
Abstract
The mature nervous system relies on the polarized morphology of neurons for a directed flow of information. These highly polarized cells use their somatodendritic domain to receive and integrate input signals while the axon is responsible for the propagation and transmission of the output signal. However, the axon must perform different functions throughout development before being fully functional for the transmission of information in the form of electrical signals. During the development of the nervous system, axons perform environmental sensing functions, which allow them to navigate through other regions until a final target is reached. Some axons must also establish a regulated contact with other cells before reaching maturity, such as with myelinating glial cells in the case of myelinated axons. Mature axons must then acquire the structural and functional characteristics that allow them to perform their role as part of the information processing and transmitting unit that is the neuron. Finally, in the event of an injury to the nervous system, damaged axons must try to reacquire some of their immature characteristics in a regeneration attempt, which is mostly successful in the PNS but fails in the CNS. Throughout all these steps, glycans perform functions of the outermost importance. Glycans expressed by the axon, as well as by their surrounding environment and contacting cells, encode key information, which is fine-tuned by glycan modifying enzymes and decoded by glycan binding proteins so that the development, guidance, myelination, and electrical transmission functions can be reliably performed. In this chapter, we will provide illustrative examples of how glycans and their binding/transforming proteins code and decode instructive information necessary for fundamental processes in axon physiology.
IN MEMORIAM Professor Hans-Joachim Gabius
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Axon
- Neuron
- Differentiation
- Glycoconjugate
- Glycosylation
- Ganglioside
- Axon guidance
- Axon outgrowth
- Axon regeneration
- Axon initial segment
- Ion channels
- Microbiota-Gut-Brain axis
1 Introduction
Glycans play crucial roles in the nervous system by their influence in neuronal development, plasticity, differentiation or signal transduction, intervening also in the onset and evolution of neurodegenerative pathologies. However, only recent technological advances in bio-physicochemical and imaging analyses are permitting researchers to unveil and define the precise functions of specific glycan structures associated with glycoproteins and glycolipids during neuronal differentiation. In this chapter we will present and discuss the state-of-the-art and future perspectives of carbohydrate-related processes involved in neuron physiology, focusing on axon determination, development, structure and function.
2 Axon Determination/Early Neuronal Differentiation
Neuronal precursors go through several differentiation stages before turning into neurons, and it is becoming clear that this progression is precisely regulated by glycosylation and glycan interactions.
An instructive example of this is bisecting GlcNAc, a branch structure in N-glycans, most highly expressed in the nervous system where, besides other physiological and pathological functions (Kizuka and Taniguchi 2018), it plays a central role in neuronal differentiation. The expression of bisect-type N-glycans is up-regulated in human iPSC-derived neurons (Tateno et al. 2011), as shown by lectin microarray analyses. These kind of glycan was found to be up-regulated in both iPSCs and ESCs-derived murine neurons analysed by glycoblotting-based cellular glycomics (Terashima et al. 2014). These quantitative analyses revealed that bisecting GlcNAc glycans range from 1% to 2% of total glycans in iPSCs and ESCs, increasing up to 4% in derived NSCs, and reaching values between 11% and 12% in differentiated neurons, similar to those of cultured primary neurons, and very different to those of differentiated astrocytes shown to have between 1% and 4% of bisecting GlcNAc glycans (Terashima et al. 2014). However, these studies did not link glycan structures with their related glycoproteins, impeding detailed structural/functional pairing. New methods have been developed to address this drawback.
Using human-induced pluripotent stem cells (iPSCs) and iPSC-derived neuronal cells as a model of neuronal differentiation, and applying sequential LC/MS/MS analyses of protein extracts upon tryptic digestion, glycopeptide enrichment, and peptide deglycosylation, an altered production of a series of N-glycoproteins and their glycoforms has been shown during the differentiation into iPSC-derived dopaminergic neurons. In particular, there is an increase of N-glycoproteins bearing a bisected biantennary glycan structure with five N-acetylglucosamines, three mannoses, and a fucose. These dopaminergic and site-selective modifications are independent of the level of protein synthesis. These modifications target proteins functionally involved in neural cell adhesion (L1CAM, NCAMs, etc.) and in axon guidance/semaphorin-plexin signalling (PLXNs, SEMAs, etc.) (Kimura et al. 2021). These strategies that integrate glycoproteome analyses and resultant glycoprotein profiles with their evolving glycoforms, provide detailed information on the role of (N-)glycosylation in neuronal differentiation and could be the basis to a more precise understanding of neurodegenerative diseases and neural regeneration.
Neuron function requires different processes occurring in separate parts of the cell from the very early stages of differentiation (Dotti et al. 1988). Starting from the symmetric neuroblast stage, such a functional polarization is preceded by a precise molecular segregation that provides a single early neurite with axonal features. Underlining the relevance of glycans in early neuronal polarization, the plasma membrane ganglioside neuraminidase Neu3 (also known as PMGS) is located in a single neurite of non-polarized neurons (stages 1–2) before their morphological polarization (stage 3), becoming a polarity landmark at the pre-axon plasma membrane.
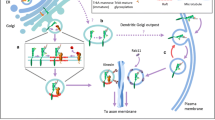
The low filamentous actin (F-actin) content in the terminal segment of a neurite has been considered an early marker for the formation of fast-advancing growth cones occurring in axons (Bradke and Dotti 1999). In fact, Neu3 localizes at the neurite with lowest F-actin content specifying its axonal fate, and its local enzymatic inhibition triggers the polymerisation of actin filaments blocking axon development (Da Silva et al. 2005). Although the mechanism for the early polarized segregation of Neu3 is still unknown, its enzymatic activity locally removes neuraminic acid moieties from gangliosides GD1a or GT1b to produce GM1 bearing a pentasaccharide with a single sialic acid (Kopitz 2017; Ledeen and Wu 2015; Schengrund 2015). GM1 accumulation spatially-restricts neurotrophin receptor TrkA by direct interaction and stimulates signalling (Mutoh et al. 1995; Rabin and Mocchetti 1995) for phosphoinositide 3-kinase (PI3K)-induced inactivation of RhoA small-GTPase pathway proceeding through the disassembly of the complex RhoA/Rock (RhoA-associated kinase) from the brain actin-binding protein profillin-IIA (PIIA). This provokes a localized subcortical F-actin depolymerisation allowing microtubule protrusion and axon elongation (Da Silva et al. 2005; Da Silva et al. 2003) (Fig. 7.1). During subsequent stages of neuronal differentiation, Neu3 remains enriched in the axon membrane and its local enzymatic activity is required for normal axonal growth (Da Silva et al. 2005; Rodriguez et al. 2001).

Neu3/GM1-induced axon outgrowth. Membrane neuraminidase Neu3 locally removes neuraminic acid moieties (purple diamonds) from gangliosides GD1a or GT1b to produce GM1, which spatially-gathers neurotrophin receptor TrkA by direct interaction, stimulating signalling for the inactivation of RhoA small-GTPase pathway, and disassembling the RhoA/Rock complex from the brain actin-binding protein profillin-IIA (PIIA). The subsequent localized subcortical F-actin (red long rods) depolymerisation allows microtubule to protrude supporting axon outgrowth (indicated by the green big arrow)
Besides local F-actin regulation, GM1 also influences axon development by facilitating the trafficking of glycosylated neurotrophin receptors along the axon, and by its interaction with integrins and galectins such as Gal-1.
TrkA displays up to eleven putative N-glycosylation sites and only N-glycosylated TrkA forms bearing fully mature N-glycans can be targeted to the axon membrane (Watson et al. 1999). GM1 specifically binds to the mature form of TrkA, gathering it to transport vesicle membrane rafts for an efficient anterograde transport to the axon plasma membrane (Abad-Rodriguez and Díez-Revuelta 2015; Mutoh et al. 1995; Rabin and Mocchetti 1995). Cells lacking GM1, or expressing deficiently glycosylated TrkA, fail to target the receptor to the membrane precluding any neurotrophic effect (Mutoh et al. 2000; Mutoh et al. 2002). GM1 also interacts with glycosylated α5β1-integrin (Wu et al. 2007) and both molecules are stabilized in the complex by the specific cross-linking with homobivalent dimers of Gal-1. This membrane process stimulates axon growth by triggering the signalling cascade of focal adhesion kinase (FAK)/phospholipase Cγ/phosphoinositide-3 kinase (PI3K), and the opening of TRPC5 channels leading to a transient raise of intracellular Ca2+ (Wu et al. 2016).
In all, variations in the glycosylated heads of gangliosides can induce local changes in the membrane, alone or in combination with glycan-crosslinking proteins such as galectins, gathering distinct molecular machineries and stimulating diverse signalling cascades that control axon outgrowth.
3 Axon Guidance
The correct anatomical and functional development of the nervous system requires the precise navigation of growing axons towards their specific targets. Axon guidance is based on different sets of tissue attractive/repulsive molecular cues whose disposition changes as the nervous system develops, and that are detected and interpreted by sensing systems at the axon membrane. Among others, classic axonal cues (netrins, semaphorins, slits, and ephrins) [4,5], neurotrophins (NGF, BDNF, NT3) and morphogens (sonic hedgehog, Wnt, BMP7) have been implicated in axonal guidance and their molecular mechanisms have been thoroughly studied (Seiradake et al. 2016; Ye et al. 2019). Playing a central role in these mechanisms, glycosylation confers a superior level of complexity to axon guidance pathways, allowing the refinement of their regulation for an optimized biological function. Within the next sections we will discuss the role of glycosylation of two major molecular groups involved in axon guidance, namely cell adhesion molecules and glycosaminoglycans. We will also explain the role of endogenous galectins and their interactions with glycosylated guidance cues in the navigation of olfactory axons in the adult olfactory system, as a paradigmatic case of physiological neuron regeneration within the central nervous system.
3.1 Axon Guidance by Glycosylated Cell Adhesion Molecules (CAMs)
Neuronal cell adhesion molecule (NCAM) presents a characteristic glycosylation with terminal polysialic acid (PSA), a polymer of α2–8 linked neuraminic acid. These polymers are formed by ST8Sia II and ST8Sia IV polysialyltransferases acting on hybrid, bi- to tetra-antennary complex glycans bearing, preferably, a terminal α2–6-linked neuraminic acid (Bhide and Colley 2016). Supporting the role of PSA-NCAM in guidance, impaired N-glycosylation or polysialylation by enzymatic ablation produces defective development of the mouse olfactory bulb and failed neuronal migration (Hildebrandt et al. 2009; Weinhold et al. 2005), projection errors in the chicken optic fiber layer due to the blockade of axon exit from the retina, and increased fasciculation of chicken spinal ganglia (Monnier et al. 2001). Similarly, impaired PSA synthesis affects granular cell projections (mossy fibers) along the hippocampal stratum lucidum. Their axons become misrouted due to over-adhesion to the pyramidal cell layer eliminating the lamination of the supra- and infra-pyramidal mossy fiber bundles (Fig. 7.2) (Koyama and Ikegaya 2018; Weinhold et al. 2005). There is a structural reason that explains these effects. In fact, PSA in NCAM confers numerous negative charges on its structure creating a strong electrostatic repulsion and increasing intercellular space that hampers adhesion (Rutishauser and Landmesser 1996). In all, PSA regulates NCAM-mediated neuronal migration and axonal fasciculation and guidance, becoming essential for the development of the nervous system.

