
Abstract
Intermediate and active site identification and quantification are crucial for understanding the structure-activity relationships and designing efficient catalysts. We overview how time-resolved XAS (TR-XAS) with the emerging use of multivariate data analysis allows the isolation of the pure species and the description of their evolution over time. Special emphasis is placed on the concepts of multivariate curve resolution with alternating least square (MCR-ALS) fitting. Investigation of catalyst preparation steps highlights the strength of TR-XAS to solve fast chemical transformations in liquid and solid states, including nanoparticle nucleation and growth, metal dispersion and redistribution, formation of undesirable phases, and metal-support interaction. Moreover, recent operando monitoring of the direct methane to methanol conversion, CO oxidation, methanol synthesis by CO2 hydrogenation, and ethanol steam reforming are presented illustrating the great potential of TR-XAS to quantify active sites and reaction intermediates. The main achievements to identify deactivation mechanisms including particle sintering, atom redistribution in bimetallic particles, and oxidation-state change are discussed. The combination of TR-XAS with complementary techniques is outstanding to fully describe deactivation by carbon deposition and regeneration. It is expected that TR-XAS combined with MCR-ALS analysis will be groundbreaking in research in many catalysis science areas.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Time-resolved X-ray absorption spectroscopy
- Quick-EXAFS
- Multivariate analysis
- MCR-ALS
- Catalysis
- Nucleation and growth
- Active species
- Reaction
- Deactivation
- Regeneration
1 Introduction
In the last decade, several new time-resolved X-ray absorption spectroscopy (TR-XAS) beamlines [1,2,3,4], totally or partly dedicated to operando characterization of catalysts, have offered the time resolution to unravel complex mechanisms involved in catalyst synthesis, activation, reaction, or deactivation. The time resolution improvement was strongly related to the access to third-generation synchrotron radiation facilities offering the mandatory monochromatic flux of 1011 (or more) photons per second on the sample, to record high-quality sub-second absorption spectra [5]. Moreover, as beneficial “side effect” of the high brightness of new-generation synchrotron sources, more sophisticated sample environments can be used for in situ characterizations. In this framework, all the aspects of the catalyst life span can be scrutinized with deeper details by XAS. New strategies can be proposed for the design of more efficient catalysts which encompass the development of challenging synthesis routes and optimization of activation treatments. The study of the structure-performance relationships using operando characterizations under realistic conditions allows the identification of active species and finally the development of new regeneration strategies of deactivated catalysts through the in-depth understanding of their performance loss.
The larger volume of XAS data obtained with the current sub-second time resolution gives rise to more complex and precise information on all steps of the catalyst life span but also to an apparent greater workload to analyze in depth the huge number of spectra. To overcome this difficulty, multivariate data analysis has been explored by several groups to reduce the data complexity of time-resolved studies. Sometimes qualified as an unsupervised machine learning method, the multivariate data analysis allows reducing the deep analysis of thousands of measured spectra to one of a few spectra representative of the evolving pure species. Therefore, the data analysis has been largely simplified thanks to fast-handling tools for automatic energy calibration/normalization [6,7,8,9,10] and thanks to those methods isolating the chemical species and describing their evolution upon a reaction parameter (temperature, time, and so on) [11,12,13,14,15,16,17]. Further, using conventional EXAFS data treatment and/or comparison with spectra of known phases, the chemical nature of those pure species can be identified.
The chapter is organized as follows: the first part will present the basic concepts underlying the multivariate data analysis of TR-XAS including a discussion of their limitations and strategies to overcome them. In the second part, selected TR-XAS studies illustrating the state of the art of catalyst characterizations at different stages of their life span will be discussed. We will focus on the use of the technique for unravelling dynamic processes which are of paramount importance in the preparation, use, and regeneration of heterogeneous catalysts.
2 Multivariate Curve Resolution with Alternating Least Square (MCR-ALS) Analysis
2.1 Basic Concepts
The catalytic steps are hardly a two-phase transformation process. The temperature programmed reduction (TPR) of supported Cu(II) species reveals, for instance, a two-stage process with the formation of Cu(I) and Cu(0) species [11]. Supported cobalt catalysts, which find wide applications in many catalytic processes such as Fischer-Tropsch [18,19,20,21,22] or ethanol steam reforming reactions [23,24,25], must be firstly reduced into metallic active species. This activation, simple at first glance, often involves a mixture of oxidic species. Alumina-supported calcined cobalt catalysts presenting Co3O4 and CoAl2O4 phases go through the formation of CoO before Co(0) species but with a well-known different reducibility. Those systems stress out that XAS investigations of the activation face a problem of phase mixtures. The knowledge gained on the identification and quantification of those phases evolving under activation is of prime importance for improving catalyst performances.
Multivariate curve regression methods are highly relevant to solve the problem of mixture of phases observed by analytical techniques in which the measure is the sum of the signal contributions coming from each one of the mixture components [26]. XAS is governed by the Beer-Lambert’s law and thus totally fulfils the aforementioned condition for the use of the MCR-ALS method. The rows of the experimental matrix D, gathering the q-XAS spectra collected during a reaction with k energy points, can be described by a bilinear model according to the following relation:
where the columns of matrix C contain the relative proportions of the n pure species in the mixture, the rows of the matrix ST are the spectra of those n pure species (ST meaning the transpose of matrix S), and E is the matrix expressing the error or variance unexplained by the C.ST product which should be in principle not larger than the experimental noise (Fig. 29.1). The data presented herein correspond to the Cu K-edge quick-XAS monitoring of the self-reduction of an alumina-supported Cu catalyst [27]. In this example, the minimization considering three components leads to the isolation of spectra which are characteristic of pure Cu(II)-Cp1, Cu(I)-Cp2, and Cu(0)-Cp3 species and to their related concentration profiles.

Schematic representation of the MCR-ALS analysis of the matrix D built from the Cu K-edge quick-XAS monitoring of the self-reduction of a Cu-based catalyst under C2H5OH/He atmosphere [27]. LoF = 0.115% and 99.9999% of variance explained
The principle of the MCR-ALS analysis is to proceed to a least square minimization of E based on the alternative optimization of C and S at each iterative cycle. For this scope, physically and chemically meaningful constraints on the C and S matrices are used to help the convergence but also to limit the pitfalls related to rotational ambiguities inherent to the method leading to non-unicity of the solutions [26]. Those constraints depend on the structure of the D matrix of the process which is under analysis and of preliminary knowledge that one can have on the species in presence. However, two constraints are always used: the concentration matrix contains only positive values, and for the analysis of normalized XAS data, the sum of the species concentrations is equal to 100% at any time of the process (closure relation). Additional constraint on the S matrix is the nonnegativity of absorbance when the D matrix is built from normalized XAS spectra, as presented in Fig. 29.1. This constraint is obviously no longer respected when EXAFS spectra are considered. Details about the constraints used in the MCR-ALS program implemented on the MATLAB® platform by the group of Roma Tauler can be found in [26, 28]. Minimization carried out under the action of those constraints is stopped when the convergence criterion, defined by the difference in fit improvement between consecutive iterations, is fulfilled. One of the parameters used to measure the fit quality of the MCR-ALS solution is the lack of fit, LoF (%), defined by the following relation:
where dij is an element of D and eij is the related residual value obtained from the difference between D and the rebuilt data over the n pure components calculated as follows:
where Cip is an element of C and Spj an element of S. The subindexes i, j, and p are related to the index of the spectrum, of the energy, and of the pure species isolated by MCR-ALS, respectively.
Essential steps of the MCR-ALS method reside in (i) the determination of the number of components and (ii) the initial guess of C or S matrix used for starting the first iteration loop. We will examine in the next sections strategies to afford this information.
2.2 Rank Determination of Matrix D
Principal component analysis (PCA) allows the transformation of a correlated data set with high dimensionality into a smaller-dimensional subspace expressing most of the variance of the original data set and based on non-correlated data. Computationally speaking, singular value decomposition (SVD) method is often used for calculating the principal components (PC) of a data matrix. PCA-SVD algorithm allows the factorization of the D matrix into the form:
where U is a m × n unitary matrix (i.e., orthogonal matrix satisfying U.UT = UT.U = I, where I is the identity matrix), S is a n × n diagonal matrix with nonnegative real numbers on the diagonal called eigenvalues, and VT is a n × n unitary matrix (i.e., orthogonal matrix satisfying V.VT = VT.V = I). The U.S matrix product is a diagonal matrix also called score matrix, whereas VT is the loading matrix and corresponds to the PC vectors. The main difficulty is not the PCA-SVD calculation itself insofar as this method can be found in most of the computer programs widely used by the scientific community for XAS analysis, but its use for the correct determination of number of PC, which corresponds to the so-called rank of the D matrix. Several plots can be used as skills for this purpose. Figure 29.2 illustrates how the rank of D corresponding to the TR-XAS data shown in Fig. 29.1 has been determined.
-
(a)
Scree plot , displaying in log scale the eigenvalues determined by PCA-SVD for each PC arranged in descending order as a function of the number of spectra in D, is one of those skills (Fig. 29.2a). The rank of D is obtained with the number of components describing the steep curve before the first point that starts the asymptotic trend of the curve. Herein the asymptotic behavior of the eigenvalues describing the PCs starts at the fourth component which is aligned with the higher-order components of lower eigenvalues and considered as belonging to the noise of the data.
-
(b)
2D score plots , displaying the projection of the data into the plane defined by the pairs of PC axes, can be also advantageously used for evolutionary analytical data, as those measured for phase transformations monitored by TR-XAS. In that case, as illustrated in Fig. 29.2b, the projections of data display trajectory-like evolutions only for pairs involving PCs explaining the variance of D, herein PC1, PC2, and PC3, whereas noisier projections are obtained for the other PCs, such as PC4.
-
(c)
1D score trajectory plots , displaying along the reaction coordinates the projection of the data onto the different PCs, are also a convenient skill for exploring the rank of evolutionary data. As shown in Fig. 29.2c, no-noise trajectory-like curves are expected for PCs describing significantly the variance, herein the first 3 PCs. The 1D score trajectory plots offer additional information regarding the detection of reaction coordinates at which strong chemical transformation occurs. Herein it clearly appears that significant changes of the evolution of trajectories occur at the spectrum index around 300. This location in reaction coordinates of data, presented in Fig. 29.1 as a temperature scale, corresponds to the maximum concentration of the intermediate species.
In summary, regardless of the graphs displayed in Fig. 29.2, interpretations converge towards the same conclusion: three components must be used for explaining most of the variance contained in the TR-XAS data set presented in Fig. 29.1.


