
Abstract
Primary vitreoretinal lymphoma (PVRL) is rare. Confirming the diagnosis can be challenging, but cytokine analysis and sequencing can improve diagnostic accuracy. The risk for relapse is high and relapse into the CNS can be fatal. The optimal treatment for PVRL is yet to be established. Therapeutic options are limited by drug penetration through the blood–retinal and blood–brain barriers, but different routes of delivery attempt to circumvent these barriers.
In the chapter, we describe the management for PVRL and review current therapeutic options for local control and prevention of CNS progression. We discuss the role of focal therapies, systemic therapies, and the evolving role of whole-brain radiation therapy. We also discuss the treatment strategies under investigation to avoid toxicity and improve survival. Finally, we look to the future and discuss emerging therapies, including upstream and downstream targets of the Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, immunomodulatory drugs, and immune checkpoint blockade.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction and Nomenclature
Primary vitreoretinal lymphoma (PVRL) is a subgroup of primary intraocular lymphoma (PIOL) and a subtype of primary central nervous system lymphoma (PCNSL) [1, 2]. PVRL can arise in different eye compartments, including the vitreous and retina. The eye is considered a sanctuary site for neoplastic cells. Like PCNSL, the majority of PVRL (>90%) are non-Hodgkin diffuse large B-cell lymphoma (DLBCL). The remainder are T-cell and natural killer (NK)-cell lymphomas.
First recognized in 1951 by Cooper and Riker, “ocular reticulum cell sarcoma” was named in a patient with disease occurring in the retina and uvea [3]. Ocular reticulum cell sarcoma was then renamed to “primary intraocular lymphoma” (PIOL) after the discovery that the tumor cells originate from only ocular tissues [4, 5].
PIOL can be further subdivided [6, 7]. PVRL, the most common form of intraocular lymphoma (IOL), is a high-grade B-cell malignancy that originates from the posterior eye segments (vitreous and retina). In contrast, primary uveal lymphoma involves the choroid, iris, ciliary body, or a combination of these structures. Choroidal lymphomas are usually low-grade B-cell lymphomas (BCLs), not associated with central nervous system (CNS) disease, and are most frequently extranodal marginal zone lymphomas (EMZLs). Primary iridal lymphomas are exceedingly rare and tend to be B-cell or T-cell lymphomas. Secondary intraocular lymphoma occurs in patients with advanced systemic disease. The terms IOL and vitreoretinal lymphoma (VRL) are sometimes used interchangeably in various studies, which adds to confusion with nomenclature. For the purpose of this chapter, we are consistently using the term VRL.
2 Epidemiology
PVRL is approximately 1% of non-Hodgkin’s lymphomas, 1% of intracranial tumors, and <1% of intraocular tumors [8]. Given its rarity, the true incidence of PVRL is unknown, but 300–380 new cases of PVRL are reported annually [7]. PVRL typically presents in patients of median age in the 60s.
More epidemiologic data exist for PCNSL. The rising incidence of PCNSL, which peaked in 1995, was partially driven by the acquired immune deficiency syndrome (AIDS) epidemic and susceptibility in the elderly population [9, 10]. Since the 2000s, the incidence has decreased in younger adults with the decreased incidence of AIDS, whereas the incidence in the elderly population is increasing as the overall population ages [11]. The incidence of PCNSL is slightly higher in men. There is an association between PCNSL and iatrogenic immunosuppression and autoimmune diseases such as rheumatoid arthritis, Sjögren syndrome, systemic lupus erythematosus, and sarcoidosis.
3 Prognostic Factors
There are no identifiable prognostic factors for PVRL, but age and performance status have been proposed based on data from PCNSL studies [12, 13]. Significantly improved outcomes occur in newly diagnosed PCNSL patients that are younger and with a Karnofsky performance score (KPS) of >70. Due to the high incidence of CNS progression, the prognosis of PVRL is poor. It is not clear whether isolated PVRL at presentation carries a better prognosis than VRL with CNS involvement as most patients eventually develop brain or leptomeningeal disease. In a series of 22 patients with PVRL with or without CNS involvement, there was a trend toward longer survival in patients with isolated PVRL compared to those with concurrent central nervous system lymphoma (CNSL), 43.4 vs. 30.3 months, respectively [14]. A 20-year retrospective review of 19 PVRL patients, 7 of whom had concurrent CNS lymphoma, reported a median progression-free survival (PFS) of 11 months, and median survival of 33 months [15]. In a large multicenter retrospective study of patients with isolated PVRL, 29% (23/78) died; of these, 30% of patients died from CNSL, 13% died from indirect complications of CNSL (including treatment complications), and 13% died from unrelated causes [16]. The five-year cumulative survival rate was also lower in patients with CNSL compared to patients without CNSL (35% and 68%, respectively; p = 0.003) [16]. Local relapse typically occurs within 2 years of initial diagnosis and CNS dissemination is the most common cause of fatality [17,18,19].
4 Clinical Presentation
PVRL patients can initially present with unilateral disease, but approximately 80–90% develop bilateral disease [7, 20,21,22,23,24]. Ocular involvement does not necessitate ocular symptoms and patients can present with a broad range of visual acuities, ranging from normal vision to blindness [25, 26]. Other presenting symptoms are blurry vision, floaters, and less commonly photophobia and ocular pain [2, 22, 27,28,29,30]. These non-specific symptoms often lead to diagnostic delay [31] of over 1 year on average from symptom onset to diagnosis [7, 25, 31].
5 Diagnosis and Pathophysiology
PVRL can be a diagnostic challenge and is known as a masquerade syndrome, mimicking other diseases, such as chronic uveitis and other inflammatory processes, including endophthalmitis, sarcoidosis, multiple sclerosis, rheumatoid arthritis, Behcet’s and Vogt-Koyanagi-Harada disease, and other inflammatory processes [4, 6, 20, 27, 30, 32,33,34]. Other neoplastic processes can also imitate PVRL, including amelanotic choroidal melanoma, multiple myeloma, and metastatic systemic non-Hodgkin lymphoma [34,35,36].
Patients with suspected PVRL should be referred for thorough ophthalmic examination and for consideration of diagnostic vitrectomy, or retinal biopsy in appropriate cases. Further diagnostic evaluation for CNS involvement with magnetic resonance imaging (MRI) brain and spine, cerebrospinal fluid (CSF) analysis, and staging evaluation for systemic lymphoma with computed tomography or positron emission tomography (PET) of the chest, abdomen, and pelvis should also be performed.
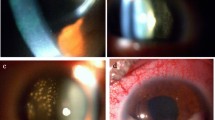
Slit lamp and dilated fundus examination can reveal cells in the anterior chamber, which are non-specific as they can also be seen in the setting of non-neoplastic inflammatory disease [4, 7]. Infiltrating lymphomatous cells are classically observed in the vitreous cavity and are characterized by hallmark sheets or clumps of cells; retinal and subretinal pigment epithelium (RPE) infiltrates can also be visualized in some cases [2, 4, 6, 7, 30, 36,37,38,39].
Non-invasive ancillary imaging including optical coherence tomography (OCT), and ultrasonography in some cases, can be helpful. OCT may show hyper-reflective infiltrates containing discrete nodules or confluent bands with a predilection for the sub-RPE space [40, 41]. Fluorescein angiography can demonstrate punctate hyper-fluorescent foci corresponding to lymphoma infiltrates; however, findings of inflammation (petaloid leakage consistent with cystoid macular edema and perivascular staining with or without leakage) are frequently suggestive of a diagnosis other than PVRL [39, 42].
