial oxidative phosphorylation (Nair et al., 2019). In various aging models, an exaggerated cytokine response (increased serum TNFα) is associated with the development of cognitive, behavioral, and physiological impairments typical of neurodegenerative diseases (Holmes et al., 2009).
Chronic low-level neuroinflammation accompanies brain aging. Microglia-derived inflammatory factors can damage local tissue and can further increase inflammation and glial activation, leading to a vicious inflammatory cycle (Cherry et al., 2014). However, it is still unclear which factors trigger this aging-associated chronic process.
5 The Aged Microglia in Neurodegenerative Diseases
Inflammation and the age-associated processes described earlier represent potential pathogenic factors for many CNS pathologies, including chronic neurodegenerative diseases such as AD or Parkinson’s disease (PD). Activation of microglia is an early sign that allows a persistent local pro-inflammatory response and often precede neuronal death. Upon activation, aged microglia acquire a series of pro-inflammatory functions, and increased oxidative stress (von Bernhardi et al., 2015), constituting the main cellular source of both inflammatory mediators and high levels of reactive oxygen species (Pawate et al., 2004; Qin et al., 2005; Hayashi et al., 2008; von Bernhardi et al., 2015), and participating actively in the genesis of neuronal damage like the one observed in neurodegenerative diseases (von Bernhardi et al., 2015).
Many authors point to the sustained pro-inflammatory activation of microglia as the main cause of the neurotoxicity associated with neurodegenerative diseases, among which AD stands out. Others emphasize “dysregulated” microglia, more than their inflammatory over-activation, as responsible for neurodegenerative changes (von Bernhardi et al. 2007, 2015). By not responding adequately to regulatory feedback mechanisms and/or through the impairment of their ability to eliminate harmful agents, microglia lose their ability to handle potentially harmful compounds and become cytotoxic due to their persistent inflammatory and oxidative activation (von Bernhardi et al., 2015).
Thus, how harmful is a constantly activated microglia to the brain? The most likely answer is that microglial regulation is not an activation-inactivation-binary mechanism, but rather an unbalanced response. From this point of view, it appears that microglia are neuroprotective against harmful stimuli in the early stages of activation, but under sustained activation, they could become cytotoxic from the moment they lose their homeostatic regulation (Eyüpoglu et al., 2003).
Synaptic impairment is an early hallmark of aging and neurodegeneration, preceding the loss of neurons. The complement has been involved in chronic inflammatory responses that contribute to neurodegeneration (Benoit & Tenner, 2011). Activation of the complement protein C1q has been reported in AD patients and murine models (Benoit & Tenner, 2011), revealing the role played by complement proteins in the cell dynamics of the adult and aging brain, and suggesting their role in neurodegenerative pathology. A significant increase in the C1q level has been reported in the aging human and mouse brains, mostly localized in synapses in brain areas that are vulnerable to neurodegenerative processes (Stephan et al., 2013). On the contrary, adult C1q KO mice show an activity-dependent enhancement of synaptic potentiation (Stephan et al., 2013). A common factor of both developmental synaptic pruning and synapse loss in disease is that microglia adopt a highly phagocytic state. Thus, inflammatory signals that promote reactive microglia may in turn stimulate the elimination of complement-mediated aged neuronal synapses. It remains to be elucidated whether complement-dependent microglia-mediated synaptic pruning in neurodegeneration and inflammation represents a true reactivation of the pruning program occurring during development. This mechanism may provide insight into the use of complement proteins as biomarkers in early diagnosis (Hakobyan et al., 2016) or as new therapeutic strategies for neurodegenerative diseases.
6 Conclusion
Aging involves several morphological and metabolic changes of the brain. Many of those changes are a consequence of the impairment of the normal functioning of brain cells and how they deal with stressor stimuli, leading to genetic and epigenetic changes. Neuron-microglia interactions in a context of chronic inflammation may promote changes in microglial cell activation and regulation, promoting their increased reactivity and a cytotoxic activation.