PCA-SVD determination of the number of components to be used in the MCR-ALS analysis of the data set presented in Fig. 29.1 using (a) the scree plot for the eigenvalues associated to the first 20 principal components, (b) the 2D score plots corresponding to the projection of the data along different PC axes, and (c) 1D score trajectory plots corresponding to the projection of the data along the reaction coordinates, herein represented by the spectrum index, for the first 6 PCs
2.3 Initial Estimates
The MCR-ALS algorithm optimizes alternatively C and S until the convergence is reached. Before starting the iterative optimization (and after setting the rank of D), it requires the construction of an initial guess of either the matrix of pure spectra or the one of concentrations. Initial estimates of the S or C matrices can be obtained using methods based on the detection of “purest” variables in the experimental data set using, for instance, the so-called SIMPLISMA (SIMPLe to use Interactive Self-modeling Mixture Analysis) method or with techniques still based on PCA with the so-called EFA (Evolving Factor Analysis) method [26]. SIMPLISMA is a general method which does not require the analysis of evolutionary or process-like data. Taking into account the rank of D determined by PCA-SVD, the SIMPLISMA algorithm provides an initial estimate of S on the basis of standard deviations, under the assumption that the most dissimilar spectra will have larger standard deviations than less significant ones. In contrast, EFA is a method which requires a data set with a structure in the concentration direction and consequently well suited to be applied to evolutionary processes where spectra in D have been ordered (in time, temperature, and so on). EFA performs subsequent PCA-SVD runs on gradually enlarged sub-matrices of D by successive addition of one spectrum starting from the beginning to the end of the process for an evaluation in the forward direction of the data and from the end to the beginning in the backward direction [26]. The appearance (resp. disappearance) of a new pure species during the forward (resp. backward) examination of the data is indicated by the emergence (resp. decay) of a PC with a significant eigenvalue. The overlay of the forward and backward analyses allows for the derivation of estimates of concentration profiles for species involved in the process.
In the emerging MCR-ALS application to XAS data, SIMPLISMA is the most often used method, in particular, to determine the initial S matrix estimate. The reason for this success relates to instantaneous calculation of the initial S matrix using SIMPLISMA against several minutes of calculations of the initial C matrix using EFA with a computing time increasing with the number of spectra building D. However, for few chemical systems, SIMPLISMA or EFA methods can lead to very different outcomes. This is illustrated in Fig. 29.3 with the initial estimates obtained by SIMPLISMA and EFA for two quick-XAS data sets recorded either at the Co K-edge during the TPR of a Co catalyst [25] or at the Mo K-edge during the temperature programmed sulfidation (TPS) of a Mo catalyst [12]. PCA-SVD results show that the rank of D is 3 for the Co K-edge data and 4 for the Mo K-edge ones. Irrespective of the method used for the determination of the initial estimate matrix, the income of the MCR-ALS analysis for the Co K-edge data set has been found equivalent starting from the S guessed matrix or the C one. Both methods extract similar initial estimates: more dissimilar spectra have been found for spectrum indexes 1, 235, and 441 (over 441 spectra) corresponding to the concentration maxima of the 3 components (Fig. 29.3a). A different conclusion arises for the MCR-ALS analysis of the Mo K-edge data set (built from 404 spectra) for which only the solution from the C guessed matrix from EFA is satisfactory with acceptable chemical identification of the isolated pure species. SIMPLISMA builds the guessed S matrix from spectra with indexes 2, 63, 150, and 399 (Fig. 29.3b). The report of those reaction coordinates on the guessed concentration profiles determined by EFA clearly evidences that the intermediate species do not present a maximum of concentrations at coordinates 63 and 150. The reason for the discrepancy between both methods for this system is related to the structure of the XAS data composing D. The changes on Mo K-edge XAS spectra are only related to the shift of the rising-edge position to lower energies coming from both the oxidation-state change and the exchange of oxygen by sulfur ligands upon TPS [29]. Herein the isolation of “pure” components by SIMPLISMA fails due to the strong overlapping signals related to the different chemical species [26]. In contrast, for the Co K-edge data, the shift of the rising-edge position is also accompanied with a strong modification of the white line intensity during the reduction of cobalt species, allowing SIMPLISMA to properly isolate the “pure” components.

Initial estimates of S and C matrices provided by SIMPLISMA and EFA methods, respectively, for (a) the Co K-edge quick-XAS data recorded during the TPR of a calcined Co oxidic precursor catalyst and (b) the Mo K-edge quick-XAS data recorded during the TPS of a Mo oxidic precursor catalyst. Spectra are presented as processed by the algorithm, i.e., along a spectral range axis; since the spectral range is a non-equidistant spaced energy grid, it gives a strong weight on the XANES part
In summary, the MCR-ALS outcome is far from being insensitive to the method used for guessing the initial estimate matrix. The latter must be well thought in relation with the structure of the XAS data. EFA is highly advisable whenever an evolutionary process is under analysis [26]. It is noteworthy that initial estimates for the S matrix can be also imposed providing that the pure spectra of the chemical components involved in the process-like data have been previously recorded.
2.4 Limitations: Deviation of the Bilinearity Model for Evolutionary Data Set Recorded in Temperature
The intrinsic bilinearity of D supported by the Beer-Lambert’s law is the fundamental property justifying the use of the MCR-ALS algorithm. This assumes that the D matrix can be written as a sum of single component data matrices, where each of them decomposes into the product of two vectors containing the concentration and spectrum profiles. For XAS, temperature variation affects to a certain extent the bilinearity property of D by introducing a damping factor of the measured resonance intensity of the recorded spectra. This effect, which traduces the increase of thermal vibrations of atoms upon temperature rise, is supported by the temperature dependence of the Debye-Waller factor σ [30]. The damping effect, which is taken into account in the EXAFS formula through the term exp(−2σ2k2) (k is the photoelectron wavevector), mostly affects the signal at the extended energy range corresponding to the EXAFS spectrum (k > 2.6 Å−1) rather than the XANES spectrum (k ≤ 2.6 Å−1). Consequently, MCR-ALS analysis of a data set built from temperature-evolving EXAFS spectra can potentially suffer from an overestimation of the number of components, some of them being chemically identical but affected by the Debye-Waller damping. This is, for instance, illustrated in Fig. 29.4 with the MCR-ALS analysis of the matrix composed of the EXAFS spectra recorded during the TPR of a Ni catalyst [31]. Chemical rank analysis concludes to the presence of three components (Fig. 29.4b, c). The outcome of the MCR-ALS analysis (Fig. 29.4d) reveals that the first two components display EXAFS oscillations in phase, both characteristic of the same NiO-based species but with amplitude for the second component suffering from damping when the temperature is increased. The transformation of the NiO species into Ni(0) species occurs only above 300 °C, which corresponds to a spectrum index greater than 420 (Fig. 29.4d).


MCR-ALS analysis of Ni K-edge quick-EXAFS data recorded during the TPR of a calcined Ni oxidic precursor catalyst. (a) Quick-EXAFS normalized data. Rank analysis using (b) the 1D score trajectory plot and (c) the scree plot. (d) Outcome of the MCR-ALS optimization considering 3 components. It is noteworthy that the MCR-ALS resolution has been performed with the addition of EXAFS spectra of fully reduced Ni(0) supported species recorded at 500 °C (data with indexes higher than 599 in (d)) and corresponding to the yellow-colored EXAFS spectrum in (d)
Considering the elegant demonstration done by Martini et al. [15] on the negligible temperature dependence of the XANES spectra, a first strategy to overcome the bilinearity deviation associated to the temperature increase can be the MCR-ALS minimization considering the XANES region only. This strategy, presented in Fig. 29.5 (same data set as Fig. 29.4), allows obtaining accurate determination of the number of “pure” chemical species involved in the reaction process and their associated concentration profiles. Only two components are significant in the scree plot. Identification of the chemical species involved in the reaction is therefore obtained from the analysis of the XANES spectra of pure species, using comparison with spectra of already recorded known references or the use of ab initio XANES spectrum calculations [32]. The minimization based on XANES data set has the drawback that no pure EXAFS spectra can be isolated and further used for identifying the pure chemical species by least square fittings. However the knowledge of concentrations determined by MCR-ALS at a given process temperature can be used for a better simulation of the experimental EXAFS data, thus without temperature limitation [25], taking into account the right contributions of the different species in the EXAFS signal.

MCR-ALS analysis of Ni K-edge quick-XANES data recorded during the TPR of a calcined Ni oxidic precursor. (a) Quick-XANES normalized data, (b) rank analysis using the scree plot, (c and d) outcome of the MCR-ALS optimization considering two components. It is noteworthy that the MCR-ALS resolution has been performed with the addition of XANES spectra of fully reduced Ni(0) supported species recorded at 500 °C. LoF = 0.46% and 99.9979% of variance explained
A second strategy for temperature-dependent XAS data set is to consider that most of the variance related to structure modifications (by phase transformation and not by temperature increase) is contained in the XANES part and that a minimization of a normalized data set containing both the XANES and the EXAFS energy regions is a good compromise to overcome the temperature-damping effect, while keeping the possibility to isolate the EXAFS spectra of pure chemical species. This approach has been successfully used for a wide number of catalyst activation and reaction processes and has demonstrated its efficiency to further identify the chemical species [6, 12, 24, 29, 33,34,35,36,37]. However, it is noteworthy that for a given species, the EXAFS spectrum extracted by MCR-ALS and rebuilt from the sum of the contributions of this species over the full process is affected by a damping contribution related to the temperature window of existence of this component. Therefore, this strategy allows for EXAFS fitting and chemical identification but in no case for any fine discussion on Debye-Waller modifications occurring during the chemical process.
2.5 Limitations: Rank Deficiency by Existing Correlated Data
One of the main issues of the multivariate analysis is related to the so-called rank deficiency of the data, an issue which occurs when large similarity among the spectra of pure species or coevolving concentration profiles exist [26]. In that case, the number of components detected by PCA-SVD is lower than the number of chemical species present in the mixture. This is, for example, illustrated in Fig. 29.6a with the Zn K-edge quick-XAS monitoring of the thermal decomposition of the same Zn-Cu-Al layered double hydroxide (LDH) sample with a heating ramp of 5 °C/min (D1 matrix) and 10 °C/min (D2 matrix) in static air. Both data sets are described individually with a chemical rank of 2, a value which is underestimated considering the extra information gained by wide-angle X-ray scattering (WAXS) and the final XANES spectra. Indeed, WAXS monitoring of the thermal decomposition of the same sample points out the formation of nanocrystalline ZnO phase while a careful examination of the XANES spectra obtained at the end of both decompositions [38] does not reveal characteristic XANES fingerprint of nanocrystalline ZnO. The outcome of the decompositions is actually a mixture of species, one of them being ZnO. In order to overcome the rank deficiency characterizing each individual data set, the so-called column-wise augmented (CWA) data strategy can be used. It consists in analyzing simultaneously both data sets with the assumption that irrespective of the heating ramp, D1 and D2 matrices share common chemical components, expressed in the single ST matrix. C1 and C2 matrices contain the concentration profiles related to each data set. SVD analysis of the augmented data set detected three components. Figure 29.6b, c displays the concentration profiles and the spectra obtained within the CWA strategy. The thermal decomposition of the pristine LDH gives rise to the formation of nanocrystalline ZnO and ZnAl2O4 phases which have been identified by comparing their spectra isolated by MCR-ALS with those of synthesized references. From the comparison of the concentration profiles, the reason for rank deficiency of each data set is clearly related to coevolving concentrations of ZnO and ZnAl2O4 nanophases. The simultaneous resolution of both data sets allows breaking this rank deficiency because the relative concentrations of both phases obtained by thermal decomposition vary sufficiently from D1 to D2.

