
Abstract
Years ago gastrointestinal motility was thought to be due to interactions between enteric nerves and smooth muscle cells (SMCs) in the tunica muscularis. Thus, regulatory mechanisms controlling motility were either myogenic or neurogenic. Now we know that populations of interstitial cells, c-Kit+ (interstitial cells of Cajal or ICC), and PDGFRα+ cells (formerly “fibroblast-like” cells) are electrically coupled to SMCs, forming the SIP syncytium. Pacemaker and neurotransduction functions are provided by interstitial cells through Ca2+ release from the endoplasmic reticulum (ER) and activation of Ca2+-activated ion channels in the plasma membrane (PM). ICC express Ca2+-activated Cl− channels encoded by Ano1. When activated, Ano1 channels produce inward current and, therefore, depolarizing or excitatory effects in the SIP syncytium. PDGFRα+ cells express Ca2+-activated K+ channels encoded by Kcnn3. These channels generate outward current when activated and hyperpolarizing or membrane-stabilizing effects in the SIP syncytium. Inputs from enteric and sympathetic neurons regulate Ca2+ transients in ICC and PDGFRα+ cells, and currents activated in these cells conduct to SMCs and regulate contractile behaviors. ICC also serve as pacemakers, generating slow waves that are the electrophysiological basis for gastric peristalsis and intestinal segmentation. Pacemaker types of ICC express voltage-dependent Ca2+ conductances that organize Ca2+ transients, and therefore Ano1 channel openings, into clusters that define the amplitude and duration of slow waves. Ca2+ handling mechanisms are at the heart of interstitial cell function, yet little is known about what happens to Ca2+ dynamics in these cells in GI motility disorders.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- SIP syncytium
- Ca2+ stores
- Voltage-dependent Ca2+ channels
- ANO1 channels
- Enteric nervous system
- GI motility
22.1 Introduction
Interstitial cells of Cajal (ICC or c-Kit+) and platelet-derived growth factor receptor alpha (PDGFRα+) cells provide important regulatory behaviors in the generation of motility patterns in the gastrointestinal (GI) tract. ICC and PDGFRα+ cells are electrically coupled to smooth muscle cells (SMCs) [32, 44, 66], so changes in membrane conductances in these cells influence the excitability and contractility of SMCs. Together these cells make up an electrical syncytium known as the SIP syncytium [64]. The functions of interstitial cells were suggested by their anatomical characteristics and associations, such as formations of networks, abundant close proximity to varicose processes of motor neurons, and gap junction connectivity with SMCs. Thus, it was proposed that ICC might be pacemaker cells and innervated by motor neurons [24, 69, 79]. The role of PDGFRα+ cells, referred to as “fibroblast-like” by anatomists, was assumed to be only structural in nature. Recent work has begun to dissect the mechanisms of action of interstitial cells and reveal the many important functions of these cells in GI motility.
Major breakthroughs for understanding the functions of interstitial cells came when (i) mutants in which ICC failed to develop were used to investigate the role of ICC [36, 73, 74]: (ii) specific immunolabels for ICC and PDGFRα+ cells were discovered [42, 51, 52, 56, 70]; (iii) reporter strains were developed [31, 63, 83], allowing observation of specific types of interstitial cells in intact muscles and unequivocal identification of cells after enzymatic dispersion of muscle tissues [47, 51, 52, 70, 83]; (iv) Cre-loxP technology was used to allow highly specific deletion or expression of genes in ICC and allowed cell specific expression of genetically encoded Ca2+ sensors to monitor Ca2+ dynamics [3, 7, 39, 48, 68]. Evaluations of genes expressed in cellular components of the SIP syncytium showed highly specific expression of genes important for the functions of these cells such as Ano1 (originally called Tmem16a) in ICC [12] and Kcnn3 in PDGFRα+ cells [52]. Electrophysiological experiments on isolated cells using the patch clamp technique confirmed the importance of the highly expressed membrane conductances in pacemaker activity and responses to neurotransmitters [52, 83]. Activation of Ano1 channels results in an inward current, and activation of SK3 channels activates outward current. Thus, ICC and PDGFRα+ cells provide opposing regulatory inputs in the SIP syncytium.
The two major ionic currents of ICC and PDGFRα+ cells are activated by intracellular Ca2+, suggesting that Ca2+ dynamics regulate the electrophysiological behaviors of these cells. This chapter summarizes some of the current understanding about Ca2+ handling mechanisms in ICC and PDGFRα+ cells. Realization of the importance of Ca2+ dynamics has coincided with the ability to create transgenic mice with expression of genetically encoded Ca2+ sensors in ICC. Ca2+ dynamics in ICC can be monitored in these mice in situ, making it possible to follow the activation of cells, whether spontaneous or in response to neurotransmitters and other bioagonists. Cell-specific expression of genetically encoded sensors has not been accomplished for PDGFRα+ cells due to the lack of highly selective Cre recombinase strains for these cells. However, PDGFRα+ cells can be distinguished in intact muscles due to nuclear expression of eGFP in a reporter strain [52] and the ability to monitor Ca2+ transients in cell cytoplasms after loading with membrane-permeable Ca2+ sensors [5, 6].
22.2 Basal Ca2+ Transients in Interstitial Cells
There are two major classes of ICC, intramuscular cells and networks of interconnected cells. Intramuscular ICC (ICC-IM and ICC in the region of the deep muscular plexus of the small intestine, ICC-DMP) lie in close association with enteric motor nerve processes in most smooth muscle regions of the GI tract. In mice expressing GCaMPs exclusively in ICC, we have found that all intramuscular ICC studied generate spontaneous Ca2+ transients that originate from multiple sites within individual cells [3, 7, 16, 17]. These Ca2+ transients occur in a stochastic and localized manner and show no coupling between firing sites only a few microns from each other in the same cell or in cells nearby. The concept of coupled oscillators, used to describe the behaviors of ICC in the past [35, 72], is not apparent at this level of organization.