Numerous studies have identified the role of the complement system in the elimination of weak, immature, or unused synapses through microglia-mediated phagocytosis during development, and its participation in chronic inflammatory responses contributing to neurodegeneration has also been determined, although the cellular and molecular mechanisms involved are still not entirely clear. It is proposed that, in aging, inflammatory microglia lead to complement-mediated loss of neurons, revealing the role played by complement proteins in the cell dynamics of the adult and aging brain, and suggesting their role in neurodegenerative pathology.
Emerging evidence suggests that under disease conditions, dysregulated microglia may induce structural and functional changes in glial cells along with neuronal dysfunction, revealing the importance of neuron-glia interactions in the pathophysiology of neurological disorders (Ruqayya et al., 2020).
Abbreviations
- ATP:
-
Adenosine triphosphate
- bFGF:
-
Basic fibroblast growth factor
- C3a:
-
Anaphylatoxin originating from activation and cleavage of complement component 3
- C3b:
-
By-products of the classical pathway of complement activation (C3)
- C3d:
-
Breakdown products of C3b
- C5a:
-
Anaphylatoxin originating from activation and cleavage of complement 5
- C5b:
-
By-products of the classical pathway of complement activation (C5)
- CNS:
-
Central nervous system
- CR3; CD11b/CD18:
-
Complement receptor 3
- CX3CL1:
-
CX3C chemokine subclass (fractalkine)
- CX3CR1:
-
Fractalkine receptor
- IFNγ:
-
Interferon gamma
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- IL-10:
-
Interleukin-10
- LPS:
-
Lipopolysaccharides
- MHC II:
-
Class II molecules of major histocompatibility complex
- NGF:
-
Nerve growth factor
- NMDA:
-
N-Methyl-D-aspartate
- NO:
-
Nitric oxide
- TGFβ:
-
Transforming growth factor β
- TNFα:
-
Tumor necrosis factor α
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
References
Beltrán-Castillo, S., Eugenín, J., & von Bernhardi, R. (2018) Impact of aging in microglia mediated D-serine balance in the CNS. Mediators of Inflammation, 7219732. https://doi.org/10.1155/2018/7219732
Benoit, M. E., & Tenner, A. J. (2011). Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. The Journal of Neuroscience, 31, 3459–3469.
Bialas, A., & Stevens, B. (2013). TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature Neuroscience, 16, 1773–1782. https://doi.org/10.1038/nn.3560
Biber, K., de Jong, E. K., van Weering, H. R. J., & Boddeke, H. W. (2006). Chemokines and their receptors in central nervous system disease. Current Drug Targets, 7, 29. https://doi.org/10.2174/138945006775270196
Biber, K., Neumann, H., Inoue, K., & Boddeke, H. W. (2007). Neuronal ‘On’ and ‘Off’ signals control microglia. Trends in Neurosciences, 30(11), 596–602. https://doi.org/10.1016/j.tins.2007.08.007
Bruttger, J., Karram, K., Wörtge, S., Regen, T., Marini, F., Hoppmann, N., Klein, M., Blank, T., Yona, S., Wolf, Y., Mack, M., Pinteaux, E., Müller, W., Zipp, F., Binder, H., Bopp, T., Prinz, M., Jung, S., & Waisman, A. (2015). Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity, 43(1), 92–106. https://doi.org/10.1016/j.immuni.2015.06.012
Chekeni, F. B., Elliott, M. R., Sandilos, J. K., Walk, S. F., Kinchen, J. M., Lazarowski, E. R., Armstrong, A. J., Penuela, S., Laird, D. W., Salvesen, G. S., Isakson, B. E., Bayliss, D. A., & Ravichandran, K. S. (2010). Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature, 467(7317), 863–867.
Cherry, J. D., Olschowka, J. A., & O’Banion, M. K. (2014). Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. Journal of Neuroinflammation, 11, 98. https://doi.org/10.1186/1742-2094-11-98
Cornejo, F., Vruwink, M., Metz, C., Muñoz, P., Salgado, N., Poblete, J., Andrés, M. E., & von Bernhardi, R. (2018). SR-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer’s disease pathophysiology. Brain, Behavior, and Immunity, 69, 336–350. https://doi.org/10.1016/j.bbi.2017.12.007
Crews, F. T., Zou, J., & Coleman Jr., L. G. (2021). Extracellular microvesicles promote microglia-mediated pro-inflammatory responses to ethanol. Journal of Neuroscience Research, n/a. https://doi.org/10.1002/jnr.24813.
