
Abstract
This chapter focuses on the mechanosensitive properties of epithelial tissues. Epithelia experience a range of mechanical forces arising both intrinsically from their constituent cells and extrinsically from forces such as touch or alveolar inflation. We discuss how cell-cell junctions, such as adherens junctions and tight junctions, play key roles in the mechanobiology of epithelial tissues. At these sites, forces are generated through contraction of the actomyosin cytoskeleton and transmitted between neighbouring cells and across tissues by adhesion systems within the junctions. We also consider other potential cellular mechanisms that can allow epithelia to respond to mechanical stresses: mechanosensitive ion channels which are implicated in homeostatic control of cell density via modulation of cell proliferation and live-cell extrusion and caveolae, membrane invaginations that can buffer epithelia in response to change in membrane tension.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Mechanical forces are fundamental determinants of tissue integrity, morphogenesis and homeostasis in epithelia and endothelia. Here, we focus on epithelia because, as principal tissue barriers of the body, epithelia experience a diverse range of mechanical stresses that derive both from their constituent cells and from extrinsic forces. Epithelia are capable of detecting such changes in force and can respond in ways that can preserve tissue integrity and homeostasis. Cell-cell junctions play central roles in the mechanobiology of epithelia. They can be sites where active cellular forces are generated by the contractile actomyosin cytoskeleton. The cell-cell adhesion molecules in junctions can serve to transmit forces from cells to cells and/or integrate the transmission of force at the tissue level. As well, epithelial cell-cell junctions possess mechanosensing and signal transduction mechanisms that allow them to detect changes in mechanical force and elicit homeostatic responses. In this chapter, we discuss both well-defined and emerging topics relating to the mechanisms that allow epithelia to use mechanical force as a currency of biological information.
Tissue Forces in Epithelia
Tissues are subjected to constant mechanical forces which have diverse impacts on their integrity and function. These forces can be either compressive or tensile, which we define as forces that may result in tissue compaction or fracture, respectively [1].
Compressive Forces. These are most obviously a consequence of extrinsic physical stimuli, such as the pressure applied to the skin from touch. Although compressive forces are less studied compared to their tensile counterpart, they play important roles in directing, regulating and maintaining various processes during development and adult homeostasis. For example, compressive forces generated by morphogenetic events at the onset of gastrulation in the Drosophila melanogaster embryo influence the expression of Twist, a key regulator of epithelial-to-mesenchymal transitions (EMT) during gastrulation. Here, 10% of uniaxial lateral compression was reported to induce ectopic expression of Twist around the entire dorsal-ventral axis of Drosophila melanogaster, resulting in ventralisation of the embryo [2]. Furthermore, mechanical compression forces exerted in early Xenopus laevis embryos provoke phosphorylation of components of focal adhesions and tight junctions, resulting in tissue strengthening [3]. Additionally, compressive forces increase the abundance of mesenchymal-to-epithelial transition (MET) proteins, while suppressing proteins involved in EMT, resulting in MET-like phenotype [3].
Apart from externally applied stimuli, compressive forces may arise from overcrowding of epithelial tissues due to cell proliferation [4, 5]. This tissue-intrinsic compression provides information to maintain cell density through several mechanisms. For example, overcrowding can halt cell cycle progression at the G1-S checkpoint [6], though this does not, by itself, correct the elevated cell density. Instead, such compressive forces initiate the process of live-cell extrusion, where surplus cells are physically expelled from the epithelium to moderate cell density [4].
Compressive forces exerted on epithelia can also be a consequence of tensile forces, often occurring tangential to the plane of tension [7]. To simulate the morphological heterogeneity of epithelial tissue in vivo, Gjorevski and Nelson [7] engineered epithelial tissues with various geometric features. They found that with curved, duct-like geometries, regions of compression occurred both tangentially to tensile forces and within the concave regions where the opposing convex region was under tension. Interestingly, these negative forces appeared to be passive and dependent on active generation of cellular tension, as regions of compression did not show elevated cell proliferation, and blocking myosin activity curtailed both tensile and compressive forces within the tissue.
Tensile Forces. These forces have been far more extensively studied compared to compressive forces. Though tensile forces are often considered as extrinsic to tissues, such as pulmonary inflation or physiologic gastric distension, intrinsic tensile forces are also significant and contribute to homeostasis. These intrinsic forces arise from the contraction of the actomyosin cytoskeleton. In the case of epithelial and endothelial cells, force generation by actomyosin contraction requires a physical connection between the contractile machinery and the cell-cell junctions. This enables transmission of tension between adjacent cells and thus facilitates propagation of the mechanical signal across epithelial tissues. Components of both adherens and tight junction complexes have been shown to experience and respond to changes in cortex-derived tension. Key examples of such proteins include adherens junction (AJ)-based α-catenin and tight junction (TJ)-based ZO-1, both of which were shown to sense increased tension and undergo a conformational change in response to cortical tension [8,9,10,11,12,13,14]. Detailed mechanisms of force-sensing across junctions will be discussed later.