Definitive diagnosis requires vitreal or retinal biopsy as the gold standard [43]. If the initial biopsy is non-diagnostic, repeat biopsy should be considered when diagnostic suspicion is high. Since malignant cells are typically found within the vitreous, it is the preferred sampling site. The retina and subretinal space are alternative sites for biopsy. Vitrectomy is associated with fewer complications compared to chorioretinal biopsy and may provide added therapeutic benefits of decreasing cell load or inducing remission [44, 45].
The numerous reactive lymphocytes that are admixed with the malignant PVRL cells can compromise the diagnosis [4, 7, 39, 46]. Additionally, scant cellularity resulting from the fragility of the lymphoma cells and the cytotoxic effects of corticosteroids on lymphoma cells can necessitate multiple biopsies [4, 7, 29, 47]. Prompt processing of the samples minimizes cytolysis and optimizes cytologic analysis.
Cytologic analysis shows large pleomorphic atypical lymphoid cells with irregular nuclei and prominent nucleoli with rare mitoses. Monoclonal B-cells stain positive for Cluster of Differentiation 19 (CD19), CD20, and CD22. Detection of CD79BY196 and expression of transcriptional factors B-cell lymphoma (BCL)-6 and Multiple Myeloma Oncogene (MUM)-1 in vitreous DNA may support the diagnosis of PVRL [48, 49]. Flow cytometry assesses for monoclonality for both exclusively kappa-bearing or lambda-bearing B- and T-lymphocytes [39, 50, 51].
Molecular analysis and polymerase chain reaction (PCR) are beneficial adjuncts in diagnosing PVRL [4, 52]. Next-generation sequencing of vitrectomy fluid can detect mutations of Myeloid Differentiation Factor 88 (MYD88)L265P gain, and CDKN2A and PTEN losses [53]. MYD88 mutations are common in PVRL and can increase the diagnostic yield for PVRL [53,54,55,56]. A retrospective review found that 69% (20/29) of 75 vitrectomy samples in 69 PVRL patients harbor MYD88 mutations [56]. Hotspot somatic mutation L265P is the most frequently observed mutation, while 103 and 143 position mutations are less frequent [56, 57]. Similar findings were reported in a proof-of-concept study of 23 VRL patients, with 74% of patients harboring the MYD88L265P mutation [58]. Similar to interleukin (IL)-10 levels, MYD88 mutations are detectable in both the vitreous fluid and aqueous humor (sensitivity 75% and 67%, respectively). When clinical presentation is highly suggestive of PVRL, a positive MYD88-allele-specific PCR in combination with CD20+ vitreous cells could potentially be adequate for diagnosis in absence of definitive diagnosis on cytopathologic analysis. The aqueous humor is more easily accessible than vitreous fluid and MYD88 mutations in the aqueous humor can also be informative. MYD88 mutations may have higher concordance rates than IL-10 in the vitreous compared to aqueous fluid. One study reported a concordance rate of 89% in 12-paired samples. Patients with PVRL have IgH rearrangements similarly found in systemic non-Hodgkin lymphoma, but can be falsely negative [43]. T-cell receptor gene rearrangements have been reported in T-cell lymphoma [4].
Finally, intraocular cytokine analysis of interleukin (IL)-10 and IL-6 can be informative in the diagnosis of PVRL. B-lymphoma cells secrete IL-10, an immunosuppressive TH2-cytokine of growth and differentiation [59,60,61]. IL-10 simultaneously stimulates tumor cell proliferation and shields tumor cells from the immune response. Elevated IL-10 levels correlate with shorter event-free survival (EFS) and high levels are found in the vitreous in PVRL patients. As a result, IL-10 levels can be used for PVRL screening to support its diagnosis or for indirect biomarker analysis to determine remission [34, 60, 62,63,64,65]. Whereas PVRL B-lymphoid cells produce high levels of IL-10, normal lymphocytes and macrophages produce the proinflammatory cytokine IL-6 during inflammatory states, including uveitis [4, 60, 66]. An IL-10 level >100 pg/mL in combination with IL-10:IL-6 ratio of >1 is beneficial for diagnosing PVRL [31]. Vitreal IL-10:IL-6 ratio less than 1.0 may be due to early stage disease [67]. A retrospective study comparing IL-10/IL-6 ratio greater than 1.0, IgH rearrangements, and cytology in the vitreous specimens of 217 patients found that IL-10/IL-6 ratio had the highest detection rate (91.7%, 80.6%, and 44.5%, respectively) [68]. Both vitreous and aqueous humor levels can be used for IL-10 detection, but aqueous IL-10 levels are variable and cannot alone be used to confirm a PVRL diagnosis.
Patients diagnosed with PVRL should undergo further evaluation with magnetic resonance imaging (MRI) to assess for CNS involvement and a lumbar puncture with CSF studies to assess for leptomeningeal involvement [4, 7, 66]. Since PVRL is a subgroup of PCNSL, diagnosis can be made with a less invasive lumbar puncture with CSF findings of lymphoma cells, sparing the patient from a more invasive diagnostic biopsy or vitrectomy [4, 48, 62, 69]. Oculocerebral lymphoma with a positive brain biopsy may also preclude the requirement for an intraocular biopsy [64]. Brain MRI should then be performed every 3 months for 2 years, then every 6 months [43].
6 Treatment
Management of PVRL requires close surveillance and a multidisciplinary team approach consisting of an ophthalmologist, a neuro-oncologist or hemato-oncologist, and a pathologist [4]. Due to its rarity, there is no consensus on the optimal treatment for PVRL [4, 25]. In 2011, the International PCNSL Collaborative Group (IPCG) recommended dedicated local therapy with intravitreal (IVT) chemotherapy (CT) or irradiation for unilateral isolated PVRL, local or systemic therapy for bilateral PVRL, and systemic with local therapy for concurrent CNS disease [7]. Others have also recommended deferring systemic treatment for CNS involvement to minimize toxicities [16, 70]. At the same time, the British Neuro-Oncology Society recommended systemic therapy with high-dose methotrexate (HD-MTX) followed by ocular and cranial irradiation (www.bnos.org.uk, June 2011). The French Oculocerebral Lymphoma (LOC) Network, which was established in 2011, recommended treating patients with isolated PVRL in good medical condition with intravenous HD-MTX-based therapy and those in poor condition with local therapy. More recently in 2021, they published results from a European survey and updated their guidelines to include additional consolidative low-dose bilateral ocular radiation [71].
There are challenges to the treatment of PVRL. Two blood-ocular barriers, the blood-aqueous and blood-retinal barriers, can limit drug penetration and lead to insufficient drug levels within the eye. The blood-retinal barrier is of key importance given that intraocular lymphoma primarily affects the posterior eye, including optic nerve, vitreous, and retina. To circumvent these barriers, numerous delivery methods have been developed, including the use of both local and systemic treatment modalities [17]. Local therapy includes intravitreal (IVT) chemotherapy (CT) and ocular radiation therapy (ORT), whole-brain radiation therapy (WBRT), and intrathecal (IT) CT. Systemic therapy consists of intravenous CT and autologous stem cell transplantation (ASCT).
6.1 Local Therapy
For isolated PVRL restricted to one eye, local therapies with external beam radiotherapy (EBRT), intravitreal methotrexate (IVT-M), or intravitreal rituximab (IVT-R) are effective therapies [7, 14, 43, 72, 73]. No studies have been conducted to compare these local therapies, but there are no apparent significant differences in local control or visual outcomes among these therapies. Often, the choice of first-line therapy is driven by the specialist involved [6, 24, 43].