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., Littman, D. R., Dustin, M. L., & Gan, W. B. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience, 8, 752–758.
Dissing-Olesen, L., LeDue, J. M., Rungta, R. L., Hefendehl, J. K., Choi, H. B., & MacVicar, B. A. (2014). Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. The Journal of Neuroscience, 34, 10511–10527.
Elliott, M. R., Chekeni, F. B., Trampont, P. C., Lazarowski, E. R., Kadl, A., Walk, S. F., Park, D., Woodson, R. I., Ostankovich, M., Sharma, P., Lysiak, J. J., Harden, T. K., Leitinger, N., & Ravichandran, K. S. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature, 461(7261), 282–286.
Eyüpoglu, L. Y., Bechmann, L., & Nitsch, R. (2003). Modification of microglial function protects from lesion-induced neuronal alteration and promotes sprouting in the hippocampus. The FASEB Journal, 17, 1110–1111.
Frank, M. G., Barrientos, R. M., Biedenkapp, J. C., Rudy, J. W., Watkins, L. R., & Maier, S. F. (2006). mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiology of Aging, 27, 717–722.
Frank, M. G., Baratta, M. V., Sprunger, D. B., Watkins, L. R., & Maier, S. F. (2007). Microglia serve as a neuroimmune substrate for stress induced potentiation of CNS pro-inflammatory cytokine responses. Brain, Behavior, and Immunity, 21, 47–59.
Godbout, J. P., Chen, J., Abraham, J., Richwine, A. F., Berg, B. M., Kelley, K. W., & Johnson, R. W. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. The FASEB Journal, 19, 1329–1331.
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., Cruchaga, C., Sassi, C., Kauwe, J. S., Younkin, S., Hazrati, L., Collinge, J., Pocock, J., Lashley, T., Williams, J., Lambert, J. C., Amouyel, P., Goate, A., Rademakers, R., … Hardy, J. (2013). Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. The New England Journal of Medicine, 368(2), 117–127. https://doi.org/10.1056/NEJMoa1211851
Hakobyan, S., Harding, K., Aiyaz, M., Hye, A., Dobson, R., Baird, A., Liu, B., Harris, C. L., Lovestone, S., & Morgan, B. P. (2016). Complement biomarkers as predictors of disease progression in Alzheimer’s disease. Journal of Alzheimer’s Disease, 54(2), 707–716. https://doi.org/10.3233/JAD-160420
Hayashi, Y., Yoshida, M., Yamato, M., Ide, T., Wu, Z., Ochi-Shindou, M., Kanki, T., Kang, D., Sunagawa, K., Tsutsui, H., & Nakanishi, H. (2008). Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. The Journal of Neuroscience, 28, 8624–8634. https://doi.org/10.1523/jneurosci.1957-08.2008
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W. B., & Julius, D. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nature Neuroscience, 9(12), 1512–1519.
Hefendehl, J. K., Neher, J. J., Sühs, R. B., Kohsaka, S., Skodras, A., & Jucker, M. (2014). Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell, 13(1), 60–69. https://doi.org/10.1111/acel.12149
Henry, C. J., Huang, Y., Wynne, A. M., & Godbout, J. P. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain, Behavior, and Immunity, 23, 309–317.
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., Culliford, D., & Perry, V. H. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology, 73(10), 768–774.
Huang, Y., Xu, Z., Xiong, S., Sun, F., Qin, G., Hu, G., Wang, J., Zhao, L., Liang, Y., Wu, T., Lu, Z., Humayun, S. M., So, K. F., Pan, Y., Li, N., Yuan, T. F., Rao, Y., & Peng, B. (2018). Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nature Neuroscience, 21, 530–540. https://doi.org/10.1038/s41593-018-0090-8
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., Bjornsson, S., Huttenlocher, J., Levey, A. I., Lah, J. J., Rujescu, D., Hampel, H., Giegling, I., Andreassen, O. A., Engedal, K., Ulstein, I., Djurovic, S., Ibrahim-Verbaas, C., Hofman, A., … Stefansson, K. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. New England Journal of Medicine, 368(2), 107–116. https://doi.org/10.1056/NEJMoa1211103
Li, Y., Du, X. F., Liu, C. S., Wen, Z. L., & Du, J. L. (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell, 23, 1189–1202.