Anomalous lamination of hippocampal mossy fibers in PSA-deficient mice. (a) PSA chains are synthesized on 5th and 6th N-glycosylation sites in immunoglobulin (Ig) domain 5 of NCAM, and are formed by repeated α2–8 N-acetylneuraminic acid units (degree of polymerization –DP- from 8 to 400 units). (b) Schematic design of wild type mouse dentate gyrus (DG) and hippocampus CA3 zone illustrating the mossy fibers, axons of granule cells (gcs) that project along the stratum lucidum (sl) in parallel to the pyramidal cell layer (pcl). These axons send collaterals to contact pyramidal cells (pc), constituting a fully laminated structure (ml, molecular layer; pl, polymorphic layer; gcl, granular cell layer). This laminated structure is partially maintained by PSA through the repulsive field provided by its bulky, polyanionic properties (b, bottom panel). Such an electrostatic repulsion regulates cell-cell interspace distance, avoiding the direct interaction of NCAM. (c) In PSA-deficient mice lacking the sialyl-transferases that synthesize PSA (ST8Sia II−/− /ST8Sia IV−/−) mossy fiber bundling and lamination is severely perturbed and axons become misrouted. The absence of PSA negatively charged “clouds” reduces the distance between axon membranes, and permit the direct interactions of NCAM with itself and other molecules in cis or trans (c, bottom panel) that provoke the over-adhesion among granular axons and to the pyramidal cell layer
Another member of the immunoglobulin cell adhesion molecule family (IgCAM) that is polysialylated is the synaptic cell adhesion molecule SynCAM. It functions as a guidance cue in the central and peripheral nervous system (Frei and Stoeckli 2017) by the expression of its PSA-SynCAM form in the NG2 glial subpopulation that integrates in different murine neuronal networks (Galuska et al. 2010).
Besides the regulation of axon growth and guidance by PSA-induced electrostatic hindrance, other adhesion molecules act through heterophilic interactions with glycosylated molecules on the membranes of adjacent cells. Such is the case of L1CAM that binds in trans to the highly glycosylated mucin glycoprotein CD24 carrying α2–3 linked sialic acid and Lewisx terminals on its structure. Interestingly, L1CAM/CD24 interaction can exert axon outgrowth stimulation in mouse cerebellar neurons or inhibition in DRG neurons (Kleene 2001). Neuronal L1 interacts in trans with α2–3 linked sialic acid in glial CD24 through a siglec homologous sequence, while CD24 Lewisx terminals interact with TAG1 and contactin in cerebellar neurons to assemble a neurite-growth stimulating complex. In contrast, TAG1 and contactin in DRG neurons, interacting in trans with CD24 Lewisx terminals, form complexes by cis interactions on neuronal surfaces with Caspr1 and Caspr2, respectively, triggering outgrowth inhibitory signalling (Lieberoth et al. 2009). Noteworthy, contactin binding to Caspr1 is also regulated by contactin glycosylation state. This will be discussed below within the Sect. 4.
Besides CAMs, other transmembrane glycoproteins such as endoglycan and dystroglycan play central roles in axon guidance. Endoglycan belongs to the CD34 family of sialomucins. It displays one membrane spanning peptide and heavily glycosylated extracellular domains. Similarly to PSA-bearing proteins, extracellular domains of sialomucins display highly negative net charge due to multiple N- and O-linked glycosylations with sialylated terminals. This arrangement maintains a low adhesion level between the floor plate and the commissural axons, allowing them to exit the floor plate and navigate in the rostral direction (Baeriswyl et al. 2021). Dystroglycan is also a transmembrane protein, with a heavily glycosylated extracellular domain that is required for the precise patterning of different guidance proteins. Such is the case for slit, whose binding to glycosylated dystroglycan is required for its correct localization at the floor plate. As a matter of fact, genetic down regulation of dystroglycan or of its glycosylating enzymes produces misguidance of spinal commissural axons comparable to that in slit and slit receptor roundabout (robo) mutants. Other guidance-related molecules such as laminin, perlecan, etc. are also arranged in the basement membrane by scaffolding glycosylated dystroglycan, which is required, for instance, for the correct axonal sorting of contralateral and ipsilateral optical tracts in the optic chiasm by organizing guidance cues at the basement membrane (Clements et al. 2017; Lindenmaier et al. 2019; Wright et al. 2012). Underlining the relevance of glycosylated dystroglycan, though it is unknown whether guidance failures are involved, defective dystroglycan glycosylation produces a group of muscular dystrophies known as dystroglycanopathies, associated with abnormal development of the nervous system (Godfrey et al. 2011; Kanagawa 2021; Paprocka et al. 2021).
3.2 Glycosaminoglycans in Axon Guidance
Glycosaminoglycans (GAGs) are long linear polysaccharides built out of repeated disaccharides most usually containing GlcNAc or GalNAc, combined with uronic acid or galactose. There are several GAGs subfamilies, i.e. heparan sulfate (HS), chondroitin sulfate (CS), keratan sulfate (KS), that present sulfate groups at different positions, and the non-sulfated glycosaminoglycan hyaluronan (HA) (Higuero et al. 2017). Detailed information on GAG biosynthesis and structure is described in Chap. 5 by Schwartz and Domowicz. CS, HS, and KS covalently bound to proteins render the proteoglycans (PGs), CSPG, HSPGs, and KSPGs, respectively. HA is the only GAG that does not form PGs. GAGs are particularly enriched in the ECM where they modulate axonal directional outgrowth through the spatial patterning of many guidance cues. Consistently, deficient GAG biosynthesis induces failed nerve system formation and mistaken connectivity (Saied-Santiago and Bülow 2018).
HA is a key constituent of the nervous system ECM that surrounds neurons and glial cells. So far it has been found to function in axon guidance at the optic chiasm, where it colocalizes and binds CD44, a major local guidance cue. As a matter of fact, multiple guidance errors at the midline in the mouse optic chiasm are observed upon administration of exogenous HA or by genetic-driven impaired HA biosynthesis, demonstrating the regulation of axonal guidance through HA interaction with CD44 (Haupt and Huber 2008; Lin et al. 2007). Similar functions of HA have been suggested in other parts of the brain, such as the innervation of entorhinal fibers to the hippocampus that is perturbed by hyaluronidase in vitro (Förster et al. 2001). Nevertheless, it is not known whether HA plays this role by modulating guidance cues, as it does in the optic chiasm.
HSPGs are major components of the ECM and are essential for axon guidance, as shown in basic studies using Drosophila (Inatani et al. 2003; Smart et al. 2011). Importantly, HSPGs organize the patterning of most of the relevant guidance cues during CNS development.
Syndecan is the major family of transmembrane HSPGs that regulates the localization of slit in the ECM, a repulsive cue located at the ventral midline that repels the growth of axons expressing slit receptor robo (Johnson et al. 2004b). Slit-mediated repulsion is also regulated by membrane-linked glypicans, whose extracellular domain bears multiple HS chains (De Pasquale and Pavone 2019). Perturbation of HS, either by enzymatic hydrolysis with heparinase III or by genetic down regulation of the Ext1 gene (coding for the enzyme that polymerizes HS sugar chains), disrupts chemorepulsion dependent on slit-robo interaction (Piper et al. 2006) leading to severe defects in the development of the olfactory bulb and the cerebellum, and to major defects in the commissural tracts (Inatani et al. 2003). In contrast to the severe phenotypes of Ext1 mutants, variations in the sulfation of HSPG (i.e. mutants for heparan sulfate sulfotransferases hs2st and hs6st) give rise to normal brain morphology with moderate and localized axon guidance issues at the optic chiasm and corpus callosum (Pratt et al. 2006), even though slit-mediated repulsion is reduced
Netrin-1 is another critical guidance molecule regulated by HSPGs. It shows chemoattraction or chemorepulsion depending on the axon projections (Boyer and Gupton 2018), and its deletion provokes embryonic lethality in mice by failed formation of the corpus callosum, and mistaken projections of spinal and hippocampal commissures (Yung et al. 2015). HS interacts with netrin-1 and genetic ablation of Ext-1, induces similar phenotypes to those of netrin-1 knockouts (Finci et al. 2014; Matsumoto et al. 2007). In addition, as shown in C. elegans, glypican modulates UNC6/netrin mediated axonal guidance by interacting with the netrin receptor UNC40/DCC (Blanchette et al. 2015). The fact that netrin and its receptors are evolutionarily conserved in mammals makes the regulation of netrin-1 by glypican feasible also in these organisms (Mutalik and Gupton 2021).
Like netrins, semaphorins are relevant cues in axonal guidance with both repulsive and attracting functions. Semaphorin 3A (sema 3A) is a secreted protein that binds to components of ECM (i.e. GAGs), regulating its patterning and interactions with its axonal co-receptors neuropilin and plexin for a correct directionality.
Similar to other guidance molecules mentioned above, down regulation of HS by genetic deletion of Ext1 also affects axon guidance-related ephrins (Klein 2004). The growth cone collapse mediated by ephrin A (type-A ephrins are GPI-anchored proteins) is precluded in Ext1 knockout mice, highlighting the role of HS in the regulation of ephrin A, though the underlying molecular mechanisms remain unknown (Irie et al. 2008). In the case of ephrin B (type-B ephrins are transmembrane proteins) the raise in specific N-glycosylation after crossing of the corpus callosum axons diminishes their response to semaphorins compared to pre-crossing axons, which proves the capacity of HSPG to modulate the reaction to guidance cues in a precise manner (Mire et al. 2018).
CSPGs found at the ECM of the nervous system exert mainly repulsive functions. Their down regulation by chondroitinase enzymatic hydrolysis caused different axon pathfinding mistakes: (i) retinal projections at the midline of the optic chiasm grow axons in regions that normally do not allow their outgrowth (Chung et al. 2000); (ii) abnormal growth of the ventral motor nerve in the zebrafish embryo (Bernhardt and Schachner 2000), and (iii) axon navigation and targeting defects in RGC projections of Xenopus and chick embryos (Ichijo and Kawabata 2001; Walz et al. 1997). A recent in vitro study showed the direct inhibitory effects of CSPGs on the growth cones of mouse cerebellar granule neurons (Jin et al. 2018). Of note, semaphorin 5A is regulated and interacts with CSPGs and also with HSPGs. Interaction with the former confers sema5A a repulsive character, while interaction with the latter turns it into an axon attractive cue (Kantor et al. 2004), showing how different GAGs can re-define the signals for axonal pathfinding.
3.3 Galectins and Their Interactions with Guidance Cues in the Adult Olfactory System
Olfactory sensory neurons are periodically replaced with neuroblasts arisen from a stem cell niche in the olfactory mucosa. Those new neurons must project axons from the nasal neuroepithelium to the olfactory bulb (OB) in the brain, and they are guided by glycosylated cues interacting with endogenous galectins. Galectin-1 (Gal-1) notably contributes to this guidance, which is lost in Gal-1 knockout mice where axons fail to reach their targets at the OB(Puche et al. 1996). Non-covalent dimers of Gal-1 are proposed to associate ECM components such as laminin with unknown glycoconjugates on the axon membrane (likely GM1 or NAcLac-bearing glycoproteins) to guide the axon elongation, or to stimulate it by crosslinking different axons to generate fascicles (Tenne-Brown et al. 1998). Primary olfactory axons also express other galectins (Gal-3, -7, and -8), and the presence of different lactosamine-bearing guidance cues (i.e. NCAM), suggests the implication of these galectins in the axon guidance and targeting associated with the continuous renovation of the olfactory receptors (Storan et al. 2004).
Besides sensory olfactory neurons, in the subventricular zone (SVZ) neuroblasts derived from immature precursors migrate along the so-called “rostral migratory stream” (RMS) up to the OB, where they differentiate into granular and periglomerular interneurons (Luskin 1993). Along the RMS, ependymal cilia and astrocytes that form glial tubes to direct neuroblast migration (Peretto et al. 1997) express Gal-3. Gal-3-knockout mice display altered ependymal cilia and SVZ astrocytes with associated reduction of neuroblast migration and defective pathfinding. These effects correlate with the increased phosphorylation of epidermal growth factor receptor (EGFR) pointing to modulation of neuroblast migration either by direct (Díez-Revuelta et al. 2010) or indirect (Comte et al. 2011) Gal-3 interactions with N-glycosylated EGFR.