(a) Zn K-edge quick-XAS monitoring of the thermal decomposition of Zn-Cu-Al LDH samples at 5 °C/min (D1 matrix) and 10 °C/min (D2 matrix). (b) Concentration profiles of each data set and (c) MCR-ALS isolated spectra of pure species obtained in the CWA data resolution, considering a rank for D1 + D2 equal to 3, and identified by comparison with spectra of known references
This example emphasizes how powerful is the multi-data set analysis to overcome the rank deficiency issue of individual data sets. It points out also that the users must take a critical look at the PCA-SVD analysis regarding the estimation of the number of components for evolutionary process-like data. MCR-ALS is often described as a “blind-source separation” method [13]. “Blind” is only justified by the fact that MCR-ALS does not require a priori the use of any standards to isolate the chemical species involved in the reaction. In that sense, the method is really unique. In particular in catalysis, it allows the identification of chemical species from process-like data set, for which references are not available either due to the nanocrystalline nature of the species leading to spectra different from their bulk counterparts [11] or due to the difficulty to synthesize them as, for instance, for metallic species in interaction with zeolite framework [16]. However, the way to properly perform the MCR-ALS optimization often requires other information obtained by complementary techniques [14, 35, 38,39,40]. For instance, additional equality constraints associated with the knowledge of the presence of pure species can be introduced in the minimization, either as the spectra of those species or as their presence windows in the concentration profiles [26]. Finally, it is noteworthy that the CWA method has been successfully used in numerous recent publications related to MCR-ALS analysis of time-resolved data [29, 34, 37,38,39,40,41]. This clearly points out that any rank deficiency of a data set should be detected at the beamline during the data collection to allow the user to immediately perform additional measurements by changing a parameter of evolution (heating ramp, gaseous atmosphere, and so on) to break the rank deficiency. This justifies the strong effort carried out nowadays by beamline scientists to offer more automated data analysis strategies.
Until recently, linear combination fitting (LCF) of XAS spectra was the common methodology for the determination of species concentration provided that the spectra of appropriate reference compounds were available. Currently, the more sophisticated and powerful MCR-ALS analysis is widely used with the advantage that not only the concentration profiles are determined but also the XAS spectra of those species allowing their further identification by EXAFS fitting.
3 How Time Resolution Can Give Insights on “Birth, Life, and Death” of Solid Catalysts
As pointed out in a perspective article [42], the developments of time-resolved operando and in situ methods are crucial to study the “birth, life, and death” of catalysts operating under harsh conditions.
3.1 Preparation of Catalysts: From Solution Processes to Solid-State Reactions
Among the different stages of the catalyst life span, the “birth” stage, which refers to the catalyst preparation, is probably the one which greatly took advantages of the last decade of development of TR-XAS beamlines, at least for the genesis of multicomponent nanoparticles from solution processes [14]. Taking into account the incisive influence of the morphology and size of metallic nanoparticles on the catalytic activity [43], the understanding of the mechanisms and kinetics of precursor transformation is highly demanded to achieve a comprehensive control of the morphological parameters. Besides those bottom-up preparation routes of colloidal particles in liquid media, the solid-state preparation route, encompassing support impregnation and subsequent thermal treatment, is also crucial to control and rationalize. This section will be focused on a few selected results gained by TR-XAS: first on nucleation and growth of colloidal nanoparticles and then on thermal decomposition routes for better controlling the dispersion and nature of active phases.
3.1.1 Solution Preparation of Colloidal Particles
The study of bottom-up particle preparation routes gives rise to tremendous efforts to develop appropriate in situ characterizations due to the short time span of nucleation processes [44] but also to explore new preparation strategies. The nucleation and growth mechanisms are not unique and, depending on the chemical systems, can be supported either by the classical nucleation theory (CNT), where nucleation and growth are conceptually separated in time, or the nonclassical nucleation theory, where growth occurs partially simultaneously with the nucleation [45]. Further, various growth laws can be highlighted for ripening or oriented attachment growth models. Irrespective of the nucleation pathways, in situ TR-XAS has been recognized as being unique for shedding light on the nucleation stage. For instance, considering the LaMer model for nanoparticle generation which is the basis of the CNT, nucleation spontaneously occurs when the concentration of free monomers formed by the chemical transformation of pristine precursor reactants reaches a level of supersaturation (Fig. 29.7). During the nucleation encompassing stage I and stage II of Fig. 29.7, chemical reactions of precursors with solvent or reducing species can induce ligand exchange around the metal ions and/or change of formal oxidation state of metal ions. Both can be dynamically well studied by TR-XAS, revealing the formation of the free monomers and when nucleation occurs.

Schematic illustration of the LaMer model describing the three stages of colloidal nanoparticle formation in solution according to the CNT. The generation of precursors encompasses the transformation of precursor reactant (M+ in orange) into nanoparticle precursors also called free monomers (M(0) in blue). (Reprinted with permission from Ref. [45]. Copyright (2016) Elsevier)
Due to their relevant applications in catalysis, noble metal nanoparticles have been considerably studied using various preparation routes in solution with, as quite common characteristic, a fast nucleation rate in the second or millisecond timescale when using strong reducing chemical agents [46,47,48,49]. For instance, the formation of gold nanoparticles by reduction of AuCl3 by BH4− ions required the use of sub-second timescale available at dispersive XAS [47] or quick-EXAFS beamlines [46]. Comparison of Au L3-edge XANES spectra recorded with 100 ms time resolution upon nanoparticle formation with references of Au(III), Au(I), and Au(0) species reveals that after 104 ms, the pristine Au(III) ions have been reduced all into the Au(I) form (Fig. 29.8a). Then gradually Au(I) is transformed into Au(0), and the complete reduction is achieved in less than 20 s. Figure 29.8b compares the concentration profiles of Au(0) after the reactant mixing (red curve) with the amount of Au(0) specifically in nanoparticles derived from the analysis of time-resolved small-angle X-ray scattering (SAXS) data (blue curve). It is noteworthy that the red curve determined by XAS is equal to all the Au(0) species in solution, i.e., nanoparticles but also free monomers formed upon reduction of Au(I) species. The subtraction of both curves is consequently equal to the amount of Au(0) free monomers, and their variation upon time is a perfect illustration of a nucleation governed by the CNT [45] as presented in Fig. 29.7.

(a) Au L3-edge XANES spectra recorded at different times of the gold nanoparticle synthesis and (b) concentrations of the different species during the reaction: Au(0) as determined by LCF of the time-resolved XANES data recorded upon synthesis (red symbols); Au(0) contained in the nanoparticles measured by SAXS (blue symbols); Au(0) free monomers (green symbols) deduced by subtraction of the red and blue curves, responsible beyond the supersaturation point to spontaneous aggregation to form nuclei. (Reprinted with permission from Ref. [47]. Copyright (2010) American Chemical Society)
CNT model assumes that the self-formation of seeds occurs through the successive accretion of free monomers and through dissolution of unstable pre-nucleation clusters characterized by a subcritical size. Smart experiments were recently carried out for the study of the onset of nucleation of palladium nanoparticles [49], in which the concept of reaction at the interface of two immiscible liquids has been used in order to confine the nucleation in space but also to control the onset of nucleation through electrochemical trigger. TR-XAS measurements revealed that at peculiar PdCl42− concentrations, spontaneous reduction of aqueous PdCl42− occurred at the interface with an organic solution of ferrocene acting as a reducing agent, according to a stochastically fluctuating equilibrium. The Pd(0) nuclei concentration fluctuated over a number of hours suggesting random oscillations between PdCl42− reduction (leading to the formation of pre-nuclei with subcritical size) and their subsequent dissolution (Fig. 29.9b). At the opposite, under potential control, the oscillatory growth is eliminated, and a continuous increase of the XAS edge jump is observed attesting to the increase of concentration of stable Pd(0) nuclei (Fig. 29.9a).

Pd K-edge XAS monitoring of the nucleation of Pd nanoparticles at the liquid-liquid interface (a) under electrochemical triggering and (b) in the absence of applied potential. For (a), [PdCl42−] = 5 mM = [reducing agent], whereas for (b), [PdCl42−] = 10 mM and [reducing agent] = 20 mM. (Reprinted with permission from Ref. [49]. Copyright (2017) Elsevier)
XAS spectroscopy can be used beyond the only oxidation-state characterization of the multivalent precursors before nanoparticle nucleation. For some systems, XAS provides also important information about the chemical nature of the ligands and intermediates before nucleation, which enlightens their crucial role in tailoring the final shape of the nanoparticles [50, 51]. The versatile use of oleylamine (OLAM) reagent has been recently explored to tailor the composition, size, shape, and crystalline structure of various nanoparticle systems. Using TR-XAS measurements, the role of oleylamine in combination with trioctylphosphine (TOP) or trioctylphosphine oxide (TOPO) has been comprehensively discussed rationalizing the shape control of copper nanoparticles prepared from CuBr under high temperature [51]. Thanks to the combination of XAS, nuclear magnetic resonance, and mass spectrometry (MS), it has been shown that the choice of reagents first impacts the structure of the Cu(I) precursor (Fig. 29.10a). Then, the subsequent heating and aging at 260 °C lead to the formation of nanospheres for the CuBr-OLAM-TOP system and nanocubes for the CuBr-OLAM-TOPO as measured by transmission electron microscopy (TEM) (Fig. 29.10a). The in situ TR-XAS monitoring reveals very different kinetics of Cu nanoparticle nucleation driven by the structural differences of the pristine complexes. Actually, under temperature, a mixture of equal proportions of Cu(I), Cu(II), and Cu(0), resulting from a disproportionation equilibrium (2 Cu(I) → Cu(II) + Cu(0)), is evidenced from the presence of characteristic XAS features related to each oxidation state (Fig. 29.10b, e). For the CuBr-OLAM-TOPO system, the three species evolve with the same kinetics upon heating to 260 °C (Fig. 29.10d), in agreement with the disproportionation. Under the same conditions, a deviation from the equilibrium is observed for the CuBr-OLAM-TOP system with the faster appearance of Cu(0) species (Fig. 29.10c), indicating that Cu(0) nuclei are formed and no longer participate to the disproportionation equilibrium. The nucleation occurs continuously during the heating ramp for the CuBr-OLAM-TOP system, whereas it appears abruptly after 10 minutes at 260 °C for the CuBr-OLAM-TOPO system, as evidenced with the rising of the signal characteristic of Cu(0) at the detriment of Cu(I) and Cu(II) signals (Fig. 29.10e). For the CuBr-OLAM-TOP system, the gradual flux of Cu(0) monomers favors the thermodynamically controlled shape, i.e., nanospheres, while for the CuBr-OLAM-TOPO, the sudden increase of Cu(0) monomers leads to the formation of particle shapes dictated by the kinetic regime, i.e., nanocubes.