A number of studies have revealed the importance of intrinsic forces in the maintenance of epithelial homeostasis and tissue development. For example, Acharya et al. [15] demonstrated that the formin mammalian diaphanous 1 (mDia1) is crucial for contractility at the zonula adherens and stability of tight junctions in epithelial monolayers. Interestingly, as monolayers depleted of mDia1 exhibited increased transepithelial permeability, mDia1-dependent intrinsic tension appeared to be essential for the maintenance of epithelial barrier function. Further, mechanical tension may balance cell proliferation patterns, leading to uniform levels of local cell division, despite inhomogeneous distribution of growth factors across the developing tissues. Using a developing Drosophila melanogaster wing as a model for a homogenously growing tissue, Pan et al. [16] demonstrated that local hyperproliferation is accompanied by reduction in junctional tension. This results in increased activity of the Hippo signalling pathway and downregulation of the growth-promoting Yorkie, thus leading to a decrease in local growth rates. These observations, supported by computational simulations, suggested existence of a mechanical feedback mechanism, where the gradual reduction in intrinsic tension increases Hippo pathway activity to mediate evenly distributed tissue growth [16]. Seminal work by Dupont et al. [17] show that the localisation of the Yes-associated protein (YAP) can be influenced by substrate stiffness. Further, as Irvine and Harvey [18] have reviewed, mechanical signals emanating from cell-cell interactions can modulate the Hippo pathway to control organ growth.
Mechanical Forces as Cellular Information
Importantly, mechanical forces can be transmitted and detected by cells as a mode of communication. This can potentially be elicited in response to both compressive and tensile forces, although here we will focus on mechanotransduction of tensile forces in epithelia. The cellular mechanisms that support communication by mechanical forces involve mechanisms that transmit forces (within and between cells) and mechanisms that sense changes in mechanical forces. Broadly, examples of mechanosensors include proteins which undergo conformational changes in response to tension, ion channels which can be forced open and plasma membrane invaginations which unfold with increased tension. These mechanosensitive responses may elicit recruitment of effector proteins, induction of ion influx/efflux, or induction of gene transcription/silencing due to translocation of proteins to the nucleus. In this chapter, we will discuss a range of various mechanisms that enable cells to respond to mechanical cues, focusing on both junction-based and membrane-based mechanosensitivity, specifically with respect to epithelial tissues.
Mechanosensing at Cell-Cell Junctions
Epithelial cells are joined together by specialised cell-cell junctions: tight junctions (TJs) found at the most apical region of the cell-cell interface; E-cadherin-based AJs, located under TJs; as well as more basally located desmosomes and gap junctions. Cell-cell junctions are fundamental for the development and maintenance of tissues and have a variety of functions. AJs initiate cell-cell contacts and are primary sites of mechanical force sensing; TJs regulate transepithelial permeability; desmosomes provide epithelial tissues with mechanical resistance; and gap junctions permit chemical communication between adjacent cells. Here, we will discuss selected developments in the knowledge of mechanosensing at cell-cell junctions.
AJ-Based Mechanosensing
AJs are characterised by their enrichment of classical cadherins, which can reach concentrations of ~2000 molecules/μm2 at these cell contacts [19]. Cadherins traverse the plasma membranes (PM) of neighbouring cells, ligating in a homophilic fashion to bridge across the intercellular space (Fig. 1). Like other classical cadherins, epithelial (E)-cadherin 1 (cadherin 1) comprises three distinct regions: an N-terminal extracellular region, a single transmembrane region and a C-terminal intracellular region. The extracellular region of E-cadherin facilitates the adhesion of adjacent cells, a function reliant on its five extracellular cadherin domains (EC1–5) interspaced by Ca2+-binding motifs. Here, the β-barrel structure of distal EC1 domains from adjacent cells lies antiparallel, exchanging β-strands in trans for the primary adhesion site in a Ca2+-dependent manner. Interestingly, the homophilic nature of cadherin ectodomain subtypes can allow selective adhesion between cell types providing a powerful, though not ubiquitous, means of directing morphogenesis [20, 21]. However, such cell sorting can also occur in cell populations expressing quantitative differences of the same cadherin [22]. It has been suggested that the loss of E-cadherin increases the metastatic potential of tumourigenic cells [23, 24] as loss of E-cadherin correlates with increased migration in vitro [25]. However, it has recently become evident that the role of E-cadherin in cancer is much more complicated than previously appreciated. For example, most patients with invasive ductal carcinomas express E-cadherin in both the primary tumour and metastases. Further, Padamanaban et al. [26] show that the loss of E-cadherin, while increasing invasiveness, decreases metastatic potential, as well as cell proliferation and survival of circulating tumour cells.