6.2 Ocular Radiation Therapy
ORT was previously the mainstay treatment for PVRL [74]. Given the high incidence of bilateral eye involvement, it is typically delivered to both eyes [7, 20]. EBRT to a total of 35–40 Gy delivered in 15 fractions provides good local control rates [20, 22, 75, 76]. One retrospective study showed no relapse in 58% (7/12) of PVRL patients who received radiation therapy (RT; six patients with combined CT and one patient with RT alone). Nevertheless, disease often recurs and progresses to CNSL. Radiation toxicities occur in up to 70% of patients including retinopathy, optic neuropathy, cataract, dry eye, conjunctivitis, and vitreous hemorrhage [77, 78]. While unusual in doses less than 36 Gy, radiation retinopathy can occur even at reduced levels of 20 Gy, particularly in patients with comorbidities such as diabetes and other systemic vascular diseases [79]. Re-treating recurrent disease is a challenge with RT due to the issue of overlapping treatment fields, which may limit the future use of WBRT for CNS disease [74, 80, 81]. CyberKnife has some advantages over EBRT, including shorter treatment time and decreased risk of radiation retinopathy, but experience with this technique is limited by short follow-up [82].
6.3 Intravitreal Therapy
Intravitreal treatment (IVT-M and IVT-R) offers an alternative therapy to overcome the disadvantages of ocular RT [80]. Turaka et al. reported that 58.3% (7/12) PVRL patients treated with either IVT-M or rituximab (RTX) monotherapy achieved complete response (CR) at a median follow-up of 33.5 months [72]. However, as with ocular RT, intravitreal therapy is a focal treatment that does not address concurrent brain disease and is associated with a CNS recurrence risk [70].
Methotrexate (MTX), a dihydrofolate reductase inhibitor, used intravitreally, has shown long-term success in studies and provides good local control without severe toxicity [26, 72, 80, 83]. One 10-year single-institution study of 44 eyes in 26 patients treated with IVT-M showed no intraocular recurrence in the 8 surviving patients after >3 years [26]. MTX can also provide prolonged local remission in recurrent disease after RT and systemic CT [27, 84]. Resistance to IVT-M in PVRL patients may be caused by the simultaneous increased efflux of MTX by multidrug resistance-related protein and decreased influx of MTX by reduced expression of reduced folate carrier and folate binding protein [85]. One disadvantage to IVT-M is the number of injections that must be administered over an extended period of time [43, 83, 86]. In addition, toxicities can develop, including cataract (73%), corneal epitheliopathy (58%), maculopathy (42%), vitreous hemorrhage (8%), optic atrophy (4%), and sterile endophthalmitis (4%) [83]. Nevertheless, IVT-M remains a frequent frontline option for PVRL in the newly diagnosed or recurrent setting.
Rituximab (RTX), a humanized chimeric anti-CD20 monoclonal antibody, is used for treatment of PCNSL and PVRL [87]. RTX induces apoptosis, complement-mediated cytolysis, and antibody-dependent cytotoxicity although its exact role in PCNSL is unclear. IVT-R has been investigated both as a single agent and in combination with IVT-M [72, 87,88,89,90]. Kitzmann et al. found no toxicities in five treated eyes [87]. In a prospective case series of 20 patients, Hashida et al. showed improvement with a single course of IVT-R in PVRL patients who previously developed toxicity to IVT-M [88]. Similar to recurrence rates with ocular RT and IVT-M, disease recurrence rates with IVT-R were relatively high and occurred in 55% (11/20) of patients [88]. However, a second remission can be achieved with an additional course of injections [88]. In a retrospective study conducted by Larkin et al., 48 eyes of 34 PVRL patients treated with IVT-R achieved 64.6% (31/48) CR and 22.9% (11/48) partial response (PR) [90]. RTX toxicities include transient elevated intraocular pressure (IOP), keratic precipitates, cataract, vitreous hemorrhage, and retinal detachment [88, 90].
6.4 Systemic Chemotherapy
Some experts follow the PCNSL treatment strategies for PVRL patients [43, 91]. Systemic chemotherapy penetrates the blood-ocular barrier and achieves therapeutic intraocular fluid levels, but the levels in the vitreous humor can be unreliable [48, 92].
Ara-C is a pyrimidine analogue used in the treatment of PCNSL. In 1986, Bauman et al. were the first to report CR with high-dose Ara-C (HiDAC) in a case report of PVRL treated with 3 g/m2 every 12 h [93]. Therapeutic intraocular levels with 2 g/m2 were sustained. While promising results were observed in a subsequent study of 3 patients with isolated PVRL, another case series of 6 PVRL patients showed CR in only 20% (1/5) of patients treated with single-agent HiDAC [94, 95]. Toxicities associated with HiDAC include conjunctivitis, keratitis, ocular irritation, myelosuppression, nausea, and vomiting.
Similar to HiDAC, HD-MTX (8 g/m2) produces sustained drug levels in the vitreous and aqueous humor [24, 92, 96, 97]. In a case series of 9 patients (2 patients with isolated PVRL and 7 patients with PCNSL with vitreoretinal (VR) involvement), Bachelor et al. demonstrated 78% CR in the eye and 100% CR in the brain with HD-MTX 8 g/m2 every 2 weeks until CR followed by consolidation MTX for 2 doses then maintenance every 4 weeks for 11 doses [92]. In a phase 2 study conducted by the New Approaches to Brain Tumor Therapy (NABTT) 96–07 CNS Consortium, 20% (5/25) of patients with newly diagnosed PCNSL had concurrent ocular lymphoma [98]. Following HD-MTX, CR and partial response (PR) were achieved in 80% and 20%, respectively, suggesting HD-MTX may be effective for patients with PCNSL and concomitant PVRL. Deficient levels or short duration of cytotoxic levels may contribute to HD-MTX resistance [92]. HD-MTX carries a risk of delayed neurotoxicity (NT) and leukoencephalopathy [99]. The optimal dose of HD-MTX and the best combination of chemotherapy agents for PVRL have not been defined.
Temozolomide, an oral alkylating agent, has been shown to be effective in a retrospective study of 21 patients with relapsed/refractory (R/R) PVRL with a CR rate of 71% and median PFS of 12 months [100]. CNS relapses were noted in 24% (5) of the patients.
7 Combined Systemic and Local Treatment
Treatment goals of combined therapy center on both eradication of lymphoma in the eye and preventing subsequent CNS relapse, but evidence for prophylactic therapy to prevent CNS dissemination is conflicting given various modalities have been used. Margolis et al. reported CNS dissemination in 89% (8/9) patients during their disease course and clinical improvement after 7 of the affected patients were treated with CNS irradiation [20]. De la Fuente et al. reported in a retrospective study that combined bilateral ocular RT followed by MTX-based chemotherapy provides effective local control, prevention of CNS relapse, and prolonged survival [101]. Of 12 patients with isolated bilateral PVRL, 10 patients were treated with ORT followed by systemic CT, 1 patient was treated with RT, and 1 patient was treated with IVT-M [101]. Complete response (CR) was achieved in all patients and PFS and overall survival (OS) were not reached at a median follow-up of 68 months. The cumulative incidence of CNS relapse was 37.5%, lower than the previously reported incidence, ranging from 56 to 85%. Moreover, in a prospective study, Kaburaki et al. reported that CNS prophylaxis with combination systemic CT and reduced dose (rd)-WBRT (rituximab, methotrexate, procarbazine, vincristine (R-MPV), rd-WBRT, and HiDAC) significantly decreased CNS relapse in 17 patients, of whom 65% (11/17) had isolated intraocular disease [17]. CNS relapse was noted in 1 (of 11) patient with isolated PVRL and ocular relapse occurred in 3. Four-year PFS and OS were 72.4% and 88.9%, respectively, in isolated PVRL patients. In a retrospective study of 31 isolated PVRL patients, Hormigo et al. reported that CNS prophylaxis with systemic CT, combined CT and RT, or RT alone significantly improves survival compared to treatment after CNS progression [22]. Of these 31 patients, 17 patients received CNS prophylaxis and 14 patients received CNS treatment at the time of progression [22]. The prophylactically treated patients had significantly improved median survival of 60 months compared to the non-prophylactically treated group with a median survival of 35 months. However, CNS progression rates were similar, 53% (9/17) in the prophylactically treated patients and 50% (7/14) in the non-prophylactically treated group, thus raising the question of whether or not treatment modality compromises risk of CNS progression.