(a) Proposed structures for the complexes formed from reactions of CuBr(OLAM)3 with TOPO and TOP (TOP, TOPO, and OLAM ligands are, respectively, represented by their donor atoms P, O, and N) and representative shapes of nanoparticles after heating and aging at 260 °C. (b) displays some reference spectra and stack plots of Cu K-edge spectra recorded upon heating from 80 to 260 °C for both systems and obtained by subtracting the XANES spectrum at 80 °C from each subsequent spectrum. (c) and (d) display the intensities of the Cu(0), Cu(I), and Cu(II) pre-edge structures normalized by the highest value observed during the heating ramp. (e) is the stack plots of Cu K-edge spectra recorded upon aging at 260 °C for 60 min. (Reprinted with permission from Ref. [51]. Copyright (2019) American Chemical Society)
The bottom-up preparation of colloidal particles is specially used for the preparation of photocatalysts, electrocatalysts, or heterogeneous catalysts for selective hydrogenation of organics in autoclaves. Such catalysts do not require severe pretreatments before their use, except the deposition of the preformed particles on the adequate support [22]. The removal of capping ligands remains nevertheless a challenge for obtaining active and stable heterogeneous catalysts, which is probably insufficiently addressed by in situ and/or TR-XAS characterizations. The landscape is very different regarding conventional preparation of heterogeneous catalysts, for which XAS has merely contributed to better understand the impact of the catalyst pretreatment on the structure-activity relationship.
3.1.2 Synthesis of Supported Catalysts
The industrial preparations of supported catalysts often use solid-state routes based on the simple and inexpensive impregnation of metal salts on supports followed by drying, calcination, and activation [22]. Irrespective of the catalytic reaction, synthesis parameters and support characteristics are of crucial importance. They directly alter the catalyst activity by affecting the metal dispersion but also its distribution. This section will examine some elementary preparation steps of heterogeneous catalysts impacting the catalyst properties and for which TR-XAS provides real-time knowledge of phase transformations.
3.1.2.1 Active-Phase Dispersion: Particle Size and Particle Density
Dispersion herein encompasses two different concepts: the first one is the ratio between the number of atoms exposed at the surface of the particle and the total number of atoms in the particle. The smaller the particles, the larger their surface contributions, the greater the dispersion values. The second is the density of particles, which is related to the concept of distance between adjacent particles on the support. Both concepts have a direct bearing on catalytic activity and stability. The recent progress in in situ TEM under reactive environments leads to unique opportunities for accessing the sizes, shapes, particle size distribution, and density [52]. XAS offers complementary advantages, such as its high time resolution for in situ and operando characterizations, to highlight a specific key parameter of the preparation protocol, influencing the particle dispersion.
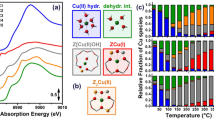
For instance, in-depth investigations of preparation parameters of cobalt-based Fischer-Tropsch (FT) catalysts were carried out to optimize the dispersion of the active metallic Co(0) particles. Among them, the calcination atmosphere for the thermal activation of supported cobalt hydrated species has been scrutinized by several groups [18,19,20,21,22]. In particular, it was reported that the calcination under NO-containing atmosphere enabled the formation of smaller Co3O4 particles on porous silica supports [53] than the one carried out under air. The Co K-edge quick-XAS monitoring of the calcination revealed different decomposition pathways depending on the reactive atmosphere (Fig. 29.11) [20]. Starting from the Co(H2O)6(NO3)2 species, the calcination under air first led to the formation of the anhydrous Co(NO3)2 species, which was further gradually transformed into Co3O4, as evidenced by the characteristic energy shift of the rising edge associated to the cobalt oxidation and by the second neighbor contributions above 160 °C in the Fourier transform moduli (Fig. 29.11a1 to c1). NO-assisted decomposition gave rise to the formation of a supplementary intermediate species before the formation of Co3O4, identified as a cobalt hydroxynitrate Co(OH)x(NO3)y phase (Fig. 29.11a2 to c2). It was underlined that the brutal O2 release for the transformation under air of Co(NO3)2 into Co3O4 (3 Co(NO3)2 ↔ Co3O4 + 6NO2 + O2) is detrimental to oxide particle dispersion and that similar O2 release is not in the equilibrium balance during the transformation of Co(OH)x(NO3)y into Co3O4. De Jong et al. [21] further explained the role of the hydroxynitrate as an immobile phase preventing redistribution of the pristine Co(H2O)6(NO3)2 species and yielding small cobalt oxide nanoparticles by its further decomposition at higher temperature. Unfortunately for the catalytic activity, smaller Co3O4 particles (8.4 nm) obtained upon NO-assisted calcination are more difficult to reduce than larger (10.7 nm) ones prepared by calcination under air [20]. This behavior, observed in many studies involving NO-assisted calcination [54], was ascribed to a stronger interaction of smaller cobalt oxide particles with the support leading to the formation of cobalt silicate-like species.

Co K-edge quick-XAS monitoring of the thermal activation of a Co/SiO2 FT catalyst with heating from RT to 240 °C: (a1 to c1) under air and (a2 to c2) under 5% NO/He with (a) XANES spectra, (b) EXAFS k3-weighted Fourier transform moduli, and (c) cobalt-phase speciation determined by LCF of the TR-XAS spectra. (Reprinted with permission from Ref. [20]. Copyright (2012) Wiley)
The use of noble metal promoters (Ru, Pt, Re, and so on) for preparing cobalt FT catalysts has been also the subject of intense researches aiming to relate the noble metal reduction to cobalt reducibility improvement [19]. The element selectivity of XAS can be used to determine not only the local environment of cobalt but also of the promoter. In a comprehensive study combining Co and Ru K-edge quick-XAS monitoring with MCR-ALS analysis, the impact of Ru promoter on the active Co(0) site density has been evaluated for CoRu/SiO2 catalysts prepared in the presence of sorbitol [33]. The transformation of Co3O4 into CoO under H2 is characterized by an increase of the white line intensity of the XANES spectra associated to the conversion of the tetrahedral cobalt sites in the spinel-like Co3O4 into octahedral ones in CoO. This transformation occurs at significantly lower temperatures for CoRu-sorbitol/SiO2 oxidic precursors (Fig. 29.12b) compared to non-promoted ones (Fig. 29.12a) in total agreement with the TPR profiles (Fig. 29.12d) [18, 20]. But more remarkably is the quasi absence of Co(0) species detected at 500 °C for the non-promoted sample compared to the promoted one (Fig. 29.12a, b). This is ascribed to the formation of cobalt silicate species which are reduced at high temperatures, as displayed in Fig. 29.12d with the peak at 855 °C, whereas the strong peak of the TPR profile around 369 °C for the CoRu-sorbitol/SiO2 catalysts is ascribed to the complete reduction of CoO into Co(0) observed by XAS. The MCR-ALS analysis of the Ru K-edge data reveals also a two-stage mechanism (Fig. 29.12e). First, the initial oxidic species obtained after calcination, identified by EXAFS fitting as characteristic of a Ru(IV) ion substituting Co(III) ion in an octahedral cationic site of the spinel-like Co3O4 species, is transformed into a Ru(III) ion in substitution of Co(II) in the NaCl structure of CoO. In a second step, the reduction to the metallic state takes place from 150 to 500 °C and yields Ru embedded in a bimetallic Co-Ru nanoparticle, as revealed by EXAFS fitting. The size of the metallic nanoparticles determined by TEM emphasizes the beneficial impact of sorbitol for enhancing the dispersion of the nanoparticles with sizes lower than 5 nm compared to the CoRu/SiO2 catalysts with 25% of the nanoparticles of sizes up to 12 nm. The TPR and Co K-edge quick-XAS monitoring highlight also the beneficial role of the promoter for improving the reduction of cobalt and thus leading to a higher density of active particles. The Ru K-edge quick-XAS characterization of the promoted catalysts rationalizes the synergetic effect between the reduction steps of both metals since they occur at approximately the same temperatures due to the close vicinity of Ru inside the cobalt oxidic network, first at the end of the calcination stage into Co3O4 then as belonging to the CoO network and finally through an autocatalytic process forming bimetallic CoRu particles.


Quick-XAS monitoring of the reduction (a) at the Co K-edge of Co-sorbitol/SiO2 and (b) at the Co and (c) Ru K-edges of CoRu-sorbitol/SiO2 and related MCR-ALS speciations. (d) TPR profiles of Co-sorbitol/SiO2 and CoRu-sorbitol/SiO2. (e) Representation of the Ru species involved during the reduction of CoRu-sorbitol/SiO2 identified by EXAFS fitting of the different Ru MCR-ALS components: red, ruthenium; black, cobalt; gray, oxygen. (Reprinted with permission from Ref. [33]. Copyright (2015) American Chemical Society)
3.1.2.2 Metal Distribution: Formation of Undesirable Phases and Interaction with the Support
The formation of phases involving one of the chemical elements of the support at the impregnation step could have strong impact in the further catalyst preparation stages, such as calcination or activation. For instance, the Anderson-like heteropolyanion (HPA) Al(OH)6Mo6O183− is a well-known species reported during the preparation of alumina-supported Mo catalysts used in hydrodesulfurization (HDS) process for the removal of sulfur in crude oils [34]. The formation of this HPA observed for high Mo loading solutions results from the support lixiviation due to the solution acidity and further reactions with soluble Mo species during the maturation in solution and drying stages. The impact of its formation has been described in the literature as leading to MoO3 upon calcination, which is detrimental for the dispersion of the further active MoS2 phase formed under sulfiding atmosphere. The formation of the Anderson-like HPA species can be also observed upon further exposure under moisture conditions of calcined catalysts, as illustrated in Fig. 29.13 [55, 56].

Simultaneous monitoring by Mo K-edge XAS (a) and Raman spectroscopy (b) of the RT rehydration of a calcined 5 wt. % Mo catalyst supported on amorphous silica-alumina. The spectra of the Al(OH)6Mo6O183− ion, labeled AlMo6, are presented for comparison. Stars in (b) indicate the position of characteristic MoO3 lines. (Reprinted with permission from Ref. [55]. Copyright (2005) IOP Publishing)
3.1.2.3 Metal Distribution Within Bimetallic Particles
Besides the addition of promoters for improving the formation of active species, the use of bimetallic systems is also frequently considered. Taking advantage of an interplay of electronic and geometric characteristics of both metals, the catalytic properties are improved. The knowledge and mastering of the atomic distribution of the two metals inside those systems are of paramount importance for the design of efficient catalysts. With selected examples, we will examine herein the use of TR-XAS for highlighting processes leading to the formation of bimetallic particles and to composition adjustments passing from core-shell structures to alloys and vice versa.
The thermal decomposition of LDH-like compounds is an effective synthesis route for the preparation of highly dispersed multimetallic catalysts. Quick-XAS monitoring combined with MCR-ALS analysis has been used for unravelling and quantifying the intermediate species involved during the transformation of NiCuAl LDH-based oxidic precursors into NiCu/Al2O3 catalyst [37]. The use of a direct activation route, starting from the as-prepared NiCuAl LDH-based oxidic gel and involving an oxidative treatment up to 210 °C followed by reduction under H2 up to 500 °C, displays very different speciation patterns for nickel and copper compared to conventional activation [31], as illustrated in Fig. 29.14. During the reductive step of the direct activation, the nickel and copper phases are completely transformed into metallic species before reaching 500 °C at the opposite of the conventional route where only 75% of Ni(0) is formed after 3 h of H2 treatment at 500 °C. As the onset of reduction of the oxidic nickel phases formed upon LDH oxidative decomposition is coincident with the completion of Cu reduction, it was assumed that Cu(0) catalyzes the decomposition of adsorbed H2, supplying active H species for promoting the reduction of nickel. The comparison with the monometallic Ni LDH gel evidences a decrease of the temperature for complete nickel reduction, from 495 °C in the monometallic sample to 450 °C for the bimetallic one. This was explained by a heterogeneous nucleation of Ni(0) particles over the Cu(0) ones early formed, leading to the formation of NiCu alloyed particles [31, 37].