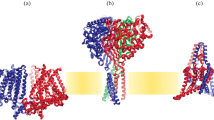
Adherens junctions (AJs). Schematic representations of the main components of the adherens junction. Cell adhesion is mediated by the EC1 domains of the E-cadherin extracellular regions. Under normal physiological conditions (resting state), the mechanosensitive protein, α-catenin, is in a closed conformation, with a weak affinity for F-actin. Under periods of high tension (>5 pN, stressed state), α-catenin unfolds to expose an F-actin-binding domain, greatly increasing its affinity for F-actin, strengthening the cell’s cortex. Additionally, the unfolding of α-catenin exposes a vinculin-binding domain, providing further reinforcement to the cortex by recruiting and binding vinculin
AJ Mechanosensing Through α-Catenin and Vinculin. AJs are subjected to constant mechanical stress from both extrinsic and intrinsic sources. Accordingly, AJs have evolved to cope with, and become sensitive to, these stresses. As tensile forces are generated within cells and tissues, E-cadherin molecules work as mechanotransducers to bear and transmit forces to the underlying molecular complex (Fig. 1). In order to propagate tissue tension, E-cadherin adhesions need to be physically coupled to the contractile actomyosin machinery of the cells. Indeed, the intracellular region of E-cadherin binds p120-catenin and β-catenin, with β-catenin associating with the actin-binding protein (ABP) α-catenin, thus bridging the adhesive and force-generating components of the AJs [27]. Importantly, the role of α-catenin is not limited to constituting a passive link between E-cadherin and actin. Instead, molecular properties of α-catenin are altered upon increased tension, making it an active, key AJ-based mechanosensor.
The actin-binding and actin-bundling protein α-catenin contains two distinct mechanosensitive domains for interacting with vinculin and F-actin (Fig. 1). Application of mechanical tension to α-catenin can alter its conformation to expose a cryptic vinculin-binding domain (VBD) and promote vinculin recruitment and binding [8, 10, 11, 13, 14]. The association of vinculin with F-actin strengthens the interactions between cadherin-catenin complexes and the cytoskeleton. During homeostasis, intrinsic tissue tension due to actomyosin results in ~5 pN of force across each E-cadherin molecule [1]; approximately the same level of force is required to unfold α-catenin for recruitment and binding of vinculin [13]. Additionally, the actin-binding domain of α-catenin can also undergo a tension-sensitive conformational change. Here, mechanically induced unfolding of the α1 helix of α-catenin enhances the binding affinity between α-catenin and F-actin (Fig. 1). Under periods of high tension, the ratio of open to closed α-catenin molecules is increased, providing significant reinforcement to the AJ, largely due to this increased association with cortical F-actin. Of note, vinculin and F-actin are not the only binding partners of α-catenin. Other partners include α-actinin [28, 29], afadin [11], EPLIN [30] and ajuba [31, 32]; however their specific contributions to α-catenin-based mechanosensitivity has yet to be well-defined.
In addition to α-catenin, vinculin is itself an important mechanosensitive protein. Cytoplasmic, inactive vinculin exists in an autoinhibited conformation, with the tail domain interacting with the head domain. Abl-dependent phosphorylation at the Tyr822 residue and/or actomyosin contractility, combined with association with α-catenin, leads to unfolding of vinculin [33, 34]. In this active, unfolded state, vinculin strengthens the AJs by binding directly to both α-catenin and β-catenin via the vinculin-head domain (Vh) [35, 36]. Furthermore, vinculin directly associates with F-actin and the actin regulatory protein MENA, an interaction essential for vinculin-dependent F-actin polymerisation in epithelial tissue under mechanical stress [37]. Vinculin at AJs has been shown to potentiate the mechanosensing response of the E-cadherin-catenin complex. le Duc et al. [38] showed that in vinculin KO cells, cell attachment to E-cadherin-coated coverslips was impaired due to compromised cell spreading. This study also demonstrated that both mechanical force-dependent cell stiffening and reinforcement of adherens junctions were compromised in vinculin KO cells when compared to vinculin-positive cells. Furthermore, vinculin KO cells display perturbed HGF-mediated recruitment of phosphorylated myosin light chain II (pMLCII), suggesting vinculin may play an important role in the recruitment of NMII to AJs following mechanical force [38].