Ca2+ transients in ICC result from brief release events from intracellular Ca2+ stores. Drugs that block the uptake of Ca2+ into stores via SERCA pumps (CPA or thapsigargin) inhibit the generation of spontaneous Ca2+ transients within a few minutes [3, 16, 17]. Drugs that inhibit Ca2+ release through IP3 receptor (IP3R) channels or ryanodine (RyR) receptor channels also inhibit Ca2+ transients; however, the dependence upon IP3Rs and RyRs varies between different types of ICC [3, 16, 17]. Ca2+ release appears to occur in microdomains (now known as endoplasmic reticulum–plasma membrane junctions; ER/PM junctions) formed by close associations between the ER and the PM [65]. In the excluded volumes of ER/PM junctions, Ca2+ released from the ER reaches high concentrations and activates Ca2+-dependent conductances, such as Ano1, in the plasma membrane. Indeed, ICC-DMP generate spontaneous transient inward currents (STICs) due to transient activation of Ano1 channels by Ca2+ release [85]. In this manner, the stochastic release of Ca2+ from many sites within individual cells and from thousands of ICC within tissues can generate a net inward current that is conducted to SMCs via gap junctions. Thus, ICC exert a net excitatory influence in the SIP syncytium (Fig. 22.1a).

Schematics showing fundamental Ca2+ dynamics in ICC (a) and PDGFRα+ cells (b). These cells display localized Ca2+ transients due to release of Ca2+ from the endoplasmic reticulum (ER). The ER forms close associations with the plasma membrane (PM). These junctions are referred to as ER/PM junctions. High concentrations of [Ca2+]i are achieved during Ca2+ transients in the excluded volumes of the ER/PM junctions. (a) The rise in [Ca2+]i activates Ca2+-activated Cl− channels in the PM (encoded by Ano1) of ICC, producing spontaneous transient inward currents (STICs) and depolarization. STICs activated by Ca2+ transients in ICC conduct to adjoining SMCs electrically coupled via gap junctions (not shown). Generation of Ca2+ transients is highly localized within cells and independent of Ca2+ transients even at nearby ER/PM junctions or events occurring in nearby cells (i.e., purpose of showing two discrete ER/PM junctions in the schematic). Coupling between active Ca2+ release sites has not been observed, even after addition of excitatory neurotransmitters. (b) The rise in [Ca2+]i activates SK3 in the PM (encoded by Kcnn3) of PDGFRα+ cells, producing spontaneous transient outward currents (STOCs) and hyperpolarization or stabilization of membrane potential. STOCs activated by Ca2+ transients in PDGFRα+ cells conduct to adjoining SMCs that are electrically coupled via gap junctions (not shown)
When Ca2+ is released from stores into an ER/PM junction, some of the Ca2+ can be recovered via the SERCA pump, but some is lost through general diffusion to the bulk cytoplasm or to the extracellular space via plasma membrane Ca2+ ATPase (PMCA) or Na+/Ca2+ exchange, both of which are expressed by ICC [3, 16, 81]. Therefore, mechanisms to recover Ca2+ must exist to maintain the large ER-to-cytoplasm gradient and sustain the ability of the ER to release Ca2+. A major contributor to Ca2+ recovery in ICC occurs by the process of store-operated Ca2+ entry (SOCE) [60,61,62, 67, 71]. The apparatus for SOCE consists of a protein, stromal interaction protein (STIM) that spans the ER membrane. The portion of this protein within the ER lumen contains a Ca2+ sensor. When ER Ca2+ is depleted by release events, STIM molecules oligomerize, and the cytoplasmic portion of STIM binds to and activates ORAI, a highly selective Ca2+ channel in the PM. Thus, STIM and ORAI form a complex that senses a reduction in ER Ca2+ and activates Ca2+ entry to facilitate store refilling. Drugs that block ORAI can reduce or stop spontaneous Ca2+ transients in ICC [80].
While much less is known about Ca2+ dynamics in PDGFRα+ cells, these cells also have the ability to generate spontaneous Ca2+ transients [5, 6]. These events, however, couple to SK3 channels, that are also Ca2+ activated but cause the development of outward currents [52]. As a result of Ca2+ transients and activation of SK3 channels, isolated PDGFRα+ cells can generate spontaneous transient outward currents (STOCs). Generation of STOCs by many PDGFRα+ cells within GI muscles exerts a net hyperpolarizing or membrane-stabilizing effect, thereby reducing the excitability and contractility of muscles (Fig. 22.1b).
22.3 Neurotransduction by Interstitial Cells
Both types of interstitial cells in GI muscles contribute to neural regulation of motility. Although this topic has been somewhat controversial, some investigators have clung to the notion that SMCs are the cells innervated and responsible for post-junctional responses. Monitoring of Ca2+ signaling in ICC and PDGFRα+ cells clearly shows that these cells are innervated and display appropriate responses and temporal characteristics, which suggests that they mediate significant components of post-junctional responses. Blocking the ionic conductances specific to these cells, Ano1 or SK3, can reduce or block post-junctional electrical and mechanical responses to motor nerve stimulation [2, 4, 6, 19].
ICC form very close associations with the varicosities of motor neurons. This is not to say that such junctions are never found between varicosities and SMCs [57], but a morphometric study of esophageal muscles revealed a high propensity of these synaptic-like junctions between motor neurons and ICC [15]. These junctions may facilitate the availability of high concentrations of neurotransmitters near receptors of ICC. Expression of the appropriate receptors by ICC is another indication that these cells are involved in transduction of neural inputs. In the case of excitatory neurotransmission, ICC express type 3 muscarinic (M3) receptors and neurokinin type 1 (NK1) receptors [4, 12, 19, 43]. These receptors dominate responses to neurotransmitters released from enteric excitatory motor neurons (Fig. 22.2). The metabolic enzyme for ACh (acetylcholine esterase (AChE)) is expressed by enteric motor neurons [78], so it is likely that ACh is broken down rapidly in junctional spaces and sufficiently high concentrations of the transmitter may not reach muscarinic receptors expressed by SMCs. Our experiments have shown this to be the case in the murine gastric fundus, but after inhibition of AChE or when gastric ICC-IM fail to develop in the fundus, as in W/WV mutants, thus removing the synaptic-like junctions, new post-junctional mechanisms are recruited, suggesting that in these conditions ACh reaches SMC receptors [10]. Enteric inhibitory responses due to release of nitric oxide (NO) are compromised in parts of the GI tract where ICC-IM are depleted in W/WV mutants. ICC express soluble guanylate cyclase (sGC), the receptor for NO, suggesting these cells mediate at least a portion of post-junctional nitrergic responses [40, 41]. However, diffusion of NO may not be so heavily confined, and it may spread to other components of the SIP syncytium. Cell-specific knockdown of sGC suggests that nitrergic, post-junctional responses are mediated by ICC and SMCs [9, 28, 29, 54, 55]. Pathways for nitrergic inhibitory regulation are shown in Fig. 22.2.