Simultaneous Ni and Cu K-edge quick-XAS monitoring of the activation of a NiCu/Al2O3 oxidic precursor derived from LDH. (a) Conventional activation, carried out after stagnant air calcination, under H2 from RT to 500 °C. The MCR-ALS speciation revealed the transformation of NiO into Ni(0) and a two-stage transformation for CuO into Cu2O and Cu(0) [31]. (b) Direct activation route. Due to the presence of chloride anions as charge compensating of the positively charged LDH layers, CuCl2- and atacamite-like species, first formed at the early stage of the oxidative treatment, are promptly reduced into Cu(0) species at 300 °C, whereas Cu-depleted LDH is transformed into a first oxidic nickel species, which is a mixture of NiO and NiAl2O4 evolving with a similar formation rate before being reduced into Ni(0) species at the completion of Cu reduction. (Reprinted with permission from Ref. [37]. Copyright (2020) The Royal Society of Chemistry)
The emergence of environmental TEM cells provides unique opportunities for imaging at the atomic level the particle composition and the dynamic restructuration of bimetallic nanoparticles under reaction conditions [52]. However, information afforded by TR-XAS still remains very complementary to the ones obtained by TEM. The element sensitivity of XAS offers the clear advantage of local-order information around each of the elements composing the materials. It provides a complete description of degree of element mixing and oxidation state of the atoms. Furthermore, the so-obtained information are averaged over a larger field of view than TEM and consequently over a large number of multimetallic nanoparticles, providing some advantages such as a higher statistical relevance of the observation but also mitigation to a certain extent of artifacts induced by electron beams for sensitive materials.
Quick-XAS was applied to unravel, with one-second time resolution, the reaction mechanism of the dynamic restructuring of Pt13In9 nanocrystals during high-temperature O2-H2 redox cycling [57]. Similar redox cycling treatments are used for reactivating dehydrogenation Pt-In nanocatalysts after coking in high-temperature alkane flows. In situ Pt L3-edge quick-XAS measurements were used to probe the Pt oxidation state during nanoalloy segregation dynamics induced by the repetitive H2-O2 redox cycling. The reversible change of the white line (WL) intensity and of the energy position of the WL maximum upon cycling (Fig. 29.15a) is interpreted, by comparison with the XANES spectra of references (Fig. 29.15b), as being related to the reversible transformation between a reduced state, in which Pt is alloyed with In, and an oxidized state where PtOx is formed. This interpretation is clearly deduced from the analysis of the wavelet-transformed quick-EXAFS magnitude measured at specific points of a cycle (Fig. 29.15c) where at the end of the H2 pulse, Pt is surrounded by In and Pt first neighbors in a Pt-In alloy. Upon O2 exposure, the Pt-In alloy has decomposed by segregation into a PtOx phase in which Pt is surrounded by O in the first coordination shell and Pt as second nearest neighbors. The segregation is fully confirmed by microscopy (Fig. 29.15e, f) showing a metallic Pt core particle surrounded by an In2O3 crust. It is noteworthy that the WL intensity characteristic of the PtOx species significantly decreases upon successive cycles as the result of the Pt nanoparticle sintering. Namely, electron microscopy highlights the particle growth from ca. 2 nm at the end of the first cycle (Fig. 29.15d) to ca. 10 nm after 60 cycles (Fig. 29.15g). The in-depth analysis of the results performed at different temperatures upon cycling allowed the identification of the reaction mechanism describing the dynamic restructuring of Pt-In nanocatalysts and the calculation of activation energy of alloying and dealloying processes.

Pt L3-edge quick-XAS monitoring of the H2-O2 redox cycling performed at variable temperatures to induce structural changes in the Pt-In nanocatalysts: (a) WL intensity and energy position of the WL maximum as a function of the number of H2-O2 redox cycles, (b) XANES reference spectra, (c) zoom of the WL intensity and WL energy position within the 21st redox cycle. On the top, Pt L3-edge wavelet-transformed quick-EXAFS magnitude maps at specific points of the 21st redox cycle. The plots reveal the element type (k-axis) and position (R-axis) of neighbors around Pt, namely, In, O, or Pt. (d to g) Electron microscopy images of Pt-In nanoparticles after the 1st (d and e) and the 60th (f and g) redox cycles. (Reprinted with permission from Ref. [57]. Copyright (2018) Wiley VCH)
3.2 Catalysts in Operation: From Active Phases to Spent Catalysts
The ability to visualize a catalytic transformation in real time under real conditions using TR-XAS has motivated studies of many catalytic reactions due to the potential for unprecedented understanding of reaction mechanisms. The structure-performance relationship can be either indirectly established by comparison of in situ XAS measurements, i.e., under gas, temperature, and pressure, with catalytic activity measured independently or directly via TR-XAS and simultaneous monitoring of the products of the catalytic reaction by an analytic method, through the operando approach. This section is focused on a few recent studies which illustrate the insights gained by TR-XAS into the modification of catalyst structure and composition in relation to the catalytic performances. Studies carried out in order to optimize the process conditions regarding catalytic activity will be distinguished to those specifically performed to better understand the origin of deactivation.
3.2.1 Insights of Catalyst Structure and Composition vs Activity: A Lever for Process Optimization
Because of major concerns in relation to climate change, the mitigation of greenhouse gases in the atmosphere has been the subject of intense researches in the last decade. In particular, an abundant literature can be found about the nature of active sites in Cu-based zeolites used for the direct methane to methanol (DMTM) conversion. DMTM is considered as a potential alternative route to the conventional and costly indirect ones, involving steam reforming of methane to produce syngas further converted into methanol by hydrogenation. As reviewed in [58] and [41], considerable debates about the basic mechanisms and structure of active sites in those systems still exist. However, those reviews agree on the importance of TR-XAS to provide dynamical information on the structural and electronic changes impacting copper atoms in Cu-based zeolites during the DMTM conversion cycling process. For instance, Lomachenko et al. [59] tested on Cu mordenite (Cu-MOR) catalysts different O2 activation and CH4 loading duration before performing the last step of cycling dealing with the steam-assisted CH3OH extraction. For the 0.18Cu-HMOR(7) composition presented in Fig. 29.16a–d, it was observed that the abundance of the Cu(I) species is strongly dependent of the O2 protocol duration with more than 60% at the onset of methanol extraction for long protocol (LP). Rapidly, after CH4 introduction, the fraction of Cu(I) species in interaction with the zeolite is stabilized. This behavior indicates a saturation by CH4 of the Cu(II) active species interacting with the zeolite framework formed during the O2 activation at a level which is nearly two times larger for LP than for SP (short protocol). Adding the results obtained for the 0.36Cu-HMOR(11), a clear linear correlation between the methanol productivity per Cu versus the fraction of Cu(I) determined at the end of the CH4 loading step was established (Fig. 29.16e): the higher the Cu(I) fraction, the better the performance. This study highlights the correlation between Cu speciation and performances thanks to a spectroscopic descriptor of the CH3OH productivity per Cu represented by the Cu(I) fraction formed after CH4 exposure. It is noteworthy that the full description of Cu speciation during activation required to go beyond the description gained by the common LCF method from XANES of standard compounds using the CWA-MCR-ALS strategy detailed in Sect. 29.2.5, with the analysis of a data set built from the matrix concatenation of several high-energy resolution fluorescence detected (HERFD) XANES data sets collected on the same zeolite systems activated under O2 and He but with a higher-energy resolution [41]. This strategy revealed that the Cu(II)-MOR reference used for LCF is actually a mixture of three different Cu(II) intermediates, which are characterized in each individual data set by close HERFD-XANES spectra and nearly correlated concentration profiles during activation.

Cu K-edge time-resolved operando XANES recorded for Cu-MOR zeolite (Cu/Al = 0.18 and Si/Al = 7) during CH4 loading and subsequent CH3OH extraction (a, b) for the SP and (c, d) for the LP processes. (b) and (d) display the fraction of Cu species during the DMTM reaction obtained by LCF using as references Cu(II)-hydrated, Cu(II)-MOR, and Cu(I)-MOR. (e) Correlation of the normalized productivity to the total fraction of Cu(I) per Cu after methane loading for the experiments presented for Cu/Al = 0.18 and Si/Al = 7 and for Cu/Al = 0.36 and Si/Al = 11. (Adapted with permission from Ref. [59]. Copyright (2019) Elsevier)
The understanding of the dynamic evolution under reaction conditions of promising single-atom catalysts (SACs), considering the relationship between ultimate active site dispersion and catalytic efficiency, is of prime importance for the design of industrially relevant catalysts. Furthermore, when catalysts involve precious metals, like in many industrial applications, e.g., petrochemistry, the decrease of the catalyst metal loading is a parameter of importance due to its impact on cost production. This quest of highly efficient lowly loaded noble metal catalysts motivates many studies involving operando/in situ characterization of the archetype catalytic CO oxidation reaction [60, 61]. Recently, the operando monitoring of the thermal treatments on Pt/Al2O3 catalysts used for CO oxidation (Fig. 29.17) was discussed at the light of TR-XAS and DRIFTS results obtained under cycling reaction conditions with CO conversion measured simultaneously by MS [60]. From TR-XAS, it was concluded that the oxidized single Ptm+ atoms (m > 2) resulting from impregnation-calcination procedure were stable under O2 heating and presented a moderate activity for CO oxidation with a half-conversion temperature around 230 °C (Fig. 29.17c). Upon cooling under reaction conditions, a clustering of SACs occurred. It is revealed by both TR-XAS (appearance of Pt-Pt contribution at the same time than Pt-O ones and decrease of the WL intensity) and DRIFTS (signature of CO adsorption on oxidized clusters together with remaining fingerprint of CO linearly adsorbed on Ptm+ atoms). The formation of partially oxidized Ptδ+ clusters (δ < 2), with size ≈ 1 nm, was observed in a high CO conversion temperature window suggesting outperformance of Ptδ+ clusters for CO oxidation than single Ptm+ atoms (m > 2). The hypothesis was fully confirmed by the second part of the experiments dealing with heating under H2, during which reduced Pt clusters were mostly formed (Fig. 29.17a, d). Upon further exposure to CO/O2 atmosphere, those reduced clusters were found stable until reaching the light-off temperature around 150 °C (Fig. 29.17c) at which a sudden disappearance of the 2070–2050 cm−1 DRIFTS band ascribed to Pt(0) clusters and a Pt-O contribution and increase of WL intensity characteristic of oxidation were observed. Actually, the 100% CO conversion observed at this stage led to a highly oxidative atmosphere in the catalytic bed inducing the formation of amorphous PtOx clusters. This set of results, in particular with the significant decrease of the light-off temperature after reductive treatment favoring cluster formation (Fig. 29.17c), clearly highlights that clusters are more active than SACs for CO oxidation.

Operando Pt L3 TR-XAS and DRIFTS monitoring of CO oxidation on a Pt/γ-Al2O3 catalyst, throughout the calcination/reaction/reduction/reaction procedure. (a) EXAFS color map showing the temporal evolution of the phase-corrected FT with two FT signals shown in classical view. The white points represent the XANES WL intensity. (b) DRIFTS color maps showing the υ(C−O) absorption band evolution and post-reduction reaction steps. Three spectra corresponding to the horizontal lines (at 150 °C during heating and 100 °C during cooling) on the DRIFTS maps are plotted in classical view. (c) CO oxidation light-off and extinction curves. (d) Evolution of XANES WL intensities and Pt-Pt coordination numbers derived from EXAFS fitting for the catalyst monitored in (a) and for other Pt loadings and CO:O2 gas composition [60]. (Copyright (2019) American Chemical Society)
Those examples illustrate how powerful is TR-XAS operando characterization to assess the structure-activity relationships. They also emphasize that its combination with state-of-the-art multivariate analysis or complementary techniques is mandatory for unravelling the complexity of the dynamic transformations of working catalysts. Similar strategy involving operando XAS-XRD monitoring assisted by MCR-ALS was recently used to understand the driving force leading to catalyst stabilization and further deactivation during methanol synthesis by CO2 hydrogenation under pressure (Fig. 29.18) [17]. In agreement with the In-O and In-In coordination numbers derived from EXAFS fitting and XANES rising-edge energy position, the MCR-ALS analysis isolates during the reaction process three phases involving the pristine In2O3 nanoparticles, an In2O3-x phase resulting from the formation of oxygen vacancy in In2O3 and a molten In(0) species. The comparison of the catalytic activity with the MCR-ALS profiles associated to those species highlights that the active species for CO2 hydrogenation into CH3OH is the oxygen-vacant In2O3-x phase, accounting for about 60% of the indium speciation at the reaction stage where outperformance is observed. The onset of increase of In(0) proportion corresponding to the reductive amorphization of In2O3 observed by XRD is linked to the gradual deactivation observed upon time on stream (TOS). The higher the proportion of In(0), the lower the crystallinity of the bcc-In2O3 crystallites, the lower the catalytic activity. It is noteworthy that upon activation or deactivation stages, no sintering of the In2O3 phase has been revealed by XRD. As the crystallinity and particle sizes remain nearly constant during the activation stage and only a slight crystallite size decrease is observed during the deactivation stage, it can be concluded that the change of indium speciation is mainly responsible for the catalyst stabilization and further deactivation.