In contrast to the reports above, multiple studies have found that treatment modality does not appear to compromise disease control or risk of CNS progression or OS [7, 16, 22, 64, 70, 102]. In a large multicenter retrospective study conducted by Riemens et al., 78 isolated PVRL patients were evaluated. Seventy-five patients were treated with either local therapy alone (31 patients), extensive systemic therapy (21 patients), or combination local and extensive therapy (23 patients). Three patients were not treated. At median follow-up of 49 months, CNSL developed in 36% (28/78) of patients. Progression to CNSL did not differ between patients receiving local therapy alone compared to aggressive systemic therapy with CNSL developing with local therapy alone in 32% (10/31), extensive systemic therapy in 43% (9/21), and combination ocular and extensive treatment in 39% (9/23) [16]. Moreover, in a retrospective study of 26 isolated PVRL patients conducted by Hashida et al., prophylactic therapy did not prevent progression to CNS disease [102]. Fifteen patients received local therapy (IVT-M with 8 patients additionally receiving IVT-R) and 11 patients received prophylactic therapy (6 received HD-MTX and 5 received IT-MTX). CNSL developed in 54% (14/26) of patients at mean follow-up of 44.0 ± 18.7 months, with 40% (6/15) in the local therapy alone group and 73% (8/11) in the prophylactic treatment group. However, the time to onset of CNS progression was significantly prolonged in the prophylactically treated patients compared to the patients who did not receive prophylactic treatment, 42.8 ± 13.8 months and 10.2 ± 2.0 months, respectively. HD-MTX in particular had significantly prolonged time to CNS progression compared to IT-MTX or local therapy. In an IPCG (International PCNSL Collaborative Group) Report, Grimm et al. compared local therapy (23 patients) to extensive therapy (53 patients treated with combinations of systemic CT, ocular CT, WBRT, and ocular RT) [70]. Local therapy alone did not increase relapse risk compared to extensive therapy: 56% and 60%, respectively. Relapse occurred in 47 patients at a median of 19 months, with 47% (22/47) in the brain, 30% (14/47) in the eyes, 15% (7/47) in the eyes and brain, and 8% (4/47) systemically. Further, more toxicities have been reported with systemic CT [16]. Therefore, in the absence of CNS disease, systemic therapy or combination intravitreal and systemic chemotherapy remains controversial [7, 27, 64]. Local therapy remains a reasonable approach for patients with contraindications to systemic chemotherapy, elderly patients, or local recurrence.
8 High-Dose Chemotherapy (HDT) and Autologous Stem Cell Transplant (ASCT)
Thiotepa-based high-dose chemotherapy (HDT) and ASCT is used for consolidation in newly diagnosed PCNSL. In PVRL, it has been investigated in the R/R setting. A prospective pilot study, 11 refractory PVRL patients were treated with HD-MTX and HiDAC; 5 of these patients were treated with ASCT and demonstrated promising results [103]. The conditioning regimen for ASCT consisted of thiotepa 750 mg/m2, busulfan 10 mg/kg, and cyclophosphamide 120 mg/kg (TBC), all of which are good brain penetrants. All 5 patients had a CR, and 3 remained in CR between 14 and 15 months. Two patients relapsed at 6 months. Encouraged by these results, a study was conducted by Soussain et al. investigating the feasibility of the cytarabine and etoposide (CYVE) followed by TBC-ASCT in R/R patients [104]. In this study, 12 (out of 22) patients had eye involvement (isolated or in conjunction with brain or CSF involvement). Eleven patients had a response after ASCT; ten CR, one PR, and one had systemic progression, although two in CR progressed at 3 and 5 months, respectively. Apparent longer survival times were observed in patients with isolated VRL compared to patients with brain parenchyma involvement, with median survival time not reached and 5.5 months, respectively [104]. However, in a phase 2 prospective study of CYVE and TBC-ASCT in patients with recurrent or primary refractory IOL or PCNSL, there was no significant difference in median PFS or OS in those patients who had VRL (isolated or with brain/CSF disease) compared to those who did not [105]. Soussain et al. also reported on the largest retrospective series of 79 R/R PCNSL and VRL patients [106]. All patients had VRL with or without brain or CSF involvement and CR was achieved in 83.5% (66/79), PR in 5% (4/79), stable disease (SD) in 2.5% (2/79), and progressive disease (PD) in 4% (3/79) of patients. Following ASCT, 35% (28/79) patients relapsed; 68% in the CNS, 11% in the eye, 7% in both the eye and CNS, 3.5% in the CSF, 7% systemically, and 3.5% unknown. Durable CR between 9 and 124 months was achieved in 4 patients. In general, the role of HDT-ASCT is at present restricted to younger and fit R/R patients.
9 Emerging Therapies
Bruton tyrosine kinase (BTK) inhibitors and immunomodulatory drugs (IMiDs) have been investigated in prospective trials in R/R PVRL. Additionally, novel agents including other targeted therapies and immunotherapies are currently under investigation for PCNSL, although many of these have not specifically included PVRL. A checkpoint inhibitor, pembrolizumab, has been studied in a phase 2 trial in R/R PCNSL and PVRL, although the final results are pending (NCT03012620).
9.1 BTK Inhibitors
Constitutive Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activity is thought to contribute to the poor prognosis of patients with activated B cell-like (ABC) subtype of DLBCL, including PCNSL [107]. PCNSL patients harbor mutations in B-cell antigen receptor (BCR) and toll-like receptor (TLR) signaling pathways that enhance NF-κB activity [108]. Cluster of Differentiation (CD)79B and MYD88 activate the BCR and TLR signaling pathways, respectively. CD79B is a BCR-associated protein, and gene alterations are found in 64% of PCNSL tumors [109]. MYD88 is a cytosolic adapter protein, and gene alterations are found in 71.7–86.7% of PCNSL including PVRL tumors [109, 110]. Drugs targeting the NF-κB pathway both upstream and downstream are under investigation. Upstream, Bruton tyrosine kinase (BTK) links BCR and TLR signaling and BTK inhibition is one of the emerging therapies under investigation.
