
Abstract
Electrochemical promotion of catalysis (EPOC) is an electrochemically induced catalytic effect which corresponds to in situ reversible modification of the catalytic behavior of metal or metal oxide catalyst-electrodes deposited on solid electrolytes or mixed ionic-electronic conductors (MIEC) upon polarization of the electrode/electrolyte or MIEC interface. This work highlights the key landmarks in EPOC starting from its discovery. Firstly, a brief history of the experimental work that resulted in the rather unexpected discovery of EPOC when using as catalysts metal film electrodes deposited on ZrO2(Y2O3), an O2− conductor, is presented. Secondly, the efforts towards validation of the general nature of EPOC, by investigating this phenomenon in a very large number of combinations of electrolytes or MIEC, electrodes, and catalytic reactions, are briefly described in chronological order. A short reference is then made to the experimental and theoretical works that led to understanding of the mechanistic origin of EPOC and its functional equivalence to metal-support interactions. The focus is then shifted to current activities and efforts towards developing configurations for practical applications of EPOC, with emphasis on bipolar designs, monolithic electropromoted reactors, EPOC with nanodispersed catalysts, and wireless self-driven and self-sustained EPOC systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Electrochemical promotion of catalysis (EPOC)
- Electrochemical activation of catalysis
- Non-faradaic electrochemical modification of catalytic activity (NEMCA)
- Spillover-backspillover
- Ionic promoters
- Solid ionic and mixed conductors
1 Introduction
The terms electrochemical promotion of catalysis (EPOC) or non-faradaic electrochemical modification of catalytic activity (NEMCA) both refer interchangeably to an electrochemically induced catalytic effect which corresponds to in situ or in operando reversible modification of the activity and selectivity of catalysts interfaced to solid electrolytes or mixed ionic-electronic conductors (MIEC), upon polarization of the catalyst/solid electrolyte or MIEC interface [1, 2]. This effect was first observed by M. Stoukides and C.G. Vayenas in 1981, as detailed in Chap. 1, in the reaction of ethylene oxidation on Ag deposited on yttria-stabilized zirconia or ZrO2(Y2O3), an O2− conducting solid electrolyte [3], who reported polarization-induced non-faradaic changes in the catalytic rates of CO2 and C2H4O production as well as in selectivity to C2H4O. However, this behavior was then attributed to peculiarity of the specific catalytic system and was explained by a kinetic model which was based on the hypothesis of formation of surface silver oxide, more active than reduced silver, on sites adjacent to chemisorbed oxygen [3]. It was some years later, in 1988, when C. G. Vayenas and his coworkers reported results showing conclusively that EPOC, then referred to as NEMCA effect, is a general effect in heterogeneous catalysis, which they attributed to electrochemically induced change in catalyst work function [4].
The distinguishing feature of EPOC is that the induced catalytic rate change Δr = r – rο upon catalyst polarization (via current or potential application), where r and ro denote the observed reaction rates under closed- circuit (current I ≠ 0) and open-circuit (I = 0) conditions, is higher, even by orders of magnitude, than the electrocatalytic rate dictated by Faraday’s law, which is equal to the rate of ion transport through the electrolyte. The electrocatalytic rate is equal to I/(nF), where I denotes the applied current, n denotes the number of exchanged electrons in the charge transfer reaction at the catalyst-electrode/solid electrolyte or MIEC interface (e.g., n = 2 for an O2− conductor), and F is Faraday’s constant. Thus, under EPOC conditions the absolute value of the enhancement factor or faradaic efficiency, Λ, of the process, defined from [1, 4],
is larger than 1 (|Λ| > 1), which can be explained only by induced changes in the catalytic properties of the electrode. Another important parameter for quantifying the EPOC effect is the rate enhancement ratio, ρ, defined from [1, 4]
as the ratio of the catalytic rates in the presence and absence of polarization.
EPOC has been reported for a large number of catalytic reactions, involving various combinations of metal or metal oxide catalysts and solid electrolytes or MIEC as active catalyst supports, whereas it has been also demonstrated for catalytic reactions in aqueous electrolytes and inorganic melts. Work on EPOC prior to 2001 is summarized in a book [1] whereas more recent progress in several book chapters and review articles [2, 5,6,7,8,9,10,11,12,13,14,15]. This chapter concerns a historical tracing of the progress in research related to EPOC, describing briefly the important steps from its discovery to the fundamental understanding of its origin and the exploration of the underlying rules and principles, as well as its relation to other catalytic effects, and, finally, the key steps towards technological applications.
2 The Discovery of EPOC: An Electrochemically Induced Catalytic Effect
2.1 EPOC with O2− Conductors
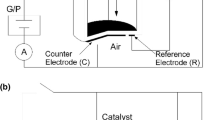
In 1981, M. Stoukides and C.G. Vayenas were the first to report that the catalytic activity and selectivity of a silver catalyst-porous electrode deposited on ZrO2 (8 mol% Y2O3) solid electrolyte in a two-electrode asymmetric set-up (Fig. 2.1) could be significantly affected by polarization of the Ag electrode under conditions of ethylene epoxidation [3]. Specifically, they observed that electrochemical pumping of O2− to the Ag catalyst via positive current application, under oxidative conditions, could induce reversible increases in both the rates of ethylene oxide and CO2 production which exceeded by up to ca. 400 times (for the C2H4O production rate) the ion pumping rate and were accompanied by an increase in selectivity to ethylene oxide (by more than 20%) [3, 16]. The opposite effect, i.e., a decrease in catalytic rates and in selectivity to C2H4O, was observed upon negative current application [3, 16]. A typical example of this behavior is shown in Fig. 2.2 [16]. The non-faradaic increase of the rates upon O2− pumping to the Ag catalyst, corresponding to enhancement ratio ρ values up to 3 [16] and faradaic efficiency Λ values up to 400 [3], was attributed by the two authors to peculiarity of the specific catalytic system and was explained by a kinetic model which was based on the hypothesis of formation of a surface silver oxide, more active than reduced silver, on sites adjacent to chemisorbed oxygen [3]. However, a decade later it became clear that the observed changes in catalytic activity and selectivity were manifestation of the EPOC effect in this important catalytic system [17].

Schematic diagram of the solid electrolyte cell reactor for the first EPOC experiments. The Ag catalyst-electrode film (working electrode) was deposited on the inner bottom side of a ZrO2(8 mol% Y2O3) tube and was exposed to the reaction mixture. A similar Ag film deposited on the outside bottom side of the tube and exposed to ambient air served as the counter electrode. (Reprinted from ref. [3], with permission from Elsevier)

Transient effect of electrochemical oxygen pumping during C2H4 epoxidation on Ag interfaced to ZrO2(8 mol %Y2O3); r1, rate of epoxidation; r2, rate of CO2 formation. Conditions: \( {P}_{C_2{H}_4} \) = 1.6 kPa, \( {P}_{O_2} \) = 9.5 kPa, T = 400 °C. Open-circuit rates: r10 = 7.6 × 10−8 mol/s, r2,0 = 8.9 × 10−8 mol/s. O2− pumping rate: \( {G}_{O^{2-}} \) = i/2F = 1.3 × 10−10 mol/s. (Reprinted with permission from ref. [16]. Copyright (1982) American Chemical Society)
A qualitatively identical effect of O2− pumping on rates and selectivity, considered in consistence with the surface silver oxide model, was reported in 1984 by Stoukides and Vayenas [18] for the reaction of propylene epoxidation on porous Ag interfaced to ZrO2 (8 mol% Y2O3) in a similar two-electrode asymmetric set-up (Fig. 2.1). O2− pumping to the Ag catalyst resulted in increase of both the epoxidation rate (by up to 45%) and deep oxidation rate (by up to 11%) and in a concomitant increase of selectivity to C3H6O (by more than 30%), which however did not exceed 4% [18]. Enhancement in the total rate of C3H6 consumption exceeding the O2− pumping rate by more than two orders of magnitude was reported [18].
In 1988, the electrochemical promotion effect was reported for the first time by C. G. Vayenas and his coworkers as a general effect in heterogeneous catalysis, having been observed until then for a number of different reactions on porous Pt and Ag catalyst-electrodes (5–10 μm thick) interfaced to ZrO2 (8 mol% Y2O3) or YSZ [4]. The term “non-faradaic electrochemical modification of catalytic activity” (acronym NEMCA) was also introduced to describe it [4]. Specifically, electrochemical promotion had been observed in C2H4 epoxidation on Ag/YSZ [3, 16], in C3H6 epoxidation on Ag/YSZ [18], in C2H4 combustion on Pt/YSZ [4, 19], in CO oxidation on Pt/YSZ [4, 20], in CH3OH oxidation to CO2 and H2CO on Pt/YSZ [4, 21], and in CH3OH dehydrogenation to H2CO and decomposition to CO and H2 on Ag/YSZ [4, 22], with faradaic efficiency absolute values up to 3 × 105 [19], rate enhancement ratios up to 55 [19], and significant changes in selectivity [4, 21, 22] and in oscillatory behavior [20], where appropriate. Results from these studies, published from 1988 to 1991, are shown in Figs. 1.5 and 1.6 of Chap. 1 for C2H4 oxidation on Pt/YSZ [19] and in Fig. 2.3 for methanol oxidation on Pt/YSZ [21], respectively.

Methanol oxidation on Pt/YSZ: Effect of dimensionless catalyst potential Π ≡ F UWR/RT on the selectivity to CO2 and H2CO; \( {P}_{C{H}_3 OH} \) = 0.9 kPa, \( {P}_{O_2} \) = 19 kPa. (Reprinted from ref. [21], with permission from Elsevier)
With the exception of C2H4 and C3H6 epoxidation on Ag/YSZ, the aforementioned studies were performed in a three electrode set-up [19] where two porous metal (Pt or Ag) films were deposited on the outer bottom side of the YSZ tube (Fig. 2.1), exposed to ambient air and serving as counter and reference electrode, respectively. This allowed for measurement of the catalyst-working electrode (W) potential UWR with respect to the reference electrode (R) and for its association with the observed rate change. Interestingly, over specific UWR range for each reaction, the catalytic rate, r, was found to depend exponentially on ΔUWR = UWR –UWR* according to the equation
where ro is the open-circuit rate (current I = 0), F is the Faraday’s constant, R is the universal gas constant, T is the absolute temperature, and α and UWR* are reaction and catalyst specific constants, with |α| ranging typically from 0.1 to 1.0 [19, 21,22,23]. Reactions for which α > 0, i.e., exhibiting rate increase by O2− pumping to the catalyst (I > 0, ΔUWR > 0, Λ > 0), were termed electrophobic and reactions for which α < 0, i.e., exhibiting rate increase upon O2− removal from the catalyst (I < 0, ΔUWR < 0, Λ < 0), were termed electrophilic [1, 22]. An example of electrophilic behavior is shown in Fig. 2.4 for H2CO and CO formation by CH3OH dehydrogenation and decomposition on Ag/YSZ [22]. For dimensional catalyst potential Π ≡ FUWR/RT values below −16 for H2CO and − 18 for CO, the corresponding formation rates increase exponentially with decreasing Π, in agreement with Eq. (2.3), with α values equal to −0.14 and − 0.30, respectively.

CH3OH dehydrogenation and decomposition on Ag/YSZ: Effect of dimensionless catalyst-electrode potential Π ≡ FUWR/RT on the formation rates of H2CO and CO. \( {P}_{C{H}_3 OH} \) = 5 kPa. ▲: T = 620 °C, ■: T = 643 °C, ●: T = 663 °C. (Reprinted (adapted) from ref. [22], with permission from Elsevier)
In the first EPOC studies with O2− electrolyte where EPOC is reported as a general effect in heterogeneous catalysis [4, 19, 21, 22], the observed non-faradaic rate changes as well as the selectivity changes upon polarization were attributed to work function change ΔΦ of the gas-exposed catalyst-electrode surface due to electrochemical adsorption of partially charged oxygen species Oδ- created at the gas-metal-solid electrolyte three-phase boundaries (tpd) and then migrating (backspillovering) onto the catalyst surface. The first indication of this electrochemically controlled migration of oxygen promoting species came out from the observation that the catalytic rate relaxation time constant, τ, during galvanostatic transients (imposition of constant current I), i.e., the time required to reach 63% of the final rate change at steady state, was on the order of the time required to form a monolayer of Oδ- species on the catalyst surface when O2− are supplied at the metal-gas-electrolyte three-phase boundaries (tpb) at a rate I/(2F), as dictated by Faraday’s law, i.e.,
where NG is the independently measured catalyst surface area, expressed as reactive oxygen uptake (in mol O) [3, 16, 18, 20,21,22]. The concomitantly induced work function change ΔΦ was predicted theoretically on the basis of the spatial uniformity of the Fermi level throughout the conductive electrode and the consideration that the Volta potential (Ψ) difference at the electrode/gas interface is equal to zero (ΔΨ = 0), because of the presence of an overall neutral double layer at the metal-gas interface formed by the electrochemically adsorbed species paired with their compensating image charges in the metal, i.e., [Oδ- – δ+] for O2− conductors [19, 21, 22]. Specifically, it was predicted that a change ΔUWR in the (ohmic-drop free) catalyst-electrode (working electrode W) potential vs. the reference electrode (R) induces a change ΔΦ in the work function of the gas-exposed catalyst-electrode surface, given by the equation (e: electron charge) [19, 21, 22]
However, this theoretical prediction needed experimental validation.
This validation came after 1 year, in 1990, when C. G. Vayenas and his collaborators used the Kelvin probe vibrating condenser method to measure in situ the work function of a Pt catalyst-electrode interfaced to YSZ, both under open- and closed-circuit conditions [24]. These measurements (Fig. 2.5) showed conclusively that Eq. (2.5) describes the relation between the change ΔUWR in catalyst-electrode potential, caused either by polarization or by variation of the gaseous composition, and the induced change ΔΦ in the work function of the gas-exposed catalyst-electrode surface. Eq. (2.5), which was also confirmed experimentally for Pt catalyst-electrodes interfaced to β″-Al2O3 [25, 26], a Na+ conductor, allows to write Eq. (2.3) as:
where Φ* ≡ e UWR* and kb is Boltzmann constant.

