
Abstract
Guillain-Barré syndrome (GBS) and Miller Fisher syndrome (MFS) are acute immune-mediated neuropathies, often preceded by an infection. Anti-glycolipid antibodies are frequently detected in patients’ sera in the acute-phase. In particular, IgG anti-GQ1b antibodies are positive in as high as 90% of MFS cases. Anti-glycolipid antibodies are useful for the diagnosis of GBS and MFS. In addition, those antibodies may be directly involved in the pathogenetic mechanisms by binding specifically to the regions where the target glycolipid antigen is densely localized. This was proven by the development of animal models of anti-glycolipid antibody-mediated neuropathies. The presence of antibodies that specifically recognize a new conformational epitope formed by two gangliosides (ganglioside complex) in the acute-phase sera of some GBS patients suggested existence of a carbohydrate-carbohydrate interaction between glycolipids. Further intensive research is needed to clarify this point.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Guillain-Barré syndrome
- Miller Fisher syndrome
- Ganglioside
- Neuroimmunology
- Peripheral nerve
- Autoantibody
1 Introduction
There are a group of neurological diseases caused by autoimmune mechanisms. Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP) and IgM paraproteinemic neuropathy affect the peripheral nervous system (PNS) through autoimmune mechanisms and are therefore categorized as autoimmune neuropathies.
GBS is an acute neuropathy, in which weakness in the four limbs is the predominant symptom. Miller Fisher syndrome (MFS) is a variant of GBS, in which ophthalmoplegia and ataxia are the predominant symptoms. GBS and MFS share several characteristics: frequent presence of antecedent infection involving the respiratory or gastrointestinal tract, an acute and self-limited clinical course, and albuminocytological dissociation in cerebrospinal fluid.
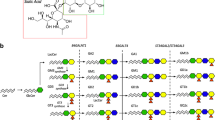
Anti-glycolipid antibodies are frequently present in the acute phase sera from patients with GBS and MFS (Kusunoki 2000; Kaida and Kusunoki 2010). There are diversities in the carbohydrate sequences of the glycolipids (Fig. 16.1). Gangliosides are the glycolipids with sialic acid(s) included in the carbohydrate sequence. Each glycolipid has a unique distribution within the PNS. Anti-glycolipid antibodies can be useful markers for diagnosing both GBS and MFS. Because glycolipids are localized in the plasma membrane with their carbohydrate portions extended to the extracellular space, anti-glycolipid antibodies may bind to those epitopes and cause neuropathy. The antibody titers are highest in the acute phase and decrease with clinical improvement (Kusunoki 2000). It indicates that the elevation of the anti-glycolipid antibody titer may not be a result of damage to the peripheral nerve but may be directly associated with the pathogenetic mechanisms.

Carbohydrate sequences of glycolipids
This chapter focuses primarily on the anti-glycolipid antibodies in GBS and MFS. In addition, the anti-glycolipid antibodies in CIDP and diseases affecting the central nervous system (CNS) are briefly described in the last part.
2 Antibodies to Glycolipid Antigens in GBS and MFS
The effectiveness of plasma exchange in the treatment of GBS was established in the 1980’s. It suggested that autoantibodies binding to the peripheral nervous system are probably involved in the pathogenetic mechanisms of GBS.
Antibodies to the carbohydrate epitopes in autoimmune neuropathies were first reported for the IgM paraproteinemic neuropathy (Braun et al. 1982). The IgM M-protein was shown to bind to myelin-associated glycoprotein (MAG). After that, a unique glycolipid was also reported to be recognized by the same IgM M-protein because the sugar chains containing the sulfated glucuronyl epitope were shared by MAG and the glucolipid, later identified as sulfated glucuronyl paragloboside (SGPG) (Ariga et al. 1987). Subsequently, the frequent presence of anti-GM1 IgM antibody in multifocal motor neuropathy (MMN), considered as a clinical variant of CIDP, was reported (Pestronk et al. 1988). These reports indicated that the antibodies against carbohydrate epitopes are involved in the pathogenesis of autoimmune neuropathies.
Following the above reports, the presence of anti-glycolipid antibodies in the acute-phase sera from GBS patients was reported in 1988 (Ilyas et al. 1988). In that paper, anti-glycolipid antibodies were detected in 5 of 26 cases of GBS. IgG anti-LM1 antibodies were detected in sera from one patient, IgG anti-GD1b antibodies in sera from two, and IgM antibodies to GD1a and GT1b from two. Since then, the association between anti-glycolipid antibodies and GBS has been extensively investigated (Kusunoki 2000; Kaida and Kusunoki 2010).
2.1 IgG Anti-GM1 Antibody
IgG anti-GM1 antibodies in GBS were first reported in 1990 (Yuki et al. 1990), and subsequently many papers reporting their presence in GBS were published (Kusunoki 2000; Kaida and Kusunoki 2010).
PNS myelin was originally considered to be the only target affected in GBS and acute inflammatory demyelinating polyneuropathy (AIDP) had been used as a synonym for GBS before the 1980s. Around 1990, the presence of a GBS subtype in which the primary target is the axonal membrane, rather than myelin, became recognized and that subtype was referred to as acute motor axonal neuropathy (AMAN). The patients with IgG anti-GM1 antibodies had predominantly AMAN type phenotype and frequently had a preceding Campylobacter jejuni infection.