AJ Mechanosensing Through Myosin VI. Recently, the F-actin-binding motor protein Myosin VI was identified as another mechanosensitive protein associated with AJs [39]. Intriguingly, the function of Myosin VI is dependent on the force it is experiencing. Under loads of ~ <2 pN, Myosin VI behaves as a typical motor protein, but under loads exceeding ~2.5 pN, Myosin VI briefly reverses direction along the actin filament and behaves as an anchor [40]. Under mechanical stress in epithelial monolayers, the association between E-cadherin and Myosin VI is rapidly increased [41]. Next, the heterotrimeric Gα12 protein is recruited to the E-cadherin-Myosin VI complex where it activates p114 RhoGEF to enhance RhoA activation, thus reinforcing the cell-cell contact through increasing the pool of cortical actomyosin. However, signalling through Myosin VI also increases epithelial integrity and protects against rupture by increasing the tensile strength of junctions: this is achieved via mDia1-dependent stabilisation of E-cadherin at multicellular vertices [41].
TJ-Based Mechanosensing
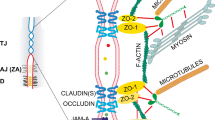
While much work has focused on how adherens junctions (AJs) can influence tissue mechanics and mediate mechanotransduction, recent studies have revealed that tight junctions (TJs) can also represent parallel sites of mechanotransduction at cell junctions. TJs are canonically responsible for establishment of a paracellular semipermeable diffusion barrier that limits free passage of ions and solutes through epithelial and endothelial layers [42, 43]. Tight junctions are composed of at least 40 different proteins, including (1) transmembrane proteins (e.g. claudins, occludin and JAM-A), (2) cytoplasmic proteins (e.g. ZO, cingulin) and (3) cytoskeletal filaments (actin, myosin, microtubules) [44] (Fig. 2a).

Tight junctions (TJs). (A) Schematic representation of the basic components of tight junctions. Extracellular part of the TJ transmembrane proteins (occludin, claudins and JAMs) interact at cell-cell contacts existing between the plasma membranes of two neighbouring cells. Cytoplasmic tails of the transmembrane components of TJs connect to actin filaments via scaffolding proteins (ZO proteins, cingulin and afadin). (B) Schematic representation of ZO-1 structure and its interacting partners. (C) ZO-1 conformational change: in the absence of binding partners and/or under low tension, ZO-1 exists in an autoinhibited conformation, preventing its interaction with occludin; upon interaction with a binding partner and/or under high tension, ZO-1 transits to a stretched (active) conformation permitting interactions with occludin and actin
TJs Regulate Actomyosin Cytoskeleton. Even though TJs and AJs are present in both epithelial and endothelial cells, epithelial junctions are well-defined – TJs are located more apically than AJs. However, in less polarised endothelial cells, AJs and TJs are intermingled, and their spatial separation is less obvious [45, 46]. This different junctional organisation between the cell types may reflect different interplay between tight and adherens junctions. Indeed, downregulation of ZO-1 in epithelial cells results in increased tension on AJs [47,48,49,50], suggesting that ZO-1 exerts an inhibitory effect on junctional tension. By contrast, ZO-1 depletion in endothelial cells leads to decreased tension on VE-cadherin [51]. Despite this discrepancy, ZO-1 does not affect cadherin localisation/expression in either epithelial or endothelial cells, indicating that changes in tension on cadherins originate from changes in the actomyosin network. Indeed, components of TJs, including ZO proteins, regulate cells’ contractile cytoskeleton in various ways. ZO proteins may influence cell contractility by their interactions with actomyosin-associated proteins – α-catenin [52, 53], vinculin [54], shroom2 [55] or cortactin [56]. Furthermore, TJ components regulate multiple aspects of actomyosin by interacting with Rho family GTPases, thus carrying the capacity to support the activation or inactivation of major regulators of actomyosin – primarily RhoA, Rac1 and Cdc42. Among others, ZO-1 interacts with PDZ-RhoGEF [57] and Cdc42 GEF Tuba [58], and cingulin binds p114RhoGEF, GEF-H1 as well as MgcRacGAP [59,60,61]. Taken together, one way that TJs may influence cell mechanotransduction is by regulating actomyosin contractility, thus modulating AJs and E-cadherin-based mechanotransduction.