Elements and mechanisms of the SIP syncytium and how enteric and sympathetic motor neurons regulate the output of the SIP syncytium. The SIP syncytium is composed of SMCs, ICC (intramuscular and pacemaker types of ICC), and PDGFRα+ cells. Ca2+ release regulates the open probabilities of Ca2+-activated conductances in ICC and PDGFRα+ cells (see Fig. 22.1). Ca2+ release in ICC is regulated by excitatory and inhibitory neurotransmission by release of ACh and neurokinins from enteric excitatory enteric motor neurons (EEN) and NO from enteric inhibitory motor neurons (EIN). Pathways activated either increase (excitatory neurotransmission) or inhibit (nitrergic neurotransmission) the release of Ca2+ and therefore the activation of Ano1 channels in the PM. Ca2+ release in PDGFRα+ cells is regulated by release of purines from enteric inhibitory motor neurons and norepinephrine (NE) from sympathetic neurons. These neurotransmitters increase release of Ca2+ from the ER and activate SK3 channels, producing an outward current. Currents generated by ICC and PDGFRα+ cells conduct to SMCs and regulate the excitability of the SMC component of the SIP syncytium. SMCs also receive input from pacemaker ICC, such as ICC-MY. The pacemaker cells generate slow waves that cause periodic depolarizations of SMCs, activation of L-type Ca2+ channels (VDCC in SMCs), and phasic contractions of these cells. Active propagation of slow waves in the network of ICC-MY coordinates contractions of the muscles, producing behaviors such as gastric peristalsis and intestinal segmentation. Active propagation of slow waves depends upon T-type Ca2+ channels in ICC-MY (shown as VDCC in ICC). SMCs also receive direct stimulation by neurotransmission from, at a minimum, NO and NE. These neurotransmitters regulate smooth muscle contractions through electrophysiological responses (not shown) and by altering the Ca2+ sensitivity of contractile elements (CE). Based on the expression of receptors in SIP cells, stimulation of PDGFRα+ cells by inhibitory neuropeptides (e.g., VIP) is likely to occur
Effector pathways in ICC, including Ca2+ handling mechanisms and dominant ion channels, transduce neural inputs. Stimulation of intrinsic motor neurons with electrical field stimulation (EFS) under conditions favoring excitatory neurotransmission (i.e., by blocking inhibitory pathways) causes significant enhancement in the frequency and amplitude of Ca2+ transients in ICC-IM in the colon and ICC-DMP in the small intestine. Stimulation of ICC-DMP appears to be dominated through tachykinin release and binding of NK1 receptors [4], while these receptors are hardly functional and responses are dominated by M3 receptors in the colon [19]. M3 and NK1 receptors couple through G proteins (Gq/11) and activation of phospholipase Cβ, causing generation of IP3 [1, 76]. Enhanced production of IP3 causes dramatic increases in Ca2+ release from ER via IP3Rs. These events, like spontaneous Ca2+ transients, activate Ano1 channels in the PM and elicit a depolarizing trend in the SIP syncytium, enhancing SMC excitability and contraction (Fig. 22.2). Although Ca2+ transient frequency increases in ICC, entrainment of Ca2+ release or coupled oscillations between discrete Ca2+ release sites have not been observed. Thus, no evidence for coupled oscillators is substantiated at this level of organization.
Opposite effects on Ca2+ transients are observed in ICC-IM and ICC-DMP in response to enteric inhibitory neural stimulation (i.e., when excitatory pathways are blocked). The inhibitory effects are mediated by NO and transduced through activation of sGC and generation of cGMP. Downstream from production of cGMP, the mechanisms for the inhibitory effects of NO on Ca2+ release in ICC are less well understood, but may occur through cGMP-dependent protein kinase I and phosphorylation of IP3R-associated cGMP kinase substrate (IRAG) [26, 75]. Initiation of enteric inhibitory responses blocks Ca2+ transients for the duration of stimulation (Fig. 22.2). Inhibition of Ca2+ release causes deactivation of Ano1 channels and cessation of the basal inward current in the SIP syncytium. In colonic muscles, this resulted in a 9 mV hyperpolarization of muscles [17]. Hyperpolarization reduces the likelihood that the threshold for Ca2+ action potentials is reached in SMCs, so this is a mechanism through which nitrergic neurotransmission reduces SMC excitability and inhibits contractions.
A long-acknowledged phenomenon in GI muscles is tonic inhibition [77]. In some muscles, excitability and contractions are suppressed by ongoing release of inhibitory neurotransmitter, and SMC excitability and contractions of the muscles are greatly enhanced by TTX or by inhibition of nNOS. We found that tonic inhibition is linked to ongoing partial suppression of Ca2+ transients in ICC. TTX and antagonists of nNOS or sGC greatly increased the occurrence and amplitude of Ca2+ transients in ICC concomitantly with the increase in contractions [18]. The increased contractions resulting from suppression of tonic inhibition were reversed by an antagonist of Ano1 channels, supporting the idea that tonic inhibition in the proximal colon is mediated by nitrergic effects on ICC.
Enteric inhibitory neurons exhibit co-transmission whereby single neurons release multiple neurotransmitters. The synthetic enzyme for NO (nNOS) and the peptide VIP co-localize in enteric motor neurons [14, 45]. It is likely that these neurons also release purine neurotransmitters; however, this has never been shown definitively. A somewhat controversial story exists around the identity of the purine neurotransmitter in the GI tract. Classically, the transmitter was believed to be ATP [11], but more recent studies demonstrate that β-NAD+ fulfills the criteria for a neurotransmitter much better than ATP in mouse, monkey, and human GI muscles [38, 58]. Actually, enteric inhibitory neurons may release a cocktail of purines that include β-NAD+, ADPR, and Up4A [21, 22]. This story is detailed and not among the main topics of this review.