(a) Setup used for operando In K-edge TR-XAS and XRD experiments performed for a model In2O3 catalyst under CO2 hydrogenation conditions (300 °C, 20 bar) with presentation of selected TR-XANES and Fourier transforms of EXAFS signals, selected XRD patterns (λ = 0.506 Å) with indexed reflection peaks for bcc-In2O3, and selected chromatographs showing the methanol amounts with TOS. The arrows indicate the direction of changes with TOS. (b) Summary of the results gained upon TOS by GC for methanol production; by EXAFS with the fitted In-O and In-In coordination numbers (CN), XANES edge energy shift (ΔE0) with respect to In2O3 reference, and MCR-ALS concentration profiles; and by XRD with the average crystallite size and fraction of crystalline bcc-In2O3. (Reprinted with permission from Ref. [17]. Copyright (2019) American Chemical Society)
3.2.2 Catalyst Deactivation and Regeneration
Besides the in-depth identification of active sites leading to the best performances, operando TR-XAS provides also unique opportunity to address the deactivation topic since, at the same time, the technique provides the monitoring of the evolution of the active site structure and of catalytic activity. Bartholomew et al. [62] reviewed the mechanisms of catalyst deactivation. Some of them can be readily characterized by XAS insofar as they affect the same catalyst characteristics than those already evaluated by XAS during activation studies detailed in section “Synthesis of Supported Catalysts”, such as active-phase dispersion, metal distribution, or catalyst composition. In this last section, we will examine some deactivation mechanisms with the emphasis to propose regeneration strategies.
Particle sintering , which reduces the active site dispersion, is reported using TR-XAS as the main deactivation mechanism for supported Pt catalysts used in the harsh temperature conditions of automotive converters with high temperature and fluctuation of exhaust gas composition between oxidative and reductive conditions [63]. Redistribution of atoms in bimetallic particles , which changes the active site nature, has been also identified by TR-XAS for Pd-Pt/Al2O3 catalyst deactivation during total oxidation of methane, with segregation in oxidized state of Pt and Pd in core-shell bimetallic particles [64]. Another deactivation route well characterized by TR-XAS is the change in the oxidation state of the active phase induced by the presence of reactive gases in the feed, some of them being present as contaminants [65], whereas others as result of the catalytic reaction [19, 66]. For the latter case, many publications aimed to study the fast oscillatory behaviors induced by peculiar reaction conditions, leading to bistability of the catalyst structure, passing from active to inactive states driven by changes in the oxidation of the metal [66, 67]. Understanding catalyst evolution under oscillatory conditions is of prime importance for the development of diesel oxidation catalysts, which suffer similar ignition and extinction conditions. For instance, TR-XAS monitoring of the catalytic partial oxidation of methane over Pd/Al2O3 catalysts is presented in Fig. 29.19a [66]. The in-depth characterization of the Pd oxidation state along the catalyst bed together with the reactant conversion measured by MS at the reactor outlet allowed for proposing the scheme shown in Fig. 29.19b. It explains the cycling behavior of the catalyst upon reaction conditions, based on four stages: (1) PdOx first promotes the complete CH4 oxidation; (2) upon total O2 consumption, catalytic partial oxidation occurs leading to the reduction of PdOx into Pd(0) from the reactor outlet; (3) propagation of the reduction occurs toward the inlet driven by temperature hot spots; (4) when Pd is fully reduced, deactivation occurs dropping the CH4 oxidation activity leading to oxidative conditions and reoxidation of Pd(0).

(a) Pd K-edge TR-XAS monitoring of oscillations with a Pd/Al2O3 catalyst loaded in a fixed bed catalyst and heated at 410 °C under 6% CH4 : 3% O2:He gas mixture. (b) Suggested scheme for the oscillatory behavior of the catalytic partial oxidation (CPO) of CH4. (Reprinted with permission from Ref. [66]. Copyright (2012) American Chemical Society)
The poisoning by contaminants leads to active site occupation by extraneous species blocking the reactant access. Deactivation and regeneration processes are often addressed by operando steady-state XAS characterization, e.g., with sulfur poisoning of noble metal catalysts used for methane oxidation under typical conditions of lean burn gas engines [68] or for methanation from dry biomass [69]. However, experiments carried out under high frequency of gaseous pulses of contaminants or regenerative reactants can require TR-XAS, as illustrated with the use of modulation excitation spectroscopy (MES) [70] for studying the stability of catalysts for hydrogenation of ethylene glycol. As already mentioned, MoS2-based catalysts find applications for HDS treatment. Their optimization makes a great use of TR-XAS for understanding the impact of the sulfiding activation processes [6, 12, 29, 36] on Mo and promotor (Ni, Co) speciation. The use of Mo-based catalysts for biomass conversion into upgraded bio-oil by catalytic hydrodeoxygenation (HDO) requires to optimize their tolerance against water and to develop strategies for their stabilization in oxygen-rich feedstock (20–50 wt. %). The catalytic active sites being located at the corners and edges of MoS2 slabs only constitute a small fraction of all the Mo atoms in the slabs letting the bulk XAS technique mainly dominated by the contribution of the Mo spectator atoms which are not involved in the reaction. To overcome the poor sensitivity of XAS toward minority species, MES can be used to separate the spectroscopic answer of active species from spectator ones [71]. The principle is to periodically (and reversibly) perturbate the catalytic system by external stimulation, such as the alternate use of different gaseous pulses, which only influences the active species. The further mathematical treatment, called demodulation or phase-sensitive detection, allows for isolating only signals of the active species, which respond to the external simulation with the same frequency, and their further identification. MES allows also a better differentiation of signals with different kinetics. Readers are invited to refer to the dedicated literature for a deep understanding of the mathematical treatment [71, 72]. In order to understand the influence of H2O and H2S partial pressures on the stability of active MoS2 slab edges used for HDO, MES experiments were carried out by cycling the catalysts to pulses of H2O/H2 and H2S/H2 [70]. Figure 29.20a shows that no modification of the Mo K-edge XANES data can be observed during pulse cycling. The match of the demodulated spectra with the difference between the XANES spectra of MoS2 and MoO3 references (Fig. 29.20b) demonstrates a reversible oxidation-sulfidation process in which 1% of Mo(IV) atoms undergo oxidation to Mo(VI) with the replacement of S atoms at the edge of MoS2 by O atoms. Similar behaviors are reported for MES of Co- and Ni-promoted MoS2 catalysts (Fig. 29.20c), however with a reduced amplitude of the demodulated spectra at the energy position P centered on the shoulder H characteristic of MoS2, suggesting a decrease in S-O exchange upon promotion. Further information on the kinetics of oxidation-sulfidation can be obtained by plotting the variation of the shoulder H intensity with time (Fig. 29.20d). The decrease of absorption during the H2O pulse and its increase during the H2S pulse are delayed by about 30 s for CoMo compared to Mo (not shown) or NiMo. Mo and CoMo catalysts suffer from oxidation-sulfidation at the first period of pulses, whereas nothing is happening for the NiMo one before the fifth period. All these results emphasize a different response of the promoted catalysts compared to the non-promoted one with as general conclusion an improvement of the stability of the promoted catalyst against H2O exposure explaining their better activity for ethylene glycol HDO and the benefit of small quantity of H2S to regenerate the catalysts at each cycle.

(a) Mo K-edge TR-XAS monitoring of unpromoted Mo catalyst during 10 periods of 180 s pulses of 3% H2O/H2 and 0.1% H2S/H2 and demodulated spectra at selected values of phase angle. (b) Experimental XANES spectra for MoS2 and MoO3 references and comparison of their difference with the maximum amplitude signal obtained after demodulation for the Mo catalyst. (c) Comparison of the maximum amplitude demodulated signal obtained for the Mo, CoMo, and NiMo catalysts and (d) kinetics of the phase transition of Mo observed for the promoted catalysts considering the intensity of the shoulder H versus time. The dashed line is the sine curve with the phase shift revealed by demodulation. (Reprinted with permission from Ref. [70]. Copyright (2019) American Chemical Society)
Besides, the fruitful combination of TR-XAS with Raman spectroscopy can address the impact on catalyst deactivation of carbonaceous deposits. Raman spectroscopy enables the specific characterization of the carbon species at the surface of the catalysts under reaction conditions and highlights the onset of coke formation inside the catalytic bed [31, 37]. Recently, the reaction, deactivation, and regeneration of a Co/Al2O3 catalyst used for ethanol steam reforming (ESR) have been studied through operando monitoring by TR-XAS, MS, and Raman spectroscopy. Figure 29.21 displays the main results gained during the monitoring of successive ESR and oxidative conditions aiming to regenerate the catalyst after coking. Figure 29.21c evidences that the decrease of ethanol conversion during the first ESR reaction is coincident with the increase of coke deposits at the surface of the catalyst, for which the abundance is measured by the Raman G-line intensity. MCR-ALS analysis of the TR-XAS data recorded simultaneously with Raman shows nearly invariant proportions of Co(0) (≈ 89%) and CoO (≈ 11%) after passing the first 10 minutes of reaction, and EXAFS fitting reveals that no sintering occurs. These results indicate that the fouling of the catalyst surface by coke is responsible for the observed deactivation after 90 min of reaction. In order to regenerate the catalyst, 5% O2/He is then added to the reactant flow aiming to oxidize the coke deposits. The duration of the oxidative ESR treatment (OX) is determined from the careful in-line examination of the recorded Raman spectra. As soon as a total vanishing of the Raman G-line is observed (Fig. 29.21a), the O2 flow is stopped, and the catalyst is exposed back to the original ESR conditions for 60 min. Due to the removal of coke deposits, the metallic cobalt species in contact to O2 at the end of OX conditions have been partially oxidized (Fig. 29.21b). However, ESR restarts immediately at the contact of ethanol steam with regenerated catalyst, which produces H2 and gives rise to a self-reactivation of the catalyst with 74% of Co(0) after 60 min of the second ESR. Again, Raman indicates that the deactivation occurring in the second ESR is related to the coke formation. Finally at the end of the third ESR, 67% of Co(0) are obtained. It is noteworthy that despite a decrease of the concentration of active species upon the successive regenerative treatments, the ethanol conversion is still observed at the same level around 77–80%, when it reaches its maximum. This behavior is explained as the result of the fragmentation of the coke-encapsulated Co(0) particles upon O2 exposure and induced by the so-called Kirkendall effect. Due to the encapsulation of particles by carbon shells, the lattice stress associated with the volume expansion during particle oxidation cannot be released, except by the rupture of the particles into smaller fragments at the Kirkendall nanovoids (Fig. 29.21e). The fragmentation of the Co(0) nanoparticles has been fully confirmed by microscopy, showing a decrease of the mean particle size (Fig. 29.21d), and by EXAFS fitting (Fig. 29.21c), which displays a decrease of the coordination number NCo-Co. This study clearly demonstrates how powerful is the combination of TR-XAS/MS with Raman spectroscopy for understanding the deactivation mechanisms related to coke deposits but also to propose strategies to regenerate catalysts.

Monitoring of successive ESR and oxidative (OX) conditions over a Co/Al2O3 catalyst by (a) Raman spectroscopy and (b) Co K-edge quick-XAS. (c, top) Evolution of ethanol conversion and intensity of the Raman G-line (diamond symbols). (c, bottom) MCR-ALS quick-XAS speciation of Co phases and NCo–Co coordination numbers rescaled by the percentage of Co(0) species during ESR-OX cycles. (d) STEM images of the catalyst after the first ESR and the first OX, together with the respective size distributions. (e) Scheme for the oxidative regeneration of a coke-encapsulated Co(0) particle. (Reprinted with permission from Ref. [25]. Copyright (2018) The Royal Society of Chemistry)
4 Conclusion
This chapter clearly demonstrates how TR-XAS, available with state-of-the-art quick-EXAFS or energy-dispersive beamlines, is essential for an in-depth description of the structural and electronic modifications suffered by catalysts. The technique can be used in very versatile experimental conditions to solve the nucleation and growth of nanoparticles in liquid media, eventually at high temperature, or to monitor liquid or gas-solid reactions leading to the activation of catalysts and its further evolution in reaction conditions encompassing high pressure and temperature. The time-resolved data collected with sub-second time resolution are successive snapshots of the mixture of species representative of the evolution of the reaction. To describe the precise movie of the catalyst life span, multivariate data analysis methods such as MCR-ALS are nowadays emerging. They lead to the isolation of the species involved in the reaction together with the description of their evolution as function of the reaction coordinates. Beyond the speciation provided by MCR-ALS or LCF from known references, X-ray absorption spectroscopy, by EXAFS fittings, offers also unique opportunity to identify the species participating to the reaction. Not yet fully used, the speciation available by MCR-ALS analysis of TR-XAS data offers also solid bases to investigate the kinetic laws driving the phase formation with the determination of growth exponent and the effective activation energy. Many of the examples presented herein also use complementary techniques, such as simultaneous Raman spectroscopy or SAXS to cite only a few, in order to obtain a full and detailed description of the complexity of the reactions in which catalysts are involved and eventually guide the multivariate data analysis toward a more complete description of the chemical transformation. Finally, the next step for the precise description of the catalyst life span will be to routinely combine sub-second time resolution of XAS data with spatial resolution at the micro- or nanometer scales relevant for the investigated processes .
References
Müller, O., Nachtegaal, M., Just, J., Lützenkirchen-Hecht, D., Frahm, R.: Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10ms time resolution. J. Synchrotron Radiat. 23, 260–266 (2016)
Briois, V., La Fontaine, C., Belin, S., Barthe, L., Moreno, T., Pinty, V., Carcy, A., Girardot, R., Fonda, E.: ROCK: the new quick-EXAFS beamline at SOLEIL. J. Phys. Conf. Ser. 712, 012149–012155 (2016)
Palomino, R.M., Stavitski, E., Waluyo, I., Chen-Wiegart, Y.c.K., Abeykoon, M., Sadowski, J.T., Rodriguez, J.A., Frenkel, A.I., Senanayake, S.D.: New in-situ and operando facilities for catalysis science at NSLS-II: the deployment of real-time, chemical, and structure-sensitive X-ray probes. Synchrotron Radiat. News. 30, 30–37 (2017)
Caliebe, W.A., Murzin, V., Kalinko, A., Görlitz, M.: High-flux XAFS-beamline P64 at PETRA III. AIP Conf. Proc. 2054, 060031 (2019)
Van Bokhoven, J.A., Lamberti, C. (eds.): X-Ray Absorption and X-Ray Emission Spectroscopy. Wiley, Chichester, UK (2016)
Lesage, C., Devers, E., Legens, C., Fernandes, G., Roudenko, O., Briois, V.: High pressure cell for edge jumping X-ray absorption spectroscopy: applications to industrial liquid sulfidation of hydrotreatment catalysts. Catal. Today. 336, 63–73 (2019)
Clark, A.H., Imbao, J., Frahm, R., Nachtegaal, M.: ProQEXAFS: a highly optimized parallelized rapid processing software for QEXAFS data. J. Synchrotron Radiat. 27, 551–557 (2020)
Figueroa, S.J.A., Prestipino, C.: PrestoPronto: a code devoted to handling large data sets. J. Phys. Conf. Ser. 712, 12012 (2016)
Newville, M.: Larch: an analysis package for XAFS and related spectroscopies. J. Phys. Conf. Ser. 430, 12007 (2013)
Stötzel, J., Lützenkirchen-Hecht, D., Grunwaldt, J.-D., Frahm, R.: T-REX: new software for advanced QEXAFS data analysis. J. Synchrotron Radiat. 19, 920–929 (2012)
Cassinelli, W.H., Martins, L., Passos, A.R., Pulcinelli, S.H., Santilli, C.V., Rochet, A., Briois, V.: Multivariate curve resolution analysis applied to time-resolved synchrotron X-ray Absorption Spectroscopy monitoring of the activation of copper alumina catalyst. Catal. Today. 229, 114–122 (2014)
Rochet, A., Baubet, B., Moizan, V., Pichon, C., Briois, V.: Co-K and Mo-K edges Quick-XAS study of the sulphidation properties of Mo/Al2O3 and CoMo/Al2O3 catalysts. C. R. Chim. 19, 1337–1351 (2016)
Voronov, A., Urakawa, A., van Beek, W., Tsakoumis, N.E., Emerich, H., Rønning, M.: Multivariate curve resolution applied to in situ X-ray absorption spectroscopy data: an efficient tool for data processing and analysis. Anal. Chim. Acta. 840, 20–27 (2014)
Staniuk, M., Hirsch, O., Kränzlin, N., Böhlen, R., van Beek, W., Abdala, P.M., Koziej, D.: Puzzling mechanism behind a simple synthesis of cobalt and cobalt oxide nanoparticles: in situ synchrotron X-ray absorption and diffraction studies. Chem. Mater. 26, 2086–2094 (2014)
Martini, A., Borfecchia, E., Lomachenko, K.A., Pankin, I.A., Negri, C., Berlier, G., Beato, P., Falsig, H., Bordiga, S., Lamberti, C.: Composition-driven Cu-speciation and reducibility in cu-CHA zeolite catalysts: a multivariate XAS/FTIR approach to complexity. Chem. Sci. 8, 6836–6851 (2017)
Clark, A.H., Nuguid, R.J.G., Steiger, P., Marberger, A., Petrov, A.W., Ferri, D., Nachtegaal, M., Kröcher, O.: Selective catalytic reduction of NO with NH3 on Cu−SSZ-13: deciphering the low and high-temperature rate-limiting steps by transient XAS experiments. ChemCatChem. 12, 1429–1435 (2020)
Tsoukalou, A., Abdala, P.M., Stoian, D., Huang, X., Willinger, M.-G., Fedorov, A., Müller, C.R.: Structural evolution and dynamics of an In2O3 catalyst for CO2 hydrogenation to methanol: an operando XAS-XRD and in situ TEM study. J. Am. Chem. Soc. 141, 13497–13505 (2019)
Rochet, A., Moizan, V., Diehl, F., Pichon, C., Briois, V.: Quick-XAS and Raman operando characterisation of a cobalt alumina-supported catalyst under realistic Fischer–Tropsch reaction conditions. Catal. Today. 205, 94–100 (2013)
Jacobs, G., Ma, W., Gao, P., Todic, B., Bhatelia, T., Bukur, D.B., Davis, B.H.: The application of synchrotron methods in characterizing iron and cobalt Fischer–Tropsch synthesis catalysts. Catal. Today. 214, 100–139 (2013)
Hong, J., Marceau, E., Khodakov, A.Y., Griboval-Constant, A., La Fontaine, C., Briois, V.: A time-resolved in situ quick-XAS investigation of thermal activation of Fischer–Tropsch silica-supported cobalt catalysts. Chem. – A Eur. J. 18, 2802–2805 (2012)
Munnik, P., Krans, N.A., de Jongh, P.E., de Jong, K.P.: Effects of drying conditions on the synthesis of Co/SiO2 and Co/Al2O3 Fischer–Tropsch catalysts. ACS Catal. 4, 3219–3226 (2014)
Munnik, P., de Jongh, P.E., de Jong, K.P.: Recent developments in the synthesis of supported catalysts. Chem. Rev. 115, 6687–6718 (2015)
Passos, A.R., Martins, L., Pulcinelli, S.H., Santilli, C.V., Briois, V.: Effect of the balance between Co(II) and Co(0) oxidation states on the catalytic activity of cobalt catalysts for Ethanol Steam Reforming. Catal. Today. 229, 88–94 (2014)
Passos, A.R., Martins, L., Pulcinelli, S.H., Santilli, C.V., Briois, V.: Correlation of sol-gel alumina-supported cobalt catalyst processing to cobalt speciation, ethanol steam reforming activity, and stability. ChemCatChem. 9, 3918–3929 (2017)
Passos, A.R., La Fontaine, C., Martins, L., Pulcinelli, S.H., Santilli, C.V., Briois, V.: Operando XAS/Raman/MS monitoring of ethanol steam reforming reaction–regeneration cycles. Cat. Sci. Technol. 8, 6297–6301 (2018)
de Juan, A., Jaumot, J., Tauler, R.A.: Multivariate Curve Resolution (MCR). Solving the mixture analysis problem. Anal. Methods. 6, 4964–4976 (2014)
Cassinelli, W.H., Martins, L., Magnani, M., Pulcinelli, S.H., Briois, V., Santilli, C.V.: Time-resolved XAS/MS/Raman monitoring of mutual copper self-reduction and ethanol dehydrogenation reactions. RSC Adv. 6, 20453–20457 (2016)
Jaumot, J., De Juan, A., Tauler, R.: Chemometrics and intelligent laboratory systems MCR-ALS GUI 2.0: new features and applications. Chemom. Intell. Lab. Syst. 140, 1–12 (2015)
Rochet, A., Baubet, B., Moizan, V., Devers, E., Hugon, A., Pichon, C., Payen, E., Briois, V.: Intermediate species revealed during sulfidation of bimetallic hydrotreating catalyst: a multivariate analysis of combined time-resolved spectroscopies. J. Phys. Chem. C. 121, 18544–18556 (2017)
Fornasini, P., Grisenti, R.: On EXAFS Debye-Waller factor and recent advances. J. Synchrotron Radiat. 22, 1242–1257 (2015)
Passos, A.R., Pulcinelli, S.H., Santilli, C.V., Briois, V.: Operando monitoring of metal sites and coke evolution during non-oxidative and oxidative ethanol steam reforming over Ni and NiCu ex-hydrotalcite catalysts. Catal. Today. 336, 122–130 (2019)
Bugaev, A.L., Skorynina, A.A., Braglia, L., Lomachenko, K.A., Guda, A., Lazzarini, A., Bordiga, S., Olsbye, U., Lillerud, K.P., Soldatov, A.V., Lamberti, C.: Evolution of Pt and Pd species in functionalized UiO-67 metal-organic frameworks. Catal. Today. 336, 33–39 (2019)
Hong, J.P., Marceau, E., Khodakov, A.Y., Gaberova, L., Griboval-Constant, A., Girardon, J.S., La Fontaine, C., Briois, V., Gaberová, L., Griboval-Constant, A., Girardon, J.S., La Fontaine, C., Briois, V.: Speciation of ruthenium as a reduction promoter of silica-supported Co catalysts: a time-resolved in situ XAS investigation. ACS Catal. 5, 1273–1282 (2015)