Evidence of Mechanotransduction on TJs. As discussed above, for the transduction of mechanical force between cells to mediate cell-cell communication, there must be adhesive junctional proteins which can transmit forces to mechanosensitive proteins. Further, propagation of the intrinsic forces across a tissue requires coupling of junctional mechanotransmitters to the cell contractile cytoskeleton [62, 63]. There are many potential ways for molecular elements of TJs to connect to the contractile apparatus of cells. The N-termini of ZO proteins interact with claudins, occludin and JAM-A, whereas the C-terminal region of ZO-1 and ZO-2 contains an actin-binding region (ABR) [64,65,66]; afadin links JAM-A and nectins to the actin cytoskeleton [67, 68], and the globular head domain of cingulin binds to ZO-1 and actin, while its coiled-coil region binds to myosin [69, 70] (Fig. 2a). Of note, among the transmembrane TJ proteins, JAM-A has emerged as a potential mechanotransducer. Using a magnetic tweezers approach, Scott et al. showed that, in single epithelial and endothelial cells, tension imposed on non-junctional JAM-A activates RhoA via GEF-H1 and p115 RhoGEF to increase cell stiffness [71]. There is no evidence, however, that JAM-A works as a mechanotransducer in confluent, junction-forming monolayers. Instead, the search for TJ-based proteins that may participate in mechanotransduction has focused on ZO-1 as a force-bearing protein [9] whose molecular properties may be altered upon increased tension.
ZO-1 as a Mechanosensor. The molecular structure of ZO-1 consists of an N-terminal region containing three PSD-95/DLG/ZO-1 (PDZ) domains (PDZ1–3) that can bind to claudins (PDZ1), ZO-2 or ZO-3 (PDZ2) and JAM (PDZ3). These are followed by Src homology-3 (SH3), unique-5 (U5), occludin-binding guanylate kinase (GUK) and unique-6 (U6) domains. The C-terminal region of ZO-1 consists of actin-binding region (ABR) and ZU5 domain [12, 66, 72] (Fig. 2b). Importantly, Spadaro et al. have recently demonstrated that ZO-1 can exist in either an autoinhibited (folded) or stretched (unfolded) conformation [12]. ZO-1 assumes a folded conformation as a result of intramolecular interactions between the C-terminal ZU5 domain and ZO-1 central region (from PDZ3 to GUK domain). Unfolding of ZO-1 has been attributed to tension generated by actomyosin contractility, as magnetic tweezers experiments showed that physiological pN-scale tension is sufficient to unfold ZO-1 and maintain the ‘active’ stretched conformation. However, the stretched conformation can also be maintained by heterodimerisation with ZO-2: folding of ZO-1 could only be achieved when inhibition of the contractile network is combined with depletion of ZO-2 [12] (Fig. 2c).
Importantly, this tension-induced change in ZO-1 conformation can affect its intermolecular interactions. The folded, autoinhibited conformation of ZO-1 prevents the GUK domain from binding and recruiting occludin to TJs. It also blocks association of the central domain of ZO-1 with the transcription factor DbpA (Fig. 2c). Thus, tension-induced unfolding of ZO-1 may modulate its molecular interactions and downstream signalling [12]. It remains to be established whether ZO-1 conformational change influences its interactions with other binding partners including α-catenin, afadin and vinculin.
Recent evidence suggests that ZO-1 and many other TJ cytosolic and membrane proteins undergo cytosolic phase separation prior to arrival at the junctional membrane [73, 74]. During early zebrafish development, components of TJs accumulate at the boundary between the yolk syncytial layer (YSL) and the enveloping cell layer (EVL) . Interestingly, accumulation of TJ proteins depends on the actomyosin tension within the YSL. Here, cytoplasmic, phase-separated ZO-1 is transported towards junctions by tension-dependent retrograde actomyosin flow, indicating existence of yet another mechanosensitive TJ-based event. Stinkingly, non-junctional ZO-1 must be unfolded and undergo multimerisation to allow phase separation to drive formation of TJ junctions. However, the ABR domain is not required for phase separation, suggesting that cytosolic unfolding of ZO-1 is not induced by tension, but rather by association of another molecule. As a consequence, conformational changes of cytosolic ZO-1 may not be powered by the actomyosin-generated tension, as phase-separated ZO-1 seems to arrive at the junction in already opened ‘active’ conformation. Nevertheless, while the ABR domain of ZO-1 may be dispensable for ZO-1 phase separation, it is essential to efficiently integrate ZO-1 into junctions and confer mechanosensitivity upon TJs [74]. What then can facilitate cytoplasmic unfolding of ZO-1? Among many TJ proteins, the ZO-1 binding partner, cingulin, may be present in the highly concentrated phase-separated compartments. Cingulin supports efficient accumulation of ZO-1 at TJs, and its interaction with ZO-1’s ZU5 domain may be sufficient to unfold ZO-1. Recent work suggests that ZO-1 exists in a ‘double-folded’ conformation with its ZU5 domain interacting with the central region of ZO-1 (as described above), as well as with the ABR domain. Binding of ZU5 to the ABR domain is predicted to prevent ZO-1 from forming efficient interactions with F-actin. Binding of cingulin to ZU5 promotes ZO-1 unfolding and may disrupt the interactions between ABR and ZU5, therefore allowing actin binding [75].