Effect of change in ohmic-drop-free catalyst potential UWR on the work function Φ of the gas-exposed catalyst-electrode surface; Pt catalyst film deposited on ZrO2(8 mol% Y2O3) solid electrolyte; T = 300 °C. Filled symbols: Closed-circuit operation with the Pt catalyst exposed to air. Open symbols: Open-circuit operation with the Pt catalyst exposed to air, C2H4/O2/He and NH3/O2/He mixtures. The straight line of slope unity corresponds to Eq. (2.5). (Reprinted from ref. [24], with permission from Springer Nature)
According to Eq. (2.6), electrophobic (α > 0) and electrophilic (α < 0) reactions are accelerated respectively by an increase or decrease in the catalyst work function, Φ, and by the concomitant decrease or increase in the availability of electrons from the catalyst for chemisorptive bond formation [22]. Equation (2.6) implies that catalytic rates depend exponentially on catalyst work function, which in many EPOC studies, with O2− and cationic conductors, has been found to hold over wide ranges of experimental conditions and work function (typically 0.2–0.8 eV) on the basis of the corresponding catalyst potential change [1, 8, 13, 14, 19, 21, 22, 26,27,28,29,30]. More generally, it directly attributes the induced changes in catalytic activity and selectivity under EPOC conditions to changes in the catalyst-electrode work function, which alter the chemisorptive bond strengths and surface coverages of the adsorbed reacting species.
From a practical perspective, Eq. (2.5) shows that the work function of metal catalysts interfaced to solid electrolytes can be varied at will to influence their catalytic properties in desirable directions. Moreover, on the basis of Eq. (2.5), it can be anticipated [31] that in solid electrolyte cells the potential difference UWR reflects the difference in the actual, adsorption and spillover modified, work functions ΦW and ΦR of the working and reference electrodes, respectively, i.e.,
Equation (2.7) shows that solid electrolyte cells can be used as work function probes for their gas-exposed electrode surfaces. Moreover, it allows establishment of an experimentally accessible absolute electrode potential scale in solid-state electrochemistry [32]. In 2001, Eq. (2.7) was experimentally validated by Tsiplakides and Vayenas [32] in YSZ cells with various combinations of porous Pt, Ag, and Au working and reference electrodes exposed to O2-He, O2-H2, and H2-He mixtures, above 600 K and typically over 0.8 to 1 V wide UWR ranges, using two Kelvin probes to measure ΦW and ΦR in situ and practically at the same time.
Up to 2001, electrochemical promotion with ZrO2(Y2O3) (or YSZ) as well as with mixed oxygen ion-electronic conductors (TiO2, CeO2, TiO2-doped YSZ) had been studied in a large number of catalytic reactions using a variety of porous metal and metal oxide catalyst-electrodes (Pt, Rh, Pd, Ag, Ag-Au, Au, Ni, IrO2, RuO2) [1, 23, 31, 33]. These reactions included not only oxidation of CO, deep oxidation of light hydrocarbons (CH4, C2H4, C2H6, C3H6) and CH3OH, and epoxidation of C2H4 and C3H6 but also NO reduction (by C2H4 and by CO or C3H6, in the presence of O2), N2O reduction by CO, hydrogenation of CO and CO2, CH3OH dehydrogenation and decomposition, H2S decomposition, and CH4 steam reforming, corroborating the general nature of EPOC [1, 23, 31, 33]. Rate enhancement ratios ρ up to 150 [34] and faradaic efficiencies Λ up to 3 × 105 [19] were reported. CH4 steam reforming on Ni-YSZ and Ni-YSZ cermet/YSZ [35] was the first EPOC study where non-noble metal catalyst-electrodes were used. Concerning the r vs. UWR or, equivalently, r vs. Φ behavior over the entire experimentally accessible range (global behavior), four types of behavior were observed depending on the catalytic system and the experimental conditions: purely electrophobic behavior (∂r/∂UWR > 0, ∂r/∂Φ > 0), as in C2H4 oxidation on Pt/YSZ [19]; purely electrophilic behavior (∂r/∂UWR < 0, ∂r/∂Φ < 0), as in NO reduction by C2H4 on Pt/YSZ [36]; volcano-type behavior, i.e., a maximum in the reaction rate r with varying catalyst potential UWR or work function Φ, as in CO oxidation on Pt/YSZ (under reducing conditions) [20]; and inverted volcano-type behavior, i.e., a minimum in r with varying UWR or Φ, as in NO reduction by C3H6 on Rh/YSZ in the presence of excess O2 [34].
An interesting aspect of EPOC discovered in 1997 by Comninellis and coworkers in the reaction of C2H4 oxidation on IrO2/YSZ [37] is the “permanent NEMCA” or “permanent EPOC” (P-EPOC) effect, i.e., the remaining rate enhancement after current interruption following prolonged anodic polarization of the catalyst-electrode. Permanent EPOC, which is potentially important for practical applications (e.g., utilizing EPOC during catalyst preparation), has been observed also in C2H4 combustion on RuO2/YSZ [38] and Pt/YSZ [39] and in NO reduction by C3H6 [34, 40, 41] or CO [41] in the presence of O2 on Rh/YSZ. Permanent EPOC with YSZ has been explained by storage of promoting oxygen species at the catalyst-electrode/YSZ interface and subsequent migration towards the catalyst/gas interface after positive current interruption, through the catalyst-YSZ-gas three-phase boundaries [39, 42].
2.2 EPOC with Cationic Conductors and Aqueous Electrolytes
Studies concerning the discovery of EPOC and its first report as a general, electrochemically induced, catalytic effect were carried out using YSZ, an O2− conducting solid electrolyte. In 1991 C.G. Vayenas and his coworkers reported for the first time EPOC using a Na+ conductor, in the reaction of ethylene complete oxidation on Pt interfaced to β″-Al2O3 solid electrolyte in a fuel cell type reactor (β″-Al2O3 tube) [26]. An important conclusion of this study was that the observed EPOC features were the same as those for ethylene complete oxidation on Pt/YSZ, in particular the exhibited electrophobic behavior and the exponential dependence of rate on catalyst potential (Eq. 2.3) [26], which supported the explanation of the EPOC effect on the basis of an electrochemically induced alteration of the catalyst surface work function, as given by Eq. (2.5) [19]. Also, the fact that a very small sodium coverage, equal to 0.015, was found sufficient to cause a pronounced 70% decrease in the rate of ethylene oxidation provided strong evidence for “long-range” electronic interactions, ruling out any interpretation of EPOC based on geometric factors [26]. The main features observed in this work were confirmed some years later by Harkness et al. [43] who studied the same system in a single pellet reactor [44] and over a wider range of conditions, in parallel with kinetic and spectroscopic experiments with Pt(111)/Na model catalysts. In this type of reactor, the Pt catalyst-working electrode and two inert Au reference and counter electrodes were deposited on the opposite sides of a β″-Al2O3 disk, all exposed to the reaction mixture. Besides the application of a different reactor type, a new aspect of their work was the observation of a volcano-type behavior (a maximum in the ethylene combustion rate) with decreasing catalyst potential UWR (increasing sodium coverage), corresponding to a very sharp cutoff of the rate below sufficiently negative values of UWR which was attributed to extensive blocking of the Pt surface with sodium surface compound(s) and to strongly enhanced competitive adsorption of oxygen. Moreover, over the same range of sodium coverages, they observed agreement between the kinetic behavior of the electropromoted Pt catalyst and that of the Pt(111)/Na model catalyst, providing strong evidence for the role of sodium as the key promoting species under conditions of EPOC with Na+ conducting solid electrolytes.
From 1991 to 2003, EPOC using alkali ion conductors, specifically β″-Al2O3, K-β′′-alumina and NASICON (Na3Zr2Si2PO12), was studied by the groups of C. G. Vayenas in Patras, R.M. Lambert in Cambridge, and G. Haller in Yale in many other reactions [1], including complete and partial oxidations (C2H4 oxidation on Pt/NASICON [45]; CO oxidation on Pt/β″-Al2O3 [46]; C2H4 epoxidation on Ag/β″-Al2O3 [47]); NO reduction on Pt/β″-Al2O3 by C2H4 [48], CO [49], H2 [50], or C3H6 [51], on Rh/β″-Al2O3 by CO [52, 53] or C3H6 [52, 54], and on Cu/β″-Al2O3 by CO [55]; hydrocarbon hydrogenations (C6H6 hydrogenation on Pt/β″-Al2O3 [56] and selective C2H2 hydrogenation on Pd/β″-Al2O3 [57], Pt/β″-Al2O3 [58], and Pt/K-β′′-alumina [59]); CO2 hydrogenation on Pd/β″-Al2O3 [28, 60]; and Fischer-Tropsch synthesis on Ru/β″-Al2O3 [14, 61] and Rh/K-β′′-alumina [62]. The results of these studies confirmed that EPOC is not restricted to O2− conductors and to oxidation reactions but can be also induced with alkali ion conductors in many reaction types providing a means of in situ controlled alkali promotion with numerous potential applications. Since 2003, a large number of important new contributions to EPOC with alkaline conductors have appeared, as summarized in recent reviews [5, 7, 63, 64], including the electrochemically assisted NOx storage/reduction on porous Pt/K-β-Al2O3 [65] under negative/positive polarization, respectively. EPOC with alkaline conductors for emissions control catalysis is fully described in Chap. 4. We can also mention the K+-promoted (negative polarization) H2 production by CH3OH steam reforming on nanocolumnar Ni films deposited on K-β-Al2O3 and its simultaneous reversible storage in the presence of potassium species, mainly via spillover of H atoms from Ni onto graphene oxide (GO) produced in situ via electropromoted methanol decomposition [66]. This study is detailed in Chap. 9.
An attractive feature of using an alkali ion conductor for electrochemical promotion is that the coverage of alkali electrochemically introduced onto the gas-exposed catalyst surface can be accurately measured coulometrically. This allows easy comparison with classical promotion studies and evaluation of the promotion index PIi, which is an important phenomenological parameter for quantification of the promoting or poisoning effect of a given species i (e.g., Na+) co-adsorbed on a catalytic surface during a reaction. The promotion index PIi is defined from:
where ro is the catalytic reaction rate in the absence of species i and r – ro is the induced rate change by a change Δθi in the coverage θi of the promoting (or poisoning) species i. For the aforementioned reactions, promotion index values up to 6000 have been reported for the sodium species supplied electrochemically onto the catalyst surface [1]. An example of electrochemical promotion using a Na+ conductor is shown in Fig. 2.6 for ethylene epoxidation on Ag/β″-Al2O3 in the presence of 1,2-C2H4Cl2 as moderator, studied in a single pellet reactor [47]. Negative current application, i.e., Na+ pumping to the Ag catalyst-electrode, resulted in enhancement of the ethylene epoxidation rate without affecting the ethylene combustion rate and, concomitantly, in substantial increase of the selectivity to C2H4O. The latter reached a maximum value of 88% for a sodium coverage of 0.03 and 1 ppm of 1,2-C2H4Cl2 in the gas phase. The strong promotional effect of the electrochemically supplied Na species is reflected in the high values, up to 40, of the promotion index PINa.

C2H4 epoxidation on Ag/β″-Al2O3: Transient effect of a negative applied current (Na+ supply to the catalyst-electrode) and corresponding sodium coverage on the rates of C2H4O (▲) and CO2 (●) formation and on catalyst potential UWR. The dotted line is constant promotion index line. T = 260 °C, total pressure P = 5 atm, \( {P}_{O_2} \) = 17.5 kPa, \( {P}_{C_2{H}_4} \) = 49 kPa, 0.6 ppm 1,2-C2H4Cl2. (Reprinted (adapted) from ref. [47], with permission from Elsevier)
Proton conductors are another class of solid electrolytes which has been used to induce electrochemical promotion of catalyst-electrodes [1, 11]. In 1990 Politova, Sobyanin, and Belyaev in Novosibirsk reported reversible non-faradaic rate changes in the reaction of ethylene hydrogenation on Ni interfaced to CsHSO4, a H+ conducting solid electrolyte, upon electrochemical H+ pumping to or from the catalyst surface, corresponding to electrophobic EPOC behavior with enhancement factor ρ and faradaic efficiency |Λ| values up to ca. 2 and 300, respectively [67]. This work was the first EPOC study using a proton conductor and also the first EPOC study of a hydrogenation reaction. In the same decade, from 1993 to 2000, EPOC was studied in several other reactions using a variety of H+ conductors. In 1993, M. Stoukides and his coworkers studied for the first time the non-oxidative coupling of methane to ethane and ethylene on Ag interfaced to SrCe0.95Yb0.05O3 in a one-chamber cell [68]. The reaction exhibited electrophobic behavior with ρ values up to 8 and total selectivity to C2 hydrocarbons near 100%; however, no faradaic efficiency values were reported. The first report of the use of a proton conductor for electrochemical promotion of an oxidation reaction appeared in 1996 by Makri et al. [30] who investigated the effect of electrochemical pumping of H+ on the rate of ethylene combustion on Pt interfaced to CaZr0.9In0.1O3-δ, a solid electrolyte with predominantly proton conductivity over the temperature range 380–450 °C that was used in this study. Electrophilic EPOC behavior was observed, corresponding to reversible increase of the rate of C2H4 combustion by up to 500% upon proton supply to the catalyst-electrode film (negative current application) and to faradaic efficiency |Λ| values up to 2 × 104.
In 2000, Yiokari et al. [69] demonstrated for the first time electrochemical promotion under high pressure (50 atm) and using a dispersed industrial non-precious metal catalyst. Specifically, they achieved up to 1300% non-faradaic (|Λ| = 6) enhancement of the rate of NH3 synthesis on a state-of-the-art iron-based industrial catalyst (BASF S6-10RED) by depositing it on proton conductor CaIn0.1Zr0.9O3-δ pellets, using a mixture of catalyst powder and commercial Fe paste, and electrochemically pumping H+ to its surface. This work was also the first demonstration of scale-up of an EPOC reactor as a multi-pellet configuration (twenty-four CaIn0.1Zr0.9O3-δ pellets) was used (Fig. 2.7). Some years later Stoukides and coworkers [70] also reported a similar weak EPOC effect (|Λ| and rate enhancement ratio ρ less than 3 and 2, respectively) for NH3 synthesis on an industrial Fe-based catalyst-electrode interfaced to the H+ conductor SrZr0.95Y0.05O3-δ at 450–700 °C and atmospheric pressure. Recent advances on the electrochemical promotion of ammonia synthesis are reviewed in Chap. 8.

Multi-pellet EPOC reactor for NH3 synthesis at high pressure: (a) Machinable ceramic holders and two CaIn0.1Zr0.9O3-δ pellets showing the location of the electrodes. (b) Twenty-four pellet unit. (Reprinted with permission from ref. [69]. Copyright (2000) American Chemical Society)
The use of a solid polymer proton conductor in EPOC was reported for the first time in 1997 by Tsiplakides et al. [71] who studied the oxidation of H2 by O2 at room temperature on a Pt black catalyst-electrode deposited on Nafion 117 membrane, with the other side of the membrane being in contact with a 0.1 M KOH aqueous solution with a Pt wire counter electrode immersed in it. It was observed that positive current application reversibly increased the rate of H2 oxidation by up to a factor of 20, whereas the induced rate increase was up to 300 times larger than the electrochemical rate of H2 oxidation, implying change in the chemisorptive bond strength of adsorbed reactants with changing catalyst potential and work function (Eq. 2.5). Smotkin and coworkers, also using Nafion as solid electrolyte, demonstrated in 1997 for the first time electrochemical promotion for a unimolecular and non-redox catalytic reaction, specifically for the isomerization of 1-butene on a high surface area Pd/C cathode deposited on Nafion 117 in a membrane electrode assembly (MEA) with an essentially nonpolarizable Pt black/H2 counter electrode [72]. Under galvanic cell operation at 70 °C, the rates of cis- and trans-2-butene formation increased dramatically, prior to a significant increase in butane formation by 1-butene hydrogenation, passing through a maximum at a cell voltage of 0.16 V and 0.1 V, respectively (Fig. 2.8). The enhancement factor ρ values at these maxima were approximately 38 and 46, respectively, whereas the absolute value of the faradaic efficiency for both isomers was approximately 28, denoting a strong non-faradaic electrophilic behavior. A mechanistic study of this electropromoted isomerization revealed that it is an acid-catalyzed reaction at the Pd surface facilitated by the superacidic Nafion electrolyte [73].