Ibrutinib is an oral irreversible inhibitor of BTK with good blood–brain barrier penetration. Ibrutinib has demonstrated activity in PVRL and PCNSL [111, 112]. In a retrospective case series of 14 patients with R/R PCNSL treated with ibrutinib monotherapy, Chamoun et al. reported 50% ORR and 2 patients with CR >8 months [113]. Of these 14 patients, 4 patients had VRL. Two phase 1 studies in R/R PCNSL patients showed objective responses to ibrutinib monotherapy and in combination with chemotherapy [111, 114]. The promising results of these retrospective and prospective phase 1 studies led to the first large, multicenter phase 2 proof-of-concept study of ibrutinib (560 mg/day in 28-day cycle) in R/R PVRL and PCNSL patients conducted by the Lymphoma Study Association (LYSA) and the French LOC Network [112]. Of these 52 patients, 14 patients had PVRL, 2 of which had CSF involvement. Responses were achieved in all compartments; 19% (10/52) CR, 33% (17/52) PR, and 10% (5/52) SD. Of the 14 PVRL patients, CR and PR were achieved in 71% (10/14) and 29% (4/14), respectively. The median PFS was longer for patients with PVRL with or without CSF involvement compared to patients with brain or spinal cord lymphoma: 22.7 months vs. 2 months, respectively. Patients without brain involvement also had longer median OS (not reached) compared to with brain involvement (4.3 months). Minimal or undetectable IL-10 anterior chamber levels correlated with clinical CR in 15 evaluable patients and increasing IL-10 levels correlated with progressive disease (PD).
In a phase 1/2 study of second-generation BTK inhibitor, tirabrutinib, partial responses were noted in 3/3 patients [115].
9.2 Immunomodulatory Drugs
Lenalidomide and pomalidomide are IMiDs that suppress NF-κB activity and simultaneously augment interferon beta (IFNβ) production [116]. ABC DLBCL maintains its viability through the regulation of the BCR signaling pathway by SPIB/interferon regulatory factor (IRF4) heterodimers. IMiDs target both IRF4 and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) in the NF-κB pathway [108]. IMiDs have demonstrated activity in R/R PCNSL and PVRL.
Lenalidomide has been studied in PVRL and PCNSL in combination with RTX in the R/R setting and as maintenance therapy [117, 118]. Lenalidomide has been shown to have good CNS penetration, and detectable CSF drug levels are observed without blood brain barrier (BBB) disruption [117]. The addition of lenalidomide to RTX may aid in overcoming RTX resistance by augmenting the antibody-dependent cytotoxicity effects of RTX. In a phase 1 study, 14 R/R PCNSL and secondary central nervous system lymphoma (SCNSL) patients (5 with intraocular involvement) were treated with lenalidomide in combination with intraventricular or intravenous RTX [117]. Durable responses were observed in 6 patients for ≥9 months and in 4 for ≥18 months. The PVRL patient had a prolonged CR of ≥25 months. The LYSA and French LOC Network conducted a phase 2 proof-of-concept clinical trial that evaluated lenalidomide and RTX REVlimid RItuximab (REVRI) followed by maintenance lenalidomide in R/R PVRL or PCNSL patients [118]. Induction therapy consisted of lenalidomide at 20 mg daily for 21/28-day cycle combined with RTX at 375 mg/m2 on day 1. Of a total 45 patients, 17 patients had VRL involvement (9 with isolated PVRL, 2 with PVRL and CSF involvement, and 6 with both CNS and VRL involvement). Responders were treated with maintenance lenalidomide at 10 mg daily for 21/28-day cycle. The median PFS of the PVRL and PCNSL patients was 9.2 and 3.9 months, respectively, and 35% (6/17) of patients with VRL had a CR. Both studies showed clinical activity in the eye, brain, and CSF. The median PFS and OS for the entire group were 7.8 months and 17.7 months, respectively.
Pomalidomide was studied in combination with dexamethasone in a phase 1 study and was found to have activity in R/R PCNSL and PVRL [119].
10 Perspective
PVRL is rare and commonly mimics other diseases. In suspected cases, particularly in elderly or immunocompromised individuals with new onset “uveitis,” evaluation by an experienced ophthalmologist is important in order to avoid diagnostic delay. It can be a challenge to diagnose due to its rarity and also as vitreous fluid samples can be paucicellular due to lymphoma cell fragility. Recent advances in cytokine assays and genomic testing have led to increased diagnostic sensitivity of the vitreous fluid. The Study Group for the VitreoRetinal Lymphoma Diagnostics recommends diagnostic vitrectomy as the gold standard for diagnosis [120]. In the absence of positive cytology or flow cytometry, IL-10:IL-6 ratio >1, positive mutation for MYD88, or monoclonality by PCR to identify IgH gene rearrangement is highly suggestive of VRL. Multimodal imaging including optical coherence tomography (OCT), autofluorescence, and angiography can also be helpful in establishing diagnosis. Determining the extent of disease involvement is essential. Additionally, if vitrectomy or retinal biopsy is not revealing/possible, CSF sampling, brain biopsy, or biopsy of systemic lesions, if present, should be utilized for diagnosis.
Treatment modalities include both local and systemic therapies, although there are no standard of care guidelines. While there have been recommendations over the years from various groups, including the IPCG, British Neuro-Oncology Society, and more recently the LOC Network and European Reference Network (ERN)- EuroBloodNet, the treatment typically offered in clinical practice depends on the favored approach of the treating physician and varies between ophthalmologists, oncologists, and neuro-oncologists. Collaboration from a multidisciplinary team of an ophthalmologist, oncologist/neuro-oncologist, and pathologist is critical for diagnostic accuracy and initiation of appropriate treatment. It is important to evaluate the risk of the specific therapeutic modality while considering the best option. In general, we recommend an approach similar to treatment of newly diagnosed PCNSL and consideration of systemic chemotherapy in conjunction with local therapy (with IVT-M or IVT-R and/or ORT) in a young, healthy patient who is able to tolerate systemic treatment with the goal of achieving CR and reducing the risk of CNS relapse. In elderly frail patients, local treatment is suggested to prevent morbidity from systemic chemotherapy. The choice between IVT-M and ORT should depend on patient characteristics and feasibility as there does not appear to be a difference in efficacy. For persistent disease following systemic and/or IVT treatment, ORT is recommended. The role of ASCT in newly diagnosed PVRL remains to be elucidated, although it is a reasonable approach in the R/R setting in an appropriate patient. Comparative studies with ASCT are lacking. If possible and available, clinical trials should be offered to patients with R/R PVRL. Re-treatment with IVT-M can be considered in R/R PVRL. Novel agents like ibrutinib and lenalidomide can be considered as well. Assessment of therapeutic response is an area that also requires standardization. At this time, assessment of ocular response is performed by a clinical ophthalmological examination. Per IPCG guidelines, absence of vitreous cells and resolution of previously seen retinal or optic nerve infiltrates are indicative of ocular CR. However, in clinical practice CR can be difficult to determine on a clinical examination. Biomarker evaluation is ongoing, but there is no definitive correlation between levels of IL-10 and presence or absence of MYD88 mutation in the context of response, progression, or overall survival. It is clear that international and multidisciplinary collaborations are essential. Given the rarity of this disease, much remains to be elucidated, but progress has been made in the last decade and future therapies appear promising.
References
Cassoux N. Primary vitreous and retinal lymphoma (PVRL): risk factor and prognosis. Acta Ophthalmol. 2019;97(S263)
Chukwueke UN, Nayak L. Central nervous system lymphoma. Hematol Oncol Clin North Am. 2019;33(4):597–611.
Cooper EL, Riker JL. Malignant lymphoma of the uveal tract. Am J Ophthalmol. 1951;34(8):1153–8.
Chan CC, Sen HN. Current concepts in diagnosing and managing primary vitreoretinal (intraocular) lymphoma. Discov Med. 2013;15(81):93–100.
Char DH, Ljung BM, Miller T, Phillips T. Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology. 1988;95(5):625–30.