Li, Q., Cheng, Z., Zhou, L., Darmanis, S., Neff, N. F., Okamoto, J., Gulati, G., Bennett, M. L., Sun, L. O., Clarke, L. E., Marschallinger, J., Yu, G., Quake, S. R., Wyss-Coray, T., & Barres, B. A. (2019). Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron, 101(2), 207–223.e10. https://doi.org/10.1016/j.neuron.2018.12.006
Maggi, L., Scianni, M., Branchi, I., D’Andrea, I., Lauro, C., & Limatola, C. (2011). CX3CR1 deficiency alters hippocampal-dependent plasticity phenomena blunting the effects of enriched environment. Frontiers in Cellular Neuroscience, 5, 22.
Marín-Teva, J. L., Dusart, L., Colin, C., Gervais, A., van Rooijen, N., & Mallat, M. (2004). Microglia promote the death of developing Purkinje cells. Neuron, 41(4), 535–547. https://doi.org/10.1016/S0896-6273(04)00069-8
Moriyama, M., Fukuhara, T., Britschgi, M., Yingbo, H., Narasimhan, R., Villeda, S., Molina, H., Huber, B. T., Holers, M., & Wyss-Coray, T. (2011). Complement receptor 2 is expressed in neural progenitor cells and regulates adult hippocampal neurogenesis. Journal of Neuroscience, 31(11), 3981–3989.
Nair, S., Sobotka, K. S., Joshi, P., Gressens, P., Fleiss, B., Thornton, C., Mallard, C., & Hagberg, H. (2019). Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia, 67, 1047–1061. https://doi.org/10.1002/glia.23587
Nguyen, P. T., Dorman, L. C., Pan, S., Vainchtein, I. D., Han, R. T., Nakao-Inoue, H., Taloma, S. E., Barron, J. J., Molofsky, A. B., Kheirbek, M. A., & Molofsky, A. V. (2020). Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell, 182(2), 388–403. https://doi.org/10.1016/j.cell.2020.05.050
Nimmerjahn, A., Kirchhoff, F., & Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308, 1314–1318.
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., Giustetto, M., Ferreira, T. A., Guiducci, E., Dumas, L., Ragozzino, D., & Gross, C. T. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333(6048), 1456–1458. https://doi.org/10.1126/science.1202529
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R., Lafaille, J. J., Hempstead, B. L., Littman, D. R., & Gan, W. B. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155, 1596–1609.
Pawate, S., Shen, Q., Fan, F., & Bhat, N. R. (2004). Redox regulation of glial inflammatory response to lipopolysaccharide and interferon gamma. Journal of Neuroscience Research, 77, 540–551. https://doi.org/10.1002/jnr.20180
Peterson, S. L., Nguyen, H. X., & Mendez, O. A. (2017). Complement protein C3 suppresses axon growth and promotes neuron loss. Scientific Reports, 7, 12904. https://doi.org/10.1038/s41598-017-11410-x
Qin, L., Li, G., Qian, X., Liu, Y., Wu, X., Liu, B., Hong, J. S., & Block, M. L. (2005). Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia, 52, 78–84. https://doi.org/10.1002/glia.20225
Rahpeymai, Y., Hietala, M. A., Wilhelmsson, U., Fotheringham, A., Davies, I., Nilsson, A. K., Zwirner, J., Wetsel, R. A., Gerard, C., Pekny, M., & Pekna, M. (2006). Complement: A novel factor in basal and ischemia-induced neurogenesis. The EMBO Journal, 25(6), 1364–1374.
Rogers, J. T., Morganti, J. M., Bachstetter, A. D., Hudson, C. E., Peters, M. M., Grimmig, B. A., Weeber, E. J., Bickford, P. C., & Gemma, C. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. The Journal of Neuroscience, 31, 16241–16250.