Isomerization of 1-butene on Pd/C interfaced to Nafion 117: Effect of cell voltage on the rates of cis- and trans-2-butene and butane formation. T = 70 °C. (Reprinted (adapted) with permission from ref. [72]. Copyright (1997) American Chemical Society)
An interesting EPOC study with Pb2+ ion conductor appeared in 2002, when Lambert and coworkers reported the first demonstration of the use of electrochemical promotion to in situ control, in a reversible and reproducible manner, the composition and catalytic performance of bimetallic surface Pt/Pb alloy for hydrogenation of acetylene, via interfacing a porous Pt film with a Pb-β″-Al2O3 electrolyte (a Pb2+ conductor) and electrochemically pumping of Pb2+ to the Pt film [74, 75]. Increase in selectivity to ethylene from 20% on pure Pt to 85% on a ∼ 26% Pb alloy surface formed via Pb2+ pumping was observed, accompanied by a decrease in C2H2 conversion, which was attributed to weakening of the ethylene and acetylene chemisorptive bonds.
In 1993 Anastasijevic et al. [76] were first to report a non-faradaic effect in aqueous electrochemistry, specifically a current efficiency above 100% for H2 evolution during HCHO oxidation on Cu and Ag electrodes in 0.1 M KOH solution. In 1994, Neophytides et al. [77] reported electrochemical promotion for H2 oxidation on a graphite-supported Pt electrode immersed in 0.1 M KOH solution and, some years later, on a Pt black electrode [78], obtaining identical results. The Pt/graphite or Pt black working electrode was deposited on a Teflon-frit through which the reactant mixture was sparged, while a Pt counter electrode was positioned in a separate compartment. Figure 2.9 shows a characteristic galvanostatic transient obtained with the Pt/graphite electrode. As seen in the figure, the observed rate changes in H2 and O2 consumption are reversible and non-faradaic, corresponding, for I = 15 mΑ, to faradaic efficiencies equal to ca. 7 and 5, respectively, while the rate relaxation time constant is on the order of 2FNG/Ι, as in EPOC studies with O2− conductors (Eq. 2.4). Application of positive overpotentials in this system, i.e., OH− supply to the Pt electrode, increased the rate of H2 oxidation by up to 500% in a non-faradaic manner with faradaic efficiencies up to 100 [77, 78]. Qualitatively similar results were obtained using a 0.1 M LiOH solution. Similarly to electrochemical promotion with solid electrolytes, the observed EPOC behavior was attributed to polarization-induced changes in the work function of the Pt surface and to concomitant changes in the coverages and binding strengths of dissociatively chemisorbed oxygen and hydrogen [1, 77, 78].

EPOC in H2 oxidation on Pt/graphite in 0.1 M KOH: Transient effect of applied positive and negative current I (15 mA and − 10 mA) on the rates of consumption of oxygen (rO, mol O/s) and hydrogen (\( {r}_{H_2} \), mol H2/s); \( {P}_{H_2} \) = 0.75 kPa, \( {P}_{O_2} \) = 1.06 kPa, gas flow rate Fv = 280 cm3/min at STP. Total Pt surface area NG = 3.0 × 10−6 mol Pt. (Reprinted from ref. [77] with permission from Springer Nature)
3 The Physicochemical Origin of EPOC: Rules of Promotion
Since the early 1990s, EPOC had been conclusively associated with polarization-induced changes of the catalyst-electrodes, and Eq. (2.5) had been experimentally validated for oxygen and sodium ion conductors [24,25,26]; however, a detailed understanding of the EPOC effect at the molecular level was missing. Starting essentially from 1993, a large number of surface science, catalytic and electrochemical techniques, as well as theoretical calculations and thermodynamic considerations, mentioned in early and recent reviews [1, 2, 6, 8, 13, 14, 79,80,81], were employed to determine the physicochemical origin of electrochemical promotion, in particular as it concerns the nature of the promoting species and the catalyst surface state under electrochemical promotion conditions.
In 1993, X-ray photoelectron spectroscopy (XPS) was employed for the first time to study the effect of polarization on Pt catalyst-electrode films interfaced with YSZ [82]. Backspillover of oxygen species from the solid electrolyte onto the Pt surface upon electrochemical oxygen pumping to the catalyst was evidenced by an almost 60% increase in the total O1s spectrum area compared to that under open-circuit, which was accompanied by a shift of the peak maximum to lower binding energy by ca. 1.8 eV (Fig. 2.10). The difference O1s spectrum revealed that two distinct oxygen species existed on the Pt surface following oxygen pumping, specifically chemisorbed atomic oxygen, at a binding energy of 530.4 eV (peak γ), and a more anionic oxygen species, at a binding energy of 528.8 eV (peak δ). The latter oxygen species, which was less reactive than normally chemisorbed oxygen under the reducing UHV conditions, was identified as the promoter oxygen species responsible for electrochemical promotion of catalytic oxidations using O2− solid electrolytes [1, 82].

Effect of electrochemical O2− pumping on the O 1 s spectrum of Pt/YSZ. XPS spectra at 673 K: (A) open-circuit conditions; (B) constant overpotential ΔUWR = 1.2 V (steady state current I = 40 μA); (C) O 1 s difference spectrum. (Reprinted with permission from ref. [82]. Copyright (1993) American Chemical Society)
In 1995, Neophytides and Vayenas used for the first time temperature-programmed desorption (TPD) under high-vacuum conditions to investigate the state of adsorbed oxygen on Pt catalyst-electrode films interfaced to YSZ as a function of catalyst potential [83]. A more detailed TPD study of the same system followed 3 years later by Neophytides et al. [84]. As shown in Fig. 2.11, gaseous oxygen adsorption (t = 0) resulted in a single adsorption state, while mixed gaseous-electrochemical oxygen adsorption via subsequent electrochemical supply of O2− for various times (t = 125–3900 s) led to two distinct adsorbed oxygen states, specifically a weakly bonded state, shifted to lower peak desorption temperature Tp (by ca. 50 K) compared to that for oxygen adsorption from the gas phase only (Tp ≈ 738 K), and a strongly bonded state (Tp from 743 to 773 K) gradually developing with increasing time of current application. Considering that the time required for complete development of the strongly bonded oxygen state and the relaxation time for the catalytic rate to reach its promoted state during galvanostatic transients in EPOC experiments were both comparable to 2FNG/I (Eq. 2.4), equal to 2500 s, the conclusion drawn was that the strongly bonded oxygen state corresponds to partially charged ionic oxygen species created on the catalyst surface upon electrochemical pumping of O2− to the catalyst-electrode, which forces normally chemisorbed oxygen to a more weakly bonded and more reactive adsorption state acting as “sacrificial promoter” as it is also consumed reacting with the oxidizable species present (e.g., C2H4, CO), at a rate I/(2F) at steady state [83, 84]. In this context, the faradaic efficiency Λ (Eq. (2.1)) can be considered to express at steady state the ratio of the turnover frequencies TOFr and TOFp for the electrochemically promoted reaction and for the consumption of the sacrificial promoting species, respectively, or, equivalently, the ratio of the average lifetimes τp and τr of the promoting species and of the adsorbed gas-supplied reactants on the catalyst surface, the former estimated from the time decay of the reaction rate upon current interruption [1, 13].

Oxygen TPD spectra after gaseous oxygen adsorption on Pt/YSZ at 673 K and \( {P}_{O_2} \) = 4 × 10−6 Torr (oxygen exposure equal to 7.2 kilolangmuirs) followed by electrochemical O2− supply (I = 15 μA) for various time periods; heating rate: 1 K/s. (Reprinted (adapted) with permission from ref. [83], copyright (1995) American Chemical Society and from ref. [84], copyright (1998) Elsevier)
The aforementioned XPS [82] and TPD [83, 84] studies of the Pt/YSZ system provided for the first time evidence for creation of a strongly bonded oxygen state on the Pt catalyst surface upon positive polarization, which does not form via adsorption from the gas phase and can act as a sacrificial promoting species in catalytic oxidations under EPOC conditions by weakening the binding strength of the more reactive normally adsorbed oxygen. Since these first studies and up to 2001 the electrochemically induced creation of promoting oxygen species on metal catalyst-electrodes and the sacrificial promoter mechanism of EPOC with O2− conductors had been corroborated by the results of many spectroscopic (including XPS, ultraviolet photoelectron spectroscopy (UPS), photoemission electron spectroscopy, and surface enhanced Raman spectroscopy) and electrochemical (AC impedance, cyclic voltammetry, potential programmed reduction) techniques [1]. Direct confirmation of the sacrificial promoter mechanism came in 2004 by Katsaounis et al. [85, 86] who studied under high-vacuum conditions both the adsorption of 18O2 [85] and the oxidation of CO by 18O2 [86] on electropromoted catalyst-electrode Pt films interfaced to YSZ. Their results showed clearly that under anodic polarization 16O from the YSZ lattice migrates onto the Pt catalyst surface, acting there as a sacrificial promoter since the 16Oδ− species both react with CO and promote the catalytic reaction between CO and adsorbed 18O from the gas phase. This is illustrated in Fig. 2.12 [86], which shows that upon positive (I = +1 μΑ) or negative (I = −1 μA) current application the induced increase or decrease, respectively, of the rate of C16O2 is very small and (sub)faradaic \( \left(\Delta {r}_{C^{16}{O}_2}/\left(I/(2F)\right)<1\right) \), whereas the corresponding increase or decrease of the rate of C16O18O formation is much larger and non-faradaic, i.e., at steady state \( \left|\Delta {r}_{C^{16}{O}^{18}O}\right| \) is ca. 11.4 and 9.6 times larger than the rate |I|/(2F) of supply or removal, respectively, of O2− to or from the Pt catalyst.

CO oxidation by 18O2 on Pt /YSZ: Low-temperature (T = 225 °C) galvanostatic transient (\( {P}_{18}{O}_2 \) = 8 × 10−8 mbar, \( {P}_{C^{16}O} \) = 2.5 × 10−8 mbar) leading to non-faradaic behavior; see text for discussion. (Reprinted from ref. [86], with permission from Elsevier)
In 2007, P. Vernoux and his coworkers validated the sacrificial promoter mechanism of EPOC with O2− conductors under real operating conditions, by performing O2-TPD in the Pt/YSZ system under atmospheric pressure in the presence and absence of propane in the gas phase [87]. In continuation of this work, isotopic labeling experiments were performed by Tsampas et al. [88, 89] to operando investigate EPOC in the reaction of propane combustion on Pt/YSZ under atmospheric pressure and distinguish the role of oxygen species originating from the gas phase and from the YSZ solid electrolyte. In agreement with the sacrificial promoter model, application of positive polarization resulted in pronounced electropromotion (faradaic efficiency Λ up to 35) of the reaction of propane with gaseous O2 (C18O2 production) and to significant surface oxygen exchange at the gas-Pt-YSZ boundaries generating active oxygen species, as evidenced by the production of C16O18O which was also promoted but to a lesser extent, whereas production of C16O2 via (sub)faradaic electrochemical oxidation of propane by O2− was also observed (Fig. 2.13) [88, 89].

Propane combustion on Pt/YSZ: Effect of electrochemical rate of O2− supply on the CO2 production rate increase. Dashed lines correspond to constant faradaic efficiency values. \( {P}_{C_3{H}_8} \) = 0.1 kPa, \( {P}_{O_2} \) = 2.5 kPa, T = 350 °C. (Reprinted from ref. [88], with permission from Elsevier)
The origin of EPOC when using alkali ionic conductors and the nature of the promoting alkali surface phases have been investigated by a large number of catalytic, spectroscopic, and surface imaging techniques [1, 6]. In 1995 and 1996 Lambert and coworkers combined for the first time kinetic and spectroscopic (postreaction XPS, Auger electron spectroscopy (AES), and TPD) data obtained with a Na-dosed Pt(111) single crystal and electrochemical promotion data for the reactions of ethylene combustion [43] and NO reduction by ethylene [48] on Pt films supported on β″-Al2O3. The observed agreement in the kinetic behavior of the Pt(111)/Na model catalyst with that of the electropromoted Pt film catalyst over the same range of sodium coverages provided strong evidence that reversible sodium backspillover is the origin of EPOC with Na+ conductors. Moreover, postreaction XP and Auger spectra showed that the chemical state of the promoting sodium depends on reaction conditions. From 1996 to 2003, Lambert and coworkers performed in situ and ex situ XPS, UPS, AES, and X-ray absorption near edge spectroscopy (XANES) studies on Pt/β″-Al2O3 [1, 51, 90], Rh/β″-Al2O3 [52, 54], Cu/β″-Al2O3 [55, 91], Pt/K-β″-Al2O3 [59], and Rh/K-β″-Al2O3 [62] under conditions simulating EPOC or after EPOC experiments (postreaction). These studies, which are summarized in a recent review [6], further confirmed that electrochemical promotion with alkali ion conductors is due to reversible backspillover of alkali species from the solid electrolyte to the catalyst-electrode. Moreover, they showed that the surface chemical state of the electrochemically pumped alkali is the same as that for alkali adsorbed on the surface via vacuum deposition and that under reaction conditions the alkali promoter is present as surface compounds with submonolayer coverage and with nature dependent on the composition of the reactive gas phase (e.g., as a mixture of NaNO2 and NaNO3 in NO reduction by propene or as Na2CO3 in propane oxidation on Pt/β″-Al2O3 [14]). Lambert and coworkers have also reported a linear correlation between catalyst potential UWR and catalyst work function Φ in the Rh/β″-Al2O3 [14, 53] and Cu/β″-Al2O3 [91] systems over an extended range of catalyst potential (ΔUWR ~ 2 V and 0.9 V, respectively) and submonolayer sodium coverages, by determining the work function of Rh and Cu via UPS (secondary electron cutoff method). After 2003, ex situ (postreaction) SEM/EDX, XRD, XPS, and FTIR analyses as well as cyclic voltammetry have been also employed by the groups of P. Vernoux, J. L. Valverde, and A. de Lucas Consuegra to study the nature and arrangement of the potassium promoter phases present on the catalyst surface under EPOC conditions in the reactions of low-temperature propene oxidation on Pt/K-β-Al2O3 [92] and partial oxidation of methanol on Pt/K-β-Al2O3 [93] and Cu/K-β-Al2O3 [94].
In 1996, Vayenas and coworkers used successfully scanning tunneling microscopy (STM) to follow the polarization-induced reversible migration of sodium across macroscopic (on the order of mm) distances from a β″-Al2O3 pellet to the surface of a Pt(111) single crystal deposited on it and exposed to air [95], also achieving for the first time imaging of an electropromoted catalyst-electrode surface with atomic-level resolution. They observed that electrochemical pumping of sodium to the Pt(111) surface caused, at low sodium coverages (less than 0.05), the creation of a Pt(111)-(12 × 12)-Na adlayer (interatomic distance of 33.2 Å) which was present over atomically huge domains of the Pt(111) surface, overlapping the well-known Pt(111)-(2 × 2)-O adlattice (interatomic distance of 5.6 Å) formed by oxygen adsorbed from the gas phase. The Pt(111)-(12 × 12)-Na adlayer disappeared upon reversal of the direction of sodium pumping, leaving the Pt(111)-(2 × 2)-O adlattice intact. Some years later, an STM study of an air-exposed Pt(111) single crystal interfaced to an YSZ pellet similarly demonstrated the reversible migration of promoting oxygen species upon polarization, i.e., under conditions simulating EPOC, which formed a (12 × 12)-O adlattice coexisting with the underlying (2 × 2)-overlayer of the normally chemisorbed oxygen from the gas phase [96]. On a distance scale larger than that in STM studies, scanning photoelectron microscopy (SPEM) has been used by Lambert and coworkers to image the spatial distribution and time dependence of electro-pumped alkali at the surface of polycrystalline Cu interfaced to β″-Al2O3 [91] and at the surface of polycrystalline Pt interfaced either to β″-Al2O3 [14] or to K-β″-Al2O3 [59]. In all cases a practically uniform spatial distribution of the alkali was observed, with its surface concentration increasing with decreasing potential of the Pt or Cu electrode.
All the experimental techniques and theoretical studies that have been used to investigate the physicochemical origin of electrochemical promotion [1, 2, 6, 8, 13, 14, 79,80,81], mostly with O2− and alkali ion conductors, have confirmed that EPOC is due to the electrochemically controlled introduction of partially charged promoting species from the solid electrolyte (or mixed ionic-electronic conductor) support to the gas-exposed catalyst-electrode surface. These electrocatalytically created species, accompanied by their compensating (image) charges in the electrode, form an overall neutral double layer on the catalyst/gas interface, as shown schematically in Fig. 2.14. The presence of this effective double layer, whose density and field strength in it vary with varying catalyst potential, affects the electronic properties (work function) of the catalyst surface and concomitantly its chemisorptive properties, thus inducing pronounced and reversible alterations in catalytic rates and selectivity. It is noted that although the nature of the promoting species under EPOC conditions has been extensively studied for O2− and alkali ion conductors, establishing the sacrificial promoter mechanism [1, 13] in the former case, much less is known about the promoting species formed on the catalyst-electrode surface in EPOC with H+ conducting electrolytes, in particular in oxidation reactions. In this case it has been proposed that adsorbed hydroxyl species formed by association of the migrating protons with chemisorbed atomic oxygen act as sacrificial promoters [1, 97].