Pe’er J, Hochberg FH, Foster CS. Clinical review: treatment of vitreoretinal lymphoma. Ocul Immunol Inflamm. 2009;17(5):299–306.
Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: a report from an international primary central nervous system lymphoma collaborative group symposium. Oncologist. 2011;16(11):1589–99.
Bardenstein DS. Intraocular lymphoma. Cancer Control. 1998;5(4):317–25.
Kadan-Lottick NS, Skluzacek MC, Gurney JG. Decreasing incidence rates of primary central nervous system lymphoma. Cancer. 2002;95(1):193–202.
Olson JE, Janney CA, Rao RD, et al. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer. 2002;95(7):1504–10.
Mendez JS, Ostrom QT, Gittleman H, et al. The elderly left behind-changes in survival trends of primary central nervous system lymphoma over the past 4 decades. Neuro-Oncology. 2018;20(5):687–94.
Abrey LE, Ben-Porat L, Panageas KS, et al. Primary central nervous system lymphoma: the Memorial Sloan-Kettering Cancer Center prognostic model. J Clin Oncol. 2006;24(36):5711–5.
Ferreri AJ, Blay JY, Reni M, et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol. 2003;21(2):266–72.
Kim MM, Dabaja BS, Medeiros J, et al. Survival outcomes of primary intraocular lymphoma: a single-institution experience. Am J Clin Oncol. 2016;39(2):109–13.
Levasseur SD, Wittenberg LA, White VA. Vitreoretinal lymphoma: a 20-year review of incidence, clinical and cytologic features, treatment, and outcomes. JAMA Ophthalmol. 2013;131(1):50–5.
Riemens A, Bromberg J, Touitou V, et al. Treatment strategies in primary vitreoretinal lymphoma: a 17-center European collaborative study. JAMA Ophthalmol. 2015;133(2):191–7.
Kaburaki T, Taoka K, Matsuda J, et al. Combined intravitreal methotrexate and immunochemotherapy followed by reduced-dose whole-brain radiotherapy for newly diagnosed B-cell primary intraocular lymphoma. Br J Haematol. 2017;179(2):246–55.
Gavrilovic IT, Hormigo A, Yahalom J, DeAngelis LM, Abrey LE. Long-term follow-up of high-dose methotrexate-based therapy with and without whole brain irradiation for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2006;24(28):4570–4.
Abrey LE, Yahalom J, DeAngelis LM. Treatment for primary CNS lymphoma: the next step. J Clin Oncol. 2000;18(17):3144–50.
Margolis L, Fraser R, Lichter A, Char DH. The role of radiation therapy in the management of ocular reticulum cell sarcoma. Cancer. 1980;45(4):688–92.
Coupland SE, Heimann H, Bechrakis NE. Primary intraocular lymphoma: a review of the clinical, histopathological and molecular biological features. Graefes Arch Clin Exp Ophthalmol. 2004;242(11):901–13.
Hormigo A, Abrey L, Heinemann MH, DeAngelis LM. Ocular presentation of primary central nervous system lymphoma: diagnosis and treatment. Br J Haematol. 2004;126(2):202–8.
Nussenblatt RB, Chan CC, Wilson WH, Hochman J, Gottesman M, Group CaOLW. International central nervous system and ocular lymphoma workshop: recommendations for the future. Ocul Immunol Inflamm. 2006;14(3):139–44.
Plotkin SR, Batchelor TT. Advances in the therapy of primary central nervous system lymphoma. Clin Lymphoma. 2001;1(4):263–75; discussion 276-267.
Grimm SA, McCannel CA, Omuro AM, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008;71(17):1355–60.
Frenkel S, Hendler K, Siegal T, Shalom E, Pe'er J. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br J Ophthalmol. 2008;92(3):383–8.
Davis JL. Intraocular lymphoma: a clinical perspective. Eye (Lond). 2013;27(2):153–62.
Mochizuki M, Singh AD. Epidemiology and clinical features of intraocular lymphoma. Ocul Immunol Inflamm. 2009;17(2):69–72.
Zaldivar RA, Martin DF, Holden JT, Grossniklaus HE. Primary intraocular lymphoma: clinical, cytologic, and flow cytometric analysis. Ophthalmology. 2004;111(9):1762–7.
Faia LJ, Chan CC. Primary intraocular lymphoma. Arch Pathol Lab Med. 2009;133(8):1228–32.
Whitcup SM, de Smet MD, Rubin BI, et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology. 1993;100(9):1399–406.
Coupland SE, Perez-Canto A, Hummel M, Stein H, Heimann H. Assessment of HOPE fixation in vitrectomy specimens in patients with chronic bilateral uveitis (masquerade syndrome). Graefes Arch Clin Exp Ophthalmol. 2005;243(9):847–52.
Vogel MH, Font RL, Zimmerman LE, Levine RA. Reticulum cell sarcoma of the retina and uvea. Report of six cases and review of the literature. Am J Ophthalmol. 1968;66(2):205–15.
Wittenberg LA, Maberley DA, Ma PE, Wade NK, Gill H, White VA. Contribution of vitreous cytology to final clinical diagnosis fifteen-year review of vitreous cytology specimens from one institution. Ophthalmology. 2008;115(11):1944–50.
Parikh AH, Khan SH, Wright JD, Oh KT. Systemic non-Hodgkin’s lymphoma simulating primary intraocular lymphoma. Am J Ophthalmol. 2005;139(3):573–4.
Salomão DR, Pulido JS, Johnston PB, Canal-Fontcuberta I, Feldman AL. Vitreoretinal presentation of secondary large B-cell lymphoma in patients with systemic lymphoma. JAMA Ophthalmol. 2013;131(9):1151–8.
Char DH, Ljung BM, Deschênes J, Miller TR. Intraocular lymphoma: immunological and cytological analysis. Br J Ophthalmol. 1988;72(12):905–11.
Chan CC, Shen D, Hackett JJ, Buggage RR, Tuaillon N. Expression of chemokine receptors, CXCR4 and CXCR5, and chemokines, BLC and SDF-1, in the eyes of patients with primary intraocular lymphoma. Ophthalmology. 2003;110(2):421–6.
Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Exp Ophthalmol. 2008;36(6):564–78.
Mashayekhi A, Shukla SY, Shields JA, Shields CL. Choroidal lymphoma: clinical features and association with systemic lymphoma. Ophthalmology. 2014;121(1):342–51.
Barry RJ, Tasiopoulou A, Murray PI, et al. Characteristic optical coherence tomography findings in patients with primary vitreoretinal lymphoma: a novel aid to early diagnosis. Br J Ophthalmol. 2018;102(10):1362–6.
Velez G, Chan CC, Csaky KG. Fluorescein angiographic findings in primary intraocular lymphoma. Retina. 2002;22(1):37–43.
Pulido JS, Johnston PB, Nowakowski GS, Castellino A, Raja H. The diagnosis and treatment of primary vitreoretinal lymphoma: a review. Int J Retina Vitreous. 2018;4:18.
Iaccheri B, Fiore T, Cerquaglia A, Lupidi M, Cagini C. Transient therapeutic effect of vitrectomy in primary intraocular lymphoma. Int Ophthalmol. 2017;37(6):1333–5.
Venkatesh P, Gogia V, Khanduja S, Gupta S, Kumar L, Garg S. Therapeutic vitrectomy for vitreal recurrence of intraocular lymphoma resistant to intravitreal methotrexate post systemic chemotherapy. J Cancer Res Ther. 2015;11(3):668.