The lipooligosaccharide from GBS patients that had an infection with C. jejuni preceding development of AMAN due to the presence of IgG anti-GM1 antibodies, was shown to have a carbohydrate sequence similar to that of GM1 (Yuki et al. 1993). The production of the anti-GM1 antibodies was therefore considered to be due to immune reactions to the carbohydrate epitopes of C. jejuni, suggesting a “molecular mimicry” mechanism.
2.2 IgG Anti-GQ1b Antibody
In 1992, we reported the detection of the IgG anti-GQ1b antibodies in sera drawn from patients in the acute-phase of MFS (Chiba et al. 1992). The antibodies were present in sera from each of the 6 MFS patients examined in the first paper. Additional studies showed that IgG anti-GQ1b antibodies were present in sera from about 90% of the patients with MFS. IgG anti-GQ1b antibodies were also frequently detected in diseases which had pathogenetic mechanisms similar to those of MFS, such as GBS with ophthalmoplegia and/or ataxia, atypical MFS showing only ophthalmoplegia or ataxia, and Bickerstaff brainstem encephalitis (BBE), which has clinical characteristics of MFS and central nervous system involvement (as described below in Sect. 7.1). The elevation of serum anti-GQ1b IgG antibody titers is therefore closely associated with acute ophthalmoplegia and/or ataxia caused by pathogenetic mechanisms similar to those underlying GBS (Chiba et al. 1993). The discovery of the anti-GQ1b antibody strongly supported the idea that the anti-glycolipid antibodies are specifically associated with the pathogenetic mechanisms of GBS and related disorders. Biochemical analyses of ganglioside fractions from human cranial nerves and spinal nerve roots (ventral and dorsal roots) showed that the cranial nerves innervating extraocular muscles (oculomotor, trochlear and abducens nerves) had a relatively higher content of GQ1b than the other cranial and peripheral nerves (Chiba et al. 1997). A monoclonal anti-GQ1b antibody specifically immunostained paranodal myelin of cranial nerves innervating ocular muscles (Chiba et al. 1992, 1993) and a subset of large primary sensory neurons (Kusunoki et al. 1999a). The binding of the anti-GQ1b antibodies to those regions may be involved in the pathogenesis of ophthalmoplegia and ataxia experienced by MFS patients.
Respiratory infections most frequently precede the neurological onset of IgG anti-GQ1b-induced symptoms in positive patients, while an antecedent infection by C. jejuni is found in some. The production of IgG anti-GQ1b antibodies in GBS and MFS preceded by C. jejuni infection was also shown to be due to the molecular mimicry mechanism. A genetic polymorphism related to the production of polysaccharides by C. jejuni was reported to determine the antibody reactivity (e.g. anti-GM1 or anti-GQ1b) (Koga et al. 2005).
The possible association of anti-GQ1b antibody and respiratory paralysis in GBS was subsequently reported (Kaida et al. 2004a). Binding of the antibody to the distal motor nerve terminals of neuromuscular junctions (Plomp et al. 1999) may be related to respiratory failure, as described in Sect. 3.
2.3 IgG Anti-GD1b Antibodies
GD1b has both a galactosyl-N-acetyl galactosaminyl (Gal-GalNAc) epitope and a disialosyl epitope (Fig. 16.1). When an antibody binds to the Gal-GalNAc epitope, that antibody can also bind to GM1 because GM1 also has the Gal-GalNAc epitope. Patients expressing an antibody to Gal-GalNAc mostly exhibit a clinical phenotype similar to that of the anti-GM1 antibody-positive patients, pure motor or AMAN type GBS. In contrast, when the patients have anti-GD1b antibody without anti-GM1 reactivity, they have different clinical features. It was reported that GBS patients having IgG anti-GD1b antibodies without other anti-glycolipid antibodies had sensory disturbances and none had the primary axonal form (Miyazaki et al. 2001). GD1b was shown to be localized in large neurons in dorsal root ganglia (DRG) and paranodal myelin of human PNS (Kusunoki et al. 1993). Clinical features of GBS patients with monospecific anti-GD1b IgG antibodies may be related to the localization of GD1b in the human PNS. The association of antibodies specific to GD1b and ataxia is described in Sects. 3 and 5.
2.4 IgG Antibodies Against GalNAc-GD1a and GM1b
Gangliosides such as GM1, GD1a, GD1b and GT1b are the major gangliosides in the nervous system and therefore antibodies against those antigens were examined in early studies. However, as GQ1b, a relatively minor component, proved to be an important target antigen, we considered that other minor unidentified gangliosides might also be targets for serum antibodies. Serum antibody activity against a crude ganglioside fraction from bovine brain was examined using thin-layer chromatogram immunostaining. As a result, the IgG antibodies in sera from some GBS patients specifically and strongly bound to an antigen that migrated just below a large amount of GD1a. Interestingly, unlike GD1a, that antigen was resistant to treatment with Clostridium perfringens neuraminidase, (catalyzes hydrolysis of GD1a to yield GM1). Therefore, after removing GD1a by treatment with Cl perfringens neuraminidase, the antigen was isolated by column chromatography and identified as GalNAc-GD1a using fast atom bombardment-mass spectrometry (Kusunoki et al. 1994).