Schematic representation of a metal electrode deposited on an O2−conducting (top) and on a Na+-conducting (bottom) solid electrolyte, showing the location of the metal-electrolyte double layer and of the effective double layer created at the metal-gas interface due to potential-controlled ion migration (backspillover). It is also depicted the interaction between the effective double layer and the adsorbed reactants during CO oxidation. (Reprinted from ref. [13], with the permission of AIP Publishing)
In 2001, a systematic and meticulous search of the electrochemical promotion literature allowed Vayenas and coworkers to classify reactions in four types (electrophobic, electrophilic, volcano, and inverted volcano-type reactions, as defined in Sect. 2.2.1) on the basis of the rate versus catalyst potential or work function behavior and, moreover, establish simple and rigorous rules which permit prediction of the electrochemical promotion behavior on the basis of the reaction kinetics under unpromoted conditions or the chemisorptive bond strengths of the electron donor (D) and electron acceptor (A) reactants on the unpromoted catalyst [1, 98]. These rules were extended also to chemical promotion [1, 98] which was validated some years later using the chemical (classical) promotion literature [99]. The four global promotional rules (R1 to R4), which predict the rate r vs. catalyst potential UWR or work function Φ behavior over the entire experimentally accessible range (typically, over 1.5 to 2 eV for Φ), are summarized schematically in Fig. 2.15 [100]. Vayenas and Brosda [100] showed that these four promotional rules can be combined in a single generalized rule which states that the r vs. Φ dependence always traces the rate vs. electron donor (D) reactant partial pressure dependence. Vayenas and coworkers also demonstrated that the electrostatic interaction between the electric field in the effective double layer and the adsorbate dipoles leads to Frumkin-type electrochemical isotherms, consistent with the experimentally observed in TPD studies linear changes of heats of adsorption with catalyst work function [1, 98], and to generalized Langmuir-Hinshelwood kinetics which successfully model the kinetics of promoted catalytic reactions, in good semiquantitative agreement with experiment, also mathematically describing the rules of promotion [1, 101].

Effect of the reaction orders αA and αD with respect to the electron acceptor, A, and electron donor, D, reactant (top) and effect of the magnitude of adsorption equilibrium constants kD and kA and corresponding partial pressures pD and pA of the electron donor and electron acceptor reactant, respectively, (bottom) on the observed rate dependence on catalyst potential or work function: electrophobic (/), electrophilic (\), volcano (∩), and inverted volcano (∪) dependence and range of validity of the corresponding four global promotional rules R1, R2, R3, and R4. (Reprinted from ref. [100], with permission from Springer Nature)
The validity of the aforementioned rules of promotion and modelling of promoted catalytic kinetics for both electrochemical promotion, where the promoter level on the catalyst surface is varied in situ by varying catalyst potential, and for chemical (classical) promotion, where the promoters are typically added ex situ in the course of catalyst preparation, underlines their functional similarity and only operational differences [1, 99] which had become evident already from the first studies of the origin of EPOC, in particular those with alkali ion conductors. However, the in situ controllable reversible promotion and the ability to create and continuously replenish short-lived but extremely effective promoter species, such as Oδ-, that are not available in classical promotion are distinct features of EPOC and important operational advantages [1, 102], highlighting its practical usefulness.
In 2001 it was also shown that EPOC with O2− conductors is functionally equivalent with metal-support interaction (MSI) promotional phenomena in dispersed nanocrystalline metal (Pt, Rh) or metal-type conducting oxide (IrO2) catalysts supported on porous O2− conducting (e.g., YSZ) or mixed ionic-electronic conducting (e.g., WO3-doped TiO2) supports [1, 103]. In both cases backspillover of oxygen ions to the catalyst surface is the dominant promoting mechanism; thus EPOC can be considered an electrically controlled MSI [1, 102, 103]. The common underlying principle is shown schematically in Fig. 2.16. In conclusion, electrochemical promotion, promotion, and MSI on ionic and mixed conducting supports are all three facets of the same phenomenon and are due to the interaction of the adsorbed reacting species with the effective double layer formed by promoting species at the gas-exposed catalyst surface [1, 102].

Schematic of a metal grain (~μm) in a metal catalyst film deposited on YSZ or TiO2 under EPOC conditions (left) and of a metal nanoparticle (~nm) deposited on a porous TiO2 support (right), showing the locations of the classical double layers formed at the metal-support interface and of the effective double layers formed at the metal-gas interface. (Reprinted (adapted) with from ref. [103], with permission from Elsevier)
4 From Fundamentals to Applications
Since its discovery, EPOC has been studied in more than 80 catalytic reactions (including oxidation, hydrogenation, and dehydrogenation, NOx reduction and storage, decomposition, isomerization, and reforming reactions) using a variety of metal (mostly noble metal) or metal-type oxide catalysts supported on many different ionic or mixed ionic-electronic conductors, as summarized in a number of early and recent reviews [1, 2, 7, 8, 11]. EPOC has also been reported in a few catalytic systems with aqueous electrolytes [1, 76,77,78, 104, 105] and inorganic melts [1, 106], including a very recent study of methanol electrolysis with methanol–KOH solution introduced in both the anode and cathode, where the observed over-faradaic H2 production was attributed to electrochemical promotion of the catalytic methanol decomposition at the Pd/C cathode by K+ ions supplied from the liquid electrolyte under electrolysis conditions [107]. Over the years, there has been a gradual shift of the interest from EPOC studies of model reactions (originally oxidation reactions of C2H4 and CO), mainly aiming to elucidation of the origin and molecular-scale mechanism of this effect, to EPOC studies of reactions of environmental and industrial importance, such as selective catalytic reduction (SCR) of NOx, H2 production by catalytic reforming or partial oxidation, and CO2 hydrogenation [1, 2, 7, 8, 11, 108,109,110,111,112]. In particular, electrochemical promotion of CO2 hydrogenation has attracted increased interest in recent years aiming to management and mitigation of CO2 emissions [2, 28, 29, 109,110,111, 113,114,115,116,117,118]. Recently, there is also growing interest for electrochemical promotion of non-noble transition metal [2, 66, 94, 109, 118,119,120,121] and metal oxide [122, 123] catalysts, driven by the need to develop sustainable and commercially viable processes, with the research efforts focused on H2 production [66, 94, 119, 120], CO2 hydrogenation to hydrocarbons and other organics [118, 121], reverse water gas shift (RWGS) reaction [109, 122], and gas phase Brønsted acid-catalyzed reactions [123]. Theoretical modelling of EPOC using quantum mechanical calculations, which started in 1996 by Pacchioni et al. [124] and continued later by Leiva et at. [125], has also drawn increased interest recently, in particular by Steinmann, Baranova, and coworkers who have used density functional theory (DFT) calculations and ab initio atomistic thermodynamics to study the origin of EPOC in C2H4 oxidation on RuO2/YSZ [79] and in CH4 oxidation on Pt/YSZ [126].
Besides its fundamental scientific importance, EPOC has a great potential for practical utilization, although its commercial application is still missing for several technical and economic reasons [1, 2, 5, 8, 12, 13, 127]. The main technical obstacles for practical utilization of EPOC have been (a) the very low dispersion (less than ca. 0.01%) of the porous metal films (0.1–5 μm typical thickness) and (b) the lack of compact, efficient, and cheap reactor designs allowing electropromotion of the catalyst with a minimum number of electrical connections [2, 11,12,13]. Efforts to address these issues and foster practical applications of EPOC have started since the mid-90s with contributions by different groups, as summarized in recent reviews [2, 5, 8, 12, 128] and concisely described below.
4.1 EPOC via Polarization in Bipolar Configuration
In 1997 Marwood and Vayenas demonstrated EPOC in C2H4 combustion on a Pt stripe catalyst deposited on YSZ between two Au electrodes in a bipolar configuration where the Pt catalyst was electronically isolated, i.e., not used as an electrode, and polarization was applied between the two catalytically inert terminal Au electrodes [129]. Although the maximum rate enhancement ratio obtained in this bipolar configuration was a factor of 2 smaller than that obtained in the conventional monopolar configuration, which was attributed to current bypass and nonuniform distribution of the work function in the bipolar Pt electrode, it was clearly shown for the first time that EPOC can be induced without direct electrical connection to the catalyst [129]. Some years later the same idea was successfully extended to multi-stripe and multi-dot bipolar Pt catalysts [130]. In 1999, Comninellis and coworkers used a bipolar configuration to demonstrate for the first time EPOC of C2H4 combustion on RuO2 catalyst deposited (by thermal decomposition of RuCl3•xH2O) on the inside of the channels (2 mm ID) of a cylindrical YSZ monolith (2.2 cm dia. × 1 cm height, 37 channels) in a plug flow reactor assembly, reporting enhancement factor and faradaic efficiency values up to 1.5 and 90, respectively, even for high (36%) open-circuit C2H4 conversions [131]. A schematic of this bipolar design is shown in Fig. 2.17a [132].

In 2010 bipolar configurations were used for the first time to electropromote isolated metal nanoparticles [133,134,135]. Comninellis and coworkers demonstrated EPOC of CO oxidation (under high vacuum) on isolated Pt nanoparticles (60 nm) sputter-deposited on a rectangular YSZ pellet by applying in plane polarization between two Au electrodes in a bipolar configuration [133, 134]. They also used for the first time isotopic 18O2 as oxidant to accurately quantify EPOC in a bipolar system of non-percolated nanoparticles [134]. Vernoux and coworkers reported non-faradaic inhibiting effect on the rate of CH4 combustion on isolated Pd nanoparticles deposited (by electroless deposition) on the inner surface of an YSZ honeycomb cylindrical monolith (3.2 cm dia. × 3.2 cm height, with ca. 600 channels 1 mm × 1 mm), upon applying polarization between a continuous Pd film deposited in the center channel and a Au film painted on the outer surface of the monolith (Fig. 2.17b) [135]. Such monolithic bipolar designs are in principle promising as it concerns scale-up of EPOC reactors; however, there are several technical issues to be addressed, including the current bypass [132] and the limited thermal stability of the metal nanoparticles as they are not deposited on a porous substrate [2].
4.2 Monolithic Electropromoted Reactors (MEPR)
A big step towards practical application of EPOC was the development of the monolithic electropromoted reactor (MEPR), whose successful operation was demonstrated for the first time in 2004 in the reactions of C2H4 combustion and NOx reduction by C2H4 in the presence of O2, on Pt and Rh catalysts [136]. The MEP reactor can be considered as a hybrid between a planar solid oxide fuel cell (SOFC) and a monolithic honeycomb reactor [12, 136]. A schematic and a picture of the MEPR are shown in Fig. 2.18. The reactor consists of several (typically >10) parallel solid electrolyte plates (flat or ribbed) positioned in properly carved grooves on the opposite internal surfaces of a ceramic casing. Thin (ca. 20–40 nm) porous catalyst films are sputter-deposited on both sides of the plates, exposed to the same reaction mixture. Suitably painted metal films on the surfaces of the grooves create two current collectors, the one providing electrical contact among all catalyst films on the top sides of the plates and the other among all catalyst films on the bottom sides of them. The entire assembly is enclosed in a properly designed stainless steel gas manifolding casing.

The MEP reactor is a simple, compact, and efficient reactor for practical utilization of EPOC. It requires only two external electrical connections to polarize simultaneously the catalyst films on all plates (with opposite polarity the two films on the two sides of each plate) and one of the plates can be used as a gas sensor element, there is only a single gas stream to and from the reactor, as in classical catalytic reactors, it is easily assembled and dismantled, permitting easy replacement of the solid electrolyte plates whenever necessary, and it exhibits very good thermal and mechanical stability, which allows its use in harsh environments [12, 136]. Equally important, the use of thin sputtered metal electrodes with metal dispersions higher than 10%, i.e., comparable with those of supported commercial catalysts, allows overcoming a major economic obstacle for practical utilization of EPOC which is the very low dispersion (less than 0.01%) and hence the poor metal utilization of the typically 0.1–5 μm-thick catalyst films used in EPOC studies [12, 136]. However, the thermal stability of the sputtered thin metal films may be low for practical long-term operation [2]. Since 2004, MEP reactors with YSZ electrolyte have been successfully implemented for EPOC of C2H4 combustion [136, 138] and NO reduction by C2H4 in the presence of O2 on Pt and Rh [112, 136], including testing with simulated and real exhaust gas of a diesel engine [139], SO2 oxidation on Pt [140], CO2 hydrogenation on Rh, Pt, and Cu [118], and, more recently, CO2 hydrogenation on Ru [137, 141], at high hourly space velocities in all cases (up to ca. 3 × 104 h−1[140]).
4.3 Electrochemical Promotion of Highly Dispersed Catalysts
The electrochemical promotion of a highly dispersed metal catalyst was first reported in 1998 by Marwood and Vayenas [142] in the reaction of C2H4 combustion on finely dispersed Pt (dispersion higher than ca. 20%) in a porous Au film electrode deposited on YSZ electrolyte. This finding pointed the way to practical applications of EPOC as supported highly dispersed metal catalysts exhibit efficient metal utilization, which is particularly important in case of noble metal catalysts, and improved thermal stability. Since this first demonstration of EPOC with highly dispersed catalysts, two main approaches have been followed in this direction, specifically dispersion of the catalytically active phase in an electronically or ionically conducting matrix and dispersion of the catalytically active phase in a mixed ionic-electronic conductive matrix, as concisely described below. Relevant work up to 2017 is summarized in recent reviews [2, 8].
4.3.1 Electrochemical Promotion of Metal Nanoparticles Dispersed in an Ionically or Electronically Conducting Matrix
In these EPOC systems, the ionically or electronically conducting matrix with the dispersed metal nanoparticles is deposited as film on a dense solid electrolyte element. Metal nanoparticles dispersed in an ionic conductor can also be deposited on a porous interlayer metal film supported on the dense solid electrolyte. The ionic promoting species are electrocatalytically created at the boundary between the gas phase, the solid electrolyte, and the electronically conductive phase and then migrate (backspillover) to the catalytic sites. The main drawback of this design is the difficulty of the ionic species to backspillover via the conductive support from the solid electrolyte to the catalytic active sites at the surface of the nanoparticles. The particular approach has been recently applied in the following catalytic systems:
-
CO2 hydrogenation (to CH4 and CO) on Ni- and Ru-impregnated carbon nanofibers (CNFs) deposited on dense YSZ, in various configurations [143]. Negative polarization resulted in weak enhancement of the CO2 consumption rate and, for the Ni-based catalyst, of the selectivity to CH4, whereas the rate enhancement ratio for CH4 (electrophilic behavior) was up to 3. The observed weak electropromotion was attributed to the limited transport of O2− along the CNF support.
-
H2 production from CH3OH via partial oxidation and steam reforming on Pt nanoparticles dispersed on a diamond-like carbon (DLC) matrix deposited on dense K-β-Al2O3 [144]. Electrochemical pumping of potassium ions to the catalyst resulted in increase of the H2 production rate up to 2.5 and 3.4 times in partial oxidation and in steam reforming of CH3OH, respectively, under optimal coverage of the potassium promoter.
-
CH4 combustion on Pd dispersed (up to 27% dispersion) on a porous interlayer YSZ film deposited on dense YSZ [145], aiming to a more intimate contact between YSZ and the palladium phases under reaction conditions. Electrophobic electrochemical promotion with faradaic efficiencies ranging from 8 to 61, depending on the CH4/O2 ratio, was reported. It is noted that CH4 combustion has been also studied on Pd dispersed on a porous interlayer CeO2 film deposited on dense YSZ [146]. This system exhibited high catalytic activity but no electropromotion was achieved.
-
CO2 hydrogenation (to CH4 and CO) on nanodispersed Ru-Co in BZY which was deposited on a porous interlayer Ru film supported on dense BZY (BaZr0.85Y0.15O3 + 1 wt.% NiO) proton conductor (Fig. 2.19a) [111]. This was the first study exploring the similarities between EPOC and MSI for the case of a H+ conducting support (BZY) and the first EPOC study of a dispersed Ru-Co catalyst. Anodic polarization (proton removal from the catalyst) resulted in reversible non-faradaic increase of the methanation rate (electrophobic behavior), with faradaic efficiency values up to 65, and in parallel decrease of the CO production rate (electrophilic behavior). The selectivity to CH4 varied between 16% and 41% with varying catalyst potential and dispersed Ru-Co/BZY loading (Fig. 2.19b). An important conclusion of this study was that both the Ru film and the Ru-Co nanoparticles were electropromoted, which implied that the polarization-imposed work function change on the Ru film is also imposed to a large extent on the metal nanoparticles of the dispersed Ru-Co/BZY catalyst.