Overall, ZO-1 emerges as a possible novel TJ-based mechanosensor; however, further studies will be needed to fully understand the interplay between tension and ZO-1 binding partners in regard to regulation of ZO-1 conformation changes and physiological consequences of these events.
Plasma Membrane-Based Mechanosensing
While we have focused on understanding how cell-cell junctions participate in epithelial mechanobiology, it is important to note that these are not the only components of epithelial cells that display mechanosensitivity. In particular, the plasma membrane (PM) forms the physical boundaries of cells and, thus, is in constant contact with both the external and internal cell environments. As such, the PM constitutes a crucial interface that mediates responses to mechanical stimuli such as external touch, changes in cell curvature or internal osmotic pressure. Here, we will focus on two well-characterised examples of PM-based force-sensing, mechanosensitive ion channels and caveolae membrane invaginations, that have recently begun to be linked to cell-cell interactions.
Piezo1. Unlike other ion channels, which are typically activated/inactivated by interactions with specific ligands or voltage gating, mechanosensitive (MS) ion channels are opened in response to the application of mechanical forces (Fig. 3). MS ion channels are key sensors of mechanical stimuli across a diverse range of living organisms [76] and are some of the fastest signal transducers in cells, translating mechanical information into intracellular signals in the order of milliseconds [77, 78]. Two prevailing mechanisms for such mechanical gating exist. Firstly, the ‘force-from-lipids’ model, where MS ion channels are opened (activated) by forces from the plasma membrane [79,80,81,82,83] (Fig. 3). Such forces can occur from extrinsic means, such as touch or intrinsically such as osmotic pressure or changes in bilayer curvature induced by factors like local lipid composition [84] or expression of curvature-generating molecules, such as BAR-domain proteins [85]. Secondly, the ‘force-from-filaments’ model (also called the tethered model) posits that MS ion channels can be activated by pulling forces from extracellular and cytoskeletal filaments attached to the channel [86,87,88].

Mechanosensitive (MS) ion channels. Piezo1 is the most characterised of the MS ion channels. MS ion channels are opened solely by means of mechanical force, rather than by ligand binding or voltage gating. The activation (opening) of MS ion channels occurs via force-from-filaments (left) and force-from-lipids (right). The former occurs by filament-mediated pulling forces from the extracellular matrix and/or the cytoplasm. The latter occurs during PM deformation, such as occurs through touch, osmotic pressure or curvature via BAR-domain proteins. Opening of Piezo1 channels allows influx/efflux of ions for modulation of processes such as live-cell extrusion and cell proliferation
Here we focus on the MS ion channel Piezo1, which influences a range of epithelial tissue phenomena, such as extrusion and control of population dynamics, that also involve cell-cell junctions. Moreover, Wang et al. [89] reported that Piezo1 can be coupled to E-cadherin, tethering the channels to the actin cytoskeleton via the cadherin-catenin complex, while others have demonstrated that Piezo channels help regulate RhoA signalling and the actin cytoskeleton and that activation of Piezo1 is dependent on integrin-related signalling [90,91,92]. It should be noted that, although Piezo1 channel is the best characterised example of a MS ion channel, there are other mammalian representants of this group, such as the Piezo isoform, Piezo2 as well as the TREK family of neuronal potassium channels, TREK-1, TREK-2 and TRAAK [93].
Piezo channels are expressed as two isoforms in mammals (Piezo1 and Piezo2), with both being highly expressed in organs where mechanosensation is functionally significant, such as the bladder, colon, kidney, lung, skin and dorsal root ganglia (Piezo2 only) [94]. Piezo channels are the principal means of sensing mechanical stimuli such as touch [94] and vascular blood flow [95]. The Piezo1 channel is a unique protein and does not bear a structural homology to any other channel [96]. Piezo1 has a trimeric, three-bladed propeller shape, which can adopt several conformations, and houses a kinked helical beam and an anchor domain. Each of the three blades contains nine repeat regions, each of which comprises four transmembrane domains, collectively passing the plasma membrane 108 times [97, 98]. This structure was shown to be fundamental in the mechanogating of the Piezo1 channel [99]. Molecular dynamics simulations suggest that increased membrane tension results in flattening and in-plane expansion of the blades, resulting in tilting of helices 37 and 38, which are then pulled away from the channel pore, leading to opening of the Piezo1 channel [100].