Another gene highly expressed by mouse and human PDGFRα+ cells is P2ry1 [5, 51, 52]. Activation of P2Y1 receptors by several purines, including ATP, ADP, β-NAD+, and the highly selective agonist MRS2365, greatly increases the frequency and amplitude of Ca2+ transients in PDGFRα+ cells in gastric fundus and colon [5, 6]. P2Y1 receptors are coupled through G proteins (Gq/11) [23], so like M3 and NK1 receptors in ICC, P2Y1 agonists increase the activity of PLCβ, production of IP3, and release of Ca2+ through IP3Rs (Fig. 22.2). Interesting to note is that MRS2500, a selective antagonist of P2Y1 receptors, blocked all of these responses except the responses of some PDGFRα+ cells to ATP. Genetic deactivation of P2ry1 ablates purinergic enteric inhibitory responses in mice [25, 37] and the increase in Ca2+ transients in PDGFRα+ cells caused by ADP, β-NAD+, and MRS2365. Responses to ATP, however, persist in some PDGFRα+ cells in GI muscles of P2ry1−/− animals [5, 6]. Similar to the Ca2+ transients enhanced by P2Y1 agonists, these compounds elicited STOCs in PDGFRα+ cells [52]. Confidence that purinergic inhibitory responses are mediated by PDGFRα+ cells increased dramatically when it was shown that large amplitude hyperpolarization responses are elicited in PDGFRα+ cells that are equivalent to purinergic inhibitory junction potentials (IJPs) elicited in whole muscles [50]. These responses, like IJPs, are blocked by apamin or MRS2365. Of importance, P2Y1 agonists failed to cause hyperpolarization of SMCs isolated from the same muscles.
Evaluation of the transcriptomes of SIP cells has revealed other receptors in these cells that might mediate regulatory effects (Fig. 22.2). An example is the expression of α adrenergic receptors (Adra1a and Adra1b) in PDGFRα+ cells [30]. Expression of these receptors was verified by real-time PCR [48]. Processes of sympathetic neurons, as labeled with antibodies to tyrosine hydroxylase, are distributed in the plane of the myenteric plexus and within circular muscles in proximity to PDGFRα+ cells. As with P2Y1 agonists, NE elicited STOCs in voltage-clamped PDGFRα+ cells that were blocked by both RS100329, a specific adrenergic α1a receptor antagonist, and apamin. In current-clamped PDGFRα+ cells, significant hyperpolarization responses were caused by NE. NE also initiated or increased Ca2+ transients in PDGFRα+ cells in situ, and these responses were also blocked by RS100329. Contractions of colonic muscles were inhibited by phenylephrine, and these responses were blocked by RS100329 and apamin. Inhibitory effects of phenylephrine did not occur in Adra1a−/− mice. A preparation with the inferior mesenteric ganglion attached to the colon via the lumbar colonic nerve was used to isolate electrical stimulation of sympathetic neurons. Sympathetic nerve stimulation (SNS) caused hyperpolarization of colonic muscles that were partially blocked by prazosin and apamin [48]. SNS also inhibited colonic migrating motor complexes (CMMCs) through the mid and distal colon. This dramatic sympathetic effect on CMMCs did not occur in colons of Adra1a−/− mice. Similar sympathetic neurotransduction in PDGFRα+ cells and regulation of contractions also occur in human colons [49].
22.4 Pacemaker Activity in Interstitial Cells
ICC generate pacemaker activity in the GI tract that is responsible for electrical slow waves [53, 83]. Networks of pacemaker cells occur along the boundaries of the muscle layers, between the circular and longitudinal muscle layers in the stomach, small bowel, and colon (ICC-MY) and at the submucosal surface of the circular muscle in the colon (ICC-SM). Recent studies have also shown that a type of pacemaker activity is also generated by the ICC along the serosal surface of the proximal colon (ICC-SS), and a type of ICC within muscle bundles (ICC-IM type II) appears to generate the high-frequency pacemaker activity responsible for tone in the internal anal sphincter [33]. A major difference between pacemaker types of ICC (i.e., ICC-MY and ICC-SM) and intramuscular types of ICC (i.e., ICC-IM and ICC-DMP) is the expression of voltage-dependent Ca2+ conductances in pacemaker cells [7, 12, 16, 27, 33, 82].
ICC-MY and ICC-SM generate spontaneous Ca2+ transients that activate Ano1 channels in the PMs of these cells (Fig. 22.2). In the small intestine, ICC-MY express T-type Ca2+ channels (CaV3.2) [12, 16, 27, 82]. These channels are activated by the small depolarizations caused by the activation of Ano1 channels (STICs). Increasing the open probability of T-type Ca2+ channels can result in development of a Ca2+ action potential that constitutes the upstroke phase of slow waves. The dV/dt of the slow wave upstroke, when recorded directly from ICC-MY, is 2 V/s in the mouse intestine and 11 V/s in the rabbit intestine [46]. As in other excitable cells connected by gap junctions, the upstroke phase of slow waves depolarizes neighboring ICC-MY, activates the T-type conductance in these cells, and regenerates the upstroke potential, facilitating active propagation of slow waves in the ICC-MY network. Propagation is seen optically as a coherent intracellular Ca2+ wave front that proceeds at about 2 mm/sec in ICC-MY networks in mouse small intestine [16, 59]. SMCs do not express the same ion channels as ICC-MY, and in spite of electrical coupling between SMCs and ICC-MY, SMCs cannot regenerate slow waves and sustain active propagation. Therefore, slow waves conduct passively and decay with distance in the SMC compartment of the SIP syncytium.
Active propagation of slow waves is an important factor in the generation of normal motility behaviors. For example, in the stomach of larger mammals, slow waves propagate without decrement for many centimeter from the dominant pacemaker in the orad corpus to the pylorus [8]. Slow wave propagation was studied in canine gastric muscles using a dual chamber apparatus where slow waves could be initiated by pacing in one chamber, and recordings could be made in the second, electrically isolated chamber at various distances from the site of initiation or after addition of antagonists of specific conductances [8]. Nicardipine had no effect on the dV/dt of the upstroke depolarization nor on the rate of propagation. Antagonists of T-type channels, however, dramatically reduced both the upstroke velocity and the propagation rate. At higher concentrations, these antagonists blocked active slow wave propagation, as did reducing extracellular Ca2+ to 0.5 mM. These experiments also showed that reducing the availability of T-type Ca2+ channels by depolarization, which causes voltage-dependent inactivation, also greatly reduced upstroke and propagation velocity. These experiments clearly showed that propagation of slow waves is dependent upon voltage-dependent activation of a Ca2+ conductance with characteristics of T-type channels.