CO2 hydrogenation on Ru-Co nanodispersed in BZY powder and deposited on a Ru film supported on a dense BZY disk: (a) Schematic of the BZY support disk and of the three electrodes. (b) Effect of catalyst potential UWR and dispersed Ru-Co/BZY catalyst loading, mRu-Co/BZY, on the selectivity to CH4 and CO. \( {P}_{H_2} \) = 7 kPa, T = 450 °C, total flowrate Ft = 200 cm3/min. (Reprinted (adapted) from ref. [111], with permission from Elsevier)
-
CO2 hydrogenation (to CH4 and CO) on nanodispersed Ru in YSZ (2 wt.% Ru/YSZ) which was deposited on a porous interlayer Ru film supported on dense YSZ [147]. Small non-faradaic enhancement of the CH4 production rate with parallel decrease in the CO production rate was observed upon positive polarization (O2− pumping to the catalyst). The weak electrochemical promotion was attributed to the fact that the catalyst was in a promoted state due to thermally induced backspillover of O2− from the YSZ support.
4.3.2 Electrochemical Promotion of Metal Nanoparticles Dispersed in a Mixed Ionic-Electronic Conducting Matrix
The idea behind this approach is that mobility of both electrons and ions in the catalytic layer can yield significant electrochemical promotion of the dispersed catalytic nanoparticles [2]. The particular approach has been recently applied in the following catalytic systems, adopting different strategies:
-
H2 production from steam reforming, partial oxidation, and autothermal steam reforming of CH4, at 500 °C, on a composite electrode Pt-Pt/YSZ electrode deposited on a dense Na-β-Al2O3 electrolyte disk, using an ink prepared by mixing a commercial Pt paste with powder of a dispersed 3 wt. % Pt on YSZ catalyst [148]. Positive polarization resulted in increase of H2 production, as chemisorption of CHx species was enhanced, but also to deactivation due to carbon deposition. However, in situ regeneration of the catalytic activity was possible, in particular under autothermal reforming conditions, by applying negative polarization, i.e., by supplying Na+ to the catalyst, due to the induced increase in the coverage of H2O and oxygen (electron acceptors) responsible for the deposited carbon removal. This behavior, which is shown in Fig. 2.20, demonstrated the possibility of cyclic operation between electropromoted H2 production from CH4 at relatively low temperature (500 °C) and in situ regeneration of the catalyst, under fixed reaction conditions.
Fig. 2.20 
Autothermal steam reforming of CH4 on a composite Pt-Pt/YSZ electrode deposited on Na-β-Al2O3: Influence of the applied cell potential (two-electrode set-up) on H2 production rate during the reproducibility experiment. T = 500 °C. Feed composition: CH4/H2O/O2: 1%/4%/0.2% in N2. Total flow rate: Ft = 6 L/h. (Reprinted (adapted) from ref. [148], with permission from Elsevier)
-
Propane combustion on Pt nanoparticles (3–20 nm) dispersed (~15% dispersion) on a porous LSCF/GDC (La0.6Sr0.4Co0.2Fe0.8O3–δ /Ce0.9Gd0.1O1.95) film (7.5 μm thick) deposited on dense GDC [149]. Electrophobic EPOC behavior was observed corresponding to increase of propane conversion up to 38% and apparent faradaic efficiency up to 85. This study demonstrated the ability to electropromote metal nanoparticles dispersed in a porous mixed ionic-electronic conducting (MIEC) electrode (LSCF/GDC), its ionic conductivity ensuring the transport of the ionic oxygen promoting species from the solid electrolyte (GDC) to the nanoparticles.
-
C2H4 combustion on Ru nanoparticles (1.1 nm) dispersed on CeO2 (a mixed electronic – O2−conductor) with the 1 wt.% Ru/CeO2 electrode deposited on dense YSZ, using a suspension of Ru/CeO2 powder in ethanol [150]. Electrophilic EPOC behavior was observed with up to 2.5 times increase in the catalytic rate and absolute values of faradaic efficiency up to 96. The increase of the C2H4 combustion rate with negative polarization was associated with partial reduction of CeO2 and with formation of a stronger metal-support interaction (MSI) between the Ru nanoparticles and the partially reduced CeO2-x, i.e., with in situ electrochemical enhancement of the MSI effect.
-
Selective partial oxidation of methanol on Au nanoparticles (2–10 nm) dispersed in an ultrafine grained YSZ matrix, with the composite Au/YSZ catalyst-electrode deposited on dense K-β-Al2O3 [151]. The Au/YSZ catalyst film, which was highly selective towards HCOOCH3 formation and stable under reaction conditions, was prepared by reactive co-sputtering of Au and Zr-Y targets. This novel preparation method resulted in Au nanoparticles confined in the YSZ matrix but in contact with the reacting mixture [2, 151]. At 280 °C, electrochemical pumping of K+ ions to the catalyst (negative polarization) resulted in enhancement of the H2 and HCOOCH3 rates by more than 9 and 5 times, respectively (electrophilic EPOC behavior), for a potassium coverage of ca. 0.5, which demonstrated the suitability of the employed configuration for electropromotion by potassium of a highly dispersed Au catalyst.
-
CH4 combustion on nanodispersed Pd supported on porous Co3O4 (5 wt.% Pd/Co3O4) with the Pd/Co3O4 electrode deposited on dense YSZ [152]. This was the first EPOC study where the catalyst consisted of metal nanoparticles supported on a semiconductor (i.e., Co3O4), the latter acting both as an electronic conductor and as a pathway for the oxygen ions to reach the Pd nanoparticles. Positive current application under reducing conditions resulted in increase of the CO2 production rate by up to 2.5 times, with faradaic efficiency up to 80.
-
CO2 hydrogenation (to CH4 and, mainly, to CO) on nanodispersed Ru (0.7–1 nm) supported on porous Co3O4 (2 wt.% Ru/Co3O4) with the Ru/Co3O4 catalyst-electrode deposited on dense BZY (BaZr0.85Y0.15O3 + 1 wt.% NiO) proton conductor [110]. Weak electrophilic EPOC behavior of the CO production rate was observed under oxidizing conditions. The low rate enhancement ratios were explained considering that the catalyst was already in a promoted state due to enhanced MSI effect (“wireless” EPOC) [103].

4.4 Wireless Self-Driven and Self-Sustained Electrochemical Promotion
In 1993, Cavalca et al. [153], in their study of electrochemical promotion of CH3OH oxidation on Pt/YSZ in a single chamber flow reactor, demonstrated for the first time that it is possible to induce electrochemical promotion in an oxidation reaction applying not an external polarization but utilizing the potential difference between the active Pt catalyst-electrode and a more inert Au counter electrode, which results from the reaction-induced lower oxygen activity on the Pt catalyst. The observed electrophobic self-driven “wireless” electrochemical promotion (wireless NEMCA), realized by short-circuiting the Pt catalyst and the Ag counter electrode, was explained by the continuous supply of promoting ionic oxygen species from the YSZ support to the Pt surface, while the spent O2− were continuously replenished by oxygen from the gas phase.
Almost 15 years later, the concept of wireless EPOC was realized by Poulidi et al. in C2H4 combustion over a porous Pt catalyst film deposited on one side of a dense pellet of a mixed ionic-electronic conductor (MIEC) in a dual chamber reactor consisting of the reaction side and a sweep side where a second Pt electrode deposited on the other side of the MIEC pellet was exposed to a sweep gas [154, 155]. In this configuration, the driving force for migration of the promoting species is the chemical potential difference across the MIEC generated by using an appropriate sweep gas, while the mixed conductivity eliminates the need for an external circuit by internally short-circuiting the system. Both La0.6Sr0.4Co0.2Fe0.8O3-δ (a mixed oxygen ion-electronic conductor) [154, 156] and Sr0.97Ce0.9Yb0.1O3-δ (a mixed protonic-electronic conductor) [155] have been used as MIEC supports, while moderate rate increases, compared to symmetric operation, were induced via exposure of the Pt electrode in the sweep side to an O2 and a H2 atmosphere, respectively. Wireless electrochemical promotion in a similar dual chamber set-up was reported recently for CO oxidation on Pt supported on BaCe0.6Zr0.2Y0.2O3-δ, a mixed protonic-electronic conductor at temperatures up to 650 °C, with a maximum rate increase of 10% observed upon introduction of H2/He flow in the sweep side [157]. Wireless EPOC has been also investigated in C2H4 combustion over Pt supported on a La0.6Sr0.4Co0.2Fe0.8O3-δ hollow fiber, where the use of this effect for in situ regeneration of the Pt catalyst was demonstrated [158].
A more recent and more promising concept for practical application of EPOC is the self-sustained electrochemical promotion (SSEP) of the catalytic activity of metal nanoparticles dispersed on ionic (O2− conducting) or MIEC supports, due to migration of ionic oxygen species from the support to the surface of the nanoparticles without applying any external polarization, even at temperatures lower than 200 °C [2, 8, 128]. The self-sustained EPOC mechanism is based on the functional similarity of EPOC and metal-support interactions (MSI) [1, 81, 102, 103], merging EPOC and MSI at the nanometric scale [8]. As summarized in recent reviews [2, 5, 8, 80, 128], the self-sustained EPOC has been studied in several catalytic systems, including propane [159, 160], toluene [161], and CO [162, 163] oxidation on Pt nanoparticles dispersed on YSZ, ethylene combustion on Pt and Ru nanoparticles dispersed on YSZ and samarium-doped ceria (SDC) [164], and ethylene combustion on Pt, PtSn, Ru, and Ir nanoparticles dispersed on CeO2 in the presence and absence of oxygen in the feed [165, 166]. These studies have elucidated the role of the ionic species in the support on the catalytic mechanism and on the promotion of the dispersed nanoparticles. Migration of ionic species from the support (backspillover) may be self-induced thermally [2, 8, 159, 163, 164] and/or by differences in the work function between the support and the metal, which affects their interaction [2, 165, 166]. In general, the driving force for migration will depend on the ionic conductivity and oxygen storage capacity of the support (e.g., for CeO2), the temperature, and the metal-support interface at the nanoscale [2, 8, 128, 166]. It is also noted that in the absence of O2 in the gas phase, Pt nanoparticles dispersed on YSZ or MIEC (SDC and CeO2) have been found much more active for CO and C2H4 combustion than Pt nanoparticles dispersed on supports not exhibiting ionic conductivity (e.g., Al2O3) [128, 164, 167]. To explain this behavior, which was observed for nanoparticles less than 5 nm implying the necessity of strong interaction with the support, Baranova and coworkers proposed the mechanism of nano-galvanic cells, according to which Pt/YSZ or Pt/MIEC interfaces act as local nano-galvanic cells where CO and C2H4 are electrochemically oxidized by O2− from the oxide support, which is reduced [167]. Thermally induced backspillover of oxygen ions to metal nanoparticles and creation of an effective double layer, accompanied by trapping of diffusing atomic metal species by the resulting surface oxygen vacancies, has been also proposed to explain the increased resistance to sintering of Ir [168] and Rh [169] nanoparticles dispersed on supports with high oxygen ion lability.
Self-sustained electrochemical promotion catalysts can also be synthesized by combining in the catalyst composition microparticles or nanoparticles of two materials with different catalytic and electrocatalytic properties which act as cathodes and anodes of microscopic galvanic cells formed if the two materials are dispersed either on a MIEC conductor or on a pure ionic conductor with an electronic conductor also added for the transfer of electrons [2]. When such a catalyst based on a mixed oxygen ion-electron conductor is used in an oxidation reaction, oxygen species generated at the cathodes of the microscopic galvanic cells via reduction of gas phase oxygen are transferred to the anodes to promote, as sacrificial promoters, the catalytic oxidation reaction on their surface, while in parallel electrons are transferred from the anodes to the cathodes. Based on this concept, Zhou and coworkers have synthesized SSEP catalysts composed of La0.9Sr0.1MnO3 as cathode, Ni-Cu-CeO2 as anode, YSZ as oxygen ion conductor, and Cu or Ni-Cu as electron conductor [170, 171]. These SSEP catalysts demonstrated improved performance for partial oxidation reforming (POXR) of n-C15H32 at 450–650 °C [170] and CH4 at 350–650 °C [171], compared both to commercial catalysts and to catalysts lacking one or more of the components of the SSEP catalyst. More recently, Vernoux and coworkers used the same concept to develop NOx storage/reduction (NSR) catalysts which exhibited improved NOx removal efficiency in synthetic diesel exhaust gas conditions [2, 172]. The NSR catalysts were developed by dispersing Pt and Rh nanoparticles on YSZ or gadolinium doped ceria (GDC) nanoparticles, to form nanometric electrodes (Pt anode and Rh cathode nano-electrodes), and then wash-coating the powder in the porosity of N-doped (electron conducting) and non-doped (insulating) SiC mini-DPFs (diesel particulate filters), respectively. A schematic illustration of these novel electrochemically assisted catalysts is shown in Fig. 2.21. The best performance was obtained with the GDC (mixed oxygen ion-electron conductor) as support of the metallic nanoparticles, whereas the reaction products analysis revealed that electrocatalytic reactions can occur on the nano-electrodes alongside the catalytic reactions.