Coupland SE. Vitreous biopsy: specimen preparation and interpretation. Monogr Clin Cytol. 2012;21:61–71.
Chan CC. Molecular pathology of primary intraocular lymphoma. Trans Am Ophthalmol Soc. 2003;101:275–92.
Chan CC, Wallace DJ. Intraocular lymphoma: update on diagnosis and management. Cancer Control. 2004;11(5):285–95.
Yonese I, Takase H, Yoshimori M, et al. CD79B mutations in primary vitreoretinal lymphoma: diagnostic and prognostic potential. Eur J Haematol. 2019;102(2):191–6.
Davis JL, Solomon D, Nussenblatt RB, Palestine AG, Chan CC. Immunocytochemical staining of vitreous cells. Indications, techniques, and results. Ophthalmology. 1992;99(2):250–6.
Davis JL, Viciana AL, Ruiz P. Diagnosis of intraocular lymphoma by flow cytometry. Am J Ophthalmol. 1997;124(3):362–72.
White VA, Gascoyne RD, Paton KE. Use of the polymerase chain reaction to detect B- and T-cell gene rearrangements in vitreous specimens from patients with intraocular lymphoma. Arch Ophthalmol. 1999;117(6):761–5.
Cani AK, Hovelson DH, Demirci H, Johnson MW, Tomlins SA, Rao RC. Next generation sequencing of vitreoretinal lymphomas from small-volume intraocular liquid biopsies: new routes to targeted therapies. Oncotarget. 2017;8(5):7989–98.
Bruno A, Boisselier B, Labreche K, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. 2014;5(13):5065–75.
Gonzalez-Aguilar A, Idbaih A, Boisselier B, et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res. 2012;18(19):5203–11.
Bonzheim I, Giese S, Deuter C, et al. High frequency of MYD88 mutations in vitreoretinal B-cell lymphoma: a valuable tool to improve diagnostic yield of vitreous aspirates. Blood. 2015;126(1):76–9.
Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011;122(6):791–2.
Hiemcke-Jiwa LS, Ten Dam-van Loon NH, Leguit RJ, et al. Potential diagnosis of vitreoretinal lymphoma by detection of MYD88 mutation in aqueous humor with ultrasensitive droplet digital polymerase chain reaction. JAMA Ophthalmol. 2018;136(10):1098–104.
Raja H, Snyder MR, Johnston PB, et al. Effect of intravitreal methotrexate and rituximab on interleukin-10 levels in aqueous humor of treated eyes with vitreoretinal lymphoma. PLoS One. 2013;8(6):e65627.
Wolf LA, Reed GF, Buggage RR, Nussenblatt RB, Chan CC. Vitreous cytokine levels. Ophthalmology. 2003;110(8):1671–2.
Benjamin D, Park CD, Sharma V. Human B cell interleukin 10. Leuk Lymphoma. 1994;12(3–4):205–10.
Merle-Béral H, Davi F, Cassoux N, et al. Biological diagnosis of primary intraocular lymphoma. Br J Haematol. 2004;124(4):469–73.
Costopoulos M, Touitou V, Golmard JL, et al. ISOLD: a new highly sensitive interleukin score for intraocular lymphoma diagnosis. Ophthalmology. 2016;123(7):1626–8.
Fend F, Ferreri AJ, Coupland SE. How we diagnose and treat vitreoretinal lymphoma. Br J Haematol. 2016;173(5):680–92.
Buggage RR, Chan CC, Nussenblatt RB. Ocular manifestations of central nervous system lymphoma. Curr Opin Oncol. 2001;13(3):137–42.
Sen HN, Bodaghi B, Hoang PL, Nussenblatt R. Primary intraocular lymphoma: diagnosis and differential diagnosis. Ocul Immunol Inflamm. 2009;17(3):133–41.
Buggage RR, Velez G, Myers-Powell B, Shen D, Whitcup SM, Chan CC. Primary intraocular lymphoma with a low interleukin 10 to interleukin 6 ratio and heterogeneous IgH gene rearrangement. Arch Ophthalmol. 1999;117(9):1239–42.
Kimura K, Usui Y, Goto H, Group JILS. Clinical features and diagnostic significance of the intraocular fluid of 217 patients with intraocular lymphoma. Jpn J Ophthalmol. 2012;56(4):383–9.
Chan CC, Buggage RR, Nussenblatt RB. Intraocular lymphoma. Curr Opin Ophthalmol. 2002;13(6):411–8.
Grimm SA, Pulido JS, Jahnke K, et al. Primary intraocular lymphoma: an international primary central nervous system lymphoma collaborative group report. Ann Oncol. 2007;18(11):1851–5.
Malaise D, Houillier C, Touitou V, et al. Primary vitreoretinal lymphoma: short review of the literature, results of a European survey and French guidelines of the LOC network for diagnosis, treatment and follow-up. Curr Opin Oncol. 2021;33(5):420–31.
Turaka K, Bryan JS, De Souza S, et al. Vitreoretinal lymphoma: changing trends in diagnosis and local treatment modalities at a single institution. Clin Lymphoma Myeloma Leuk. 2012;12(6):412–7.
Abu Samra K, Oray M, Ebrahimiadib N, Lee S, Anesi S, Foster CS. Intraocular lymphoma: descriptive data of 26 patients including clinico-pathologic features, vitreous findings, and treatment outcomes. Ocul Immunol Inflamm. 2018;26(3):347–52.
Reichstein D. Primary vitreoretinal lymphoma: an update on pathogenesis, diagnosis and treatment. Curr Opin Ophthalmol. 2016;27(3):177–84.
Berenbom A, Davila RM, Lin HS, Harbour JW. Treatment outcomes for primary intraocular lymphoma: implications for external beam radiotherapy. Eye (Lond). 2007;21(9):1198–201.
Isobe K, Ejima Y, Tokumaru S, et al. Treatment of primary intraocular lymphoma with radiation therapy: a multi-institutional survey in Japan. Leuk Lymphoma. 2006;47(9):1800–5.
Hoffman PM, McKelvie P, Hall AJ, Stawell RJ, Santamaria JD. Intraocular lymphoma: a series of 14 patients with clinicopathological features and treatment outcomes. Eye (Lond). 2003;17(4):513–21.
Witmer MT. Primary vitreoretinal lymphoma: management of isolated ocular disease. Cancer Control. 2016;23(2):110–6.
Kaushik M, Pulido JS, Schild SE, Stafford S. Risk of radiation retinopathy in patients with orbital and ocular lymphoma. Int J Radiat Oncol Biol Phys. 2012;84(5):1145–50.
Fishburne BC, Wilson DJ, Rosenbaum JT, Neuwelt EA. Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol. 1997;115(9):1152–6.
Helbig H, Cerny T, de Smet MD. Intravitreal chemotherapy for intraocular lymphoma. Ophthalmologe. 2003;100(2):145–9.
Bianciotto C, Shields CL, Lally SE, Freire J, Shields JA. CyberKnife radiosurgery for the treatment of intraocular and periocular lymphoma. Arch Ophthalmol. 2010;128(12):1561–7.
Smith JR, Rosenbaum JT, Wilson DJ, et al. Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvement. Ophthalmology. 2002;109(9):1709–16.
de Smet MD, Vancs VS, Kohler D, Solomon D, Chan CC. Intravitreal chemotherapy for the treatment of recurrent intraocular lymphoma. Br J Ophthalmol. 1999;83(4):448–51.