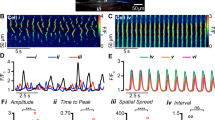
The Ca2+ waves that sweep across a network of ICC-MY are also dependent upon T-type Ca2+ channels in the murine small intestine (Fig. 22.2) [16]. Clusters of Ca2+ transients occur regularly in jejunal ICC-MY networks at a frequency of about 30 cycles per min in the mouse. The coherent spread of waves and the clustering of Ca2+ transients are blocked by NNC 55-0396 and TTA-A2, two specific T-type Ca2+ channel antagonists. However, these compounds do not block all Ca2+ transients, and in fact the occurrence of Ca2+ transients in the presence of T-type Ca2+ channel antagonists reverts to a stochastic pattern, as seen in ICC-IM. These experiments suggest that stochastic Ca2+ release is a basic behavior of ICC, and addition of a voltage-dependent Ca2+ conductance organizes Ca2+ transients into periodic clusters of events. While T-type channels are of primary importance in the stomach and small intestine, L-type channels are also important in some ICC, such as those along the submucosal surface of the circular muscle (ICC-SM) in the murine colon [7]. Muscles of the murine proximal colon are relatively more depolarized than cells in the small bowel and stomach. It would be impractical for T-type channels to be the dominant conductance providing active propagation in these muscles because T-type channels are inactivated at the depolarized potentials of colonic muscles. Instead, ICC-SM in murine colon utilize L-type Ca2+ as the dominant conductance. However, when ICC-SM are hyperpolarized, T-type channels become available and contribute to slow wave propagation. Presence of both T- and L-type channels provides a safety factor such that slow waves can persist over a broad range of membrane potentials.
Ca2+ entry due to the activation of the voltage-dependent Ca2+ conductance elicits not only depolarization but also Ca2+-induced Ca2+ release (CICR) from the ER. Multiple firing sites generate these events both in the soma of ICC and in their processes. As above, Ca2+ entry organizes the otherwise stochastic Ca2+ transients into clusters of events. Ca2+ transients and corresponding activation of Ano1 channels through the network of ICC cause sustained net activation of the Cl− conductance and clamp membrane potentials of ICC close to the equilibrium potential for the Cl− gradient (about −10 mV; [84]). Ca2+ release events persist for more than a second or until Ca2+ stores are depleted to an extent where they cannot continue to release Ca2+. The durations of the Ca2+ transient clusters define the durations of the plateau phase of slow waves. Here again, the concept of coupled oscillators is fallacious. Ca2+ release events are independent of each other and are organized into clusters by propagation of Ca2+ action potentials (i.e., the upstroke of the slow wave event), Ca2+ entry, and CICR.
A question to be considered is why the plateau phase is sustained for more than a second. Ca2+ entry through T-type Ca2+ channels is brief because the ensuing depolarization rapidly inactivates these channels. Therefore, it is unlikely that entry of Ca2+ through T-type channels is capable of sustaining CICR. The increased open probability of Ano1 channels is maintained by elevated intracellular Ca2+. Thus, a source of Ca2+ is needed to maintain openings of Ano1 channels. Several potential sources are possible. The plateau phase of slow waves recorded from ICC is in the range of potentials that generate “window currents” from L-type Ca2+ channels [13]. Therefore, openings of these Ca2+ channels could provide persistent Ca2+ entry that could sustain CICR. This is likely a mechanism for sustaining the plateau in canine gastric antrum because nicardipine dramatically decreases and shortens the plateau phase of slow waves [8]. In contrast, dihydropyridines have little to no effect on slow waves in the small intestine. In murine, small intestine reverse mode Na+/Ca2+ exchange appears to be responsible for sustained Ca2+ entry during the plateau phase [81]. During the period of Ca2+ release from stores, Ca2+ entry can also occur through ORAI [80]. When ER Ca2+ stores are depleted, Ca2+ release terminates, the open probability for Ano1 channels drops to low levels, and membrane potential repolarizes. After repolarization, membrane potential rests at negative levels, Ca2+ entry is minimal, and the stores refill via SOCE [80].
There is another ICC behavior in which membrane potential of the SIP syncytium is conditioned or tuned into a range where SMCs become rhythmic. In colonic longitudinal muscle, rhythmic intercellular Ca2+ waves occur that are due to the periodic firing of Ca2+ action potentials in longitudinal muscle cells [34]. These events are inhibited by an Ano1 antagonist [20]. Ano1 is expressed in ICC but not in SMCs, so it is likely that the inward current due to Ano1 develops in the ICC along the serosal surface, ICC-SS. This hypothesis was investigated in muscles of murine proximal colon [20]. ICC-SS form network-like structures and fire stochastic, localized Ca2+ transients. These events did not spread cell to cell as observed in ICC-MY or ICC-SM. Thus, they appear to be a hybrid type of ICC with stellate morphologies reminiscent of pacemaker ICC, but with Ca2+ dynamics similar to ICC-IM and ICC-DMP. As in all other ICC, however, Ca2+ transients in ICC-SS activate Ano1 channels in the PM. The inward currents produced summate to provide a depolarizing influence on longitudinal muscle cells, bringing membrane potential into a range where the SMCs fire Ca2+ action potentials. Thus, this example of electrical and mechanical rhythmicity represents an emergent property due to the electrophysiological characteristics of ICC-SS and SMCs and electrical coupling between these cells.
22.5 Conclusions
Ca2+ imaging has provided revelations about the mechanisms and functions of interstitial cells in GI muscles. These cells have dynamic Ca2+ handling mechanisms that utilize activation of Ano1 or SK3 channels in the PM and conduction of electrical responses to SMCs to distribute slow waves and responses to neural inputs from enteric and sympathetic motor neurons. Much has been learned about the organization and functions of ICC and PDGFRα+ cells in normal GI muscles. It is now clear that disease or genetic mutations causing loss of function in interstitial cells can result in GI motor disorders. It is also possible that responses to immune mediators and phenotypic changes in interstitial cells could be a cause of fibrosis. However, we still lack knowledge about what causes defects or alters the phenotypes of interstitial cells and have no therapeutic means of restoring their functions if damaged in disease or aging. It is possible that defects in the SIP syncytium are the primary cause of motor disorders, and additional studies are needed to learn how to manipulate and potentially repair interstitial cell networks and connectivity with motor neurons. In-depth study of the pathophysiology of interstitial cells may provide new opportunities for therapeutics.