Schematic illustration of the electrochemically-assisted NSR catalysts, with the metal nanoparticles dispersed (a) on an O2− ion conducting support (e.g., YSZ) deposited on an electronic conductor (e.g., N-doped SiC-DPF) and (b) on a mixed conducting (ionic and electronic) support (e.g., GDC). (Reprinted from ref. [172], with permission from Elsevier)
5 Conclusions
After more than 40 years, electrochemical promotion of catalysis (EPOC) remains the focus of intensive research, which highlights the scientific importance of this effect and its potential for practical applications. The physicochemical origin of EPOC has been validated via application of numerous techniques and experimental approaches, in particular for O2− and alkali ion conductors. Nevertheless, there are open research questions yet to be explored. The functional similarity of electrochemical promotion, classical promotion, and metal-support interactions has been conclusively established. Rules of promotion applying both to EPOC and classical catalysis have been derived, which allow promoter selection based on unpromoted kinetics. Current research in EPOC focuses on catalytic systems of technological and environmental importance, such as CO2 hydrogenation to value-added products or H2 production by reforming with simultaneous reversible storage, where EPOC could offer a competitive advantage, mainly aiming to electrochemical promotion of non-noble metals and nanodispersed metal/metal oxide catalysts. Development and scale-up of electropromoted reactors remains a priority, following two main directions: (a) improvement of MEPR design (durability, useful lifetime, electrolyte and stack cost minimization, scale-up) and use of nanodispersed catalysts, and (b) development of novel catalytic systems based on self-sustained EPOC. Coupling EPOC with electrolysis is also of strategic future importance as it can potentially reduce the electrolysis cost improving electrochemical technologies that are mature enough and easy to scale up.
References
Vayenas CG, Bebelis S, Pliangos C, Brosda S, Tsiplakides D (2001) Electrochemical activation of catalysis. Kluwer Academic/Plenum Publishers, New York
Vernoux P (2017) Recent advances in electrochemical promotion of catalysis. In: Spivey J, Han Y-F (eds) Catalysis. The Royal Society of Chemistry, London, pp 29–59
Stoukides M, Vayenas CG (1981) The effect of electrochemical oxygen pumping on the rate and selectivity of ethylene oxidation on polycrystalline silver. J Catal 70(1):137–146. https://doi.org/10.1016/0021-9517(81)90323-7
Vayenas CG, Bebelis S, Neophytides S (1988) Non-faradaic electrochemical modification of catalytic activity. J Phys Chem 92(18):5083–5085. https://doi.org/10.1021/j100329a007
Caravaca A, González-Cobos J, Vernoux P (2020) A discussion on the unique features of electrochemical promotion of catalysis (EPOC): are we in the right path towards commercial implementation? Catalysts 10(11):1276 (29 pages). https://doi.org/10.3390/catal10111276
González-Cobos J, de Lucas-Consuegra A (2016) A review of surface analysis techniques for the investigation of the phenomenon of electrochemical promotion of catalysis with alkaline ionic conductors. Catalysts 6(1):15 (13 pages). https://doi.org/10.3390/catal6010015
de Lucas-Consuegra A (2014) New trends of alkali promotion in heterogeneous catalysis: electrochemical promotion with alkaline ionic conductors. Catal Surv Asia 19(1):25–37. https://doi.org/10.1007/s10563-014-9179-6
Vernoux P, Lizarraga L, Tsampas MN, Sapountzi FM, De Lucas-Consuegra A, Valverde J-L, Souentie S, Vayenas CG, Tsiplakides D, Balomenou S, Baranova EA (2013) Ionically conducting ceramics as active catalyst supports. Chem Rev 113(10):8192–8260. https://doi.org/10.1021/cr4000336
Tang X, Xu X, Yi H, Chen C, Wang C (2013) Recent developments of electrochemical promotion of catalysis in the techniques of DeNOx. Sci World J:Article ID 463160 (13 pages). https://doi.org/10.1155/2013/463160
Vayenas CG (2011) Bridging electrochemistry and heterogeneous catalysis. J Solid State Electrochem 15(7–8):1425–1435. https://doi.org/10.1007/s10008-011-1336-5
Katsaounis A (2009) Recent developments and trends in the electrochemical promotion of catalysis (EPOC). J Appl Electrochem 40(5):885–902. https://doi.org/10.1007/s10800-009-9938-7
Tsiplakides D, Balomenou S (2009) Milestones and perspectives in electrochemically promoted catalysis. Catal Today 146(3–4):312–318. https://doi.org/10.1016/j.cattod.2009.05.015
Vayenas CG, Koutsodontis CG (2008) Non-faradaic electrochemical activation of catalysis. J. Chem. Phys 128:Article ID: 182506. (13 pages). https://doi.org/10.1063/1.2824944
Lambert RM (2003) Electrochemical and chemical promotion by alkalis with metal films and nanoparticles. In: Wieckowski A, Savinova ER, Vayenas CG (eds) Catalysis and electrocatalysis at nanoparticles surfaces, Ch. 16. Marcel Dekker, Inc., New York, pp 583–611
Vayenas CG, Katsaounis A, Brosda S, Hammad A (2008) Electrochemical modification of catalytic activity. In: Ertl G, Knözinger H, Schüth F, Weitkamp J (eds) Handbook of heterogeneous catalysis. Wiley-VCH, Weinheim, pp 1905–1935
Stoukides M, Vayenas CG (1982) Transient and steady-state vapor phase electrocatalytic ethylene epoxidation. In: Bell AT, Hegedus LL (eds) Catalysis under transient conditions. ACS Symp. Series 178(8), American Chemical Society, Washington, DC, pp 181–208. https://doi.org/10.1021/bk-1982-0178.ch008
Bebelis S, Vayenas CG (1992) Non-faradaic electrochemical modification of catalytic activity: 6. The epoxidation of ethylene on Ag/ZrO2 (8mol%Y2O3). J Catal 138(2):588–610. https://doi.org/10.1016/0021-9517(92)90309-6
Stoukides M, Vayenas CG (1984) Electrocatalytic rate enhancement of propylene epoxidation on porous silver electrodes using a zirconia oxygen pump. J Electrochem Soc 131(4):839–845. https://doi.org/10.1149/1.2115710
Bebelis S, Vayenas CG (1989) Non-faradaic electrochemical modification of catalytic activity: 1. The case of ethylene oxidation on Pt. J Catal 118(1):125–146. https://doi.org/10.1016/0021-9517(89)90306-0
Yentekakis IV, Vayenas CG (1988) The effect of electrochemical oxygen pumping on the steady-state and oscillatory behavior of CO oxidation on polycrystalline Pt. J Catal 111(1):170–188. https://doi.org/10.1016/0021-9517(88)90075-9
Vayenas CG, Neophytides S (1991) Non-faradaic electrochemical modification of catalytic activity: III. The case of methanol oxidation on Pt. J Catal 127(2):645–664. https://doi.org/10.1016/0021-9517(91)90189-B
Neophytides S, Vayenas CG (1989) Non-faradaic electrochemical modification of catalytic activity: 2. The case of methanol dehydrogenation and decomposition on Ag. J Catal 118:147–163. https://doi.org/10.1016/0021-9517(89)90307-2
Vayenas CG, Bebelis S, Yentekakis IV, Lintz H-G (1992) Non-faradaic electrochemical modification of catalytic activity: a status report. Catal Today 11(3):303–438. https://doi.org/10.1016/0920-5861(92)80002-5
Vayenas CG, Bebelis S, Ladas S (1990) Dependence of catalytic rates on catalyst work function. Nature 343(6259):625–627. https://doi.org/10.1038/343625a0
Ladas S, Bebelis S, Vayenas CG (1991) Work function measurements on catalyst films subject to in situ electrochemical promotion. Surf Sci 251-252:1062–1068. https://doi.org/10.1016/0039-6028(91)91151-M
Vayenas CG, Bebelis S, Despotopoulou M (1991) Non-faradaic electrochemical modification of catalytic activity: 4. The use of β″-Al2O3 as the solid electrolyte. J Catal 128(2):415–435. https://doi.org/10.1016/0021-9517(91)90300-S
Tsiakaras P, Vayenas CG (1993) Non-faradaic electrochemical modification of catalytic activity: VII. The case of methane oxidation on Pt. J Catal 140(1):53–70. https://doi.org/10.1006/jcat.1993.1068
Bebelis S, Karasali H, Vayenas CG (2008) Electrochemical promotion of the CO2 hydrogenation on Pd/YSZ and Pd/β″-Al2O3 catalyst-electrodes. Solid State Ionics 179(27–32):1391–1395. https://doi.org/10.1016/j.ssi.2008.02.043
Bebelis S, Karasali H, Vayenas CG (2008) Electrochemical promotion of CO2 hydrogenation on Rh/YSZ electrodes. J Appl Electrochem 38(8):1127–1133. https://doi.org/10.1007/s10800-008-9574-7
Makri M, Buekenhoudt A, Luyten J, Vayenas CG (1996) Non-faradaic electrochemical modification of the catalytic activity of Pt using a CaZr0.9In0.1O3-α proton conductor. Ionics 2:282–288. https://doi.org/10.1007/BF02376035
Vayenas CG, Jaksic MM, Bebelis SI, Neophytides SG (1996) The electrochemical activation of catalytic reactions. In: Bockris JO’M, Conway BE, White RE (eds) Modern aspects of electrochemistry. Springer, Boston, pp 57–202
Tsiplakides D, Vayenas CG (2001) Electrode work function and absolute potential scale in solid-state electrochemistry. J Electrochem Soc 148(5):E189–E202. https://doi.org/10.1149/1.1362547
Vayenas CG, Neophytides SG (1996) Electrochemical activation of catalysis: In situ controlled promotion of catalyst surfaces. In: Spivey JJ (ed) Catalysis, vol 12. The Royal Society of Chemistry, Cambridge, pp 199–253
Pliangos C, Raptis C, Badas T, Vayenas CG (2000) Electrochemical promotion of NO reduction by C3H6 on Rh/YSZ catalyst-electrodes. Solid State Ionics 136-137:767–773. https://doi.org/10.1016/S0167-2738(00)00549-X
Yentekakis IV, Jiang Y, Neophytides S, Bebelis S, Vayenas CG (1995) Catalysis, electrocatalysis and electrochemical promotion of the steam reforming of methane over Ni film and Ni-YSZ cermet anodes. Ionics 1:491–498. https://doi.org/10.1007/BF02375296
Marwood M, Kaloyannis A, Vayenas CG (1996) Electrochemical promotion of the NO reduction by C2H4 on Pt/YSZ and by CO on Pd/YSZ. Ionics 2:302–311. https://doi.org/10.1007/BF02376038
Nicole J, Tsiplakides D, Wodiunig S, Comninellis C (1997) Activation of catalyst for gas-phase combustion by electrochemical pretreatment. J Electrochem Soc 144(12):L312–L314. https://doi.org/10.1149/1.1838143
Wodiunig S, Patsis V, Comninellis C (2000) Electrochemical promotion of RuO2-catalysts for the gas phase combustion of C2H4. Solid State Ionics 136-137:813–817. https://doi.org/10.1016/S0167-2738(00)00505-1
Falgairette C, Jaccoud A, Fóti G, Comninellis C (2008) The phenomenon of “permanent” electrochemical promotion of catalysis (P-EPOC). J Appl Electrochem 38(8):1075–1082. https://doi.org/10.1007/s10800-008-9554-y
Fóti G, Lavanchy O, Comninellis C (2000) Electrochemical promotion of Rh catalyst in gas-phase reduction of NO by propylene. J Appl Electrochem 30:1223–1228. https://doi.org/10.1023/A:1026505829359
Pliangos C, Raptis C, Badas T, Vayenas CG (2000) Electrochemical promotion of NO reduction by C3H6 and CO on Rh/YSZ catalyst - electrodes. Ionics 6:119–126. https://doi.org/10.1007/BF02375555
Souentie S, Xia C, Falgairette C, Li YD, Comninellis C (2010) Investigation of the “permanent” electrochemical promotion of catalysis (P-EPOC) by electrochemical mass spectrometry (EMS) measurements. Electrochem Commun 12(2):323–326. https://doi.org/10.1016/j.elecom.2009.12.031
Harkness IR, Hardacre C, Lambert RM, Yentekakis IV, Vayenas CG (1996) Ethylene oxidation over platinum: In situ electrochemically controlled promotion using Na–β″ alumina and studies with a Pt(111)/Na model catalyst. J Catal 160(1):19–26. https://doi.org/10.1006/jcat.1996.0119
Yentekakis IV, Bebelis S (1992) Study of the NEMCA effect in a single-pellet catalytic reactor. J Catal 137(1):278–283. https://doi.org/10.1016/0021-9517(92)90157-D
Petrolekas PD, Brosda S, Vayenas CG (1998) Electrochemical promotion of Pt catalyst electrodes deposited on Na3Zr2Si2PO12 during ethylene oxidation. Electrochem Soc 145(5):1469–1477. https://doi.org/10.1149/1.1838506
Yentekakis IV, Moggridge G, Vayenas CG, Lambert RM (1994) In situ controlled promotion of catalyst surfaces via NEMCA: the effect of Na on the Pt-catalyzed CO oxidation. J Catal 146(1):292–305. https://doi.org/10.1016/0021-9517(94)90033-7
Karavasilis C, Bebelis S, Vayenas CG (1996) In situ controlled promotion of catalyst surfaces via NEMCA: the effect of Na on the Ag-catalyzed ethylene epoxidation in the presence of chlorine moderators. J Catal 160(2):205–213. https://doi.org/10.1006/jcat.1996.0139
Harkness IR, Lambert RM (1995) Electrochemical promotion of the NO + ethylene reaction over platinum. J Catal 152(1):211–214. https://doi.org/10.1006/jcat.1995.1075
Palermo A, Lambert RM, Harkness IR, Yentekakis IV, Marina O, Vayenas CG (1996) Electrochemical promotion by Na of the platinum-catalyzed reaction between CO and NO. J Catal 161(1):471–479. https://doi.org/10.1006/jcat.1996.0206
Marina OA, Yentekakis IV, Vayenas CG, Palermo A, Lambert RM (1997) In situ controlled promotion of catalyst surfaces via NEMCA: the effect of Na on the Pt-catalyzed NO reduction by H2. J Catal 166(2):218–228. https://doi.org/10.1006/jcat.1997.1551
Yentekakis IV, Palermo A, Filkin NC, Tikhov MS, Lambert RM (1997) In situ electrochemical promotion by sodium of the platinum-catalyzed reduction of NO by propene. J Phys Chem B 101(19):3759–3768. https://doi.org/10.1021/jp963052c
Williams FJ, Palermo A, Tikhov MS, Lambert RM (2001) Mechanism of alkali promotion in heterogeneous catalysis under realistic conditions: application of electron spectroscopy and electrochemical promotion to the reduction of NO by CO and by propene over rhodium. Surf Sci 482-485(Part 1):177–182. https://doi.org/10.1016/S0039-6028(00)01040-2
Williams FJ, Palermo A, Tikhov MS, Lambert RM (2000) Electrochemical promotion by sodium of the rhodium-catalyzed NO + CO reaction. J Phys Chem B 104(50):11883–11890. https://doi.org/10.1021/jp001689x
Williams FJ, Palermo A, Tikhov MS, Lambert RM (2001) Electrochemical promotion by sodium of the rhodium-catalyzed reduction of NO by propene: kinetics and spectroscopy. J Phys Chem B 105(7):1381–1388. https://doi.org/10.1021/jp003269d
Lambert RM, Williams F, Palermo A, Tikhov MS (2000) Modelling alkali promotion in heterogeneous catalysis: in situ electrochemical control of catalytic reactions. Top Catal 13:91–98. https://doi.org/10.1023/A:1009076720641
Cavalca CA, Haller GL (1998) Solid electrolytes as active catalyst supports: electrochemical modification of benzene hydrogenation activity on Pt/β″(Na)Al2O3. J Catal 177(2):389–395. https://doi.org/10.1006/jcat.1998.2060