Using a similar methodology, GM1b, another minor ganglioside component, was also shown to be a target antigen for IgG antibodies found in sera from acute-phase GBS patients (Kusunoki et al. 1996a). Both IgG anti-GalNAc-GD1a antibodies and IgG anti-GM1b antibodies were mostly detected in sera from patients with pure motor or AMAN type GBS.
2.5 Antiglycolipid Antibodies in AIDP
As described above, anti-glycolipid antibodies, particularly anti-ganglioside antibodies, are frequently detected in the acute-phase sera from patients with AMAN. In contrast, anti-glycolipid antibodies have been detected less frequently in sera from AIDP patients. Identification of the target molecule recognized by antibodies found in sera from patients with AIDP is a question researchers need to address. Towards that end, a few reports have been published about anti-glycolipid antibodies being associated with AIDP.
2.5.1 Anti-galactocerebroside (Gal-C) Antibody
During routine antibody assays on sera from patients with autoimmune neuropathies, we found an association between anti-Gal-C antibody and infection by Mycoplasma pneumoniae (Kusunoki et al. 1995). As described in the section about animal models, rabbits sensitized with Gal-C were reported to be affected with a demyelinating neuropathy (Saida et al. 1979). This makes it apparent that for the role of anti-Gal-C antibodies and neuropathy, an animal model was developed first and antibodies in sera from patients described later.
Pre-incubation of the patients’ sera with M pneumoniae reagent, derived from M pneumoniae and used for the serological diagnosis, significantly inhibited anti-Gal-C antibody activity in GBS patients after mycoplasma infection. M pneumoniae was shown to express several glycolipids recognized by the anti-Gal-C antibody (Kusunoki et al. 2001). The association between the presence of anti-Gal-C antibodies and the demyelinating type of GBS was reported (Samukawa et al. 2016). This observation supported the previous suggestion that when GBS is preceded by infection with M pneumoniae, the immune reaction against M pneumoniae may induce production of anti-Gal-C antibodies and that those antibodies may be involved in the pathogenesis of demyelinating disease (Kusunoki et al. 2001). This is another example of molecular mimicry in GBS.
2.5.2 Anti-LM1 Antibody
LM1 was shown to be the most abundant component in the glycolipid fraction from human peripheral nerve myelin. It was reported that some patients had IgG antibodies against LM1. GBS cases with such antibody activities had primarily AIDP symptoms (Kuwahara et al. 2011). Therefore, LM1 is among the target antigens in the sera from patients with GBS, mainly AIDP.
Anti-glycolipid antibodies in GBS and MFS, localization of glycolipid antigens and associated clinical features are listed in Table 16.1.
3 Animal Models
The antibodies to glycolipids, including gangliosides, are useful diagnostic markers of GBS. In contrast, it was an issue of controversy whether anti-ganglioside antibodies were pathogenetic autoantibodies in GBS. Now, at least some of the anti-glycolipid antibodies are considered to participate in the pathogenesis because anti-glycolipid antibody-mediated in vivo animal models have been developed.
An animal model with apparent neurological symptoms and signs was first reported by Nagai et al. They sensitized rabbits with GM1 and GD1a (Nagai et al. 1976). In that paper, descriptions of the neurological symptoms and signs, serological examinations, and pathological investigations could not be evaluated in detail. However, we can say that their work paved the way for later research of autoimmune neuropathies with anti-ganglioside antibodies.
Saida et al. sensitized rabbits with Gal-C, a major glycolipid both in the central and peripheral nervous systems. Flaccid paresis and hypesthesia of four limbs were observed in 13 of 31 sensitized rabbits (Saida et al. 1979). Some of the rabbits showed respiratory paresis. Electrophysiological studies showed multifocal conduction block and reduction of motor conduction velocities. Multifocal demyelinating lesions with macrophage infiltration in the PNS were shown pathologically. Intraneural injection of rats with rabbit anti-Gal-C serum induced demyelinating neuropathy, which was shown to be complement mediated. This animal model, reported in 1979, is the first established animal model of an autoimmune neuropathy mediated by an anti-glycolipid antibody. Although the pathogenetic roles of anti-Gal-C antibodies were clearly shown by these works, clinical relevance was not fully recognized at that time because a significant association between anti-Gal-C antibody and human diseases had not been reported. As described above, it wasn’t until 1995 when the presence of anti-Gal-C antibodies in GBS subsequent to mycoplasma infection was reported that the association was made (Kusunoki 1995).
Gal-C, which has no sialic acid, is not a ganglioside but a neutral glycosphingolipid. Even after publication of the animal model of demyelinating neuropathy caused by anti-Gal-C antibodies, there was no established animal model of anti-ganglioside antibody-mediated neuropathy. One reason for the difficulty of developing such an animal model was the difference in the distribution of individual gangliosides from species to species. It was reported that IgM M-protein binding to gangliosides with a disialosyl residue, such as GD1b, was associated with sensory ataxic neuropathy. GD1b is densely localized in the large neurons in human DRG (Kusunoki et al. 1993), suggesting that GD1b could be a target molecule for autoantibodies in sensory ataxic neuropathy. Because GD1b ganglioside is similarly localized in rabbits, we sensitized rabbits with GD1b ganglioside and were able to successfully develop an animal model with robust neurological signs (Kusunoki et al. 1996b). Muscle power was not affected but the rabbits showed awkward movements. Pathologically, there was axonal degeneration in the dorsal root and dorsal column of the spinal cord, showing involvement of large primary sensory neurons mediating deep sensation, whereas the ventral root, consisting of motor nerves, was completely intact. The affected rabbits were diagnosed with sensory ataxic neuropathy pathologically as well as clinically. Titers of anti-GD1b antibodies were increased with no lymphocytic infiltration observed in the affected areas, indicating that the autoantibodies play crucial roles. Further examinations showed that IgG antibodies monospecific to GD1b were the main causative factor in this animal model (Kusunoki et al. 1999b). Passive transfer of the antiserum from the affected rabbits to control rabbits caused neuropathological changes similar to those observed in the affected animals (Kusunoki et al. 1999c). This rabbit model of GD1b-induced sensory ataxic neuropathy (GD1b-induced SAN), was the first established animal model of an anti-ganglioside antibody-mediated autoimmune neuropathy (Kusunoki et al. 1996b). Apoptosis, identified by TUNEL assay and immunohistochemistry using an anti-caspace 3 antibody, was shown to occur in a subset of DRG neurons from affected rabbits. TUNEL positivity was found in large diameter neurons. Therefore, apoptosis of the large primary sensory neurons subsequent to the binding of the GD1b-specific antibodies was considered to be one important pathogenetic mechanism of GD1b-induced SAN (Takada et al. 2008).
Rabbits sensitized with either a bovine brain ganglioside mixture (BBG) or purified GM1 were reported to develop acute motor neuropathy (Yuki et al. 2001). The affected rabbits had anti-GM1 antibodies. Pathological findings showed axonal degeneration with neither lymphocytic infiltration nor demyelination. This study indicated that anti-GM1 antibodies are a causative factor and this model should be a useful animal model for investigation of AMAN. Immunohistochemical investigation of the nodes of Ranvier of a BBG-induced motor axonal neuropathy model showed that voltage-gated sodium channel clusters were disrupted or disappeared from nodes in the acute phase. The deposition of IgG and complement products was observed (Susuki et al. 2007). Combined the results supported the conclusion that BBG-induced motor axonal neuropathy is a complement-mediated disease.
Results obtained using the mouse hemi-diaphragm model indicated that anti-GQ1b antibodies were able to bind and disrupt presynaptic motor nerve terminals at the neuromuscular junction (NMJ) (Plomp et al. 1999). Mice injected intraperitoneally with anti-GQ1b antibody followed by an intraperitoneal injection of normal human serum, as a source of complement, showed breathing difficulties (Halstead et al. 2008). Patients with pure MFS do not develop respiratory weakness, but anti-GQ1b antibody-positive GBS patients more frequently need mechanical ventilation than anti-GQ1b-negative patients (Kaida et al. 2004a). Therefore, this mouse model can be said to be a model of GBS with respiratory insufficiency. It was also reported that eculizumab, which blocks the formation of human C5a and C5b-9, protected mice from respiratory paralysis in this model by preventing complement-mediated damage at motor nerve terminals (Halstead et al. 2008). This work provides us with the rationale for the clinical trials of anti-complement therapy for GBS. The clinical trials of eculizumab have so far presented hopeful results. Anti-complement therapy can possibly be a novel therapeutic method for severe and intractable GBS.
4 Antibodies to a Combination of Ganglioside and Phospholipid
Glycosphingolipids are localized in the plasma membrane, where they are closely surrounded by phospholipids. Therefore, possible effects of phospholipids on the activity of anti-glycolipid antibodies were examined.
The effect of phosphatidic acid (PA) on the antibodies was investigated first. The results showed that PA had an enhancing effect on the activity of IgG anti-GM1 antibodies in GBS. Some patients’ sera showed no antibody activity to GM1 alone but strong binding activity to a mixture of GM1 and PA by ELISA (Kusunoki et al. 2003). Subsequently, the effects of phospholipids other than PA on the IgG anti-GM1 antibody activities were examined. The results showed that phospholipids such as phosphatidyl inositol (PI) and phosphatidyl serine (PS) as well as PA had enhancing effects, but sphingomyelin (SM) was inhibitory. As for IgG anti-GQ1b antibodies, the enhancing effects of PA, PI, and PS were not significant while SM inhibition was seen. (Hirakawa et al. 2005).
Possible explanations for the inhibitory effect of SM on the binding activity of IgG antibodies to ganglioside epitopes is that the polar head group of SM alters the local charge thereby inhibiting antibody binding. To investigate the pathogenetic role of anti-glycolipid antibodies in autoimmune neuropathies, more attention should be focused on the effects of phospholipids that surround the glycolipids in cell membranes of the nervous system.
5 Antibodies to Ganglioside Complexes in GBS
Gangliosides tend to form clusters in the plasma membrane (Hakomori 2002). In the clusters, the carbohydrate structure of a ganglioside may interact with that of another to form a novel epitope. Some GBS patients have serum antibodies that specifically recognize novel glycoepitopes formed by two individual ganglioside molecules. We named such antibodies as “anti-ganglioside complex (GSC) antibodies” (Kaida et al. 2004a, b).
How we found the presence of anti-ganglioside complex antibodies is as follows. We investigated serum from a patient with acute severe flaccid tetraparesis by an enzyme-linked immunosorbent assay (ELISA) and thin-layer chromatogram (TLC)-immunostaining. ELISA results were negative for each of the gangliosides tested (GalNAc-GD1a, GM1, GM2, GM3, GD1a, GD1b, GD3, GT1b, and GQ1b). However, we found an unidentified immunoreactive band in the position just below GD1a on TLC of a crude ganglioside fraction from bovine brain. Upon examination of the binding activity of that serum IgG to a mixture of GD1a and GD1b by ELISA, the serum IgG was found to bind strongly to the well coated with a mixture of GD1a and GD1b (GD1a/GD1b). In TLC-immunostaining using a developing solvent of chloroform/ methanol/0.2%CaCl2–2H2O (50:45:10), positive staining was present in the lane in which both GD1a and GD1b were developed, but not in the lanes in which GD1a or GD1b were developed separately. Immunostaining was only present in the overlapping portion of GD1a and GD1b (Fig. 16.2). When another developing solvent (C/M/0.2%CaCl2–2H2O, 30/65/10) was used that completely separated the GD1a from GD1b, the immunostaining disappeared (Kaida et al. 2004b). Mixing GD1a and GD1b may produce a new conformational glycoepitope which is different from that of GD1a or GD1b alone and that the antibody specifically recognizes.

ELISA result for a serum with IgG antibodies against a GD1a/GD1b complex. The antibody activities were expressed as optical density (OD) values. IgG in this serum had strong reactivity against a mixture of GD1a and GD1b (GD1a/GD1b) but only a subtle reaction with either one alone
The anti-GD1a/GD1b-positive serum antibodies also recognized GM1/GD1a, GM1/GT1b, and GD1b/GT1b. All of the 4 antigens are combinations of a ganglioside with a terminal galactosyl epitope and one with a sialyl-galactosyl epitope (Fig. 16.1). Therefore, it can be said that the antibodies recognized the combined epitope of carbohydrate sequences.
Most of the sera from MFS patients have IgG antibodies to GQ1b, as described above. However, when binding of the anti-GSC antibodies were examined, some MFS sera had higher binding activities against a combination of GQ1b and another ganglioside than against GQ1b alone. MFS sera could be categorized into 3 patterns: those specific to (1) GQ1b alone, (2) GQ1b/GM1, and (3) GQ1b/GD1a (Kaida et al. 2006).
The reactivity of the monospecific anti-GD1b IgG against a mixture of GD1b and another ganglioside was also examined. If the interaction of two gangliosides creates new epitopes with conformational changes, the binding activity of the antibody highly specific to one ganglioside may be decreased by the addition of another ganglioside. The results showed that the reactivity of the IgG antibody monospecific to GD1b in the routine antibody assay was not affected by the addition of GM1, GM2, GM3 and GA1 (Group A), but strongly inhibited by the addition of GD1a, GD3, GT1a, GTb, GQ1b and GalNAc-GD1a (Group B) (Kaida et al. 2008). Thus, the GD1b may interact with Group B gangliosides (but not with Group A gangliosides) to form a novel epitope, not recognized by the highly specific anti-GD1b antibody. This result provides indirect evidence for the carbohydrate-carbohydrate interaction between two different gangliosides or their aggregation may provide a better ligand for antibody binding. In addition, the percent reduction of antibody binding was different between patients with ataxia and those without: binding after addition of Group B gangliosides was significantly more reduced in ataxic patients than in those without ataxia. This not only suggests that the anti-GD1b IgG antibodies in ataxic patients may be more specific to GD1b, it further confirmed the association between anti-GD1b antibody and sensory ataxic neuropathy.
The number of glycolipid complexes that are able to be examined by standard ELISA is limited. It is possible that there are antibodies to still unidentified glycolipid complexes that are present in acute-phase sera from GBS patients. To investigate antibody activities to a number of glycolipid complexes more efficiently, a combinatorial glycoarray method was recently developed (Rinaldi et al. 2013). Using this method, we found 4 of 100 patients with GBS had IgG antibodies that only recognized a GQ1b/sulfatide complex, not adhering to GQ1b or sulfatide alone. All of the four patients had ophthalmoplegia (Morikawa et al. 2016).
In another study, of 63 patients with GBS with ophthalmoplegia, 31 patients had IgG antibody activities not only to GQ1b and GSCs containing GQ1b (GQ1b-related antibodies) but also to GD1b and GSCs containing GD1b (GD1b-related antibodies). It was found that those patients with both GQ1b-related antibodies and GD1b-related antibodies required mechanical ventilation more frequently (Yoshikawa et al. 2018). Those antibody activities can possibly be used as a prognostic biomarker of GBS.
6 Anti-glycolipid Antibodies in CIDP
IgM anti-GM1 antibodies are detected in the sera from patients with multifocal motor neuropathy (MMN), a clinical variant of CIDP. In contrast, anti-glycolipid antibodies are usually not detected in sera from patients with typical CIDP.
Recently, we found some CIDP patients, as well as AIDP patients, had IgG antibodies against LM1 and LM1-contaning GSCs. Those CIDP patients with IgG antibodies to LM1-related antigens frequently had ataxia. In addition, they did not show cranial nerve involvement (Kuwahara et al. 2013). It may be explained by the localization of LM1 in the peripheral nerve; LM1 is a major glycolipid antigen in the peripheral nerve innervating extremities but is scarcely detected in cranial nerves. Nerve biopsy finding from a CIDP patient with IgG anti-LM1 antibody showed deposition of complement on the myelin (Koike et al. 2020). IgG anti-LM1 antibody could be related to the complement-mediated demyelination seen in CIDP.
It is possible that some minor unidentified glycolipid antigens or GSCs are the targets for CIDP sera. Further investigation is necessary.
7 Anti-glycolipid Antibodies in Diseases Affecting the Central Nervous System
Anti-glycolipid antibodies reported so far are mostly associated with autoimmune neuropathies that affect the PNS. Considering that glycolipids are even more abundant in the CNS than PNS, it is possible that anti-glycolipid antibodies are also involved in the pathogenesis of CNS diseases. The following are two such examples.
7.1 Anti-GQ1b Antibodies in Bickerstaff Brainstem Encephalitis
Bickerstaff brainstem encephalitis (BBE) is an acute self-limited autoimmune encephalitis. Ophthalmoplegia, ataxia and impaired consciousness are the triads of BBE. IgG anti-GQ1b antibodies are frequently detected in sera from BBE patients and BBE is considered to be a disease related to MFS. Recently, clinical characteristics of anti-GQ1b antibody-positive and –negative BBE were investigated. Results showed that preceding upper respiratory infection and sensory disturbance were more common, the cell count or protein concentration was lower in the cerebrospinal fluid, abnormal findings upon brain MRI were less, and the consciousness disturbance disappeared earlier in anti-GQ1b antibody-positive BBE (Yoshikawa et al. 2020). Therefore, BBE with IgG anti-GQ1b antibodies has homogeneous features, suggesting important pathogenetic roles of anti-GQ1b antibodies. Characteristics of the anti-GQ1b antibodies in BBE are not different from those in MFS (Yoshikawa et al. 2018), suggesting that some other factors may be needed for the development of consciousness disturbance.
7.2 Anti-Gal-C Antibodies
Gal-C is a major glycolipid in myelin in both the PNS and CNS. The spectrum of clinical characteristics and the antibodies to glycolipids including Gal-C were recently investigated in neurological diseases following M.pneumoniae infection (Kuwahara et al. 2017). Of the 46 patients studied, 30 were affected with GBS, 17 with CNS diseases (one patient was affected with both GBS and CNS disease). The patients with CNS diseases were significantly younger than those with GBS. Anti-Gal-C antibodies were the most frequently detected antibodies. In the patients with CNS diseases, IgM antibodies to Gal-C were more common than IgG antibodies. In contrast, in GBS, IgG antibodies to Gal-C were more common than IgM antibodies. It can be speculated that IgM anti-Gal-C antibodies are produced in younger patients affected for the first time with M. pneumoniae. Because the blood-brain barrier is not yet mature in younger patients, the antibodies produced might more easily access the CNS and cause inflammatory disease.
More intensive investigation of pediatric cases may be necessary to reveal the possible roles of anti-glycolipid antibodies in CNS diseases.
8 Future Perspectives
8.1 More Efficient Use of Anti-glycolipid Antibodies as Diagnostic and Prognostic Markers
The detection of a certain IgG anti-glycolipid antibodies in the acute-phase sera from patients with neuropathy strongly suggests the diagnosis of GBS. However, locations in which antibody assays can be performed is limited and hence they are not yet included in routine laboratory examinations. Development of easy and rapid assay methods is needed for the efficient use of anti-glycolipid antibodies in the daily clinical treatment of GBS.
The use of anti-glycolipid antibodies as a prognostic marker is also required in neurology. IgG antibodies to GD1a were previously shown to be associated with AMAN type GBS (Ho et al. 1999). Most recently, the association between IgG anti-GD1a antibodies and poor outcome, inability to walk independently at 6 months after disease onset, was reported (Yamagishi et al. 2020). mEGOS (modified Erasmus GBS outcome score) is a clinical score of GBS and has been reported as a useful prognostic marker. The combination of serum IgG anti-GD1a antibodies and a high mEGOS was found to provide a more accurate prediction of poor prognosis than mEGOS alone (Yamagishi et al. 2020).
8.2 To Determine the Clinical Significance of GSCs and the Anti-GSC Antibodies
Considering the characteristic formation of clusters of gangliosides in the plasma membrane (Hakomori 2002), anti-GSC antibodies might cause nerve dysfunction more efficiently than those specific to a single ganglioside. Future studies on GSCs in basic glycobiology should provide us with an understanding of the possible roles of GSCs on the cell membrane and clinical relevance of anti-GSC antibodies.
9 Conclusion
Anti-glycolipid antibodies are frequently present in sera from patients during the acute phase of GBS, MFS and related diseases. Discovery of the antibody activities to glycolipid complexes broadened the perspectives of research in these diseases. Further intensive research in this area is needed to clarify the pathogenetic mechanisms and develop useful biomarkers for the improvement of the clinical management of GBS, MFS and related autoimmune neurological diseases.
Abbreviations
- AIDP:
-
Acute inflammatory demyelinating polyneuropathy
- AMAN:
-
Acute motor axonal neuropathy
- BBE:
-
Bickerstaff brainstem encephalitis
- CIDP:
-
Chronic inflammatory demyelinating polyneuropathy
- CNS :
-
Central nervous system
- Gal-C:
-
Galactocerebroside
- GBS :
-
Guillain-Barré syndrome
- GSC:
-
Ganglioside complex
- MFS :
-
Miller Fisher syndrome
- MMN:
-
Multifocal motor neuropathy
- PNS:
-
Peripheral nervous system
- SGPG:
-
Sulfated glucuronyl paragloboside
References
Ariga T, Kohriyama T, Freddo L, Latov N, Saito M, Kon K, et al. Characterization of sulfated glucuronic acid containing glycolipids reacting with IgM M-proteins in patients with neuropathy. J Biol Chem. 1987;262:848–53.
Braun PE, Frail DE, Latov N. Myelin-associated glycoprotein is the antigen for a monoclonal IgM in polyneuropathy. J Neurochem. 1982;39:1261–5.
Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol. 1992;31:677–9.
Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology. 1993;43:1911–7.
Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Ganglioside composition of the human cranial nerves, with special reference to pathophysiology of Miller Fisher syndrome. Brain Res. 1997;745:32–6.
Hakomori S. The glycosynapse. Proc Natl Acad Sci U S A. 2002;99:225–32.
Halstead SK, Zitman FM, Humphreys PD, Greenshields K, Verschuuren JJ, Jacobs BC, et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008;131:1197–208.
Hirakawa M, Morita D, Tsuji S, Kusunoki S. Effects of phospholipids on antigangliside antibody reactivity in GBS. J Neuroimmunol. 2005;159:129–32.
Ho TW, Willison HJ, Nachamkin I, Li CY, Veitch J, Ung H, et al. Anti-GD1a antibody is associated with axonal but not demyelinating forms of Guillain-Barré syndrome. Ann Neurol. 1999;45:168–73.
Ilyas AA, Willison HJ, Quarles RH, Jungalwala FB, Cornblath DR, Trapp BD, et al. Serum antibodies to gangliosides in Guillain-Barré syndrome. Ann Neurol. 1988;23:440–7.
Kaida K, Kusunoki S. Antibodies to gangliosides and ganglioside complexes in Guillain–Barré syndrome and Fisher syndrome: mini-review. J Neuroimmunol. 2010;223:5–12.
Kaida K, Kusunoki S, Kanzaki M, Kamakura K, Motoyoshi K, Kanazawa I. Anti-GQ1b antibody as a factor predictive of mechanical ventilation in Guillain-Barré syndrome. Neurology. 2004a;62:821–4.
Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M, et al. Ganglioside complexes as new target antigens in Guillain-Barré syndrome. Ann Neurol. 2004b;56:567–71.
Kaida K, Kanzaki M, Morita D, Kamakura K, Motoyoshi K, Hirakawa M, et al. Anti-ganglioside complex antibodies in Miller Fisher syndrome. J Neurol Neurosurg Psychiatry. 2006;77:1043–6.
Kaida K, Kamakura K, Ogawa G, et al. GD1b-specific antibody induces ataxia in Guillain-Barré syndrome. Neurology. 2008;71:196–201.
Koga M, Takahashi M, Masuda M, Hirata K, Yuki N. Campylobacter gene polymorphism as a determinant of clinical features of Guillain-Barré syndrome. Neurology. 2005;65:1376–81.
Koike H, Ikeda S, Fukami Y, Nishi R, Kawagashira Y, Iijima M, et al. Complement deposition and macrophage-induced demyelination in CIDP with anti-LM1 antibodies. J Neurol Sci. 2020;408:116509.
Kusunoki S. Antiglycolipid antibodies in Guillain-Barré syndrome and autoimmune neuropathies. Am J Med Sci. 2000;319:234–9.
Kusunoki S, Chiba A, Tai T, Kanazawa I. Localization of GM1 and GD1b antigens in the human peripheral nervous system. Muscle Nerve. 1993;16:752–6.
Kusunoki S, Chiba A, Kon K, Ando S, Arisawa K, Tate A, et al. N-acetylgalactosaminyl GD1a is a target molecule for serum antibody in Guillain-Barre syndrome. Ann Neurol. 1994;35:570–6.
Kusunoki S, Chiba A, Hitoshi S, Takizawa H, Kanazawa I. Anti-Gal-C antibody in autoimmune neuropathies subsequent to mycoplasma infection. Muscle Nerve. 1995;18:409–13.
Kusunoki S, Iwamori M, Chiba A, Hitoshi S, Arita M, Kanazawa I. GM1b is a new member of antigen for serum antibody in Guillain-Barre syndrome. Neurology. 1996a;47:237–42.
Kusunoki S, Shimizu J, Chiba A, Ugawa Y, Hitoshi S, Kanazawa I. Experimental sensory neuropathy induced by sensitization with ganglioside GD1b. Ann Neurol. 1996b;39:424–31.
Kusunoki S, Chiba A, Kanazawa I. Anti-GQ1b IgG antibody is associated with ataxia as well as ophthalmoplegia. Muscle Nerve. 1999a;22:1071–4.
Kusunoki S, Hitoshi S, Kaida K, Arita M, Kanazawa I. Monospecific anti-GD1b IgG is required to induce rabbit ataxic neuropathy. Ann Neurol. 1999b;45:400–3.
Kusunoki S, Hitoshi S, Kaida K, Murayama S, Kanazawa I. Degeneration of rabbit sensory neurons induced by passive transfer of anti-GD1b antiserum. Neurosci Lett. 1999c;273:33–6.
Kusunoki S, Shiina M, Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. 2001;57:736–8.
Kusunoki S, Morita D, Ohminami S, Hitoshi S, Kanazawa I. Binding of immunoglobulin G antibodies in Guillain-Barre syndrome sera to a mixture of GM1 and a phospholipid: possible clinical implications. Muscle Nerve. 2003;27:302–6.
Kuwahara M, Suzuki S, Takada K, Kusunoki S. Antibodies to LM1 and LM1-containing ganglioside complexes in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol. 2011;239:87–90.
Kuwahara M, Suzuki H, Samukawa M, Hamada Y, Takada K, Kusunoki S. Clinical features of CIDP with LM1-associated antibodies. J Neurol Neurosurg Psychiatry. 2013;84:573–5.
Kuwahara M, Samukawa M, Ikeda T, Morikawa M, Ueno R, Hamada Y, et al. Characterization of the neurological diseases associated with Mycoplasma pneumoniae infection and anti-glycolipid antibodies. J Neurol. 2017;264:467–75.
Miyazaki T, Kusunoki S, Kaida K, Shiina M, Kanazawa I. Guillain-Barré syndrome associated with IgG monospecific to ganglioside GD1b. Neurology. 2001;56:1227–9.
Morikawa M, Kuwahara M, Ueno R, Samukawa M, Hamada Y, Kusunoki S. Serological study using glycoarray for detecting antibodies to glycolipids and glycolipid complexes in immune-mediated neuropathies. J Neuroimmunol. 2016;301:35–40.
Nagai Y, Momoi T, Saito M, Mitsuzawa E, Ohtani S. Ganglioside syndrome, a new autoimmune neurologic disorder, experimentally induced with brain gangliosides. Neurosci Lett. 1976;2:107–11.
Pestronk A, Cornblath DR, Ilyas AA, Baba H, Quarles RH, Griffin JW, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. 1988;24:73–8.
Plomp JJ, Molenaar PC, O’Hanlon GM, Jacobs BC, Veitch J, Daha MR, et al. Miller Fisher anti-GQ1b antibodies:α-latrotoxin–like effects on motor end plates. Ann Neurol. 1999;45:189–99.
Rinaldi S, Brennan KM, Kalna G, Walgaard C, van Doorn P, Jacobs BC, et al. Antibodies to heteromeric glycolipid complexes in Guillain-Barre syndrome. PLoS One. 2013;8:e82337.
Saida T, Saida K, Dorfman SH, Silberberg DH, Sumner AJ, Manning MC, et al. Experimental allergic neuritis induced by sensitization with galactocerebroside. Science. 1979;204:1103–6.
Samukawa M, Kuwahara M, Morikawa M, Ueno R, Hamada Y, Takada K, et al. Electrophysiological assessment of Guillain-Barré syndrome with both Gal-C and ganglioside antibodies; tendency for demyelinating type. J Neuroimmunol. 2016;301:61–4.
Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, et al. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci. 2007;27:3956–67.
Takada K, Shimizu J, Kusunoki S. Apoptosis of primary sensory neurons in GD1b-induced sensory ataxic neuropathy. Exp Neurol. 2008;209:279–83.
Yamagishi Y, Kuwahara M, Suzuki H, Sonoo M, Kuwabara S, Yokota T, et al. Serum IgG anti-GD1a antibody and mEGOS predict outcome in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2020;91:1339–42.
Yoshikawa K, Kuwahara M, Morikawa M, Fukumoto Y, Yamana M, Yamagishi Y, et al. Varied antibody reactivities and clinical relevance in anti-GQ1b antibody–related diseases. Neurol Neuroimmunol Neuroinflamm. 2018;5:e501.
Yoshikawa K, Kuwahara M, Morikawa M, Kusunoki S. Bickerstaff brainstem encephalitis with or without anti-GQ1b antibody. Neurol Neuroimmunol Neuroinflamm. 2020;7:e889.
Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990;40:1900–2.
Yuki N, Taki T, Inagaki F, Kasama T, Takahashi M, Saito K, et al. A bacterium lipopolysaccharide that elicits Guillain-Barré syndrome has a GM1 ganglioside-like structure. J Exp Med. 1993;178:1771–5.
Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y, et al. Animal model of axonal Guillain-Barré syndrome induced by sensitization with GM1 ganglioside. Ann Neurol. 2001;49:712–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kusunoki, S. (2023). Antibodies to Glycolipids in Guillain-Barré Syndrome, Miller Fisher Syndrome and Related Autoimmune Neurological Diseases. In: Schengrund, CL., Yu, R.K. (eds) Glycobiology of the Nervous System. Advances in Neurobiology, vol 29. Springer, Cham. https://doi.org/10.1007/978-3-031-12390-0_16
Download citation
DOI: https://doi.org/10.1007/978-3-031-12390-0_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12389-4
Online ISBN: 978-3-031-12390-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)