4 Axon/Glia Interactions
Axon myelination is required for axon homeostasis and for the transmission of fast electrical impulses along nerves. Even though the mechanisms driving myelin formation are not completely understood, myelination is based on the interaction between axons and mature oligodendrocytes (OLGs), whose membrane-associated glycans are emerging as highly relevant actors. Immature neurons express and release Gal-4 that functions as a soluble regulator of oligodendrocyte progenitor (OPC) differentiation, keeping them in a proliferative non-myelinating stage through binding to unknown glycosylated receptors. As neurons undergo differentiation, Gal-4 expression is downregulated and OLGs advance towards a myelinating stage (Stancic et al. 2012), also secreting Gal-3 that, in an autocrine manner, reinforces the differentiation of OLGs and the integrity of myelin by binding to different glycosylated sites only present on the surface of the OLGs (Pasquini et al. 2011). Final lateral extension of myelin over the axonal surface gives rise to its final discontinuous display, with myelinated segments (internodes) and non-myelinated gaps (nodes), bordered by limiting interfaces (paranodes and juxtaparanodes). Success in myelination depends on the glycosylation state of membrane molecules expressed by neurons and OLGs in the central nervous system (CNS), or Schwann cells in the peripheral nervous system (PNS), as discussed below.
Contactin1 and caspr1 bearing immature N-glycans (high mannose termini) in ER membranes associate in cis to leave the ER (Roth and Zuber 2017) using a raft-based trafficking mechanism targeted to the axon paranodes where they interact with NF-155 located at myelin terminal loop (Bonnon 2003; Faivre-Sarrailh et al. 2000; Gollan et al. 2003) (Fig. 7.3, left panels). In contrast, the maturation of contactin1 N-glycans by addition of N-acetyl-lactosamine (NAcLac) termini in the Golgi apparatus precludes the interaction with caspr1, and contactin1 is sorted to axon nodes, where it interacts with NF-186, gathering as well the Nav1.6 sodium channel involved in fast nerve impulse conduction (Fig. 7.3, right panels). In contrast, juxtaparanodes involve the pairing in cis at the axonal membrane of caspr2 and contactin2 (also known as TAG-1) that subsequently interact in trans with contactin2 at the adjacent myelin sheet. This adhesion complex recruits and clusters Kv1-type potassium channels (Poliak and Peles 2003; Susuki et al. 2016; Traka et al. 2002). Based on the parallel but diverse N-glycosylation characteristics of contactin and caspr homologs (Hasler et al. 1993; Poliak et al. 1999), a likely glycan-based mechanism for the Caspr2/Contactin2 specific sorting to juxtaparanode has been proposed though not proven so far.

Glycosylation role in the organization of nodes of Ranvier and paranodes. In the left panel it is shown how contactin1 and caspr1 bearing immature N-glycans (high mannose termini) in ER membranes associate in cis and target the axon paranodes where they interact with neurofascin-155 (NF-155) of the adjacent myelin terminal loop membrane. Alternatively, as shown in the right panels, contactin1 N-glycans can turn mature by addition of N-acetyl-lactosamine (NAcLac) termini within the Golgi apparatus. This blocks its interaction with caspr1, and targets contactin1 to the nodes of Ranvier, where it interacts with neurofascin-186 (NF-186) and recruits the Nav1.6 sodium channel required for fast nerve impulse conduction. Protein complex structures displayed in this figure are idealized for an intuitive comprehension of the processes. Protein 3D models have been obtained from www.3dproteinimaging.com
In addition, the myelin-associated glycoprotein (MAG, Siglec-4), that binds gangliosides GD1a and GT1b with high affinity in vitro (Vinson 2001; Vyas et al. 2002; Yang et al. 1996), is expressed in vivo at the membrane of myelinating OLGs and stabilizes the internode myelin by its interaction in trans with GD1a and GT1b in axonal membrane rafts (Posse De Chaves and Sipione 2010; Schengrund 2015; Schnaar 2010).
Recapitulating, lectins are involved in myelination at the level of OLG maturation and in the stabilization of myelinated internodes by MAG. This together with the strict glycosylation requirements for the correct sorting of main myelin molecules underscores the notion that glycan-related interactions are key to organize and regulate both non-myelinated and myelinated axon structures. Future research in this line promises significant insights into myelin-related physiopathology.
5 Nerve Impulse Generation/Transmission
In the nervous system, the directed flow of information relies on the polarized morphology of neurons, which typically consist of a cell body, multiple dendrites, and a single axon. A key structural and functional domain that integrates synaptic inputs received by the somatodendritic domain and in turn generates action potentials propagated by the axon is the axon initial segment (AIS) (Fig. 7.4a). As such, the AIS is a 10–60 μm long domain strategically located between the somatodendritic compartment and the axon per se. It has unique structural features such as a dense network of membrane proteins characterized by a high concentration of glycosylated voltage-gated ion channels and cell adhesion molecules, and a specialized submembranous cytoskeleton enriched in ankyrin-G and spectrin βIV anchored to both actin filaments and microtubules (Leterrier 2016; Nelson and Jenkins 2017; Rasband 2010). Some of these features are also shared by the nodes of Ranvier in myelinated axons, which are responsible for propagating the action potentials generated at the AIS (Rasband and Peles 2021).

Role of glycosylation in voltage-gated ion channels of the Axon Initial Segment (AIS). (a) Schematic representation of ion channel expressed at the AIS. Channels of the three families (sodium, potassium and calcium) have been described in the AIS, all of them present putative N-glycosylation sites, although the presence of N-glycans have been only demonstrated in some of them, denoted as (N + number). Those sites with no current evidence of attached glycans are symbolised as (N*). Updated information on channel glycosylation can be found at https://www.glygen.org. (b) The post-translational modification of voltage-gated ion channels (generic channel depicted in brown) and/or their auxiliary subunits by N-glycosylation can affect their function through different mechanisms. Lack of N-glycosylation can affect the channel folding as has been proven by directed mutagenesis of the different N-glycosylation sites. This usually induces the retention of non-glycosylayed channels at the ER precluding their trafficking to the cell surface. In contrast, intact channels exit the ER and their N-glycans undergo full maturation before reaching the plasma membrane. Once on the cell surface, mature N-glycosylated channels can perform their normal ion gating function. Nevertheless, N-glycans hydrolysis by glycosidase enzyme activities (deglycosylation) at the membrane can also alter the channel distribution and stability (altered endocytosis and recycling) and impair their ion gating function, thus affecting the transmission of the nerve impulse
5.1 Voltage-Gated Ion Channels
Voltage-gated ion channels exert important neuronal functions such as the control of cell excitability, generation and propagation of action potentials, and modulation of synaptic transmission. Of particular importance for the correct functioning of the AIS and the nodes of Ranvier are several channels that regulate membrane permeability for sodium, potassium, and calcium ions. Generally, these ion channels and their auxiliary subunits are heavily glycosylated, with some subunits having up to 36% of their mass in the form of glycans (Messner and Catterall 1985; Roberts and Barchi 1987). The post-translational modification of these channels and/or their auxiliary subunits by N-glycan attachment can affect their function through different mechanisms (Fig. 7.4b). N-glycosylation can affect the folding and trafficking of ion channels to the cell surface. Once on the cell surface, N-glycosylation can also alter the distribution and stability of these channels by modulating their endocytosis and recycling. Finally, N-glycosylation can also alter the biophysical characteristics of these channels thereby inhibiting or potentiating their function.
5.2 Voltage-Gated Sodium Channels
Voltage-gated sodium channels (VGSCs) are large integral membrane glycoproteins that regulate a fast and temporary sodium influx during the generation and propagation of an action potential. VGSCs are heterodimeric and heterotrimeric protein complexes composed of a large α subunit and either one or two β subunits, all of which are heavily glycosylated. The α subunit is the voltage-sensing pore-forming subunit, and as such is responsible for the influx of sodium ions, while the β subunits can modulate the gating kinetics (opening and closing), voltage-dependence, and surface expression of the channel (Hull and Isom 2018). In mammals, there are nine voltage-activated sodium channels (Nav1.1 – Nav1.9) encoded by a combination of nine α subunit genes and four β subunit genes (Kruger and Isom 2016). In general, Nav1.1, Nav1.2, Nav1.3 and Nav1.6 are primarily expressed in the CNS while Nav1.7, Nav1.8 and Nav1.9 are expressed in the PNS (Catterall et al. 2005; Lai and Jan 2006; Vacher et al. 2008; Wang et al. 2017; Whitaker et al. 2000). Outside of the nervous system, Nav1.4 is primarily expressed in skeletal muscle and Nav1.5 in heart (Catterall et al. 2005). Early studies identified that VGSCs were concentrated at the axon initial segment and at nodes of Ranvier in myelinated axons (Catterall 1981; Ellisman and Levinson 1982; Waxman and Ritchie 1985; Wollner and Catterall 1986). In CNS neurons, Nav1.1, Nav1.2 and Nav1.6 are localized at the AIS, with Nav1.1 and Nav1.6 being also present at mature nodes of Ranvier (Caldwell et al. 2000; Duflocq et al. 2011; Hu et al. 2009; Lorincz and Nusser 2010; Lorincz and Nusser 2008; Ogiwara et al. 2007; Van Wart and Matthews 2006). As mentioned before, these channels are highly glycosylated and, in particular, highly sialylated. Early characterization of these channels demonstrated that glycan chains can account for up to 30% of their apparent molecular mass, with sialic acid residues representing almost 50% of the total carbohydrates (Elmer et al. 1985; Messner and Catterall 1985; Miller et al. 1983; Roberts and Barchi 1987). This rapidly prompted the question of the importance of these negatively charged residues on the activity of VGSCs. When purified channels in lipid bilayers were treated with neuraminidase to remove sialic acid residues, the result was a significant depolarizing shift in the average potential required for channel activation (Recio-Pinto et al. 1990). Additional experiments using sialylation-deficient cells or mutant forms of the sodium channels that could not be glycosylated led to similar results (Bennett et al. 1997). The removal of sialic acid residues had no effect on the structure or stability of the channels, confirming the idea that the presence of negatively charged extracellular residues causes the membrane around the channel to be slightly depolarized so that only a smaller change in membrane potential is required to induce channel gating (Cronin et al. 2005). Importantly, not all channels are uniformly glycosylated. For example, cardiac sodium channels are less glycosylated that those in muscle and brain (Cohen and Levitt 1993). In accordance with the role of sialylation in the regulation of sodium channel gating, the more heavily glycosylated Nav1.4 channel produced a large depolarizing shift following desialylation compared to the less glycosylated Nav1.5 channel which was unaffected. In comparison to the muscle and cardiac channels, neuronal isoforms (Nav1.2 and Nav1.7) are moderately glycosylated and consequently showed moderate depolarizing shifts upon desialylation (Johnson et al. 2004a). Therefore, the effect of glycosylation on VGSC function is dependent not only on the specific isoform being expressed but also on the cell type in which it is expressed. Consequently, VGSC activity is regulated in a cell-specific manner by glycosylation. As mentioned before, VGSC β subunits are also extensively glycosylated (Yu et al. 2003; Zhou et al. 2012; Cortada et al. 2019). Although fewer studies have explored the role of glycosylation on their function, evidence indicates that β subunit glycosylation can regulate the trafficking of sodium channels to the cell surface. A recent study explored the functional significance of β2 subunit glycosylation on Nav1.5 channel trafficking (Cortada et al. 2019). The β2 subunit was glycosylated at three different sites (N42, N66 and N74). An unglycosylated triple mutant was mostly retained at the endoplasmic reticulum and was defective at promoting cell surface expression of the Nav1.5 α subunit.