References
Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, Martín JD, Candenas ML (2004) Tachykinins and tachykinin receptors: structure and activity relationships. Curr Med Chem 11:2045–2081
Baker SA, Drumm BT, Cobine CA, Keef KD, Sanders KM (2018a) Inhibitory neural regulation of the Ca(2+) transients in intramuscular interstitial cells of Cajal in the small intestine. Front Physiol 9:328
Baker SA, Drumm BT, Saur D, Hennig GW, Ward SM, Sanders KM (2016) Spontaneous Ca(2+) transients in interstitial cells of Cajal located within the deep muscular plexus of the murine small intestine. J Physiol 594:3317–3338
Baker SA, Drumm BT, Skowronek KE, Rembetski BE, Peri LE, Hennig GW, Perrino BA, Sanders KM (2018b) Excitatory neuronal responses of Ca(2+) transients in interstitial cells of Cajal in the small intestine. eNeuro 5(2):ENEURO.0080-18.2018. https://doi.org/10.1523/ENEURO.0080-18.2018
Baker SA, Hennig GW, Salter AK, Kurahashi M, Ward SM, Sanders KM (2013) Distribution and Ca(2+) signalling of fibroblast-like (PDGFR(+)) cells in the murine gastric fundus. J Physiol 591:6193–6208
Baker SA, Hennig GW, Ward SM, Sanders KM (2015) Temporal sequence of activation of cells involved in purinergic neurotransmission in the colon. J Physiol 593:1945–1963
Baker SA, Leigh WA, Del Valle G, De Yturriaga IF, Ward SM, Cobine CA, Drumm BT, Sanders KM (2021) Ca2+ signaling driving pacemaker activity in submucosal interstitial cells of Cajal in the murine colon. elife 10:e64099
Bayguinov O, Ward SM, Kenyon JL, Sanders KM (2007) Voltage-gated Ca2+ currents are necessary for slow-wave propagation in the canine gastric antrum. Am J Physiol Cell Physiol 293:C1645–C1659
Beck K, Friebe A, Voussen B (2018) Nitrergic signaling via interstitial cells of Cajal and smooth muscle cells influences circular smooth muscle contractility in murine colon. Neurogastroenterol Motil 30(6):e13300
Bhetwal BP, Sanders KM, An C, Trappanese DM, Moreland RS, Perrino BA (2013) Ca2+ sensitization pathways accessed by cholinergic neurotransmission in the murine gastric fundus. J Physiol 591:2971–2986
Burnstock G, Satchell DG, Smythe A (1972) A comparison of the excitatory and inhibitory effects of non-adrenergic, non-cholinergic nerve stimulation and exogenously applied ATP on a variety of smooth muscle preparations from different vertebrate species. Br J Pharmacol 46:234–242
Chen H, Ordog T, Chen J, Young DL, Bardsley MR, Redelman D, Ward SM, Sanders KM (2007) Differential gene expression in functional classes of interstitial cells of Cajal in murine small intestine. Physiol Genomics 31:492–509
Cohen NM, Lederer WJ (1987) Calcium current in isolated neonatal rat ventricular myocytes. J Physiol 391:169–191
Costa M, Furness JB, Pompolo S, Brookes SJ, Bornstein JC, Bredt DS, Snyder SH (1992) Projections and chemical coding of neurons with immunoreactivity for nitric oxide synthase in the guinea-pig small intestine. Neurosci Lett 148:121–125
Daniel EE, Posey-Daniel V (1984) Neuromuscular structures in opossum esophagus: role of interstitial cells of {Cajal}. Am J Phys 246:G305–G315
Drumm BT, Hennig GW, Battersby MJ, Cunningham EK, Sung TS, Ward SM, Sanders KM, Baker SA (2017) Clustering of Ca(2+) transients in interstitial cells of Cajal defines slow wave duration. J Gen Physiol 149:703–725
Drumm BT, Hwang SJ, Baker SA, Ward SM, Sanders KM (2019a) Ca2+ signalling behaviours of intramuscular interstitial cells of Cajal in the murine colon. J Physiol 597:3587–3617
Drumm BT, Rembetski BE, Baker SA, Sanders KM (2019b) Tonic inhibition of murine proximal colon is due to nitrergic suppression of Ca. Sci Rep 9:4402
Drumm BT, Rembetski BE, Huynh K, Nizar A, Baker SA, Sanders KM (2020a) Excitatory cholinergic responses in mouse colon intramuscular interstitial cells of Cajal are due to enhanced Ca2+ release via M3 receptor activation. FASEB J 34(8):10073–10095
Drumm BT, Rembetski BE, Messersmith K, Manierka MS, Baker SA, Sanders KM (2020b) Pacemaker function and neural responsiveness of subserosal interstitial cells of Cajal in the mouse colon. J Physiol 598:651–681
Durnin L, Hwang SJ, Kurahashi M, Drumm BT, Ward SM, Sasse KC, Sanders KM, Mutafova-Yambolieva VN (2014) Uridine adenosine tetraphosphate is a novel neurogenic P2Y1 receptor activator in the gut. Proc Natl Acad Sci U S A 111:15821–15826
Durnin L, Hwang SJ, Ward SM, Sanders KM, Mutafova-Yambolieva VN (2012) Adenosine 5-diphosphate-ribose is a neural regulator in primate and murine large intestine along with beta-NAD(+). J Physiol 590:1921–1941
Erb L, Weisman GA (2012) Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal 1:789–803
Faussone Pellegrini MS, Cortesini C, Romagnoli P (1977) Ultrastructure of the tunica muscularis of the cardial portion of the human esophagus and stomach, with special reference to the so-called Cajal’s interstitial cells. Arch Ital Anat Embriol 82:157–177
Gallego D, Gil V, Martinez-Cutillas M, Mane N, Martin MT, Jimenez M (2012) Purinergic neuromuscular transmission is absent in the colon of P2Y(1) knocked out mice. J Physiol 590:1943–1956
Geiselhoringer A, Werner M, Sigl K, Smital P, Worner R, Acheo L, Stieber J, Weinmeister P, Feil R, Feil S, Wegener J, Hofmann F, Schlossmann J (2004) IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. EMBO J 23:4222–4231
Gibbons SJ, Strege PR, Lei S, Roeder JL, Mazzone A, Ou Y, Rich A, Farrugia G (2009) The alpha1H Ca2+ channel subunit is expressed in mouse jejunal interstitial cells of Cajal and myocytes. J Cell Mol Med 13:4422–4431
Groneberg D, Konig P, Lies B, Jager R, Seidler B, Klein S, Saur D, Friebe A (2013) Cell-specific deletion of nitric oxide-sensitive guanylyl cyclase reveals a dual pathway for nitrergic neuromuscular transmission in the murine fundus. Gastroenterology 145(1):188–196
