
Abstract
Neurodegeneration refers to the progressive loss of structure and function including neuronal cell death. Neurodegenerative diseases are usually incurable and often present late in life because the greatest risk factor for neurodegeneration is aging. Neurodegeneration in the context of cerebellar ataxias is best exemplified by the cerebellar variant of multiple system atrophy (MSA-C). This is a rare but progressive disease, usually presenting in the early 50s with a combination of cerebellar ataxia and autonomic dysfunction. It is responsible for 8.5% of all ataxias. By definition, MSA-C presents with cerebellar symptoms and signs but in some cases, there is overlap with the Parkinsonian variant of MSA (MSA-P) where rigidity, bradykinesia, and other Parkinsonian features predominate. Although the pathology of MSA has been delineated, the etiology remains obscure and the disease remains untreatable leading to severe disability and premature death. Symptomatic support by a multidisciplinary team is important in the management of these patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Among progressive cerebellar ataxias, MSA-C stands out because of the rapidity of progression, early speech involvement, autonomic dysfunction, and significant disability. The historical evolution of this disease entity is worth bearing in mind: the term sporadic Olivo-Ponto-Cerebellar Atrophy (OPCA) was first introduced in 1900 (Dejerine and Thomas 1900) to describe the pathological features at PM of atrophy of the olives, pons, and cerebellum (which is the case in MSA-C). When similar clinical features were observed in some familial cases, the term familial OPCA was introduced. However, much later, it would be shown that, in fact, these familial OPCA cases were genetic ataxias due to mutations causing spinocerebellar ataxias type 1, 2, or 3 (SCA1,2,3). In 1960 another publication (Shy and Drager 1960) described two patients with progressive autonomic dysfunction causing orthostatic hypotension, and urinary and sexual dysfunction who also developed Parkinsonian features. Such cases were labeled as having Shy–Drager syndrome. In 1972, the term multiple system atrophy was introduced to describe these patients (Bannister and Oppenheimer 1972). The discovery of oligodendroglial cytoplasmic inclusion bodies in both the cerebellar and the Parkinsonian types of MSA provided conclusive evidence that OPCA and striatonigral degeneration (Shy-Drager Syndrome) are in fact the same disease entity. The terms MSA-C and MSA-P were then introduced to describe the two main phenotypes.
2 Prevalence
Among a cohort of 1500 patients with progressive ataxias studied in Sheffield, UK, MSA-C accounted for 8.5% of all ataxias and 10% of sporadic ataxias (Hadjivassiliou et al. 2016). Epidemiological studies have shown a point prevalence ranging of 3.4–4.9 per 100,000 population, increasing to 7.8 among the over 40 years of age (Schrag et al. 1999). While MSA-P cases are said to outnumber MSA-C cases by between 2:1 and 4:1, one has to be cautious about such calculations because of the way that these groups of patients are followed up. Patients with MSA-P tend to be followed up in well-established movement disorders clinics at regional neurology centers, at least in the UK. In such clinics, the majority of patients will have mainly idiopathic Parkinson’s disease and similar conditions including MSA-P. MSA-C patients who present with ataxia are less likely to be seen in such clinics and are often diagnosed with progressive ataxia and discharged to the local services. Of interest is the observation that MSA-C is by far commoner than MSA-P in Japan. Such a difference may be down to the way the neurological services are configured in Japan or may be genuine.
3 Clinical Characteristics
Patients with MSA-C, by definition, present with cerebellar ataxia. At the time of presentation, such patients may already have features suggestive of autonomic involvement including orthostatic hypotension, urinary frequency, urgency or incontinence, constipation, erectile dysfunction, and sleep disorders (e.g., REM sleep disorder). One or more of these additional autonomic features may develop at a later date or may even precede the development of cerebellar dysfunction, sometimes by years. Early speech involvement in the form of cerebellar dysarthria is characteristic. Some degree of Parkinsonian features may also be present including rigidity and bradykinesia with stooping and slowness in walking. Subtle myoclonus affecting the fingers may also be present, sometimes referred to as poly-mini myoclonus. Broken pursuit on examination of eye movements is common but gaze evoked nystagmus much less so. Generalized hyperreflexia and even extensor plantars may be present. Aside the broad-based gait and falls due to gait ataxia, patients with MSA-C may also develop anterocollis and less likely other forms of dystonia. Respiratory disturbance is also common and takes the form of inspiratory stridor, sleep apnea, and REM sleep disorder.
While patients with MSA-C do not develop cognitive decline and dementia, emotional incontinence can be seen and manifests with inappropriate laughing or crying (Fanciulli and Wenning 2015).
4 Diagnosis
For the experienced clinician and ataxologist, the diagnosis of MSA-C is usually suspected by the additional presence of autonomic features, early speech involvement, and rapidity of progression. The diagnosis can also be supported by brain imaging that may demonstrate the characteristic pattern of brainstem involvement resulting in the hot-cross bun sign. This sign is not pathognomonic of MSA-C and can be seen in SCA2 and paraneoplastic cerebellar degeneration. However, in the correct clinical context, it can be very helpful in consolidating the suspected diagnosis. It is also worth mentioning that this sign tends to develop later on in the disease course and may not be present at the presentation. MR spectroscopy of the cerebellum can be a very useful tool in the diagnosis of MSA-C at presentation. The NAA/Cr ratio measured from the vermis and the hemisphere tends to show very low values even at the early stages of the disease (Kadodwala et al. 2019). Attempts to devise neuroimaging criteria for the diagnosis of MSA have been made (Brooks et al. 2009). Along the same lines, based on the clinical features, diagnostic criteria have been developed and attempt to define three degrees of certainty for the diagnosis: definite, probable, and possible MSA. Definite diagnosis requires post-mortem evidence of widespread alpha-synuclein positive glial cytoplasmic inclusions with concomitant olivopontocerebellar atrophy (Trojanowski et al. 2007). The first clinical criteria for the diagnosis of MSA were published in 1989 (Quinn 1989) followed by consensus criteria in 1998 and a revision of the consensus criteria in 2008. Of interest is the validity of these diagnostic criteria as assessed by a clinicopathological study (Osaki et al. 2009). In this study, it was shown that after the first visit, the sensitivity for possible and probable MSA was 41% and 18%, respectively, increasing to 92% and 63% at the last visit. This is entirely expected given the progressive nature of the condition and the accumulation of more clinical features that allow a more accurate clinical diagnosis. Some have argued that the division of MSA into MSA-P and MSA-C is somewhat artificial because of a substantial overlap between the 2. It was argued that approximately half of the MSA-P patients show additional cerebellar signs and as many as 75% of the MSA-C patients develop Parkinsonian features during the disease course (Stankovic et al. 2019). While this is true later on in the disease course, as far as patients are concerned, it is the correct diagnosis at presentation that counts. Making the correct diagnosis early is helpful in outlining the prognosis and the need to address the rapidly changing circumstances (inability to work, increasing disability, etc.), something that is best addressed by a multidisciplinary team.
5 Progression and Prognosis
MSA-C is characterized by relentless worsening, sometimes with a rapid pace at the onset. The rapid progression may sometimes lead to investigations specifically looking for paraneoplastic or other immune-mediated ataxias which also tend to follow an acute/subacute course. Up to 50% of patients require walking aid within 3 years of symptom onset and 60% are wheelchair bound for 5 years (Fanciulli and Wenning 2015). The median time before the patient is bedridden is 6–8 years. Death is usually due to cardiac arrhythmia or disruption of the brain-stem cardiorespiratory drive (sudden death), pneumonia, and other sources of sepsis, particularly urinary sepsis leading to a more systemic infection. The early presence of autonomic failure is a negative prognostic factor. The course of the disease is very different from what is seen in other late-onset ataxias. For example, spinocerebellar ataxia type 6 (SCA6) is a relatively common late-onset pure ataxia that, by comparison to MSA-C, follows a much more benign course and does not usually lead to early death. Figure 98.1 compares the changes in MR spectroscopy of the vermis in patients with SCA6 and patients with MSA-C over time. Apart of demonstrating the significantly lower NAA/Cr ratio from the cerebellum in MSA-C when compared to SCA6 at the time of presentation (a useful diagnostic tool), it also shows the rapid progression when comparing the two groups.

NAA/Cr ratio from the cerebellar vermis using MR spectroscopy is depicted on the vertical axis. The graph shows the decline of the NAA/Cr over time and compares MSA-C with spinocerebellar ataxia type 6. Note the very low NAA/Cr at baseline in the MSA-C group when compared to SCA6 and the rapid decline over time for the MSA-C group when compared to SCA6. There is a very good correlation between NA/Cr and the SARA (Scale for the Assessment and Rating of Ataxia) score
6 Pathogenesis
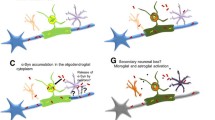
As mentioned above, a definite diagnosis of MSA requires postmortem evidence of widespread alpha-synuclein-positive glial cytoplasmic inclusions with concomitant olivopontocerebellar atrophy (Trojanowski et al. 2007). The pathology, therefore, is well described. Relocalization of an important stabilizer of myelin integrity called p25alpha into the oligodendroglial precedes alpha-synuclein aggregation. This seems to result in oligodendrocyte swelling and the overexpression of alpha-synuclein. The formation of glial cytoplasmic inclusions interferes with neuronal support and activates microglial cells. This, in turn, results in the release of misfolded alpha-synuclein into the extracellular space. Misfolded alpha-synuclein is taken up by neighboring neurons to form neuronal cytoplasmic inclusions (Fanciulli and Wenning 2015). The spreading of this process to other areas of the brain ultimately leads to neuronal death in multiple areas leading to the multisystem neuronal involvement. What leads to such pathology remains unclear. Equally unclear is the reason as to why the same pathology can affect different parts of the brain and thus result in two distinct (at least at the onset) clinical presentations of MSA-C or MSA-P.
A linkage analysis study looking at some of the very rare cases of familial MSA in Japan identified mutations in the COQ2 gene. The enzyme encoded by the COQ2 gene is essential for the biosynthesis of Coenzyme Q10. Similar findings were reported in some sporadic cases of MSA. While the proportion of such mutation-positive patients is very small, this finding raised the possibility of a role of impaired COQ2 in the pathogenesis of MSA (Tsuji et al. 2013). It also opens the possibility of a potential treatment approach by using CoQ10 supplementation. There are ongoing studies looking at this approach. Several other treatment studies have been published using various different agents, none of which has reported any benefits.
7 Management
As there is no treatment for this condition, the clinical management of patients with MSA-C is down to symptomatic treatment and support. This is best done in dedicated MSA-C clinics with a multidisciplinary team of specialist ataxia/MSA nurses, physiotherapists, and occupational therapists. Medication can be used for orthostatic hypotension (fludrocortisone, midodrine, ephedrine), urinary urgency and frequency (anticholinergics), sometimes requiring intermittent self-catheterization, and emotional lability (citalopram). For Parkinsonian symptoms (particularly rigidity), levodopa may offer temporary benefit but this is not usually sustained. Botox injections can be used for anterocollis particularly if this is associated with pain. The rapidity of progression means that these patients require regular review and support because of their rapidly changing needs. This is why an early diagnosis is important for planning for the future.
8 Conclusions
MSA-C is a common cause of late-onset progressive cerebellar ataxia. The diagnosis is based on a combination of clinical and radiological findings. MSA-C should be considered as a fatal neurodegenerative ataxia associated with significant disability and as yet without any effective treatment. Despite this, close review and monitoring by a multidisciplinary team are essential in addressing the rapidly progressive needs of these patients. Symptomatic medication can be extremely helpful for issues like bladder instability and postural hypotension.
References
Bannister R, Oppenheimer DR (1972) Degenerative diseases of the nervous system associated with autonomic failure. Brain 95:457–474
Brooks DJ, Seppi K, On behalf of the Neuroimaging Working Group on MSA (2009) Proposed neuroimaging criteria for the diagnosis of MSA. Mov Disord 24:949–964
Dejerine L, Thomas A (1900) L’atrophie olivo-ponto-cérébelleuse. Nouv iconog de la Salpêtrière 13:330
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372:249–262
Hadjivassiliou M, Martindale J, Shanmugarajah P, Grunewald RA, Sarrigiannis PG, Beauchamp N, Garrard K, Warburton R, Sanders DS, Friend D, Duty S, Taylor J, Hoggard N (2016) Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2016-314863
Kadodwala VH, Hadjivassiliou M, Currie S, Skipper N, Hoggard N (2019) Is H-MR spectroscopy useful as a diagnostic aid in MSA-C? Cerebellum Ataxias. https://doi.org/10.1186/s40673-019-0099-0
Osaki Y, Ben-Shlomo Y, Lees A, Wenning G, Quinn N (2009) A validation exercise on the new consensus criteria for multiple system atrophy. Mov Disord 71:670–676
Quinn N (1989) Multiple system atrophy-the nature of the beast. J Neurol Neurosurg Psychiatry 52(Suppl):78–89
Schrag A, Ben-Shlomo Y, Quin NP (1999) Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 354:1771–1775
Shy GM, Drager GA (1960) A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol 2:511–527
Stankovic I, Quinn N, Vignatelli L, Antonini A, Berg D, Coon E, et al, On behalf of the Movement Disorder Society Multi System Atrophy Study Group (2019) A critique of the second consensus criteria for multiple system atrophy. Mov Disord. https://doi.org/10.1002/mds.27701
Trojanowski JQ, Revesz T, On behalf of the Neuropathology Working Group on MSA (2007) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 33:615
Tsuji S, On behalf of the Multiple-System Atrophy Research Collaboration (2013) Mutations in COQ@ in familial and sporadic Multiple-System Atrophy. N Engl J Med 369:233–244
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hadjivassiliou, M. (2023). Cerebellar Variant of Multiple System Atrophy (MSA-C). In: Gruol, D.L., Koibuchi, N., Manto, M., Molinari, M., Schmahmann, J.D., Shen, Y. (eds) Essentials of Cerebellum and Cerebellar Disorders. Springer, Cham. https://doi.org/10.1007/978-3-031-15070-8_98
Download citation
DOI: https://doi.org/10.1007/978-3-031-15070-8_98
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-15069-2
Online ISBN: 978-3-031-15070-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)