5.3 Voltage-Gated Potassium Channels
Axonal voltage-gated potassium channels (VGKCs) are also involved in shaping the action potential and in controlling neuronal excitability. These types of channels are activated by membrane depolarization, which allows an outward flow of potassium ions that repolarizes the membrane and brings the action potential to an end. Following the action potential, these channels hyperpolarize the membrane and set the resting membrane potential of the cell. VGKCs are classified into 12 distinct subfamilies (Kv1 – Kv12) based on the similarity of their amino acid sequence (Gutman et al. 2005). Regarding their composition, VGKCs consist of homo- or heterotetramers of the pore-forming α subunit (usually of the same family) and may also contain auxiliary β subunits that regulate the function and distribution of the channel. They are broadly expressed in a variety of tissues including the nervous system. In neurons, they have been localized throughout the cell although those located at the AIS are especially important for action potential generation and propagation. Kv1.1, Kv1.2, Kv2.1, Kv2.2, Kv7.2 and Kv7.3 channels are localized at the AIS in different type of neurons (Battefeld et al. 2014; Devaux et al. 2004; Goldberg et al. 2008; Jensen et al. 2017; Johnston et al. 2008; King et al. 2014; Klinger et al. 2011; Kole et al. 2007; Lorincz and Nusser 2008; Ogawa et al. 2008; Pan et al. 2006; Sanchez-Ponce et al. 2012; Sarmiere et al. 2008). Kv1.1, Kv1.2, Kv3.1b, Kv7.2 and Kv7.3 are also present at the node of Ranvier and axon terminal (Chung et al. 2006; De Wit et al. 2005; Devaux et al. 2003; Devaux et al. 2004; Kim and Rutherford 2016; Pan et al. 2006; Rasband et al. 1998; Sheng et al. 1993).
The importance of VGKCs was noticed in early studies with two Drosophila melanogaster excitability mutants with a leg shaking phenotype. The shaker and hyperkinetic locus turned out to encode the α and β subunits of potassium channels (Chouinard et al. 1995; Hotta and Benzer 1972; Ikeda and Kaplan 1970; Pongs et al. 1988; Salkoff and Wyman 1981). In mice, deletion of the Kv1.1 channel causes epilepsy (Smart et al. 1998). In humans, a variety of single base missense mutations in Kv1.1 channels have been associated with episodic ataxia (Adelman et al. 1995; Browne et al. 1994). Other type of VGKCs, Kv7.2 and Kv7.3, have an important role in the pathogenesis of neonatal epilepsies (Singh et al. 1998). Mutations in these channels cause neurons to become slightly depolarized and rhythmically fire multiple action potentials. These results highlight the importance of VGKCs in regulating cell excitability.
As found for most voltage-gated ion channels, VGKCs are also extensively glycosylated and this modification regulates the cell surface expression, stability, and gating properties of the channel (Fujita et al. 2006; Hall et al. 2015; Khanna et al. 2001; Lopez-Rodriguez and Holmgren 2018; Napp et al. 2005; Petrecca et al. 1999; Sutachan et al. 2005; Thayer et al. 2016; Vicente et al. 2018; Watanabe et al. 2003; Watanabe et al. 2015; Watanabe et al. 2004; Watanabe et al. 2007) (Fig. 7.4). Regarding the role of glycosylation on the function of VGKCs, the Shaker-related Kv1 subfamily is one of the most intensely studied. When Kv1.1 channels were expressed in CHO cell lines with reduced potential for glycosylation, the voltage-dependence of activation was shifted to more positive voltages and the activation kinetics were slower than for normally glycosylated channels. Sialidase treatment of glycosylated channels reproduced the effects observed on glycosylation deficient cells. Surface expression, however, was not affected by glycosylation (Thornhill et al. 1996). Site-directed mutagenesis of the Kv1.1 channel was used to study of the effect of the complete lack of glycans on the functional properties of the channel. Kv1.1 has a single N-glycosylation site (N207) in its first extracellular loop. Expression of a N207Q mutant demonstrated that the lack of glycosylation, and in particular terminal sialylation, affected the steady-state activation and kinetic properties of the channel (Watanabe et al. 2003). Like Kv1.1, Kv1.2 channels also have a single N-glycosylation site (N207). However, the expression of Kv1.2 glycosylation mutants in hippocampal neurons revealed no apparent change in the activation and deactivation kinetics of the channel. Glycosylation at this site, on the other hand, was important for the cell surface expression and stability of the channel (Thayer et al. 2016).
Additionally, the Kv3.1 channel, which is present at the nodes of Ranvier, is also glycosylated at positions N220 and N229. Complex-type glycosylation at N229 increases trafficking of the channel to the cell surface. On the contrary, an unglycosylated mutant and a glycosylated form with high mannose-type glycans at position N220 were retained at the endoplasmic reticulum (Vicente et al. 2018). Moreover, replacement of the complex-type N-glycan with a hybrid type hindered the gating properties of Kv3.1 channels (Hall et al. 2015).
5.4 Voltage-Gated Calcium Channels
In excitable cells, voltage-gated calcium channels (VGCCs) elevate intracellular calcium concentrations after membrane depolarization. Particularly in neurons, these channels are the primary mediators of depolarization-induced calcium entry where they regulate the timing and generation of action potential bursts. VGCCs consist of a pore-forming α1 subunit and two auxiliary subunits - a cytoplasmic β subunit and an extracellular α2δ subunit. A great variety of channel subtypes can be co-assembled through the use of ten α1 subunit genes, four β subunit genes, and four α2δ subunit genes. VGCCs are classified in three channel subfamilies (Cav1, Cav2 and Cav3) according to their α1 subunit, or into five classes (L-type: Cav1.1 - Cav1.4, P/Q-type: Cav2.1, N-type: Cav2.2, R-type: Cav2.3, and T-type: Cav3.1–3.3) based on their biophysical and pharmacological properties (Catterall 2011). There is a widespread expression of VGCCs throughout the nervous system. Cav1 channels are present in dendritic spines where calcium entry regulates long-term potentiation (Grover and Teyler 1990; Navakkode et al. 2022). Cav2 channels are present in presynaptic nerve terminals where calcium entry triggers neurotransmitter release (Mochida 2019). Cav3 channels are mainly present in the soma and dendrites of neurons, and they regulate cellular excitability and oscillatory behavior as well as neurotransmitter release (Cheong and Shin 2013; Perez-Reyes 2003; Turner and Zamponi 2014; Weiss et al. 2013; Weiss et al. 2012). Importantly, several VGCCs have been localized at the AIS including Cav2.1, Cav2.2, Cav2.3, and more recently, Cav3.1 and Cav3.2 (Bender and Trussell 2009; Lipkin et al. 2021). Experiments performed on different types of neurons revealed that the activation of these channels at the AIS is essential to the generation and timing of action potential bursts (Bender and Trussell 2009).
Like other voltage-gated ion channels, mature calcium channels are also glycosylated proteins, and their glycosylation regulates the cell surface channel density and their gating properties. In silico analysis of the amino acid sequence of the ten α1 pore-forming subunits reveal canonical N-glycosylation sites (Lazniewska and Weiss 2017). Evidence pointing to the glycosylation of VGCCs have come from studies using lectin labelling to identify glycosylation sites, from biochemical analyses were the molecular mass of the channels change upon glycosidase treatment or N-glycosylation inhibition with tunicamycin, as well as from site-directed mutagenesis studies.
Results suggesting the potential glycosylation of T-type calcium channels came from experiments showing that the molecular mass of Cav3.1 and Cav3.3 channels were significantly different depending on the brain region and developmental time analyzed (Yunker et al. 2003). Furthermore, when the Cav3.3 channel was exogenously expressed in HEK293 cells and subjected to enzymatic deglycosylation with PNGase F, the differences in molecular weight were normalized, suggesting that the channel is an N-linked glycoprotein (Chen et al. 2007). A more detailed characterization of the glycosylation of T-type channels came from site-directed mutagenesis studies of the human Cav3.2 channel, which has four potential N-glycosylation sites (N192, N271, N1466 and N1710). Disruption of these sites revealed that glycosylation at asparagine N192 and N1466 increases the steady-state expression and stabilizes the channel at the cell surface (Lazniewska et al. 2016; Weiss et al. 2013). Additionally, glycosylation at both N192 and N1466 sites is essential for regulation of the gating properties of the channels as they modulate the calcium permeability and/or opening probability of the channel (Ondacova et al. 2016).
In a similar manner, site-directed mutagenesis studies showed that N-glycosylation of L-type calcium channels is also critical for their correct function. When, either one, two or the four potential N-glycosylation sites found on Cav1.2 channel were mutated, the double (N124Q/N299Q) and quadruple mutants (N124Q/N299Q/N1359Q/N1410Q) showed a positive shift in voltage-dependent gating. Furthermore, the quadruple mutant showed a decreased cell surface expression with a concomitant decrease in current density (Park et al. 2015).
In addition to the pore-forming subunit, the α2δ auxiliary subunit is also a glycosylated protein. It is highly N-glycosylated, with approximately 30 kDa of its mass in the form of glycans (Marais et al. 2001). The first studies concerning the functional significance of the glycosylation of this subunit revealed it has a role in stabilizing its interaction with the pore-forming subunit and for the functional increase in the channel’s current amplitude (Gurnett et al. 1996). Site-directed mutagenesis studies identified positions N136 and N184 as functional N-glycosylation sites necessary for enhancement of calcium current amplitudes (Andrade et al. 2009; Sandoval et al. 2004). Glycosylation of the α2δ subunit at position N633 was also identified as necessary for its surface expression and as essential for its role as a regulator of the channel’s calcium current (Tétreault et al. 2016). Additional glycosylation sites (N348, N468 and N812) also contribute to the stability of this subunit (Tétreault et al. 2016).
5.5 O-Linked β-N-Acetylglucosaminylation of AIS Proteins
O-GlcNAcylation is a posttranslational modification in which a single β-N-acetylglucosamine (O-GlcNAc) monosaccharide is attached to either serine or threonine residues of intracellular proteins. It is a unique type of intracellular glycosylation which contributes to numerous cellular functions and is dysregulated in a wide range of diseases (Chatham et al. 2021). Interestingly, O-GlcNAcylation is particularly abundant in the brain where the only two enzymes that add and remove O-GlcNAc moieties (O-GlcNAc transferase and O-GlcNAcase, respectively) are abundantly expressed (Lee et al. 2021; Liu et al. 2012; Wulff-Fuentes et al. 2021). Several studies have provided clues for the role of O-GlcNAcylation in the modulation of neuronal excitability and excitatory synaptic transmission.
Ankyrin-G is a scaffolding protein that functions as the master organizer of the AIS by clustering voltage-gated sodium and potassium channels, as well as cell adhesion molecules (e.g., neurofascin, NrCAM) (Jenkins and Bennett 2001; Pan et al. 2006; Zhou et al. 1998). It has long been known that ankyrin-G is glycosylated at its serine-rich domain with O-linked GlcNAc residues (Zhang and Bennett 1996). Additionally, immunofluorescence experiments using an antibody that recognizes single O-linked GlcNAc residues revealed that O-GlcNAc modified proteins are particularly enriched at nodes of Ranvier (Zhang and Bennett 1996). However, progress towards identifying the functional significance of O-GlcNAcylation was hampered by the inability to identify the modified residues easily and precisely. It took some time until advances in affinity chromatography and mass spectrometry allowed robust enrichment methodologies and the dissociation of peptide backbone linkages without the elimination of O-GlcNAcylated moieties from O-GlcNAcylated peptides (Chalkley et al. 2009). Now, more than 5000 proteins comprise the human O-GlcNAcome including multiple voltage-gated sodium, potassium, and calcium channels (Wulff-Fuentes et al. 2021). Many of these are key AIS proteins (e.g., Kv7.3, Cav2.1, Cav2.2, Cav3.1 and ankyrin-G) with established roles in neuronal excitability (Chalkley et al. 2009; Ruan et al. 2014; Trinidad et al. 2012). In accordance with a relevant function for O-GlcNAcylation of AIS proteins in the regulation of action potential generation, an acute increase in O-GlcNAcylation strongly suppresses the intrinsic excitability of hippocampal CA1 neurons by cooperatively modulating three different voltage gated-cation channels: voltage-gated potassium channels, voltage-gated sodium channels and HCN channels (Hwang and Rhim 2019). Furthermore, in a recent study using a murine model of temporal lobe epilepsy as well as human epileptic hippocampal tissue, a global decrease of O-GlcNAcylation and OGT expression was found in the epileptic tissue compared to non-epileptic controls. Pharmacological inhibition of OGA enzyme, which elevated protein O-GlcNAcylation decreased both seizure duration and epileptic spike events.
In summary, the importance of protein glycosylation in the nervous system is evidenced by the fact that most congenital disorders of glycosylation, caused by mutations in genes of the glycosylation machinery, exhibit numerous neurological manifestations including epileptic seizures, mental and psychomotor retardation, ataxia, hyperreflexia, hypotonia as well as structural abnormalities (Baycin-Hizal et al. 2014). It is therefore conceivable that alterations in the glycosylation of target proteins, such as the AIS-localized glycoproteins discussed here (Fig. 7.4a), can contribute to the phenotypical manifestations observed in these diseases. In accordance with this, the activity of voltage-gated ion channels and mutations in the genes that code them have been associated with neurological disorders of neuronal excitability such as epilepsy and chronic pain (Ademuwagun et al. 2021; Goodwin and Mcmahon 2021). Many of these channels are localized at the AIS, and as such, the AIS has been suggested as a “common pathogenic node” in epilepsy (Wimmer et al. 2010).
6 Axon Regeneration
PNS axons upon injury can regenerate extensively, reconnecting with their targets and recovering their function. In contrast, the axons of CNS neurons mostly fail to regenerate, in part due to their intrinsically poor regenerating capacity even in permissive environments, and also to the formation of the glial scar at the injury site, where several inhibitory molecules contribute to CNS regeneration failure. Major components of the glial scar are CSPGs that exert a strong inhibitory effect on axon growth and regeneration (Laabs et al. 2005). In fact, injury producing damage to ECM leads to upregulation of growth inhibitory CSPG (Kim et al. 2018). Logically, CSPG has been targeted to stimulate regeneration, either directly by enzymatic hydrolysis by chondroitinase (Day et al. 2020; Mountney et al. 2013), or indirectly by manipulating the expression of specific interactors that reverse the CSPG-induced inhibition, such as the heparan-binding growth-associated molecule HB-GAM, also known as pleiotrophin, that injected after experimental spinal cord injury in mice enhances axon regeneration (Rauvala et al. 2017).
Importantly, the intrinsically low regenerative capacity of CNS axons can also be overcome by glycan-related mechanisms. As discussed in Sect. 2, early spatial confinement of neuraminidase Neu3 to a single neurite increases local GM1 providing the axon fate to that neurite, where a plethora of GM1-binding proteins such as Gal-1, TrkA, integrins, etc, signal and support subsequent axon outgrowth. Nevertheless, Neu3 activity decreases in CNS neurons as they mature and it is very low in fully differentiated neurons. Actually, elevating Neu3 activity by gene transfection induces a notable enhancement of regeneration of rat hippocampal and cortical axons upon in vitro axotomy (Dotti et al. 2001). In this line, a possible explanation to the different regenerative behaviour of injured CNS and PNS axons relies on the fact that the conversion of GD1a and GT1b to GM1 induced by Neu-3 occurs only in axotomized PNS but not CNS axons, as proven when comparing injured sensory and retinal axons of adult rats (Kappagantula et al. 2014). Attempts have been made to stimulate functional regeneration in experimental CNS injuries by raising local neuraminidase activity. Infusion of Clostridium perfringens sialidase enhances spinal axon outgrowth into implanted grafts of peripheral nerve in rats after brachial plexus injury (Yang et al. 2006). Similarly, sialidase from Vibrio cholerae infused to the site of a moderate thoracic spinal cord contusion induced significant improvements of the behavioural and anatomical outcomes (Mountney et al. 2013; Mountney et al. 2010). Unexpectedly, the combination of sialidase and chondroitinase did not show an additive reinforcement of injured axon regeneration when compared with that seen when each of the enzymes was used separately (Mountney et al. 2013).
7 New Perspective in CNS Regulation Through Glycan-Lectin Interactions in the Microbiota-Gut-Brain Axis
Over the last 20 years, the concept that the gut microbiota can influence the nervous system under physiological and/or pathological conditions has gained much attention as results of an exponentially growing number of studies are published in the scientific literature. A large proportion of this body of work is based on the association of a particular condition (e.g., behavior, disease, mood disorder, etc.) with changes in different populations of bacteria composing the gut microbiota in humans and animal models or with the complete lack of a gut microbiota using germ-free animals. Advances in high throughput technologies (metagenomics, metatranscriptomics, metaproteomics and metabolomics) have started to shed light on the mechanisms responsible for this long-range regulation, thus providing a causal relationship between an “altered” gut microbiota and nervous system dysfunctions (Liu et al. 2021; Wang et al. 2019). The most common mediators of this regulation include bacterially derived metabolites that can cross the blood-brain barrier, gut derived hormones, immune cell-derived cytokines, and a direct effect through the modulation of the vagus nerve (Cryan et al. 2019). Axon physiology is by no means an exception to this type of regulation. Two processes influenced by the gut microbiota are axon growth and axonal myelination (Hoban et al. 2016; Vuong et al. 2020). Microbiota-deficient mice have hypermyelinated axons in the prefrontal cortex, coincident with a marked upregulation of neural activity-induced genes as well as genes linked to myelination. Interestingly, these gene expression changes can be reverted by colonization of germ-free mice with a conventional microbiota (Hoban et al. 2016). Moreover, during the prenatal period, the maternal gut microbiota can regulate the growth of thalamocortical axons in the fetus (Vuong et al. 2020). Depletion of maternal microbiome resulted in decreased thalamocortical projections and reduced expression of genes related to axonogenesis. Gnotobiotic colonization with selected bacteria as well as maternal supplementation with selected microbial-derived metabolites prevented the axonal defects observed in the offspring.
Quite surprisingly, considerably less attention has been given to the role played by glycan-lectin interactions between hosts and microbes, and how those interactions impact the nervous system through the microbiota-gut-brain axis. A major component of the intestinal mucosa are epithelial-derived glycans. Host glycans are critical regulators of the gut microbiota colonization since they serve not only as a source of nutrients for bacterial metabolism but also as ligands for both commensal and pathogenic bacterial lectins (adhesins) (Koropatkin et al. 2012; Kudelka et al. 2020; Poole et al. 2018; Ringot-Destrez et al. 2017). In addition, host-derived lectins with bactericidal activity can shape the microbiota composition and establish intestinal homeostasis (Lehotzky et al. 2010; Miki et al. 2017; Vaishnava et al. 2011). We therefore believe that more efforts aimed at comprehending the largely unexplored roles played by glycan-lectin interactions - not only within the nervous system but also those that shape the host-microbe symbiosis - are necessary for a better understanding of the pathophysiology of the nervous system.
Abbreviations
- AIS:
-
Axon initial segment
- CS:
-
Chondroitin sulphate
- DRG:
-
Dorsal root ganglia
- ECM:
-
Extracellular matrix
- ESC:
-
Embryonic stem cells
- GAG:
-
Glycosaminoglycan
- HA:
-
Hyaluronan
- HS:
-
Heparan sulphate
- iPSC:
-
Induced pluripotent stem cells
- KS:
-
Keratan sulphate
- NSC:
-
Neural stem cells
- OB:
-
Olfactory bulb
- OLG:
-
Oligodendrocyte
- OPC:
-
Oligodendrocyte precursor
- PG:
-
Proteoglycan
- PSA:
-
Polysialic acid
- RGC:
-
Retinal ganglion cell
- RMS:
-
Rostral migratory stream
References
Abad-Rodriguez J, Díez-Revuelta N. Axon glycoprotein routing in nerve polarity, function, and repair. Trends Biochem Sci. 2015;40(7):385–96.
Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15(6):1449–54.
Ademuwagun IA, Rotimi SO, Syrbe S, Ajamma YU, Adebiyi E. Voltage gated sodium channel genes in epilepsy: mutations, functional studies, and treatment dimensions. Front Neurol. 2021;12:600050.
Andrade A, Sandoval A, González-Ramírez R, Lipscombe D, Campbell KP, Felix R. The alpha(2)delta subunit augments functional expression and modifies the pharmacology of Ca(V)1.3 L-type channels. Cell Calcium. 2009;46(4):282–92.
Baeriswyl T, Dumoulin A, Schaettin M, Tsapara G, Niederkofler V, Helbling D, et al. Endoglycan plays a role in axon guidance by modulating cell adhesion. eLife. 2021;2:10.
Battefeld A, Tran BT, Gavrilis J, Cooper EC, Kole MHP. Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J Neurosci. Society for Neuroscience. 2014;34(10):3719–32.
Baycin-Hizal D, Gottschalk A, Jacobson E, Mai S, Wolozny D, Zhang H, et al. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem Biophys Res Commun. 2014;453(2):243–53.
Bender KJ, Trussell LO. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61(2):259–71.
Bennett E, Urcan MS, Tinkle SS, Koszowski AG, Levinson SR. Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism. J Gen Physiol. 1997;109(3):327–43.
Bernhardt RR, Schachner M. Chondroitin sulfates affect the formation of the segmental motor nerves in zebrafish embryos. Dev Biol. 2000;221(1):206–19.
Bhide GP, Colley KJ. Sialylation of N-glycans: mechanism, cellular compartmentalization and function. Histochem Cell Biol. 2016;147(2):149–74.
Blanchette CR, Perrat PN, Thackeray A, Bénard CY. Glypican is a modulator of netrin-mediated axon guidance. PLoS Biol. Public Library of Science. 2015;13(7):e1002183.
Bonnon C. The paranodal complex of F3/contactin and caspr/paranodin traffics to the cell surface via a non-conventional pathway. J Biol Chem. 2003;278(48):48339–47.
Boyer NP, Gupton SL. Revisiting netrin-1: one who guides (axons). Front Cell Neurosci. 2018;12:221.
Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283(5409):1931–4.
Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. Nature Publishing Group. 1994;8(2):136–40.
Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA. National Academy of Sciences. 2000;97(10):5616–20.
Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci. Society for Neuroscience. 1981;1(7):777–83.
Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3(8):a003947.
Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. American Society for Pharmacology and Experimental Therapeutics. 2005;57(4):397–409.
Chalkley RJ, Thalhammer A, Schoepfer R, Burlingame AL. Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc Natl Acad Sci U S A. 2009;106(22):8894–9.
Chatham JC, Zhang J, Wende AR. Role of O-linked N-acetylglucosamine protein modification in cellular (patho)physiology. Physiol Rev. 2021;101(2):427–93.
Chen Y, Sharp AH, Hata K, Yunker AMR, Polo-Parada L, Landmesser LT, et al. Site-directed antibodies to low-voltage-activated calcium channel CaV3.3 (alpha1I) subunit also target neural cell adhesion molecule-180. Neuroscience. 2007;145(3):981–96.
Cheong E, Shin H-S. T-type Ca2+ channels in normal and abnormal brain functions. Physiol Rev. 2013;93(3):961–92.
Chouinard SW, Wilson GF, Schlimgen AK, Ganetzky B. A potassium channel beta subunit related to the aldo-keto reductase superfamily is encoded by the Drosophila hyperkinetic locus. Proc Natl Acad Sci USA. National Academy of Sciences. 1995;92(15):6763–7.
Chung KY, Taylor JS, Shum DK, Chan SO. Axon routing at the optic chiasm after enzymatic removal of chondroitin sulfate in mouse embryos. Development. 2000;127(12):2673–83.
Chung HJ, Jan Y-N, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc Natl Acad Sci USA. National Academy of Sciences. 2006;103(23):8870–5.
Clements R, Turk R, Campbell KP, Wright KM. Dystroglycan maintains inner limiting membrane integrity to coordinate retinal development. J Neurosci. Society for Neuroscience. 2017;37(35):8559–74.
Cohen SA, Levitt LK. Partial characterization of the rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circ Res. 1993;73(4):735–42.
Comte I, Kim Y, Young CC, van der Harg JM, Hockberger P, Bolam PJ, et al. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J Cell Sci. 2011;124(Pt 14):2438–47.
Cortada E, Brugada R, Verges M. N-Glycosylation of the voltage-gated sodium channel β2 subunit is required for efficient trafficking of NaV1.5/β2 to the plasma membrane. J Biol Chem. 2019;294(44):16123–40.
Cronin NB, O’Reilly A, Duclohier H, Wallace BA. Effects of deglycosylation of sodium channels on their structure and function. Biochemistry. 2005;44(2):441–9.
Cryan JF, O’Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99(4):1877–2013.
Da Silva JS, Medina M, Zuliani C, Di Nardo A, Witke W, Dotti CG. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J Cell Biol. 2003;162(7):1267–79.
Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, Abad-Rodriguez J. Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat Neurosci. 2005;8(5):606–15.
Day P, Alves N, Daniell E, Dasgupta D, Ogborne R, Steeper A, et al. Targeting chondroitinase ABC to axons enhances the ability of chondroitinase to promote neurite outgrowth and sprouting. Fehlings MG, editor. PLoS One. 2020;15(1):e0221851–19.
De Pasquale V, Pavone LM. Heparan sulfate proteoglycans: the sweet side of development turns sour in mucopolysaccharidoses. Biochim Biophys Acta Mol basis Dis. 2019;1865(11):165539.
De Wit J, De Winter F, Klooster J, Verhaagen J. Semaphorin 3A displays a punctate distribution on the surface of neuronal cells and interacts with proteoglycans in the extracellular matrix. Mol Cell Neurosci. 2005;29(1):40–55.
Devaux J, Alcaraz G, Grinspan J, Bennett V, Joho R, Crest M, et al. Kv3.1b is a novel component of CNS nodes. J Neurosci. 2003;23(11):4509–18.
Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. Society for Neuroscience. 2004;24(5):1236–44.
Díez-Revuelta N, Velasco S, André S, Kaltner H, Kübler D, Gabius H-J, et al. Phosphorylation of adhesion- and growth-regulatory human galectin-3 leads to the induction of axonal branching by local membrane L1 and ERM redistribution. J Cell Sci. 2010;123(Pt 5):671–81.
Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8(4):1454–68.
Dotti CG, Hasegawa T, Rodriguez JA, Miyagi T, Piddini E. Plasma membrane ganglioside sialidase regulates axonal growth and regeneration in hippocampal neurons in culture. J Neurosci. 2001;21(21):8387–95.
Duflocq A, Chareyre F, Giovannini M, Couraud F, Davenne M. Characterization of the axon initial segment (AIS) of motor neurons and identification of a para-AIS and a juxtapara-AIS, organized by protein 4.1B. BMC Biol. 2011;9(1):66.
Ellisman MH, Levinson SR. Immunocytochemical localization of sodium channel distributions in the excitable membranes of Electrophorus electricus. Proc Natl Acad Sci USA. National Academy of Sciences. 1982;79(21):6707–11.
Elmer LW, O’Brien BJ, Nutter TJ, Angelides KJ. Physicochemical characterization of the alpha-peptide of the sodium channel from rat brain. Biochemistry. 1985;24(27):8128–37.
Faivre-Sarrailh C, Gauthier F, Denisenko-Nehrbass N, Le Bivic A, Rougon G, Girault JA. The glycosylphosphatidyl inositol-anchored adhesion molecule F3/contactin is required for surface transport of paranodin/contactin-associated protein (caspr). J Cell Biol. 2000;149(2):491–502.
Finci LI, Krüger N, Sun X, Zhang J, Chegkazi M, Wu Y, et al. The crystal structure of netrin-1 in complex with DCC reveals the bifunctionality of netrin-1 as a guidance cue. Neuron. 2014;83(4):839–49.
Förster E, Zhao S, Frotscher M. Hyaluronan-associated adhesive cues control fiber segregation in the hippocampus. Development. 2001;128(15):3029–39.
Frei JA, Stoeckli ET. SynCAMs – from axon guidance to neurodevelopmental disorders. Mol Cell Neurosci. 2017;81:41–8.
Fujita T, Utsunomiya I, Ren J, Matsushita Y, Kawai M, Sasaki S, et al. Glycosylation and cell surface expression of Kv1.2 potassium channel are regulated by determinants in the pore region. Neurochem Res. 2006;31(5):589–96.
Galuska SP, Rollenhagen M, Kaup M, Eggers K, Oltmann-Norden I, Schiff M, et al. Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proc Natl Acad Sci USA. National Academy of Sciences. 2010;107(22):10250–5.
Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev. 2011;21(3):278–85.
Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58(3):387–400.
Gollan L, Salomon D, Salzer JL, Peles E. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 2003;163(6):1213–8.
Goodwin G, Mcmahon SB. The physiological function of different voltage-gated sodium channels in pain. Nat Rev Neurosci. 2021;22(5):263–74.
Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347(6292):477–9.
Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron. 1996;16(2):431–40.
Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. American Society for Pharmacology and Experimental Therapeutics. 2005;57(4):473–508.
Hall MK, Weidner DA, Edwards MAJ, Schwalbe RA. Complex N-glycans influence the spatial arrangement of voltage gated potassium channels in membranes of neuronal-derived cells. PLoS ONE. Public Library of Science. 2015;10(9):e0137138.
Hasler TH, Rader C, Stoeckli ET, Zuellig RA, Sonderegger P. cDNA cloning, structural features, and eucaryotic expression of human TAG-1/axonin-1. Eur J Biochem. 1993;211(1–2):329–39.
Haupt C, Huber AB. How axons see their way--axonal guidance in the visual system. Front Biosci. 2008;13:3136–49.
Higuero AM, Díez-Revuelta N, Abad-Rodriguez J. The sugar code in neuronal physiology. Histochem Cell Biol. 2017;147(2):257–67.
Hildebrandt H, Mühlenhoff M, Oltmann-Norden I, Röckle I, Burkhardt H, Weinhold B, et al. Imbalance of neural cell adhesion molecule and polysialyltransferase alleles causes defective brain connectivity. Brain. 2009;132(Pt 10):2831–8.
Hoban AE, Stilling RM, Ryan FJ, Shanahan F, Dinan TG, Claesson MJ, et al. Regulation of prefrontal cortex myelination by the microbiota. Transl Psychiatry. 2016;6(4):e774.
Hotta Y, Benzer S. Mapping of behaviour in Drosophila mosaics. Nature. Nature Publishing Group. 1972;240(5383):527–35.
Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. Nature Publishing Group. 2009;12(8):996–1002.
Hull JM, Isom LL. Voltage-gated sodium channel β subunits: the power outside the pore in brain development and disease. Neuropharmacology. 2018;132:43–57.
Hwang H, Rhim H. Acutely elevated O-GlcNAcylation suppresses hippocampal activity by modulating both intrinsic and synaptic excitability factors. Sci Rep. 2019;9(1):7287.
Ichijo H, Kawabata I. Roles of the telencephalic cells and their chondroitin sulfate proteoglycans in delimiting an anterior border of the retinal pathway. J Neurosci. 2001;21(23):9304–14.
Ikeda K, Kaplan WD. Patterned neural activity of a mutant Drosophila melanogaster. Proc Natl Acad Sci USA. National Academy of Sciences. 1970;66(3):765–72.
Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302(5647):1044–6.
Irie F, Okuno M, Matsumoto K, Pasquale EB, Yamaguchi Y. Heparan sulfate regulates ephrin-A3/EphA receptor signaling. Proc Natl Acad Sci USA. National Academy of Sciences. 2008;105(34):12307–12.
Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155(5):739–46.
Jensen CS, Watanabe S, Stas JI, Klaphaak J, Yamane A, Schmitt N, et al. Trafficking of Kv2.1 channels to the axon initial segment by a novel nonconventional secretory pathway. J Neurosci. Society for Neuroscience. 2017;37(48):11523–36.
Jin J, Tilve S, Huang Z, Zhou L, Geller HM, Yu P. Effect of chondroitin sulfate proteoglycans on neuronal cell adhesion, spreading and neurite growth in culture. Neural Regen Res. 2018;13(2):289–97.
Johnson D, Montpetit ML, Stocker PJ, Bennett ES. The sialic acid component of the beta1 subunit modulates voltage-gated sodium channel function. J Biol Chem. 2004a;279(43):44303–10.
Johnson KG, Ghose A, Epstein E, Lincecum J, O’Connor MB, Van Vactor D. Axonal heparan sulfate proteoglycans regulate the distribution and efficiency of the repellent slit during midline axon guidance. Curr Biol. 2004b;14(6):499–504.
Johnston J, Griffin SJ, Baker C, Skrzypiec A, Chernova T, Forsythe ID. Initial segment Kv2.2 channels mediate a slow delayed rectifier and maintain high frequency action potential firing in medial nucleus of the trapezoid body neurons. J Physiol. John Wiley & Sons, Ltd. 2008;586(14):3493–509.
Kanagawa M. Dystroglycanopathy: from elucidation of molecular and pathological mechanisms to development of treatment methods. Int J Mol Sci. Multidisciplinary Digital Publishing Institute. 2021;22(23):13162.
Kantor DB, Chivatakarn O, Peer KL, Oster SF, Inatani M, Hansen MJ, et al. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron. 2004;44(6):961–75.
Kappagantula S, Andrews MR, Cheah M, Abad-Rodriguez J, Dotti CG, Fawcett JW. Neu3 sialidase-mediated ganglioside conversion is necessary for axon regeneration and is blocked in CNS axons. J Neurosci. 2014;34(7):2477–92.
Khanna R, Myers MP, Lainé M, Papazian DM. Glycosylation increases potassium channel stability and surface expression in mammalian cells. J Biol Chem. 2001;276(36):34028–34.
Kim KX, Rutherford MA. Maturation of NaV and KV channel topographies in the auditory nerve spike initiator before and after developmental onset of hearing function. J Neurosci. Society for Neuroscience. 2016;36(7):2111–8.
Kim J, Sajid MS, Trakhtenberg EF. The extent of extra-axonal tissue damage determines the levels of CSPG upregulation and the success of experimental axon regeneration in the CNS. Sci Rep. Nature Publishing Group. 2018;8(1):9839–10.
Kimura K, Koizumi T, Urasawa T, Ohta Y, Takakura D, Kawasaki N. Glycoproteomic analysis of the changes in protein N-glycosylation during neuronal differentiation in human-induced pluripotent stem cells and derived neuronal cells. Sci Rep. 2021;11(1):11169.
King AN, Manning CF, Trimmer JS. A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J Comp Neurol. John Wiley & Sons, Ltd. 2014;522(11):2594–608.
Kizuka Y, Taniguchi N. Neural functions of bisecting GlcNAc. Glycoconj J. 2018;35(4):345–51.
Kleene R. The neural recognition molecule L1 is a sialic acid-binding lectin for CD24, which induces promotion and inhibition of neurite outgrowth. J Biol Chem. 2001;276(24):21656–63.
Klein R. Eph/ephrin signaling in morphogenesis, neural development and plasticity. Curr Opin Cell Biol. 2004;16(5):580–9.
Klinger F, Gould G, Boehm S, Shapiro MS. Distribution of M-channel subunits KCNQ2 and KCNQ3 in rat hippocampus. NeuroImage. 2011;58(3):761–9.
Kole MHP, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55(4):633–47.
Kopitz J. Lipid glycosylation: a primer for histochemists and cell biologists. Histochem Cell Biol. Springer Berlin Heidelberg. 2017;147(2):175–98.
Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10(5):323–35.
Koyama R, Ikegaya Y. The molecular and cellular mechanisms of axon guidance in mossy fiber sprouting. Front Neurol. 2018;9:382.
Kruger LC, Isom LL. Voltage-gated Na+ channels: not just for conduction. Cold Spring Harb Perspect Biol. Cold Spring Harbor Lab. 2016;8(6):a029264.
Kudelka MR, Stowell SR, Cummings RD, Neish AS. Intestinal epithelial glycosylation in homeostasis and gut microbiota interactions in IBD. Nat Rev Gastroenterol Hepatol. 2020;17(10):597–617.
Laabs T, Carulli D, Geller HM, Fawcett JW. Chondroitin sulfate proteoglycans in neural development and regeneration. Curr Opin Neurobiol. 2005;15(1):116–20.
Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7(7):548–62.
Lazniewska J, Weiss N. Glycosylation of voltage-gated calcium channels in health and disease. Biochim Biophys Acta Biomembr. 2017;1859(5):662–8.
Lazniewska J, Rzhepetskyy Y, Zhang F-X, Zamponi GW, Weiss N. Cooperative roles of glucose and asparagine-linked glycosylation in T-type calcium channel expression. Pflugers Arch. Springer Berlin Heidelberg. 2016;468(11–12):1837–51.
Ledeen RW, Wu G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem Sci. 2015;40(7):407–18.
Lee BE, Suh P-G, Kim J-I. O-GlcNAcylation in health and neurodegenerative diseases. Exp Mol Med. Nature Publishing Group. 2021;53(11):1674–82.
Lehotzky RE, Partch CL, Mukherjee S, Cash HL, Goldman WE, Gardner KH, et al. Molecular basis for peptidoglycan recognition by a bactericidal lectin. Proc Natl Acad Sci U S A. 2010;107(17):7722–7.
Leterrier C. The axon initial segment, 50 years later: a nexus for neuronal organization and function. Curr Top Membr. 2016;77:185–233.
Lieberoth A, Splittstoesser F, Katagihallimath N, Jakovcevski I, Loers G, Ranscht B, et al. Lewis(x) and alpha2,3-sialyl glycans and their receptors TAG-1, Contactin, and L1 mediate CD24-dependent neurite outgrowth. J Neurosci. 2009;29(20):6677–90.
Lin L, Wang J, Chan C-K, Chan S-O. Effects of exogenous hyaluronan on midline crossing and axon divergence in the optic chiasm of mouse embryos. Eur J Neurosci. John Wiley & Sons, Ltd. 2007;26(1):1–11.
Lindenmaier LB, Parmentier N, Guo C, Tissir F, Wright KM. Dystroglycan is a scaffold for extracellular axon guidance decisions. eLife. 2019;8:e42143.
Lipkin AM, Cunniff MM, Spratt PWE, Lemke SM, Bender KJ. Functional microstructure of CaV-mediated calcium signaling in the axon initial segment. J Neurosci. 2021;41(17):3764–76.
Liu Y, Li X, Yu Y, Shi J, Liang Z, Run X, et al. Developmental regulation of protein O-GlcNAcylation, O-GlcNAc transferase, and O-GlcNAcase in mammalian brain. PLoS One. 2012;7(8):e43724.
Liu Z, Ma A, Mathé E, Merling M, Ma Q, Liu B. Network analyses in microbiome based on high-throughput multi-omics data. Brief Bioinform. 2021;22(2):1639–55.
Lopez-Rodriguez A, Holmgren M. Deglycosylation of Shaker KV channels affects voltage sensing and the open-closed transition. J Gen Physiol. 2018;150(7):1025–34.
Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. Society for Neuroscience. 2008;28(53):14329–40.
Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328(5980):906–9.
Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11(1):173–89.
Marais E, Klugbauer N, Hofmann F. Calcium channel alpha(2)delta subunits-structure and Gabapentin binding. Mol Pharmacol. 2001;59(5):1243–8.
Matsumoto Y, Irie F, Inatani M, Tessier-Lavigne M, Yamaguchi Y. Netrin-1/DCC signaling in commissural axon guidance requires cell-autonomous expression of heparan sulfate. J Neurosci. Society for Neuroscience. 2007;27(16):4342–50.
Messner DJ, Catterall WA. The sodium channel from rat brain. Separation and characterization of subunits. J Biol Chem. 1985;260(19):10597–604.
Miki T, Goto R, Fujimoto M, Okada N, Hardt W-D. The bactericidal lectin RegIIIβ prolongs gut colonization and enteropathy in the streptomycin mouse model for Salmonella diarrhea. Cell Host Microbe. 2017;21(2):195–207.
Miller JA, Agnew WS, Levinson SR. Principal glycopeptide of the tetrodotoxin/saxitoxin binding protein from Electrophorus electricus: isolation and partial chemical and physical characterization. Biochemistry. 1983;22(2):462–70.
Mire E, Hocine M, Bazellières E, Jungas T, Davy A, Chauvet S, et al. Developmental upregulation of Ephrin-B1 silences Sema3C/Neuropilin-1 signaling during post-crossing navigation of corpus callosum axons. Curr Biol. 2018;28(11):1768–1782.e4.
Mochida S. Presynaptic calcium channels. Int J Mol Sci. Multidisciplinary Digital Publishing Institute. 2019;20(9):2217.
Monnier PP, Beck SG, Bolz J, Henke-Fahle S. The polysialic acid moiety of the neural cell adhesion molecule is involved in intraretinal guidance of retinal ganglion cell axons. Dev Biol. 2001;229(1):1–14.
Mountney A, Zahner MR, Lorenzini I, Oudega M, Schramm LP, Schnaar RL. Sialidase enhances recovery from spinal cord contusion injury. Proc Natl Acad Sci U S A. 2010;107(25):11561–6.
Mountney A, Zahner MR, Sturgill ER, Riley CJ, Aston JW, Oudega M, et al. Sialidase, chondroitinase ABC, and combination therapy after spinal cord contusion injury. J Neurotrauma. 2013;30(3):181–90.
Mutalik SP, Gupton SL. Glycosylation in axonal guidance. Int J Mol Sci. 2021;22(10):5143.
Mutoh T, Tokuda A, Miyadai T, Hamaguchi M, Fujiki N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci U S A. 1995;92(11):5087–91.
Mutoh T, Hamano T, Tokuda A, Kuriyama M. Unglycosylated Trk protein does not co-localize nor associate with ganglioside GM1 in stable clone of PC12 cells overexpressing Trk (PCtrk cells). Glycoconj J. 2000;17(3–4):233–7.
Mutoh T, Hamano T, Yano S, Koga H, Yamamoto H, Furukawa K, et al. Stable transfection of GM1 synthase gene into GM1-deficient NG108-15 cells, CR-72 cells, rescues the responsiveness of Trk-neurotrophin receptor to its ligand, NGF. Neurochem Res. 2002;27(7–8):801–6.
Napp J, Monje F, Stühmer W, Pardo LA. Glycosylation of Eag1 (Kv10.1) potassium channels: intracellular trafficking and functional consequences. J Biol Chem. 2005;280(33):29506–12.
Navakkode S, Zhai J, Wong YP, Li G, Soong TW. Enhanced long-term potentiation and impaired learning in mice lacking alternative exon 33 of CaV1.2 calcium channel. Transl Psychiatry. Nature Publishing Group. 2022;12(1):1–10.
Nelson AD, Jenkins PM. Axonal membranes and their domains: assembly and function of the axon initial segment and node of Ranvier. Front Cell Neurosci. 2017;11:136.
Ogawa Y, Horresh I, Trimmer JS, Bredt DS, Peles E, Rasband MN. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci. Society for Neuroscience. 2008;28(22):5731–9.
Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. Society for Neuroscience. 2007;27(22):5903–14.
Ondacova K, Karmazinova M, Lazniewska J, Weiss N, Lacinova L. Modulation of Cav3.2 T-type calcium channel permeability by asparagine-linked glycosylation. Channels (Austin). 2016;10(3):175–84.
Pan Z, Kao T, Horvath Z, Lemos J, Sul J-Y, Cranstoun SD, et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26(10):2599–613.
Paprocka J, Jezela-Stanek A, Tylki-Szymańska A, Grunewald S. Congenital disorders of glycosylation from a neurological perspective. Brain Sci. Multidisciplinary Digital Publishing Institute. 2021;11(1):88.
Park H-J, Min S-H, Won Y-J, Lee J-H. Asn-linked glycosylation contributes to surface expression and voltage-dependent gating of Cav1.2 Ca2+ channel. J Microbiol Biotechnol. 2015;25(8):1371–9.
Pasquini LA, Millet V, Hoyos HC, Giannoni JP, Croci DO, Marder M, et al. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 2011;18(11):1746–56.
Peretto P, Merighi A, Fasolo A, Bonfanti L. Glial tubes in the rostral migratory stream of the adult rat. Brain Res Bull. 1997;42(1):9–21.
Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83(1):117–61.
Petrecca K, Atanasiu R, Akhavan A, Shrier A. N-linked glycosylation sites determine HERG channel surface membrane expression. J Physiol. 1999;515(Pt 1):41–8.
Piper M, Anderson R, Dwivedy A, Weinl C, van Horck F, Leung KM, et al. Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron. 2006;49(2):215–28.
Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4(12):968–80.
Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24(4):1037–47.
Pongs O, Kecskemethy N, Müller R, Krah-Jentgens I, Baumann A, Kiltz HH, et al. Shaker encodes a family of putative potassium channel proteins in the nervous system of Drosophila. EMBO J. European Molecular Biology Organization. 1988;7(4):1087–96.
Poole J, Day CJ, Itzstein von M, Paton JC, Jennings MP. Glycointeractions in bacterial pathogenesis. Nat Rev Microbiol. 2018;16(7):440–52.
Posse De Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010;584(9):1748–59.
Pratt T, Conway CD, Tian NMM-L, Price DJ, Mason JO. Heparan sulphation patterns generated by specific heparan sulfotransferase enzymes direct distinct aspects of retinal axon guidance at the optic chiasm. J Neurosci. Society for Neuroscience. 2006;26(26):6911–23.
Puche AC, Poirier F, Hair M, Bartlett PF, Key B. Role of galectin-1 in the developing mouse olfactory system. Dev Biol. 1996;179(1):274–87.
Rabin SJ, Mocchetti I. GM1 ganglioside activates the high-affinity nerve growth factor receptor trkA. J Neurochem. 1995;65(1):347–54.
Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. Nature Publishing Group. 2010;11(8):552–62.
Rasband MN, Peles E. Mechanisms of node of Ranvier assembly. Nat Rev Neurosci. Nature Publishing Group. 2021;22(1):7–20.
Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, et al. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci. Society for Neuroscience. 1998;18(1):36–47.
Rauvala H, Paveliev M, Kuja-Panula J, Kulesskaya N. Inhibition and enhancement of neural regeneration by chondroitin sulfate proteoglycans. Neural Regen Res. 2017;12(5):687–91.
Recio-Pinto E, Thornhill WB, Duch DS, Levinson SR, Urban BW. Neuraminidase treatment modifies the function of electroplax sodium channels in planar lipid bilayers. Neuron. 1990;5(5):675–84.
Ringot-Destrez B, Kalach N, Mihalache A, Gosset P, Michalski J-C, Léonard R, et al. How do they stick together? Bacterial adhesins implicated in the binding of bacteria to the human gastrointestinal mucins. Biochem Soc Trans. 2017;45(2):389–99.
Roberts RH, Barchi RL. The voltage-sensitive sodium channel from rabbit skeletal muscle. Chemical characterization of subunits. J Biol Chem. 1987;262(5):2298–303.
Rodriguez JA, Piddini E, Hasegawa T, Miyagi T, Dotti CG. Plasma membrane ganglioside sialidase regulates axonal growth and regeneration in hippocampal neurons in culture. J Neurosci. 2001;21(21):8387–95.
Roth J, Zuber C. Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochem Cell Biol. 2017;147(2):269–84.