Piezo channels have been implicated in a number of physiological processes that also involve cell-cell interactions. Piezo1 is a fundamental ion channel required for live-cell extrusion [4], a process vital for homeostatic control of cell density in epithelia. As noted earlier, a pivotal study by Eisenhoffer et al. [4] demonstrated that live-cell extrusion is triggered by compressive forces due to cell overcrowding. By growing cells to confluence on a silicone membrane stretched to 28% of its original length and then allowing the substrate to recoil, cell density was increased by ~30%, generating compressive forces across the tissue. However, within 6 h the cell density was reduced to homeostatic levels, indicating that cells had been eliminated from the monolayer. Indeed, it was apparent that the tissue was extruding living cells. This live-cell extrusion was mediated by Piezo1, as either inhibition of the channel with gadolinium (Gd3+) or morpholino-mediated knockdown of Piezo1 in zebrafish embryos limited live-cell extrusion and resulted in cell mass formation in the epithelial tissue at sites of high strain.
Further, the Piezo1 channel may promote cell proliferation when cell density is too low and cells are under positive tension [94, 101, 102]. By growing cells to confluence on a flexible substrate and applying a ~ 1.4-fold stretch, Gudipaty et al. [102] demonstrated that the rate of mitosis was increased by approximately fivefold within just 1 h. Under these conditions, treatment with Gd3+ or siRNA knockdown of Piezo1 efficiently inhibited stretch-induced proliferation, demonstrating that Piezo1 was at the apex of this phenomenon. These results were also confirmed in vivo in the zebrafish epidermis where both CRISPR-based mosaic knockout and morpholino-mediated knockdown of Piezo1 significantly decreased the number of mitotic cells. Ultimately, the activation of Piezo1 engages intracellular Ca2+ signalling to activate the ERK1/2-MEK1/2 pathway and promote the transcription of cyclin B for increased mitosis.
How tensile or compressive forces can elicit either live-cell extrusion or promote cell proliferation through the same Piezo1 channel is a significant question. Gudipaty et al. [102] suggested that the subcellular localisation of Piezo1 may be responsible. At subconfluency, Piezo1 is mainly confined to the nuclear envelope; as confluency is reached but cell density is still low, Piezo1 localises to the endoplasmic reticulum and PM. As cell density continues to increase post-confluency, Piezo1 begins to form large cytoplasmic aggregates. These changes in the subcellular localisation may reflect the adaptive mechanosensitivity of Piezo1, where its presence at the PM is optimal to sense tensile forces and its presence within the cytoplasm is optimal to sense compressive forces.
Caveolae-Based Mechanosensing. Caveolae are 60–80-nm-wide Ω-shaped invaginations of the PM [103, 104] (Fig. 4). They are enriched in cholesterol and sphingolipids, forming specialised lipid rafts that are implicated in numerous biological functions, such as endocytosis, lipid metabolism and mechanosensing. Caveolae may encompass up to 50% of the total PM surface area in cells such as myocytes [105] and their formation and stability largely dependent on two protein families, the caveolins (Cavs) and the cavins.

Caveolae as mechanosensors. Caveolae, small invaginations of the PM, are sensitive to acute increases in membrane tension from both extrinsic and intrinsic stimuli. Under normal physiologic conditions, caveolae exist as bulb-shaped pits, stabilised by a coat of caveolin and cavin proteins (left). Under periods of high tension, caveolae flatten, providing a passive means of protection by releasing a membrane reservoir, thus rapidly increasing the surface area of the cell. When caveolae flatten, cavin proteins are released into the cytosol, leaving the caveolins at the PM. Caveolae flattening also alters the cellular lipid composition, enhancing signalling through the RhoA-ROCK pathway for F-actin modulation and increasing MAPK activity
Caveolae have several characteristics which suit them well for mechanosensing. Their bulb-like morphology constitutes a membrane reservoir which is sensitive to mechanical stimuli and can be flattened by membrane stretch (Fig. 4); they are particularly abundant in tissues which experience significant mechanical forces; they are directly linked to the actin cytoskeleton and are distributed at sites of cell contact [106,107,108,109]. Indeed, caveolae have been observed to concentrate at adherens junctions [110]. The mechanical tolerance of the lipid bilayer has been measured at 4–6% areal strain before rupture [111]; thus, the ability of caveolae to passively modulate tension likely provides a significant advantage to cells and tissues affected by intrinsic or extrinsic mechanical forces. In complex living systems, cells must tolerate a range of potentially damaging mechanical stimuli, such as alveolar inflation, muscle stretching, vascular shear stress and volume expansion.