Giannikos A, Petrolekas P, Pliangos C, Frenzel A, Vayenas CG, Pütter H (1998) Electrochemical promotion of Pd for the hydrogenation of C2H2. Ionics 4:161–169. https://doi.org/10.1007/BF02375941
Tracey S, Palermo A, Vazquez JPH, Lambert RM (1998) In situ electrochemical promotion by sodium of the selective hydrogenation of acetylene over platinum. J Catal 179(1):231–240. https://doi.org/10.1006/jcat.1998.2179
Williams FJ, Palermo A, Tracey S, Tikhov MS, Lambert RM (2002) Electrochemical promotion by potassium of the selective hydrogenation of acetylene on platinum: reaction studies and XP spectroscopy. J Phys Chem B 106(22):5668–5672. https://doi.org/10.1021/jp0203954
Karasali H (1994) PhD thesis, Department of Chemical Engineering. University of Patras, Greece
Williams FJ, Lambert RM (2000) A study of sodium promotion in Fischer–Tropsch synthesis: electrochemical control of a ruthenium model catalyst. Catal Lett 70:9–14. https://doi.org/10.1023/A:1019023418300
Urquhart AJ, Keel JM, Williams FJ, Lambert RM (2003) Electrochemical promotion by potassium of rhodium-catalyzed Fischer-Tropsch synthesis: XP spectroscopy and reaction studies. J Phys Chem B 107(38):10591–10597. https://doi.org/10.1021/jp035436q
González-Cobos J, Valverde JL, de Lucas-Consuegra A (2017) Electrochemical vs. chemical promotion in the H2 production catalytic reactions. Int J Hydrog Energy 42(19):13712–13723. https://doi.org/10.1016/j.ijhydene.2017.03.085
Yentekakis IV, Vernoux P, Goula G, Caravaca A (2019) Electropositive promotion by alkalis or alkaline earths of Pt-group metals in emissions control catalysis: a status report. Catalysts 9(2):157 (74 pages). https://doi.org/10.3390/catal9020157
de Lucas-Consuegra A, Caravaca A, Sánchez P, Dorado F, Valverde JL (2008) A new improvement of catalysis by solid-state electrochemistry: an electrochemically assisted NOx storage/reduction catalyst. J Catal 259(1):54–65. https://doi.org/10.1016/j.jcat.2008.07.008
González-Cobos J, Rico VJ, González-Elipe AR, Valverde JL, de Lucas-Consuegra A (2016) Electrocatalytic system for the simultaneous hydrogen production and storage from methanol. ACS Catal 6(3):1942–1951. https://doi.org/10.1021/acscatal.5b02844
Politova TI, Sobyanin VA, Belyaev VD (1990) Ethylene hydrogenation in electrochemical-cell with solid proton-conducting electrolyte. React Kinet Catal Lett 41(2):321–326. https://doi.org/10.1007/BF02097888
Chiang PC, Eng D, Stoukides M (1993) Solid electrolyte aided direct coupling of methane. J Catal 139(2):683–687. https://doi.org/10.1006/jcat.1993.1060
Yiokari CG, Pitselis GE, Polydoros DG, Katsaounis AD, Vayenas CG (2000) High pressure electrochemical promotion of ammonia synthesis over an industrial iron catalyst. J Phys Chem A 104(46):10600–10602. https://doi.org/10.1021/jp002236v
Ouzounidou M, Skodra A, Kokkofitis C, Stoukides M (2007) Catalytic and electrocatalytic synthesis of NH3 in a H+ conducting cell by using an industrial Fe catalyst. Solid State Ionics 178(1–2):153–159. https://doi.org/10.1016/j.ssi.2006.11.019
Tsiplakides D, Neophytides SG, Enea O, Jaksic MM, Vayenas CG (1997) Non-faradaic electrochemical modification of catalytic activity (NEMCA) of Pt black electrodes deposited on Nafion 117 solid polymer electrolyte. J Electrochem Soc 144(6):2072–2088. https://doi.org/10.1149/1.1837744
Ploense L, Salazar M, Gurau B, Smotkin ES (1997) Proton spillover promoted isomerization of n-butylenes on Pt-black cathodes/Nafion 117. J Am Chem Soc 119(47):11550–11551. https://doi.org/10.1021/ja9728841
Ploense L, Salazar M, Gurau B, Smotkin ES (2000) Spectroscopic study of NEMCA promoted alkene isomerizations at PEM fuel cell Pd-Nafion cathodes. Solid State Ionics 136-137:713–720. https://doi.org/10.1016/S0167-2738(00)00567-1
Williams FJ, Palermo A, Holgado HP, Lambert RM (2002) First demonstration of in situ electrochemical control of the composition and performance of an alloy catalyst during reaction. J Catal 210(2):237–240. https://doi.org/10.1006/jcat.2002.3713
Palermo A, Williams FJ, Lambert RM (2002) In situ control of the composition and performance of a bimetallic alloy catalyst: the selective hydrogenation of acetylene over Pt/Pb. J Phys Chem B 106(39):10215–10219. https://doi.org/10.1021/jp021296t
Anastasijevic NA, Baltruschat H, Heitbaum J (1993) On the hydrogen evolution during the electrochemical oxidation of aldehydes at Ib metals. Electrochim Acta 38(8):1067–1072. https://doi.org/10.1016/0013-4686(93)80214-K
Neophytides S, Tsiplakides D, Stonehart P, Jaksic M, Vayenas CG (1994) Electrochemical enhancement of a catalytic reaction in aqueous solution. Nature 370:45–47. https://doi.org/10.1038/370045a0
Neophytides S, Tsiplakides D, Stonehart P, Jaksic M, Vayenas CG (1996) Non-faradaic electrochemical modification of the catalytic activity of Pt for H2 oxidation in aqueous alkaline media. J Phys Chem 100(35):14803–14814. https://doi.org/10.1021/jp960971u
Hajar YM, Treps L, Michel C, Baranova EA, Steinmann SN (2019) Theoretical insight into the origin of the electrochemical promotion of ethylene oxidation on ruthenium oxide. Cat Sci Technol 9(21):5915–5926. https://doi.org/10.1039/C9CY01421G
Tsampas MN, Sapountzi FM, Vernoux P (2015) Applications of yttria stabilized zirconia (YSZ) in catalysis. Cat Sci Technol 5(11):4884–4900. https://doi.org/10.1039/C5CY00739A
Vayenas CG (2013) Promotion, electrochemical promotion and metal–support interactions: their common features. Catal Letters 143(11):1085–1097. https://doi.org/10.1007/s10562-013-1128-x
Ladas S, Kennou S, Bebelis S, Vayenas CG (1993) Origin of non-faradaic electrochemical modification of catalytic activity. J Phys Chem 97(35):8845–8848. https://doi.org/10.1021/j100137a004
Neophytides SG, Vayenas CG (1995) TPD and cyclic voltammetric investigation of the origin of electrochemical promotion in catalysis. J Phys Chem 99(47):17063–17067. https://doi.org/10.1021/j100047a001
Neophytides SG, Tsiplakides D, Vayenas CG (1998) Temperature-programmed desorption of oxygen from Pt films interfaced with Y2O3-doped ZrO2. J Catal 178(2):414–428. https://doi.org/10.1006/jcat.1998.2155
Katsaounis A, Nikopoulou Z, Verykios XE, Vayenas CG (2004) Comparative isotope-aided investigation of electrochemical promotion and metal–support interactions 1. 18O2 TPD of electropromoted Pt films deposited on YSZ and of dispersed Pt/YSZ catalysts. J Catal 222(1):192–206. https://doi.org/10.1016/j.jcat.2003.10.010
Katsaounis A, Nikopoulou Z, Verykios XE, Vayenas CC (2004) Comparative isotope-aided investigation of electrochemical promotion and metal-support interactions 2. CO oxidation by 18O2 on electropromoted Pt films deposited on YSZ and on nanodispersed Pt/YSZ catalysts. J Catal 226(1):197–209. https://doi.org/10.1016/j.jcat.2004.05.009
Li X, Gaillard F, Vernoux P (2007) Investigations under real operating conditions of the electrochemical promotion by O2 temperature programmed desorption measurements. Top Catal 44(3):391–398. https://doi.org/10.1007/s11244-006-0131-5
Tsampas MN, Sapountzi FM, Boréave A, Vernoux P (2013) Isotopical labeling mechanistic studies of electrochemical promotion of propane combustion on Pt/YSZ. Electrochem Commun 26:13–16. https://doi.org/10.1016/j.elecom.2012.09.043
Tsampas MN, Sapountzi FM, Boréave A, Vernoux P (2014) Investigation of the electrochemical promotion of catalysis origins on electrochemical catalysts with oxygen ion conductive supports: isotopic labeling mechanistic studies. Solid State Ionics 262:257–261. https://doi.org/10.1016/j.ssi.2014.01.008
Filkin NC, Tikhov MS, Palermo A, Lambert RM (1999) A kinetic and spectroscopic study of the in situ electrochemical promotion by sodium of the platinum-catalyzed combustion of propene. J Phys Chem A 103(15):2680–2687. https://doi.org/10.1021/jp984186o
Williams FJ, Palermo A, Tikhov MS, Lambert RM (2000) The origin of electrochemical promotion in heterogeneous catalysis: photoelectron spectroscopy of solid state electrochemical cells. J Phys Chem B 104(3):615–621. https://doi.org/10.1021/jp993037i
de Lucas-Consuegra A, Dorado F, Valverde JL, Karoum R, Vernoux P (2007) Low-temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst: characterization, catalytic activity measurements, and investigation of the NEMCA effect. J Catal 251(2):474–484. https://doi.org/10.1016/j.jcat.2007.06.031
de Lucas-Consuegra A, González-Cobos J, García-Rodríguez Y, Mosquera A, Endrino JL, Valverde JL (2012) Enhancing the catalytic activity and selectivity of the partial oxidation of methanol by electrochemical promotion. J Catal 293:149–157. https://doi.org/10.1016/j.jcat.2012.06.016
González-Cobos J, Rico VJ, González-Elipe AR, Valverde JL, de Lucas-Consuegra A (2015) Electrochemical activation of an oblique angle deposited Cu catalyst film for H2 production. Cat Sci Technol 5(4):2203–2214. https://doi.org/10.1039/C4CY01524J
Makri M, Vayenas CG, Bebelis S, Besocke KH, Cavalca C (1996) Atomic resolution STM imaging of electrochemically controlled reversible promoter dosing of catalysts. Surf Sci 369(1–3):351–359. https://doi.org/10.1016/S0039-6028(96)00911-9
Vayenas CG, Archonta D, Tsiplakides D (2003) Scanning tunneling microscopy observation of the origin of electrochemical promotion and metal–support interactions. J Electroanal Chem 554-555:301–306. https://doi.org/10.1016/S0022-0728(03)00240-7
Thursfield A, Brosda S, Pliangos C, Schober T, Vayenas CG (2003) Electrochemical promotion of an oxidation reaction using a proton conductor. Electrochim Acta 48(25–26):3779–3788. https://doi.org/10.1016/S0013-4686(03)00511-5
Vayenas CG, Brosda S, Pliangos C (2001) Rules and mathematical modeling of electrochemical and chemical promotion: 1. Reaction classification and promotional rules. J Catal 203(2):329–350. https://doi.org/10.1006/jcat.2001.3348
Brosda S, Vayenas CG, Wei J (2006) Rules of chemical promotion. Appl Catal B 68(3–4):109–124. https://doi.org/10.1016/j.apcatb.2006.07.021
Vayenas CG, Brosda S (2014) Electron donation–backdonation and the rules of catalytic promotion. Top Catal 57(14–16):1287–1301. https://doi.org/10.1007/s11244-014-0294-4
Brosda S, Vayenas CG (2002) Rules and mathematical modeling of electrochemical and classical promotion 2. Modeling. J Catal 208(1):38–53. https://doi.org/10.1006/jcat.2002.3549
Vayenas CG, Pliangos C, Brosda S, Tsiplakides D (2003) Promotion, electrochemical promotion, and metal–support interactions: the unifying role of spillover. In: Wieckowski A, Savinova ER, Vayenas CG (eds) Catalysis and electrocatalysis at nanoparticle surfaces, Ch.19. Marcel Dekker, Inc, New York, pp 667–744
Nicole J, Tsiplakides D, Pliangos C, Verykios XE, Comninellis C, Vayenas CG (2001) Electrochemical promotion and metal–support interactions. J Catal 204(1):23–34. https://doi.org/10.1006/jcat.2001.3360
Sanabria-Chinchilla J, Asazawa K, Sakamoto T, Yamada K, Tanaka H, Strasser P (2011) Noble metal-free hydrazine fuel cell catalysts: EPOC effect in competing chemical and electrochemical reaction pathways. J Am Chem Soc 133(14):5425–5431. https://doi.org/10.1021/ja111160r
Cai F, Gao D, Zhou H, Wang G, He T, Gong H, Miao S, Yang F, Wang J, Bao X (2017) Electrochemical promotion of catalysis over Pd nanoparticles for CO2 reduction. Chem Sci 8(4):2569–2573. https://doi.org/10.1039/C6SC04966D
Petrushina IM, Bandur VA, Cappeln F, Bjerrum NJ (2000) Electrochemical promotion of sulfur dioxide catalytic oxidation. J Electrochem Soc 147(8):3010–3013. https://doi.org/10.1149/1.1393640
Ruiz-López E, Caravaca A, Vernoux P, Dorado F, de Lucas-Consuegra A (2020) Over-faradaic hydrogen production in methanol electrolysis cells. Chem. Eng. J 396: Art. No. 125217. https://doi.org/10.1016/j.cej.2020.125217
Jahromi AF, Ruiz-López E, Dorado F, Baranova EA, de Lucas-Consuegra A (2022) Electrochemical promotion of ethanol partial oxidation and reforming reactions for hydrogen production. Renew Energy 183:515–523. https://doi.org/10.1016/j.renene.2021.11.041
Zagoraios D, Tsatsos S, Kennou S, Vayenas CG, Kyriakou G, Katsaounis A (2020) Tuning the RWGS reaction via EPOC and in situ electro-oxidation of cobalt nanoparticles. ACS Catal 10(24):14916–14927. https://doi.org/10.1021/acscatal.0c04133
Zagoraios D, Panaritis C, Krassakopoulou A, Baranova EA, Katsaounis A, Vayenas CG (2020) Electrochemical promotion of Ru nanoparticles deposited on a proton conductor electrolyte during CO2 hydrogenation. Appl Catal B 276:119148. https://doi.org/10.1016/j.apcatb.2020.119148
Kotsiras A, Kalaitzidou I, Grigoriou D, Symillidis A, Makri M, Katsaounis A, Vayenas CG (2018) Electrochemical promotion of nanodispersed Ru-Co catalysts for the hydrogenation of CO2. Appl Catal B 232:60–68. https://doi.org/10.1016/j.apcatb.2018.03.031
Souentie S, Hammad A, Brosda S, Foti G, Vayenas CG (2008) Electrochemical promotion of NO reduction by C2H4 in 10% O2 using a monolithic electropromoted reactor with Rh/YSZ/Pt elements. J Appl Electrochem 38(8):1159–1170. https://doi.org/10.1007/s10800-008-9548-9
Makri M, Katsaounis A, Vayenas CG (2015) Electrochemical promotion of CO2 hydrogenation on Ru catalyst–electrodes supported on a K–β″–Al2O3 solid electrolyte. Electrochim Acta 179:556–564. https://doi.org/10.1016/j.electacta.2015.03.144
Kalaitzidou I, Makri M, Theleritis D, Katsaounis A, Vayenas CG (2016) Comparative study of the electrochemical promotion of CO2 hydrogenation on Ru using Na+, K+, H+ and O2− conducting solid electrolytes. Surf Sci 646:194–203. https://doi.org/10.1016/j.susc.2015.09.011
Kalaitzidou I, Katsaounis A, Norby T, Vayenas CG (2015) Electrochemical promotion of the hydrogenation of CO2 on Ru deposited on a BZY proton conductor. J Catal 331:98–109. https://doi.org/10.1016/j.jcat.2015.08.023
Theleritis D, Souentie S, Siokou A, Katsaounis A, Vayenas CG (2012) Hydrogenation of CO2 over Ru/YSZ electropromoted catalysts. ACS Catal 2(5):770–780. https://doi.org/10.1021/cs300072a
Ruiz E, Cillero D, Martínez PJ, Morales Á, Vicente GS, de Diego G, Sánchez JM (2013) Bench scale study of electrochemically promoted catalytic CO2 hydrogenation to renewable fuels. Catal Today 210:55–66. https://doi.org/10.1016/j.cattod.2012.10.025