Ruqayya, A., Jong-Heon, K., Habibur, R., & Kyoungho, S. (2020). Metabolic regulation of glial phenotypes: Implications in neuron–glia interactions and neurological disorders. Frontiers in Cellular Neuroscience, 14, 20. https://doi.org/10.3389/fncel.2020.00020
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., Ransohoff, R. M., Greenberg, M. E., Barres, B. A., & Stevens, B. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74, 691–705. https://doi.org/10.1016/j.neuron.2012.03.026
Sierra, A., Gottfried-Blackmorem, A. C., McEwen, B. S., & Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia, 55, 412–424.
Simon, E., Obst, J., & Gomez-Nicola, D. (2019). The evolving dialogue of microglia and neurons in Alzheimer’s disease: Microglia as necessary transducers of pathology. Neuroscience, 405, 24–34. https://doi.org/10.1016/j.neuroscience.2018.01.059
Stephan, A. H., Madison, D. V., Mateos, J. M., Fraser, D. A., Lovelett, E. A., Coutellier, L., Kim, L., Tsai, H. H., Huang, E. J., Rowitch, D. H., Berns, D. S., Tenner, A. J., Shamloo, M., & Barres, B. A. (2013). A dramatic increase of C1q protein in the CNS during normal aging. The Journal of Neuroscience, 33, 13460–13474. https://doi.org/10.1523/JNEUROSCI.1333-13.2013
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., Micheva, K. D., Mehalow, A. K., Huberman, A. D., Stafford, B., Sher, A., Litke, A. M., Lambris, J. D., Smith, S. J., John, S. W., & Barres, B. A. (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164–1178.
Subhramanyam, C. S., Wang, C., Hu, Q., & Dheen, S. T. (2019). Microglia-mediated neuroinflammation in neurodegenerative diseases. Seminars in Cell & Developmental Biology, 94, 112–120. https://doi.org/10.1016/j.semcdb.2019.05.004
Tremblay, M. È., Lowerym, R. L., & Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biology, 8(11), e1000527. https://doi.org/10.1371/journal.pbio.1000527
Ueno, M., Fujita, Y., Tanaka, T., Nakamura, Y., Kikuta, J., Ishii, M., & Yamashita, T. (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nature Neuroscience, 16(5), 543–551.
Vainchtein, I. D., & Molofsky, A. V. (2020). Astrocytes and microglia: In sickness and in health. Trends in Neurosciences, 43(3), 144–154.
Verge, G. M., Milligan, E. D., Maier, S. F., Watkins, L. R., Naeve, G. S., & Foster, A. C. (2004). Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. European Journal of Neuroscience, 20(5), 1150–1160. https://doi.org/10.1111/j.1460-9568.2004.03593.x
von Bernhardi, R., Ramírez, G., Toro, R., & Eugenín, J. (2007). Pro-inflammatory conditions promote neuronal damage mediated by amyloid precursor protein-and degradation by microglial cells in culture. Neurobiology of Disease, 26, 153–164.
von Bernhardi, R., Eugenin-von Bernhardi, L., & Eugenin, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Frontiers in Aging Neuroscience, 7, 124. https://doi.org/10.3389/fnagi.2015.00124
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., Vyssotski, A. L., Bifone, A., Gozzi, A., Ragozzino, D., & Gross, C. T. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience, 17, 400–406.
Acknowledgments
The authors acknowledge the support from the Agencia Nacional de Investigación y Desarrollo-Fondo Nacional de Desarrollo Científico y Tecnológico (ANID-FONDECYT) grant 1172647, ANID-REDES 190187, and Universia Santander to RvB.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this entry

Cite this entry
Triolo-Mieses, M., Fadic, R., von Bernhardi, R. (2022). Microglial Cell Dysregulation in the Aged Brain and Neurodegeneration. In: Kostrzewa, R.M. (eds) Handbook of Neurotoxicity. Springer, Cham. https://doi.org/10.1007/978-3-031-15080-7_180
Download citation
DOI: https://doi.org/10.1007/978-3-031-15080-7_180
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-15079-1
Online ISBN: 978-3-031-15080-7
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences