
Abstract
This chapter lifts the uncertainty quantification of the previous chapters up to the level of molecular evolution hypotheses. A relatively recent rival to the neutral theory of molecular evolution is explained in terms of how its predictions differ. The exercises at the end of the chapter give readers experience with quantifying the extent to which sequence data support one evolutionary hypothesis more than another.
In applying mathematics to subjects such as physics or statistics we make tentative assumptions about the real world which we know are false but which we believe may be useful nonetheless. – George E. P. Box1
Is it of the slightest use to reject a hypothesis until we have some idea of what to put in its place? … There has not been a single date in the history of the law of gravitation when a modern significance test would not have rejected all laws and left us with no law.
– Sir Harold Jeffreys2
“Science and Statistics,” Journal of the American Statistical Association [25], copyright Ⓒ American Statistical Association, reprinted by permission of Taylor & Francis Ltd, http://www.tandfonline.com on behalf of American Statistical Association.
Theory of Probability (Oxford University Press) [63, §7.22]. Reproduced with permission of the Licensor through PLSclear.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Maximum genetic diversity
- Epigenetic complexity
- Rapid-evolution saturation
- Outgroup
- Genetic equidistance phenomenon
- Genetic non-equidistance phenomenon
- Slow clock method
- Macroevolution
- Microevolution
- Common descent
In addition to the sources of uncertainty explained in Sects. 3.4, 5.5.1, and 4.2.1, there is uncertainty about whether the neutral theory (Sect. 1.2.2) or one of its alternatives is approximately true or at least adequate as a working hypothesis. Chapter 2 chronicles some challenges faced by the neutral theory.
The main working hypothesis of this chapter was proposed as an alternative to the neutral theory and its nearly neutral variant discussed in Sects. 2.1–2.2. That working hypothesis is summarized in Sect. 6.1. Some potential supporting evidence is then explained in Sect. 6.2. The working hypothesis suggests that the method of phylogenetic tree reconstruction that is outlined in Sect. 6.3. Finally, some open questions are raised in Sect. 6.4.
6.1 Maximum Genetic Diversity Hypothesis
The terminology and concepts of this section follow Huang [59]; see Hu et al. [58] and Huang [60] for reviews and Wang et al. [124] for a recent development. The genetic diversity of a taxon with respect to a protein or DNA sequence is the percentage of positions in the sequence that differ among members of the taxon. The upper limit of that percentage is called the maximum genetic diversity of the taxon and is achieved for rapidly changing sequences. The maximum genetic diversity measures the fraction of positions in the sequence that are free to change without negatively impacting the fitness of the members of the taxon. Such changes either improve fitness or are neutral (Shi Huang, personal communication). The positions not free to change are considered conserved.
The epigenetic complexity of a taxon is an average number of cell types of its individual members. Taxa of higher epigenetic complexity are considered more complex, whereas those of lower epigenetic complexity are considered simpler.
The maximum genetic diversity hypothesis of Huang [59] makes these claims:
-
(1)
Maximum genetic diversity tends to be higher for simpler taxa and lower for more complex taxa. The reason is that the physiology of members of more complex taxa relies on more sequence positions, which are for that reason conserved, leaving fewer positions free to change.
-
(2)
The positions that are conserved in simpler taxa tend to also be conserved in more complex taxa. In other words, the positions that are free to change in more complex taxa tend to also be free to change in simpler taxa.
-
(3)
The gradual evolution of sequences takes place at the microevolution level but cannot be extrapolated to the scale of macroevolution, as Gould [52] had concluded largely on the basis of the fossil record. Macroevolution instead involves increasing epigenetic complexity and decreasing maximum genetic diversity rather than substitutions at positions of a protein or DNA sequence.
6.2 Genetic Equidistance Phenomenon
When sequences of two taxa are compared to each other by computing their distances to the sequence of a third taxon, the third taxon is called the baseline group. If the baseline group is less related to each of the other taxa than they are to each other, then it is called an outgroup. In terms of Fig. 3.4, an outgroup is a cousin to the other two taxa, which are sisters of each other. Such comparisons to an outgroup or to another baseline group are used to test the predictions of hypothesis assuming the neutrality of substitutions.
The maximum genetic diversity hypothesis makes the same predictions as the molecular clock hypothesis (discussed in Sect. 1.2.2 and used in Sect. 3.1.2) except for sequences that evolve rapidly enough that non-conserved positions tend to experience substitutions. For such sequences, Claims 1–2 of the maximum genetic diversity hypothesis (Sect. 6.1) have the consequence of rapid-evolution saturation: the estimated number of substitutions between two sequences tends to be equal to the number of non-conserved sites in the simpler of the two taxa due to multiple substitutions at the same sites. That consequence leads to these predictions for substitutions (defined in Sec. 1.2.2) in rapidly evolving sequences [59, Fig. 3]:
-
(1)
Simpler outgroup. Considering the tip taxa at the bottom of the tree in Fig. 3.4, suppose Cousin is the simplest and that Sister 1 is the most complex. When the outgroup (Cousin) is simpler than the sister taxa (Sisters 1–2), the distances of the sisters to the outgroup are about equal. In the distance notation of Sect. 3.1.2.1,
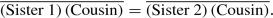
That happens because for rapidly evolving sequences, changes in the lineage leading to the outgroup (Cousin) mask any changes in the lineages leading to the other two taxa (Sisters 1–2). That in turn is a result of rapid-evolution saturation.
-
That equality of distances, called the genetic equidistance phenomenon, has often been observed and is also a prediction of the molecular clock hypothesis (Fig. 2.1). In fact, the molecular clock hypothesis was proposed in order to explain the genetic equidistance phenomenon (Sect. 2.1).
-
The genetic equidistance phenomenon was not predicted by the hypothesis that most differences between the sequences resulted from natural selection. The failure of that selection hypothesis led to several decades of debates related to how much of a role selection played in molecular evolution (Chap. 2).
-
The maximum genetic diversity hypothesis, holding that most differences between the sequences did in fact result from natural selection, explains the genetic equidistance phenomenon as a result of rapid-evolution saturation (Shi Huang, personal communication).
-
-
-
(2)
More complex outgroup. Relabeling the tips of the tree in Fig. 3.4, suppose Cousin is the most complex and that Sister 1 is the simplest. When the outgroup (Cousin) is more complex than the sister taxa, the distance from the more complex sister (Sister 2) to the outgroup (Cousin) is less than the distance from the simpler sister (Sister 1) to the outgroup (Cousin):
 (6.1)
(6.1)That is because there are fewer conserved sites in the lineage leading to the simpler of the two sisters (Sister 1) and because substitutions in the more complex outgroup (Cousin) are hidden by those of each of the two simpler taxa (Sisters 1-2) due to rapid-evolution saturation.
-
That genetic non-equidistance phenomenon is not a prediction of the molecular clock hypothesis, which instead predicts that the distance to each of the sister taxa to the outgroup (Cousin) is about equal:
 (6.2)
(6.2)
-
-
(3)
Simpler non-outgroup baseline group. Leaving the labels of Fig. 3.4 unchanged, again suppose Cousin is the most complex and that Sister 1 is the simplest. When the baseline group is not an outgroup but is the simplest of the three taxa (Sister 1) and is more closely related to the taxon of intermediate complexity (Sister 2) than to the taxon of highest complexity (Cousin), the distance between the simplest two taxa (Sisters 1-2) is approximately equal to the distance between the simplex taxon (Sister 1) and the most complex taxon (Cousin):
 (6.3)
(6.3)This results from rapid-evolution saturation: substitutions in the lineage leading to the simplest taxon (Sister 1) mask those in the lineages leading to the other two taxa (Sister 2 and Cousin).
-
That is not a prediction of the molecular clock hypothesis, which instead predicts that the distance from the simplest taxon (Sister 1) to its sister taxon (Sister 2) is smaller than the distance from the simplest taxon (Sister 1) to the most complex taxon (Cousin):
 (6.4)
(6.4)
-









Example 3
Snakes are simpler than birds, and birds are simpler than humans; snakes and birds are more similar to each other than to humans. For rapidly evolving sequences, the maximum genetic diversity hypothesis then makes these predictions:
-
More complex outgroup. With the human taxon as the outgroup (Cousin), the distance from birds (Sister 2) to humans is less than the distance from snakes (Sister 1) to humans according to formula (6.1). (The “Cousin” and “Sister” labels in parentheses are those of Fig. 3.4.)
-
The first column of Table 6.1 shows a consistent difference between those distances in the direction predicted by the maximum genetic diversity hypothesis. That observation indicates that the molecular clock hypothesis does not apply since it predicted those distances to be about equal, as seen in formula (6.2).
Table 6.1 Comparison of rapidly evolving protein sequences from snakes (simplest), birds (intermediate complexity), and humans (most complex). The last row displays mean pairwise differences. The displayed 95% confidence intervals of the mean difference in identity assume the 23%-identities of each sample are normally distributed, but the actual uncertainty is higher since only 13 of the 23 proteins were randomly selected. The maximum genetic diversity hypothesis is much better supported in this case than the molecular clock hypothesis (Example 3), though both hypotheses would be rejected at the 5% level according to null hypothesis significance testing [18]. The numbers shown were calculated from the data of Huang [59, Table S3]
-
-
Simpler non-outgroup baseline group. With the snake taxon (Sister 1) as a non-outgroup used for reference, the distance of birds (Sister 2) to snakes and the distance of humans (Cousin) to snakes are about equal according to formula (6.3).
-
The second column of Table 6.1 shows a relatively small difference between those distances. That small observed difference is not a prediction of the molecular clock hypothesis, which instead predicts that snakes (Sister 1) would be more consistently closer to birds (Sister 2) than to humans (Cousin) according to formula (6.4). \(\blacktriangle \)
-
6.3 Slow Clock Method
If Table 6.1 were used to construct a phylogenetic tree with snakes, birds, and humans, it would say that birds and humans diverged from a common ancestor more recently than the lineage of that ancestor diverged from snakes. In terms of Fig. 3.4, birds and humans are sister taxa related to the more distantly related cousin taxon of snakes, contradicting Example 3. According to Huang [59], the implausibility of those evolutionary relationships casts doubt on phylogenetic trees estimated on the basis of rapidly evolving sequences.
To solve that kind of problem, Huang [59] proposed the slow clock method of estimating phylogenetic trees from three taxa:
-
(1)
Only include slowly evolving sequences in the alignment.
-
That is recommended because the resulting estimates are not subject to rapid-evolution saturation and because slow evolution tends to be more neutral and consequently in better agreement with the molecular clock hypothesis (Shi Huang, personal communication).
-
-
(2)
Use the simplest of the three taxa as the outgroup to construct the distance matrix.
-
(3)
Use a distance-based method of estimating phylogenetic trees.
Example 4
To compare two taxa of pongids (gorillas and chimpanzees) to humans, the slow clock method suggests a simpler outgroup such as orangutans. Tables 6.2 and 6.3 summarize the results. \(\blacktriangle \)
6.4 Questions Raised by Distinguishing Macroevolution from Microevolution
In this chapter, we encountered some of the potential molecular evidence, as opposed to the better known fossil evidence (e.g., Sect. 2.4), that macroevolution is distinct from microevolution. If the gradual process of the neo-Darwinian synthesis cannot be extrapolated to macroevolution, then how should observed differences in epigenetic complexity be explained? What would be the mechanisms for evolution according to molecular versions of the punctuated equilibrium hypothesis (Sects. 2.4–2.7)? Can some kind of epigenetic inheritance [62, chapter 4] fill in the gaps?
Some maverick scientists have concluded that the evidence challenges not only the neo-Darwinian synthesis but also the theory of universal common descent [57, 105, 109]. That, however, is the foundational working hypothesis of molecular phylogenetics, at least as applied to higher level taxa. For without homology in the sense of common ancestry, tree estimation reduces to hierarchical cluster analysis (Sect. 1.2.1).
6.5 Exercises
-
(1)
(a) Why, exactly, does claim 2 of the maximum genetic diversity hypothesis (Sect. 6.1) make the predictions listed in Sect. 6.2? Hint: if there are only a few sites in a sequence that are free to change, there will be more chances for multiple substitutions at the sites that are not conserved according to claim 2. (The concept of multiple substitutions at a site was introduced in Sect. 1.1 and also appeared in Sects. 3.1.2.2 and 3.1.2.3.) (b) Why does the molecular clock hypothesis make the predictions specified in Sect. 6.2?
-
(2)
Why does the maximum genetic diversity hypothesis (Sect. 6.1) suggest working with slowly evolving sequences (Sect. 6.3)? Hint: for slowly evolving sequences, the maximum genetic diversity hypothesis makes the same predictions as the molecular clock. Why is that?
-
(3)
(a) What part of the title of Huang [59] is supported by the numbers of slowly evolving sequences in Tables 6.2 and 6.3? (b) Would that part of the title be supported by the counts of rapidly evolving sequences in Tables 6.2 and 6.3?
-
(4)
Why does the slow clock method of Sect. 6.3 require slowly evolving sequences? Hint: study the remarks in Sect. 6.3 about Table 6.1, and review your answers to Exercises 2 and 3.
-
(5)
(a) Based on your understanding of Sect. 6.3, how would you change your answers to Exercise 5b of Sect. 4.4 and to Exercise 9b of Sect. 5.6? Hint: review the remarks in Sect. 6.3 about Table 6.1. (b) Does your answer depend on whether the phylogenetic trees are based on rapidly evolving sequences? Hint: review your answer to Exercise 4.
-
(6)
(a) How would you start to answer the questions raised in Sect. 6.4? Hint: review the history of ideas sketched in Sect. 2, keeping in mind its epigraph. (b) Outline a research project that would address one of those questions.
References
Bickel, D.R. 2022b. Fisher’s disjunction as the principle vindicating p-values, confidence intervals, and their generalizations: A frequentist semantics for possibility theory. Working paper. https://doi.org/10.5281/zenodo.6590672.
Box, G.E.P. 1976. Science and statistics. Journal of the American Statistical Association 71: 791–799.
Bromham, L. 2016. An Introduction to Molecular Evolution and Phylogenetics. Oxford: Oxford University Press.
Gould, S.J. 2009. Punctuated Equilibrium. Cambridge: Harvard University Press.
Hoyle, F., and N. Wickramasinghe. 2000. Astronomical Origins of Life. New York: Springer.
Hu, T., M. Long, D. Yuan, Z. Zhu, Y. Huang, and S. Huang. 2013. The genetic equidistance result: misreading by the molecular clock and neutral theory and reinterpretation nearly half of a century later. Science China Life Sciences 56: 254–261.
Huang, S. 2012. Primate phylogeny: molecular evidence for a pongid clade excluding humans and a prosimian clade containing tarsiers. Science China Life Sciences 55: 709–725.
Huang, S. 2016. New thoughts on an old riddle: What determines genetic diversity within and between species? Genomics 108: 3–10.
Jablonka, E., M. Lamb, and A. Zeligowski. 2014. Evolution in Four Dimensions: Genetic, Epigenetic, Behavioral, and Symbolic Variation in the History of Life. Cambridge: MIT Press.
Jeffreys, H. 1948. Theory of Probability. London: Oxford University Press.
Lesk, A. 2019. Introduction to Bioinformatics. Oxford: Oxford University Press.
Schwabe, C. 2002. Genomic potential hypothesis of evolution: a concept of biogenesis in habitable spaces of the universe. The Anatomical Record: An Official Publication of the American Association of Anatomists 268: 171–179.
Steele, E., R. Gorczynski, R. Lindley, Y. Liu, R. Temple, G. Tokoro, D. Wickramasinghe, and N. Wickramasinghe, 2019. Lamarck and panspermia – on the efficient spread of living systems throughout the cosmos. Progress in Biophysics and Molecular Biology 149: 10–32.
Wang, M., D. Wang, J. Yu, and S. Huang. 2020. Enrichment in conservative amino acid changes among fixed and standing missense variations in slowly evolving proteins. PeerJ 8: e9983.
Author information
Authors and Affiliations
6.1 Electronic Supplementary Materials
Supplemental 1
ancestor uncertainty (xlsx 1021 kb)
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter
Cite this chapter
Bickel, D. (2022). Testing Hypotheses of Molecular Evolution. In: Phylogenetic Trees and Molecular Evolution. SpringerBriefs in Systems Biology. Springer, Cham. https://doi.org/10.1007/978-3-031-11958-3_6
Download citation
DOI: https://doi.org/10.1007/978-3-031-11958-3_6
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-11957-6
Online ISBN: 978-3-031-11958-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)