Sen HN, Chan CC, Byrnes G, Fariss RN, Nussenblatt RB, Buggage RR. Intravitreal methotrexate resistance in a patient with primary intraocular lymphoma. Ocul Immunol Inflamm. 2008;16(1):29–33.
Mason JO, Fischer DH. Intrathecal chemotherapy for recurrent central nervous system intraocular lymphoma. Ophthalmology. 2003;110(6):1241–4.
Kitzmann AS, Pulido JS, Mohney BG, et al. Intraocular use of rituximab. Eye (Lond). 2007;21(12):1524–7.
Hashida N, Ohguro N, Nishida K. Efficacy and complications of intravitreal rituximab injection for treating primary vitreoretinal lymphoma. Transl Vis Sci Technol. 2012;1(3):1.
Ohguro N, Hashida N, Tano Y. Effect of intravitreous rituximab injections in patients with recurrent ocular lesions associated with central nervous system lymphoma. Arch Ophthalmol. 2008;126(7):1002–3.
Larkin KL, Saboo US, Comer GM, et al. Use of intravitreal rituximab for treatment of vitreoretinal lymphoma. Br J Ophthalmol. 2014;98(1):99–103.
Hoang-Xuan K, Bessell E, Bromberg J, et al. Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: guidelines from the European Association for Neuro-Oncology. Lancet Oncol. 2015;16(7):e322–32.
Batchelor TT, Kolak G, Ciordia R, Foster CS, Henson JW. High-dose methotrexate for intraocular lymphoma. Clin Cancer Res. 2003;9(2):711–5.
Baumann MA, Ritch PS, Hande KR, Williams GA, Topping TM, Anderson T. Treatment of intraocular lymphoma with high-dose Ara-C. Cancer. 1986;57(7):1273–5.
Siegel MJ, Dalton J, Friedman AH, Strauchen J, Watson C. Ten-year experience with primary ocular ‘reticulum cell sarcoma’ (large cell non-Hodgkin's lymphoma). Br J Ophthalmol. 1989;73(5):342–6.
Strauchen JA, Dalton J, Friedman AH. Chemotherapy in the management of intraocular lymphoma. Cancer. 1989;63(10):1918–21.
DeAngelis LM, Seiferheld W, Schold SC, Fisher B, Schultz CJ. 93-10 RTOGS. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93-10. J Clin Oncol. 2002;20(24):4643–8.
Henson JW, Yang J, Batchelor T. Intraocular methotrexate level after high-dose intravenous infusion. J Clin Oncol. 1999;17(4):1329.
Batchelor T, Carson K, O'Neill A, et al. Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol. 2003;21(6):1044–9.
Herrlinger U, Küker W, Uhl M, et al. NOA-03 trial of high-dose methotrexate in primary central nervous system lymphoma: final report. Ann Neurol. 2005;57(6):843–7.
Baron M, Belin L, Cassoux N, et al. Temozolomide is effective and well tolerated in patients with primary vitreoretinal lymphoma. Blood. 2020;135(20):1811–5.
de la Fuente MI, Alderuccio JP, Reis IM, et al. Bilateral radiation therapy followed by methotrexate-based chemotherapy for primary vitreoretinal lymphoma. Am J Hematol. 2019;94(4):455–60.
Hashida N, Nakai K, Saitoh N, Nishida K. Association between ocular findings and preventive therapy with onset of central nervous system involvement in patients with primary vitreoretinal lymphoma. Graefes Arch Clin Exp Ophthalmol. 2014;252(4):687–93.
Soussain C, Merle-Béral H, Reux I, et al. A single-center study of 11 patients with intraocular lymphoma treated with conventional chemotherapy followed by high-dose chemotherapy and autologous bone marrow transplantation in 5 cases. Leuk Lymphoma. 1996;23(3–4):339–45.
Soussain C, Suzan F, Hoang-Xuan K, et al. Results of intensive chemotherapy followed by hematopoietic stem-cell rescue in 22 patients with refractory or recurrent primary CNS lymphoma or intraocular lymphoma. J Clin Oncol. 2001;19(3):742–9.
Soussain C, Hoang-Xuan K, Taillandier L, et al. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: Société Française de Greffe de Moëlle Osseuse-Thérapie Cellulaire. J Clin Oncol. 2008;26(15):2512–8.
Soussain C, Choquet S, Fourme E, et al. Intensive chemotherapy with thiotepa, busulfan and cyclophosphamide and hematopoietic stem cell rescue in relapsed or refractory primary central nervous system lymphoma and intraocular lymphoma: a retrospective study of 79 cases. Haematologica. 2012;97(11):1751–6.
Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194(12):1861–74.
Grommes C, Nayak L, Tun HW, Batchelor TT. Introduction of novel agents in the treatment of primary CNS lymphoma. Neuro-Oncology. 2019;21(3):306–13.
Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279–90.
Raja H, Salomão DR, Viswanatha DS, Pulido JS. Prevalence of MYD88 L265P mutation in histologically proven, diffuse large b-cell vitreoretinal lymphoma. Retina. 2016;36(3):624–8.
Grommes C, Pastore A, Palaskas N, et al. Ibrutinib unmasks critical role of Bruton tyrosine kinase in primary CNS lymphoma. Cancer Discov. 2017;7(9):1018–29.
Soussain C, Choquet S, Blonski M, et al. Ibrutinib monotherapy for relapse or refractory primary CNS lymphoma and primary vitreoretinal lymphoma: final analysis of the phase II ‘proof-of-concept’ iLOC study by the lymphoma study association (LYSA) and the French oculo-cerebral lymphoma (LOC) network. Eur J Cancer. 2019;117:121–30.
Chamoun K, Choquet S, Boyle E, et al. Ibrutinib monotherapy in relapsed/refractory CNS lymphoma: a retrospective case series. Neurology. 2017;88(1):101–2.
Lionakis MS, Dunleavy K, Roschewski M, et al. Inhibition of B cell receptor signaling by Ibrutinib in primary CNS lymphoma. Cancer Cell. 2017;31(6):833–843.e835.
Narita Y, Nagane M, Mishima K, et al. Phase I/II study of tirabrutinib, a second-generation Bruton’s tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro-Oncology. 2021;23(1):122–33.
Yang Y, Shaffer AL, Emre NC, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21(6):723–37.
Rubenstein JL, Geng H, Fraser EJ, et al. Phase 1 investigation of lenalidomide/rituximab plus outcomes of lenalidomide maintenance in relapsed CNS lymphoma. Blood Adv. 2018;2(13):1595–607.
Ghesquieres H, Chevrier M, Laadhari M, et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective ‘proof of concept’ phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA)†. Ann Oncol. 2019;30(4):621–8.
Tun HW, Johnston PB, DeAngelis LM, et al. Phase 1 study of pomalidomide and dexamethasone for relapsed/refractory primary CNS or vitreoretinal lymphoma. Blood. 2018;132(21):2240–8.
Carbonell D, Mahajan S, Chee SP, et al. Consensus recommendations for the diagnosis of vitreoretinal lymphoma. Ocul Immunol Inflamm. 2021;29(3):507–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yuen, C.A., Iwamoto, F.M., Nayak, L. (2022). Treatment for Primary Vitreoretinal Lymphoma: The Neuro-Oncologist’s View. In: Chawla, B.V., Aronow, M.E. (eds) Global Perspectives in Ocular Oncology. Springer, Cham. https://doi.org/10.1007/978-3-031-08250-4_9
Download citation
DOI: https://doi.org/10.1007/978-3-031-08250-4_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-08249-8
Online ISBN: 978-3-031-08250-4
eBook Packages: MedicineMedicine (R0)