Rochet, A., Baubet, B., Moizan, V., Devers, E., Hugon, A., Pichon, C., Payen, E., Briois, V.: Influence of the preparation conditions of oxidic NiMo/Al2O3 catalysts on the sulfidation ability: a quick-XAS and Raman spectroscopic study. J. Phys. Chem. C. 119, 23928–23942 (2015)
Rochet, A.A., Passos, A.R., Legens, C.C., Briois, V.V., Ribeiro Passos, A., Legens, C.C., Briois, V.V.: Sulphidation study of a dried Ni/Al2O3 catalyst by time-resolved XAS-MS combined with in situ Raman spectroscopy and multivariate Quick-XAS data analysis. Catal. Struct. React. 3, 33–42 (2017)
Nikulshina, M., Blanchard, P., Lancelot, C., Griboval-Constant, A., Marinova, M., Briois, V., Nikulshin, P., Lamonier, C.: Genesis of active phase in MoW/Al2O3 hydrotreating catalysts monitored by HAADF and in situ QEXAFS combined to MCR-ALS analysis. Appl. Catal. B Environ. 269, 118766 (2020)
Passos, A.R., La Fontaine, C., Pulcinelli, S.H., Santilli, C.V., Briois, V.: Quick-EXAFS and Raman monitoring of activation, reaction and deactivation of NiCu catalysts obtained from hydrotalcite-like precursors. Phys. Chem. Chem. Phys. 22, 18835 (2020)
Carvalho, H.W.P., Pulcinelli, S.H., Santilli, C.V., Leroux, F., Meneau, F., Briois, V.: XAS/WAXS time-resolved phase speciation of chlorine LDH thermal transformation: emerging roles of isovalent metal substitution. Chem. Mater. 25, 2855–2867 (2013)
Girard, V., Chiche, D., Baudot, A., Bazer-Bachi, D., Lemaitre, L., Moizan-Basle, V., Rochet, A., Briois, V., Geantet, C.: In situ QXAS study of sulfidation/oxidative regeneration reactions of zinc molybdate (ZnMoO4) and ZnO-MoO3 materials. Phys. Chem. Chem. Phys. 21, 8569–8579 (2019)
Rabeah, J., Briois, V., Adomeit, S., La Fontaine, C., Bentrup, U., Brückner, A.: Multivariate analysis of coupled operando EPR/XANES/ EXAFS/UV–vis/ATR–IR spectroscopy: a new dimension for mechanistic studies of catalytic gas-liquid phase reactions. Chem. – A Eur. J. 26, 7395–7404 (2020)
Kvande, K., Pappas, D.K., Borfecchia, E., Lomachenko, K.A.: Advanced X-ray absorption spectroscopy analysis to determine structure-activity relationships for Cu-zeolites in the direct conversion of methane to methanol. ChemCatChem. 12, 2385–2405 (2020)
Weckhuysen, B.M.: Studying birth, life and death of catalytic solids with operando spectroscopy. Natl. Sci. Rev. 2, 147–149 (2015)
Nørskov, J.K., Bligaard, T., Rossmeisl, J., Christensen, C.H.: Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009)
Sun, Y., Ren, Y.: In situ synchrotron X-ray techniques for real-time probing of colloidal nanoparticle synthesis. Part. Part. Syst. Charact. 30, 399–419 (2013)
You, H., Fang, J.: Particle-mediated nucleation and growth of solution-synthesized metal nanocrystals: a new story beyond the LaMer curve. Nano Today. 11, 145–167 (2016)
Tanaka, T., Ohyama, J., Teramura, K., Hitomi, Y.: Formation mechanism of metal nanoparticles studied by XAFS spectroscopy and effective synthesis of small metal nanoparticles. Catal. Today. 183, 108–118 (2012)
Abécassis, B., Testard, F., Kong, Q., Baudelet, F., Spalla, O.: Influence of monomer feeding on a fast gold nanoparticles synthesis: time-resolved XANES and SAXS experiments. Langmuir. 26, 13847–13854 (2010)
Harada, M., Kamigaito, Y.: Nucleation and aggregative growth process of platinum nanoparticles studied by in situ quick XAFS spectroscopy. Langmuir. 28, 2415–2428 (2012)
Booth, S.G., Chang, S.-Y., Uehara, A., La Fontaine, C., Cibin, G., Schroeder, S.L.M., Dryfe, R.A.W.: In situ XAFS study of palladium electrodeposition at the liquid/liquid interface. Electrochim. Acta. 235, 251–261 (2017)
Yao, T., Liu, S., Sun, Z., Li, Y., He, S., Cheng, H., Xie, Y., Liu, Q., Jiang, Y., Wu, Z., Pan, Z., Yan, W., Wei, S.: Probing nucleation pathways for morphological manipulation of platinum nanocrystals. J. Am. Chem. Soc. 134, 9410–9416 (2012)
Strach, M., Mantella, V., Pankhurst, J.R., Iyengar, P., Loiudice, A., Das, S., Corminboeuf, C., van Beek, W., Buonsanti, R.: Insights into reaction intermediates to predict synthetic pathways for shape-controlled metal nanocrystals. J. Am. Chem. Soc. 141, 16312–16322 (2019)
Pan, Y.-T., Yang, H.: Design of bimetallic catalysts and electrocatalysts through the control of reactive environments. Nano Today. 31, 100832 (2020)
Wolters, M., Munnik, P., Bitter, J.H., de Jongh, P.E., de Jong, K.P.: How NO affects nickel and cobalt nitrates at low temperatures to arrive at highly dispersed silica-supported nickel and cobalt catalysts. J. Phys. Chem. C. 115, 3332–3339 (2011)
Jacobs, G., Ma, W., Davis, B.H., Cronauer, D.C., Kropf, J.A., Marshall, C.L.: Fischer–Tropsch synthesis: TPR-XAFS analysis of Co/silica and Co/alumina catalysts comparing a novel NO calcination method with conventional air calcination. Catal. Lett. 140, 106–115 (2010)
Briois, V., Belin, S., Villain, F., Bouamrane, F., Lucas, H., Lescouzec, R., Julve, M., Verdaguer, M., Tokumoto, M.S., Santilli, C.V., Pulcinelli, S.H., Carrier, X., Krafft, J.M., Jubin, C., Che, M.: New insights for materials science characterisation using different complementary techniques combined with X-ray absorption spectroscopy. Phys. Scr. T115, 38–44 (2005)
Carrier, X., Marceau, E., Che, M.: Physical techniques and catalyst preparation: determining the interactions of transition-metal complexes with oxide surfaces. Pure Appl. Chem. 78, 1039–1055 (2006)
Filez, M., Poelman, H., Redekop, E.A., Galvita, V.V., Alexopoulos, K., Meledina, M., Ramachandran, R.K., Dendooven, J., Detavernier, C., Van Tendeloo, G., Safonova, O.V., Nachtegaal, M., Weckhuysen, B.M., Marin, G.B.: Kinetics of lifetime changes in bimetallic nanocatalysts revealed by quick X-ray absorption spectroscopy. Angew. Chem. Int. Ed. 57, 12430–12434 (2018)
Newton, M.A., Knorpp, A.J., Sushkevich, V.L., Palagin, D., van Bokhoven, J.A.: Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: a critical examination. Chem. Soc. Rev. 49, 1449–1486 (2020)
Lomachenko, K.A., Martini, A., Pappas, D.K., Negri, C., Dyballa, M., Berlier, G., Bordiga, S., Lamberti, C., Olsbye, U., Svelle, S., Beato, P., Borfecchia, E.: The impact of reaction conditions and material composition on the stepwise methane to methanol conversion over Cu-MOR: an operando XAS study. Catal. Today. 336, 99–108 (2019)
Dessal, C., Len, T., Morfin, F., Rousset, J.-L., Aouine, M., Afanasiev, P., Piccolo, L.: Dynamics of single Pt atoms on alumina during CO oxidation monitored by operando X-ray and infrared spectroscopies. ACS Catal. 9, 5752–5759 (2019)
Li, X., Yang, X., Zhang, J., Huang, Y., Liu, B.: In situ/operando techniques for characterization of single-atom catalysts. ACS Catal. 9, 2521–2531 (2019)
Argyle, D.M., Bartholomew, H.C.: Heterogeneous catalyst deactivation and regeneration: a review. Catalysts. 5, 145–269 (2015)
Nagai, Y., Dohmae, K., Ikeda, Y., Takagi, N., Hara, N., Tanabe, T., Guilera, G., Pascarelli, S., Newton, M.A., Takahashi, N., Shinjoh, H., Matsumoto, S.: In situ observation of platinum sintering on ceria-based oxide for autoexhaust catalysts using Turbo-XAS. Catal. Today. 175, 133–140 (2011)
Gremminger, A.T., Pereira de Carvalho, H.W., Popescu, R., Grunwaldt, J.-D., Deutschmann, O.: Influence of gas composition on activity and durability of bimetallic Pd-Pt/Al2O3 catalysts for total oxidation of methane. Catal. Today. 258, 470–480 (2015)
Mutz, B., Gänzler, A.M., Nachtegaal, M., Müller, O., Frahm, R., Kleist, W., Grunwaldt, J.: Surface oxidation of supported Ni particles and its impact on the catalytic performance during dynamically operated methanation of CO2. Catalysts. 7, 279 (2017)
Stötzel, J., Frahm, R., Kimmerle, B., Nachtegaal, M., Grunwaldt, J.-D.: Oscillatory behavior during the catalytic partial oxidation of methane: following dynamic structural changes of palladium using the QEXAFS technique. J. Phys. Chem. C. 116, 599–609 (2012)
Boubnov, A., Gänzler, A., Conrad, S., Casapu, M., Grunwaldt, J.-D.: Oscillatory CO oxidation over Pt/Al2O3 catalysts studied by in situ XAS and DRIFTS. Top. Catal. 56, 333–338 (2013)
Lott, P., Doronkin, D.E., Zimina, A., Tischer, R., Popescu, S., Belin, S., Briois, V., Casapu, M., Grunwaldt, J.D., Deutschmann, O.: Understanding sulfur poisonning of bimettalic Pd-Pt methane oxidation catalysts and their regeneration. Appl. Catal. B Environ. 278, 119244 (2020)
Kuzmenko, D., Nachtegaal, M., Copéret, C., Schildhauer, T.J.: Molecular-level understanding of support effects on the regenerability of Ru-based catalysts in the sulfur-poisoned methanation reaction. J. Catal. 375, 74–80 (2019)
Gaur, A., Hartmann Dabros, T.M., Høj, M., Boubnov, A., Prüssmann, T., Jelic, J., Studt, F., Jensen, A.D., Grunwaldt, J.-D.: Probing the active sites of MoS2 based hydrotreating catalysts using modulation excitation spectroscopy. ACS Catal. 9, 2568–2579 (2019)
Urakawa, A., Bürgi, T., Baiker, A.: Sensitivity enhancement and dynamic behavior analysis by modulation excitation spectroscopy: principle and application in heterogeneous catalysis. Chem. Eng. Sci. 63, 4902–4909 (2008)
Ferri, D., Newton, M.A., Nachtegaal, M.: Modulation excitation X-ray absorption spectroscopy to probe surface species on heterogeneous catalysts. Top. Catal. 54, 1070 (2011)
Acknowledgments
The development of MCR-ALS analysis at the ROCK beamline (SOLEIL) has been financially supported by a public grant overseen by the French National Research Agency (Project ANR-10-EQPX 10-45). We thank Olga Roudenko (SOLEIL) for providing fast handling calibration and normalization skills for the TR-XAS data and Ludovic Duponchel (LASIR) for allowing us the discovery of the power of the multivariate analysis applied to process-like data. The authors also acknowledge Celso V. Santilli and Sandra H. Pulcinelli (UNESP) for the collaboration and discussions about ESR catalysts.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Passos, A.R., La Fontaine, C., Rochet, A., Briois, V. (2023). Case Studies: Time-Resolved X-Ray Absorption Spectroscopy (XAS). In: Wachs, I.E., Bañares, M.A. (eds) Springer Handbook of Advanced Catalyst Characterization. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-031-07125-6_29
Download citation
DOI: https://doi.org/10.1007/978-3-031-07125-6_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-07124-9
Online ISBN: 978-3-031-07125-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)