Groneberg D, Zizer E, Lies B, Seidler B, Saur D, Wagner M, Friebe A (2015) Dominant role of interstitial cells of Cajal in nitrergic relaxation of murine lower oesophageal sphincter. J Physiol 593:403–414
Ha SE, Lee MY, Kurahashi M, Wei L, Jorgensen BG, Park C, Park PJ, Redelman D, Sasse KC, Becker LS, Sanders KM, Ro S (2017) Transcriptome analysis of PDGFRalpha+ cells identifies T-type Ca2+ channel CACNA1G as a new pathological marker for PDGFRalpha+ cell hyperplasia. PLoS One 12:e0182265
Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P (2003) Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol 23:4013–4025
Hanani M, Farrugia G, Komuro T (2005) Intercellular coupling of interstitial cells of cajal in the digestive tract. Int Rev Cytol 242:249–282
Hannigan KI, Bossey AP, Foulkes HJL, Drumm BT, Baker SA, Ward SM, Sanders KM, Keef KD, Cobine CA (2020) A novel intramuscular Interstitial Cell of Cajal is a candidate for generating pacemaker activity in the mouse internal anal sphincter. Sci Rep 10:10378
Hennig GW, Smith CB, O’Shea DM, Smith TK (2002) Patterns of intracellular and intercellular Ca2+ waves in the longitudinal muscle layer of the murine large intestine in vitro. J Physiol 543:233–253
Huizinga JD, Chen JH, Zhu YF, Pawelka A, Mcginn RJ, Bardakjian BL, Parsons SP, Kunze WA, Wu RY, Bercik P, Khoshdel A, Chen S, Yin S, Zhang Q, Yu Y, Gao Q, Li K, Hu X, Zarate N, Collins P, Pistilli M, Ma J, Zhang R, Chen D (2014) The origin of segmentation motor activity in the intestine. Nat Commun 5:3326
Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A (1995) W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 373:347–349
Hwang SJ, Blair PJ, Durnin L, Mutafova-Yambolieva V, Sanders KM, Ward SM (2012) P2Y1 purinoreceptors are fundamental to inhibitory motor control of murine colonic excitability and transit. J Physiol 590:1957–1972
Hwang SJ, Durnin L, Dwyer L, Rhee PL, Ward SM, Koh SD, Sanders KM, Mutafova-Yambolieva VN (2011) Beta-nicotinamide adenine dinucleotide is an enteric inhibitory neurotransmitter in human and nonhuman primate colons. Gastroenterology 140(608–617):e6
Hwang SJ, Pardo DM, Zheng H, Bayguinov Y, Blair PJ, Fortune-Grant R, Cook RS, Hennig GW, Shonnard MC, Grainger N, Peri LE, Verma SD, Rock J, Sanders KM, Ward SM (2019) Differential sensitivity of gastric and small intestinal muscles to inducible knockdown of anoctamin 1 and the effects on gastrointestinal motility. J Physiol 597(9):2337–2360
Iino S, Horiguchi K, Nojyo Y (2008) Interstitial cells of Cajal are innervated by nitrergic nerves and express nitric oxide-sensitive guanylate cyclase in the guinea-pig gastrointestinal tract. Neuroscience 152:437–448
Iino S, Horiguchi K, Nojyo Y, Ward SM, Sanders KM (2009) Interstitial cells of Cajal contain signalling molecules for transduction of nitrergic stimulation in guinea pig caecum. Neurogastroenterol Motil 21(542–50):e12–e13
Iino S, Nojyo Y (2009) Immunohistochemical demonstration of c-Kit-negative fibroblast-like cells in murine gastrointestinal musculature. Arch Histol Cytol 72:107–115
Iino S, Ward SM, Sanders KM (2004) Interstitial cells of Cajal are functionally innervated by excitatory motor neurones in the murine intestine. J Physiol 556:521–530
Ishikawa K, Komuro T, Hirota S, Kitamura Y (1997) Ultrastructural identification of the c-kit-expressing interstitial cells in the rat stomach: a comparison of control and Ws/Ws mutant rats. Cell Tissue Res 289:137–143
Keef KD, Shuttleworth CW, Xue C, Bayguinov O, Publicover NG, Sanders KM (1994) Relationship between nitric oxide and vasoactive intestinal polypeptide in enteric inhibitory neurotransmission. Neuropharmacology 33:1303–1314
Kito Y, Mitsui R, Ward SM, Sanders KM (2015) Characterization of slow waves generated by myenteric interstitial cells of Cajal of the rabbit small intestine. Am J Physiol Gastrointest Liver Physiol 308:G378–G388
Komuro T (2012) Atlas of interstitail cells of Cajal in the gastrointestinal tract. Springer, Dordrecht
Kurahashi M, Kito Y, Baker SA, Jennings LK, Dowers JGR, Koh SD, Sanders KM (2020a) A novel postsynaptic signal pathway of sympathetic neural regulation of murine colonic motility. FASEB J 34(4):5563–5577
Kurahashi M, Kito Y, Hara M, Takeyama H, Sanders KM, Hashitani H (2020b) Norepinephrine has dual effects on human colonic contractions through distinct subtypes of alpha 1 adrenoceptors. Cell Mol Gastroenterol Hepatol 10:658–671, e1
Kurahashi M, Mutafova-Yambolieva V, Koh SD, Sanders KM (2014) Platelet-derived growth factor receptor-alpha-positive cells and not smooth muscle cells mediate purinergic hyperpolarization in murine colonic muscles. Am J Physiol Cell Physiol 307:C561–C570
Kurahashi M, Nakano Y, Hennig GW, Ward SM, Sanders KM (2012) Platelet-derived growth factor receptor alpha-positive cells in the tunica muscularis of human colon. J Cell Mol Med 16:1397–1404
Kurahashi M, Zheng H, Dwyer L, Ward SM, Koh SD, Sanders KM (2011) A functional role for the ‘fibroblast-like cells’ in gastrointestinal smooth muscles. J Physiol 589:697–710
Langton P, Ward SM, Carl A, Norell MA, Sanders KM (1989) Spontaneous electrical activity of interstitial cells of Cajal isolated from canine proximal colon. Proc Natl Acad Sci U S A 86:7280–7284
Lies B, Beck K, Keppler J, Saur D, Groneberg D, Friebe A (2015) Nitrergic signalling via interstitial cells of Cajal regulates motor activity in murine colon. J Physiol 593:4589–4601
Lies B, Gil V, Groneberg D, Seidler B, Saur D, Wischmeyer E, Jimenez M, Friebe A (2014) Interstitial cells of Cajal mediate nitrergic inhibitory neurotransmission in the murine gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 307:G98–G106
Maeda H, Yamagata A, Nishlkawa S, Yoshinaga K, Kobayashi S, Nishi K, Nishikawa SI (1992) Requirement of c-kit for development of intestinal pacemaker system. Development 116:369–375