Ruan H-B, Dietrich MO, Liu Z-W, Zimmer MR, Li M-D, Singh JP, et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell. 2014;159(2):306–17.
Rutishauser U, Landmesser L. Polysialic acid in the vertebrate nervous system: a promoter of plasticity in cell-cell interactions. Trends Neurosci. 1996;19(10):422–7.
Saied-Santiago K, Bülow HE. Diverse roles for glycosaminoglycans in neural patterning. Dev Dyn. John Wiley & Sons, Ltd. 2018;247(1):54–74.
Salkoff L, Wyman R. Genetic modification of potassium channels in Drosophila Shaker mutants. Nature. Nature Publishing Group. 1981;293(5829):228–30.
Sanchez-Ponce D, DeFelipe J, Garrido JJ, Muñoz A. Developmental expression of Kv potassium channels at the axon initial segment of cultured hippocampal neurons. PLoS ONE. Public Library of Science. 2012;7(10):e48557.
Sandoval A, Oviedo N, Andrade A, Felix R. Glycosylation of asparagines 136 and 184 is necessary for the alpha2delta subunit-mediated regulation of voltage-gated Ca2+ channels. FEBS Lett. 2004;576(1–2):21–6.
Sarmiere PD, Weigle CM, Tamkun MM. The Kv2.1 K+ channel targets to the axon initial segment of hippocampal and cortical neurons in culture and in situ. BMC Neurosci. BioMed Central. 2008;9(1):112–5.
Schengrund C-L. Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem Sci. 2015;40(7):397–406.
Schnaar RL. Brain gangliosides in axon–myelin stability and axon regeneration. FEBS Lett. 2010;584(9):1741–7.
Seiradake E, Jones EY, Klein R. Structural perspectives on axon guidance. Annu Rev Cell Dev Biol. 2016;32:577–608.
Sheng M, Liao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. Nature Publishing Group. 1993;365(6441):72–5.
Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18(1):25–9.
Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20(4):809–19.
Smart AD, Course MM, Rawson J, Selleck S, Van Vactor D, Johnson KG. Heparan sulfate proteoglycan specificity during axon pathway formation in the Drosophila embryo. Dev Neurobiol. John Wiley & Sons, Ltd. 2011;71(7):608–18.
Stancic M, Slijepcevic D, Nomden A, Vos MJ, De Jonge JC, Sikkema AH, et al. Galectin-4, a novel neuronal regulator of myelination. Glia. 2012;60(6):919–35.
Storan MJ, Magnaldo T, Biol-N’garagba M-C, Zick Y, Key B. Expression and putative role of lactoseries carbohydrates present on NCAM in the rat primary olfactory pathway. J Comp Neurol. 2004;475(3):289–302.
Susuki K, Otani Y, Rasband MN. Submembranous cytoskeletons stabilize nodes of Ranvier. Exp Neurol. 2016;283(Pt B):446–51.
Sutachan JJ, Watanabe I, Zhu J, Gottschalk A, Recio-Pinto E, Thornhill WB. Effects of Kv1.1 channel glycosylation on C-type inactivation and simulated action potentials. Brain Res. 2005;1058(1–2):30–43.
Tateno H, Toyota M, Saito S, Onuma Y, Ito Y, Hiemori K, et al. Glycome diagnosis of human induced pluripotent stem cells using lectin microarray. J Biol Chem. 2011;286(23):20345–53.
Tenne-Brown J, Puche AC, Key B. Expression of galectin-1 in the mouse olfactory system. Int J Dev Biol. 1998;42(6):791–9.
Terashima M, Amano M, Onodera T, Nishimura S-I, Iwasaki N. Quantitative glycomics monitoring of induced pluripotent- and embryonic stem cells during neuronal differentiation. Stem Cell Res. Elsevier B.V. 2014;13(PA):454–64.
Tétreault M-P, Bourdin B, Briot J, Segura E, Lesage S, Fiset C, et al. Identification of glycosylation sites essential for surface expression of the CaVα2δ1 subunit and modulation of the cardiac CaV1.2 channel activity. J Biol Chem. 2016;291(9):4826–43.
Thayer DA, Yang S-B, Jan Y-N, Jan LY. N-linked glycosylation of Kv1.2 voltage-gated potassium channel facilitates cell surface expression and enhances the stability of internalized channels. J Physiol. 2016;594(22):6701–13.
Thornhill WB, Wu MB, Jiang X, Wu X, Morgan PT, Margiotta JF. Expression of Kv1.1 delayed rectifier potassium channels in Lec mutant Chinese hamster ovary cell lines reveals a role for sialidation in channel function. J Biol Chem. 1996;271(32):19093–8.
Traka M, Dupree JL, Popko B, Karagogeos D. The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J Neurosci. 2002;22(8):3016–24.
Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012;11(8):215–29.
Turner RW, Zamponi GW. T-type channels buddy up. Pflugers Arch. 2014;466(4):661–75.
Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88(4):1407–47.
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–8.
Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J Neurosci. Society for Neuroscience. 2006;26(27):7172–80.
Vicente PC, Kim JY, Ha J-J, Song M-Y, Lee H-K, Kim D-H, et al. Identification and characterization of site-specific N-glycosylation in the potassium channel Kv3.1b. J Cell Physiol. 2018;233(1):549–58.
Vinson M. Myelin-associated glycoprotein interacts with ganglioside GT1b. A mechanism for neurite outgrowth inhibition. J Biol Chem. 2001;276(23):20280–5.
Vuong HE, Pronovost GN, Williams DW, Coley EJL, Siegler EL, Qiu A, et al. The maternal microbiome modulates fetal neurodevelopment in mice. Nature. Springer US. 2020;586(7828):281–6.
Vyas AA, Patel HV, Fromholt SE, Heffer-Lauc M, Vyas KA, Dang J, et al. Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc Natl Acad Sci U S A. 2002;99(12):8412–7.
Walz A, McFarlane S, Brickman YG, Nurcombe V, Bartlett PF, Holt CE. Essential role of heparan sulfates in axon navigation and targeting in the developing visual system. Development. 1997;124(12):2421–30.
Wang J, Ou S-W, Wang Y-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels (Austin). 2017;11(6):534–54.
Wang Q, Wang K, Wu W, Giannoulatou E, Ho JWK, Li L. Host and microbiome multi-omics integration: applications and methodologies. Biophys Rev. 2019;11(1):55–65.
Watanabe I, Wang H-G, Sutachan JJ, Zhu J, Recio-Pinto E, Thornhill WB. Glycosylation affects rat Kv1.1 potassium channel gating by a combined surface potential and cooperative subunit interaction mechanism. J Physiol. 2003;550(Pt 1):51–66.
Watanabe I, Zhu J, Recio-Pinto E, Thornhill WB. Glycosylation affects the protein stability and cell surface expression of Kv1.4 but Not Kv1.1 potassium channels. A pore region determinant dictates the effect of glycosylation on trafficking. J Biol Chem. 2004;279(10):8879–85.
Watanabe I, Zhu J, Sutachan JJ, Gottschalk A, Recio-Pinto E, Thornhill WB. The glycosylation state of Kv1.2 potassium channels affects trafficking, gating, and simulated action potentials. Brain Res. 2007;1144:1–18.
Watanabe I, Zhu J, Recio-Pinto E, Thornhill WB. The degree of N-glycosylation affects the trafficking and cell surface expression levels of Kv1.4 potassium channels. J Membr Biol. 2015;248(2):187–96.
Watson FL, Porcionatto MA, Bhattacharyya A, Stiles CD, Segal RA. TrkA glycosylation regulates receptor localization and activity. J Neurobiol. 1999;39(2):323–36.
Waxman SG, Ritchie JM. Organization of ion channels in the myelinated nerve fiber. Science. 1985;228(4707):1502–7.
Weinhold B, Seidenfaden R, Röckle I, Mühlenhoff M, Schertzinger F, Conzelmann S, Marth JD, Gerardy-Schahn R, Hildebrandt H. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem. 2005;280(52):42971–7.
Weiss N, Hameed S, Fernández-Fernández JM, Fablet K, Karmazinova M, Poillot C, et al. A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem. 2012;287(4):2810–8.
Weiss N, Black SAG, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013;465(8):1159–70.
Whitaker WR, Clare JJ, Powell AJ, Chen YH, Faull RL, Emson PC. Distribution of voltage-gated sodium channel alpha-subunit and beta-subunit mRNAs in human hippocampal formation, cortex, and cerebellum. J Comp Neurol. John Wiley & Sons, Ltd. 2000;422(1):123–39.
Wimmer VC, Reid CA, So EY-W, Berkovic SF, Petrou S. Axon initial segment dysfunction in epilepsy. J Physiol. 2010;588(Pt 11):1829–40.
Wollner DA, Catterall WA. Localization of sodium channels in axon hillocks and initial segments of retinal ganglion cells. Proc Natl Acad Sci USA. National Academy of Sciences. 1986;83(21):8424–8.
Wright KM, Lyon KA, Leung H, Leahy DJ, Ma L, Ginty DD. Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron. 2012;76(5):931–44.
Wu G, Lu Z-H, Obukhov AG, Nowycky MC, Ledeen RW. Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with alpha5beta1 integrin initiates neurite outgrowth. J Neurosci. Society for Neuroscience. 2007;27(28):7447–58.
Wu G, Lu Z-H, André S, Gabius H-J, Ledeen RW. Functional interplay between ganglioside GM1 and cross-linking galectin-1 induces axon-like neuritogenesis via integrin-based signaling and TRPC5-dependent Ca(2+) influx. J Neurochem. 2016;136(3):550–63.
Wulff-Fuentes E, Berendt RR, Massman L, Danner L, Malard F, Vora J, et al. The human O-GlcNAcome database and meta-analysis. Sci Data. 2021;8(1):25.
Yang LJ, Zeller CB, Shaper NL, Kiso M, Hasegawa A, Shapiro RE, et al. Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc Natl Acad Sci U S A. 1996;93(2):814–8.
Yang LJS, Lorenzini I, Vajn K, Mountney A, Schramm LP, Schnaar RL. Sialidase enhances spinal axon outgrowth in vivo. Proc Natl Acad Sci USA. National Academy of Sciences. 2006;103(29):11057–62.
Ye X, Qiu Y, Gao Y, Wan D, Zhu H. A subtle network mediating axon guidance: intrinsic dynamic structure of growth cone, attractive and repulsive molecular cues, and the intermediate role of signaling pathways. Neural Plast. Hindawi. 2019;2019:1719829.
Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. Society for Neuroscience. 2003;23(20):7577–85.
Yung AR, Nishitani AM, Goodrich LV. Phenotypic analysis of mice completely lacking netrin 1. Development. 2015;142(21):3686–91.
Yunker AMR, Sharp AH, Sundarraj S, Ranganathan V, Copeland TD, McEnery MW. Immunological characterization of T-type voltage-dependent calcium channel CaV3.1 (alpha 1G) and CaV3.3 (alpha 1I) isoforms reveal differences in their localization, expression, and neural development. Neuroscience. 2003;117(2):321–35.
Zhang X, Bennett V. Identification of O-linked N-acetylglucosamine modification of ankyrinG isoforms targeted to nodes of Ranvier. J Biol Chem. 1996;271(49):31391–8.
Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143(5):1295–304.
Zhou T-T, Zhang Z-W, Liu J, Zhang J-P, Jiao B-H. Glycosylation of the sodium channel β4 subunit is developmentally regulated and involves in neuritic degeneration. Int J Biol Sci. 2012;8(5):630–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Abad-Rodríguez, J., Brocca, M.E., Higuero, A.M. (2023). Glycans and Carbohydrate-Binding/Transforming Proteins in Axon Physiology. In: Schengrund, CL., Yu, R.K. (eds) Glycobiology of the Nervous System. Advances in Neurobiology, vol 29. Springer, Cham. https://doi.org/10.1007/978-3-031-12390-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-031-12390-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12389-4
Online ISBN: 978-3-031-12390-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)