Papaioannou EI, Souentie S, Hammad A, Vayenas CG (2009) Electrochemical promotion of the CO2 hydrogenation reaction using thin Rh, Pt and Cu films in a monolithic reactor at atmospheric pressure. Catal Today 146(3–4):336–344. https://doi.org/10.1016/j.cattod.2009.06.008
de Lucas-Consuegra A, Caravaca A, González-Cobos J, Valverde JL, Dorado F (2011) Electrochemical activation of a non noble metal catalyst for the water–gas shift reaction. Catal Commun 15(1):6–9. https://doi.org/10.1016/j.catcom.2011.08.007
González-Cobos J, Ruiz-López E, Valverde JL, de Lucas-Consuegra A (2016) Electrochemical promotion of a dispersed Ni catalyst for H2 production via partial oxidation of methanol. Int J Hydrog Energy 41(42):19418–19429. https://doi.org/10.1016/j.ijhydene.2016.06.027
Ruiz E, Cillero D, Martínez PJ, Morales Á, Vicente GS, de Diego G, Sánchez JM (2014) Electrochemical synthesis of fuels by CO2 hydrogenation on Cu in a potassium ion conducting membrane reactor at bench scale. Catal Today 236(Part A):108–120. https://doi.org/10.1016/j.cattod.2014.01.016
Panaritis C, Zgheib J, Ebrahim SAH, Couillard M, Baranova EA (2020) Electrochemical in-situ activation of Fe-oxide nanowires for the reverse water gas shift reaction. Appl Catal B 269:Article 118826 (11 pages). https://doi.org/10.1016/j.apcatb.2020.118826
Khechfe AA, Sullivan MM, Zagoraios D, Katsaounis A, Vayenas CG, Román-Leshkov Y (2022) Non-faradaic electrochemical promotion of Brønsted acid-catalyzed dehydration reactions over molybdenum oxide. ACS Catal 12(2):906–912. https://doi.org/10.1021/acscatal.1c04885
Pacchioni G, Illas F, Neophytides S, Vayenas CG (1996) Quantum-chemical study of electrochemical promotion in catalysis. J Phys Chem 100(41):16653–16661. https://doi.org/10.1021/jp9612386
Leiva EPM, Vázquez C, Rojas MI, Mariscal MM (2008) Computer simulation of the effective double layer occurring on a catalyst surface under electro-chemical promotion conditions. J Appl Electrochem 38(8):1065–1073. https://doi.org/10.1007/s10800-008-9539-x
Panaritis C, Hajar YM, Treps L, Michel C, Baranova EA, Steinmann SN (2020) Demystifying the atomistic origin of the electric field effect on methane oxidation. J Phys Chem Lett 11(17):6976–6981. https://doi.org/10.1021/acs.jpclett.0c01485
Anastasijevic NA (2009) NEMCA—from discovery to technology. Catal Today 146(3–4):308–311. https://doi.org/10.1016/j.cattod.2009.02.020
Dole HAE, Baranova EA (2016) Implementation of nanostructured catalysts in the electrochemical promotion of catalysis. In: Aliofkhazraei M, Makhlouf ASH (eds) Handbook of nanoelectrochemistry. Springer International Publishing AG Switzerland, Cham, pp 1095–1124
Marwood M, Vayenas CG (1997) Electrochemical promotion of electronically isolated Pt catalysts on stabilized zirconia. J Catal 168(2):538–542. https://doi.org/10.1006/jcat.1997.1677
Balomenou S, Pitselis G, Polydoros D, Giannikos A, Vradis A, Frenzel A, Pliangos C, Pütter H, Vayenas CG (2000) Electrochemical promotion of Pd, Fe and distributed Pt catalyst-electrodes. Solid State Ionics 136-137:857–862. https://doi.org/10.1016/S0167-2738(00)00524-5
Wodiunig S, Bokeloh F, Nicole J, Comninellis C (1999) Electrochemical promotion of RuO2 catalyst dispersed on an yttria-stabilized zirconia monolith. Electrochem Solid-State Lett 2(6):281–283. https://doi.org/10.1149/1.1390811
Wodiunig S, Bokeloh F, Comninellis C (2000) Electrochemical promotion of bipolar electrodes: an estimation of the current bypass. Electrochim Acta 46(2–3):357–363. https://doi.org/10.1016/S0013-4686(00)00592-2
Xia C, Hugentobler M, Li Y, Foti G, Comninellis C, Harbich W (2010) Electrochemical promotion of CO combustion over non-percolated Pt particles supported on YSZ using a novel bipolar configuration. Electrochem Commun 13(1):99–101. https://doi.org/10.1016/j.elecom.2010.11.026
Xia C, Hugentobler M, Li Y, Comninellis C, Harbich W (2010) Quantifying electrochemical promotion of induced bipolar Pt particles supported on YSZ. Electrochem Commun 12(11):1551–1554. https://doi.org/10.1016/j.elecom.2010.08.031
Roche V, Revel R, Vernoux P (2010) Electrochemical promotion of YSZ monolith honeycomb for deep oxidation of methane. Catal Commun 11(13):1076–1080. https://doi.org/10.1016/j.catcom.2010.05.005
Balomenou S, Tsiplakides D, Katsaounis A, Thiemann-Handler S, Cramer B, Foti G, Vayenas CG (2004) Novel monolithic electrochemically promoted catalytic reactor for environmentally important reactions. Appl Catal B 52(3):181–196. https://doi.org/10.1016/j.apcatb.2004.04.007
Chatzilias C, Martino E, Katsaounis A, Vayenas CG (2021) Electrochemical promotion of CO2 hydrogenation in a monolithic electrochemically promoted reactor (MEPR). Appl Catal B 284:119695. https://doi.org/10.1016/j.apcatb.2020.119695
Hammad A, Souentie S, Balomenou S, Tsiplakides D, Figueroa JC, Cavalca C, Pereira CJ, Vayenas CG (2008) Tailor-structured skeletal Pt catalysts employed in a monolithic electropromoted reactor. J Appl Electrochem 38(8):1171–1176. https://doi.org/10.1007/s10800-008-9533-3
Balomenou SP, Tsiplakides D, Vayenas CG, Poulston S, Houel V, Collier P, Konstandopoulos AG, Agrafiotis C (2007) Electrochemical promotion in a monolith electrochemical plate reactor applied to simulated and real automotive pollution control. Top Catal 44(3):481–486. https://doi.org/10.1007/s11244-006-0140-4
Hammad A, Souentie S, Papaioannou EI, Balomenou S, Tsiplakides D, Figueroa JC, Cavalca C, Pereira CJ (2011) Electrochemical promotion of the SO2 oxidation over thin Pt films interfaced with YSZ in a monolithic electropromoted reactor. Appl Catal B 103(3–4):336–342. https://doi.org/10.1016/j.apcatb.2011.01.040
Chatzilias C, Martino E, Tsatsos S, Kyriakou G, Katsaounis A, Vayenas CG (2022) Kinetic study of CO2 hydrogenation on Ru/YSZ catalyst using a monolithic electropromoted reactor (MEPR). Chem Eng J 430 (Part 3):132967 (11 pages). https://doi.org/10.1016/j.cej.2021.132967
Marwood M, Vayenas CG (1998) Electrochemical promotion of a dispersed platinum catalyst. J Catal 178(2):429–440. https://doi.org/10.1006/jcat.1998.2156
Jiménez V, Jiménez-Borja C, Sánchez P, Romero A, Papaioannou EI, Theleritis D, Souentie S, Brosda S, Valverde JL (2011) Electrochemical promotion of the CO2 hydrogenation reaction on composite Ni or Ru impregnated carbon nanofiber catalyst-electrodes deposited on YSZ. Appl Catal B 107(1–2):210–220. https://doi.org/10.1016/j.apcatb.2011.07.016
de Lucas-Consuegra A, González-Cobos J, Carcelén V, Magén C, Endrino JL, Valverde JL (2013) Electrochemical promotion of Pt nanoparticles dispersed on a diamond-like carbon matrix: a novel electrocatalytic system for H2 production. J Catal 307:18–26. https://doi.org/10.1016/j.jcat.2013.06.012
Matei F, Jiménez-Borja C, Canales-Vázquez J, Brosda S, Dorado F, Valverde JL, Ciuparu D (2013) Enhanced electropromotion of methane combustion on palladium catalysts deposited on highly porous supports. Appl Catal B 132-133:80–89. https://doi.org/10.1016/j.apcatb.2012.11.011
Jiménez-Borja C, Brosda S, Matei F, Makri M, Delgado B, Sapountzi F, Ciuparu D, Dorado F, Valverde JL, Vayenas CG (2012) Electrochemical promotion of methane oxidation on Pd catalyst-electrodes deposited on Y2O3-stabilized-ZrO2. Appl Catal B 128:48–54. https://doi.org/10.1016/j.apcatb.2012.02.011
Makri M, Symillidis A, Grigoriou D, Katsaounis A, Vayenas CG (2018) Electrochemical promotion of CO2 reduction on a dispersed Ru/YSZ catalyst supported on YSZ solid electrolyte. Mater Today: Proc 5(14):27617–27625. https://doi.org/10.1016/j.matpr.2018.09.082
de Lucas-Consuegra A, Caravaca A, Martínez PJ, Endrino JL, Dorado F, Valverde JL (2010) Development of a new electrochemical catalyst with an electrochemically assisted regeneration ability for H2 production at low temperatures. J Catal 274(2):251–258. https://doi.org/10.1016/j.jcat.2010.07.007
Kambolis A, Lizarraga L, Tsampas MN, Burel L, Rieu M, Viricelle JP, Vernoux P (2012) Electrochemical promotion of catalysis with highly dispersed Pt nanoparticles. Electrochem Commun 19:5–8. https://doi.org/10.1016/j.elecom.2012.02.041
Dole HAE, Safady LF, Ntais S, Couillard M, Baranova EA (2014) Electrochemically enhanced metal-support interaction of highly dispersed Ru nanoparticles with a CeO2 support. J Catal 318:85–94. https://doi.org/10.1016/j.jcat.2014.07.003
González-Cobos J, Horwat D, Ghanbaja J, Valverde JL, de Lucas-Consuegra A (2014) Electrochemical activation of Au nanoparticles for the selective partial oxidation of methanol. J Catal 317:293–302. https://doi.org/10.1016/j.jcat.2014.06.022
Zagoraios D, Athanasiadi A, Kalaitzidou I, Ntais S, Katsaounis A, Caravaca A, Vernoux P, Vayenas CG (2020) Electrochemical promotion of methane oxidation over nanodispersed Pd/Co3O4 catalysts. Catal Today 355:910–920. https://doi.org/10.1016/j.cattod.2019.02.030
Cavalca CA, Larsen G, Vayenas CG, Haller GL (1993) Electrochemical modification of CH3OH oxidation selectivity and activity on a Pt single-pellet catalytic reactor. J Phys Chem 97(23):6115–6119. https://doi.org/10.1021/j100125a005
Poulidi D, Thursfield A, Metcalfe IS (2007) Electrochemical promotion of catalysis controlled by chemical potential difference across a mixed ionic-electronic conducting ceramic membrane – an example of wireless NEMCA. Top Catal 44(3):435–449. https://doi.org/10.1007/s11244-006-0136-0
Poulidi D, Mather GC, Metcalfe IS (2007) Wireless electrochemical modification of catalytic activity on a mixed protonic–electronic conductor. Solid State Ionics 178(7–10):675–680. https://doi.org/10.1016/j.ssi.2007.02.022
Poulidi D, Anderson C, Metcalfe IS (2008) Remote control of the activity of a Pt catalyst supported on a mixed ionic electronic conducting membrane. Solid State Ionics 179(27–32):1347–1350. https://doi.org/10.1016/j.ssi.2008.01.056
Stavrakakis E, West M, Johnston S, McIllwaine R, Poulidi D (2019) Hydration, CO2 stability and wireless electrochemical promotion studies on yttria-doped Ba (Ce, Zr) O3 perovskites. Ionics 25:1243–1257. https://doi.org/10.1007/s11581-019-02836-6
Poulidi D, Rivas ME, Zydorczak B, Wu Z, Li K, Metcalfe IS (2012) Electrochemical promotion of a Pt catalyst supported on La0.6Sr0.4Co0.2Fe0.8O3−δ hollow fibre membranes. Solid State Ionics 225:382–385. https://doi.org/10.1016/j.ssi.2012.03.010
Vernoux P, Guth M, Li X (2009) Ionically conducting ceramics as alternative catalyst supports. Electrochem Solid-State Lett 12(7):E9–E11. https://doi.org/10.1149/1.3122746
Fortunato MA, Princivalle A, Capdeillayre C, Petigny N, Tardivat C, Guizard C, Tsampas MN, Sapountzi FM, Vernoux P (2014) Role of lattice oxygen in the propane combustion over Pt/yttria-stabilized zirconia: isotopic studies. Top Catal 57(14–16):1277–1286. https://doi.org/10.1007/s11244-014-0293-5
Dole HAE, Isaifan RJ, Sapountzi FM, Lizarraga L, Aubert D, Princivalle A, Vernoux P, Baranova EA (2013) Low temperature toluene oxidation over Pt nanoparticles supported on yttria stabilized-zirconia. Catal Lett 143(10):996–1002. https://doi.org/10.1007/s10562-013-1071-x
Isaifan R, Dole H, Obeid E, Lizarraga L, Baranova EA, Vernoux P (2019) Catalytic CO oxidation over Pt nanoparticles prepared from the polyol reduction method supported on yttria-stabilized zirconia. ECS Trans 35(28):43–57. https://doi.org/10.1149/1.3641818
Isaifan RJ, Dole HAE, Obeid E, Lizarraga L, Vernoux P, Baranova EA (2012) Metal-support interaction of Pt nanoparticles with ionically and non-ionically conductive supports for CO oxidation. Electrochem Solid-State Lett 15(3):E14-E17. https://doi.org/10.1149/2.024203esl
Isaifan RJ, Baranova EA (2015) Effect of ionically conductive supports on the catalytic activity of platinum and ruthenium nanoparticles for ethylene complete oxidation. Catal Today 241(Part A):107–113. https://doi.org/10.1016/j.cattod.2014.03.061
Dole HAE, Costa ACGSA, Couillard M, Baranova EA (2016) Quantifying metal support interaction in ceria-supported Pt, PtSn and Ru nanoparticles using electrochemical technique. J Catal 333:40–50. https://doi.org/10.1016/j.jcat.2015.10.015
Dole HAE, Baranova EA (2016) Ethylene oxidation in an oxygen-deficient environment: why ceria is an active support? ChemCatChem 8(11):1977–1986. https://doi.org/10.1002/cctc.201600142
Isaifan RJ, Ntais S, Couillard M, Baranova EA (2015) Size-dependent activity of Pt/yttria-stabilized zirconia catalyst for ethylene and carbon monoxide oxidation in oxygen-free gas environment. J Catal 324:32–40. https://doi.org/10.1016/j.jcat.2015.01.010
Yentekakis IV, Goula G, Kampouri S, Betsi-Argyropoulou I, Panagiotopoulou P, Taylor MJ, Kyriakou G, Lambert RM (2017) Ir-catalysed nitrous oxide (N2O) decomposition: effect of Ir particle size and metal–support interactions. Catal Lett 148(1):341–347. https://doi.org/10.1007/s10562-017-2233-z
Goula G, Botzolaki G, Osatiashtiani A, Parlett CMA, Kyriakou G, Lambert RM, Yentekakis IV (2019) Oxidative thermal sintering and redispersion of Rh nanoparticles on supports with high oxygen ion lability. Catalysts 9(6):541 (16 pages). https://doi.org/10.3390/catal9060541
Wang Z, Huang H, Liu H, Zhou X (2012) Self-sustained electrochemical promotion catalysts for partial oxidation reforming of heavy hydrocarbons. Int J Hydrog Energy 37(23):17928–17935. https://doi.org/10.1016/j.ijhydene.2012.09.072
Zhou X, Huang H, Liu H (2013) Study of partial oxidation reforming of methane to syngas over self-sustained electrochemical promotion catalyst. Int J Hydrog Energy 38(15):6391–6396. https://doi.org/10.1016/j.ijhydene.2013.03.047
Hernandez WY, Hadjar A, Giroir-Fendler A, Andy P, Princivalle A, Klotz M, Marouf A, Guizard C, Tardivat C, Viazzi C, Vernoux P (2015) Electrochemically-assisted NOx storage–reduction catalysts. Catal Today 241(Part A):143–150. https://doi.org/10.1016/j.cattod.2014.03.076
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bebelis, S. (2023). Electrochemical Promotion of Catalysis: From Discovery to Fundamentals to Applications. In: Vernoux, P., Vayenas, C.G. (eds) Recent Advances in Electrochemical Promotion of Catalysis. Modern Aspects of Electrochemistry, vol 61. Springer, Cham. https://doi.org/10.1007/978-3-031-13893-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-031-13893-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-13892-8
Online ISBN: 978-3-031-13893-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)