Mitsui R, Komuro T (2002) Direct and indirect innervation of smooth muscle cells of rat stomach, with special reference to the interstitial cells of Cajal. Cell Tissue Res 309:219–227
Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, Sanders KM (2007) Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A 104:16359–16364
Park KJ, Hennig GW, Lee HT, Spencer NJ, Ward SM, Smith TK, Sanders KM (2006) Spatial and temporal mapping of pacemaker activity in interstitial cells of Cajal in mouse ileum in situ. Am J Physiol Cell Physiol 290:C1411–C1427
Prakriya M, Lewis RS (2015) Store-operated calcium channels. Physiol Rev 95:1383–1436
Putney JW (1999) “Kissin’ cousins”: intimate plasma membrane-ER interactions underlie capacitative calcium entry. Cell 99:5–8
Putney JW (1986) A model for receptor-regulated calcium entry. Cell Calcium 7:1–12
Ro S, Chanjae P, Jin J, Zheng H, Blair P, Redelman D, Ward SM, Yan W, Sanders KM (2010) A model to study the phenotypic changes of interstitial cells of Cajal in gastrointestinal diseases. Gastroenterology 138:1068–1078.e2
Sanders KM, Koh SD, Ro S, Ward SM (2012) Regulation of gastrointestinal motility--insights from smooth muscle biology. Nat Rev Gastroenterol Hepatol 9:633–645
Sanders KM, Ward SM, Koh SD (2014) Interstitial cells: regulators of smooth muscle function. Physiol Rev 94:859–907
Seki K, Komuro T (1998) Further observations on the gap-junction-rich cells in the deep muscular plexus of the rat small intestine. Anat Embryol (Berl) 197:135–141
Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M (2008) Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 135:110–122
Sung TS, Hwang SJ, Koh SD, Bayguinov Y, Peri LE, Blair PJ, Webb TI, Pardo DM, Rock JR, Sanders KM, Ward SM (2018) The cells and conductance mediating cholinergic neurotransmission in the murine proximal stomach. J Physiol 596:1549–1574
Thuneberg L (1982) Interstitial cells of Cajal: intestinal pacemaker cells? Adv Anat Embryol Cell Biol 71:1–130
Torihashi S, Ward SM, Nishikawa S, Nishi K, Kobayashi S, Sanders KM (1995) c-kit-dependent development of interstitial cells and electrical activity in the murine gastrointestinal tract. Cell Tissue Res 280:97–111
Trebak M, Zhang W, Ruhle B, Henkel MM, Gonzalez-Cobos JC, Motiani RK, Stolwijk JA, Newton RL, Zhang X (2013) What role for store-operated Ca(2)(+) entry in muscle? Microcirculation 20:330–336
Van Helden DF, Imtiaz MS (2003) Ca2+ phase waves: a basis for cellular pacemaking and long-range synchronicity in the guinea-pig gastric pylorus. J Physiol 548:271–296
Ward SM, Burns AJ, Torihashi S, Harney SC, Sanders KM (1995) Impaired development of interstitial cells and intestinal electrical rhythmicity in steel mutants. Am J Phys 269:C1577–C1585
Ward SM, Burns AJ, Torihashi S, Sanders KM (1994) Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J Physiol 480(Pt 1):91–97
Werder A, Mayr M, Schneider G, Oesterle D, Fritsch RM, Seidler B, Schlossmann J, Hofmann F, Schemann M, Allescher HD, Schmid RM, Saur D (2011) Truncated IRAG variants modulate cGMP-mediated inhibition of human colonic smooth muscle cell contraction. Am J Physiol Cell Physiol 301:C1445–C1457
Wess J (1996) Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol 10:69–99
Wood JD (1972) Excitation of intestinal muscle by atropine, tetrodotoxin, and xylocaine. Am J Phys 222:118–125
Worth AA, Forrest AS, Peri LE, Ward SM, Hennig GW, Sanders KM (2015) Regulation of gastric electrical and mechanical activity by cholinesterases in mice. J Neurogastroenterol Motil 21:200–216
Yamamoto M (1977) Electron microscopic studies on the innervation of the smooth muscle and the interstitial cell of Cajal in the small intestine of the mouse and bat. Arch Histol Jpn 40:171–201
Zheng H, Drumm BT, Earley S, Sung TS, Koh SD, Sanders KM (2018) SOCE mediated by STIM and Orai is essential for pacemaker activity in the interstitial cells of Cajal in the gastrointestinal tract. Sci Signal 11(534):eaaq0918
Zheng H, Drumm BT, Zhu MH, Xie Y, O’Driscoll KE, Baker SA, Perrino BA, Koh SD, Sanders KM (2020) Na+/Ca2+ exchange and pacemaker activity of interstitial cells of Cajal. Front Physiol 11:230
Zheng H, Park KS, Koh SD, Sanders KM (2014) Expression and function of a T-type Ca2+ conductance in interstitial cells of Cajal of the murine small intestine. Am J Physiol Cell Physiol 306:C705–C713
Zhu MH, Kim TW, Ro S, Yan W, Ward SM, Koh SD, Sanders KM (2009) A Ca(2+)-activated Cl(−) conductance in interstitial cells of Cajal linked to slow wave currents and pacemaker activity. J Physiol 587:4905–4918
Zhu MH, Sung TS, Kurahashi M, O’Kane LE, O’Driscoll K, Koh SD, Sanders KM (2016) Na+-K+-Cl- cotransporter (NKCC) maintains the chloride gradient to sustain pacemaker activity in interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol 311:G1037–G1046
Zhu MH, Sung TS, O’Driscoll K, Koh SD, Sanders KM (2015) Intracellular Ca(2+) release from endoplasmic reticulum regulates slow wave currents and pacemaker activity of interstitial cells of Cajal. Am J Physiol Cell Physiol 308:C608–C620
Acknowledgments
Writing of this paper and much of the data reviewed was supported by R01 DK120759 to KMS and SAB and R01 DK-091336 to KMS and MK.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Sanders, K.M., Baker, S.A., Drumm, B.T., Kurahashi, M. (2022). Ca2+ Signaling Is the Basis for Pacemaker Activity and Neurotransduction in Interstitial Cells of the GI Tract. In: Spencer, N.J., Costa, M., Brierley, S.M. (eds) The Enteric Nervous System II. Advances in Experimental Medicine and Biology, vol 1383. Springer, Cham. https://doi.org/10.1007/978-3-031-05843-1_22
Download citation
DOI: https://doi.org/10.1007/978-3-031-05843-1_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-05842-4
Online ISBN: 978-3-031-05843-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)