In practice, the ability of caveolae to flatten under mechanical tension has been demonstrated in both isolated cells and multicellular tissues. For example, caveolae were shown to flatten in individual HeLa and mouse lung endothelial cells (MLECs) in response to hypo-osmotic shock [112], as well as primary mouse cardiomyocytes [113]. This response by caveolae to increased membrane tension has also been demonstrated in multicellular tissues both ex vivo and in vivo. A seminal paper by Dulhunty and Franzini-Armstrong [114] showed that the abundance of caveolae on the surface of frog skeletal muscle was greatly reduced following mechanical stretch and elongation past this point resulted in rupture of the tissue. Soon after, caveolae flattening in response to mechanical tension was demonstrated in the smooth muscle [115].
More recent studies have demonstrated the mechanoprotective role of caveolae in vivo. Cheng et al. [116] used dobutamine, a β-1 adrenoreceptor agonist, to increase heart contractility and cardiac output in mice. They observed caveolar disassembly in both heart and lung tissues. Importantly, endothelial cells showed a significant damage in Cav-1 KO mice, whereas no damage was observed in WT control mice [116]. Others have shown that the notochord, which comprises cells particularly abundant in caveolae, becomes unstable when caveolae are depleted by cavin-1b KO [117, 118]. Here, the notochord of cavin-1b KO zebrafish shows a significant cellular damage, cell necrosis and the appearance of lesions. Interestingly, damage to the notochord was increased when mechanical stress was applied to the notochord during swimming, further demonstrating the importance of caveolae as mechanoprotective.
In some tissues the ability of caveolae to attenuate membrane tension is facilitated by the formation of multi-lobed, rosette-like caveolar superstructures [105]. These superstructures are composed of multiple caveolae which have fused due to membrane curvature and tension [119, 120], are more sensitive to tension, disassemble more rapidly and release a far greater pool of PM upon flattening than individual caveolae [105, 121]. This hypothesis is supported by computational modelling and experimental data which demonstrate that caveolar rosette formation is promoted by low tension [119]. Further, cells can actively respond to force by altering the abundance or properties of caveolae. For example, caveola numbers are increased by almost 50% in bovine aortic endothelial cells (BAECs) experiencing chronic shear stress [122], highlighting the importance of caveolae in protecting tissues from physical damage. Mechanical stress applied to vascular endothelial tissue, such as is experienced during hypertension, dampens the eNOS-binding capacity of Cav-1, promoting vasodilation to attenuate elevated shear stress [123]. It has also been suggested that the mechanosensitive properties of caveolae regulate cell volume, as ectopic Cav-1 expression in cells lacking caveolae can promote caveolar biogenesis, allowing cells to swell to a greater extent following hypo-osmotic exposure [124].
Interestingly, caveolae flattening also alters the microenvironment of these structures beyond gross morphology. Under acute membrane tension, caveolae flattening results in the dissociation of the cavin protein complex [112] and the EHD2 ATPase [125] but not the caveolin proteins [112], from caveolae. This suggests that caveolae may actively respond to mechanical stress using dissociated proteins as signalling intermediates. Indeed, recent studies have demonstrated that release of EHD2 upon caveolae flattening suppressed transcription of caveolins 1 and 2 and cavins 1 and 2, which are required for caveolar biogenesis [126]. Furthermore, the mechanosensitivity of caveolae, specifically the Cav-1 and cavin-4 proteins, influences the activity of RhoA [127]. Caveolae flattening promotes the phosphorylation of Cav-1 at tyrosine-14 (Y14) [128, 129]. pY14 phosphorylation of Cav-1 can enhance Cav-1-RhoA interaction to directly activate RhoA [127, 130] and negatively regulate the Src-p190RhoGAP pathway [131], suggesting that mechanotransduction via caveolae could influence RhoA for remodelling of the cytoskeleton. Furthermore, low tension at the rear of migrating cells promotes caveolar formation, activating RhoA-ROCK1-PKN2 signalling via Ect2 [132]. This drives local F-actin organisation and contractility, allowing the cell’s posterior region to complete the migration cycle.
Caveolae flattening can also influence other aspects of membrane organisation with consequences for mechanoregulatory mechanisms . Ariotti et al. [133] demonstrate that perturbation of caveolar biogenesis by either Cav-1 or cavin-1 KD alters the cellular lipid composition, particularly the distribution of phosphatidylserine at the PM. This resulted in enhanced K-Ras expression and organisation and increased MAPK activity. Interestingly, these phenomena were found to be due to the loss of intact caveolae, as the acute dissociation of cavin-1 by hypo-osmotic shock mimicked these findings. Thus, this study implies that the mechanosensitive nature of caveolae may allow cells to rapidly alter the lipid composition of the PM in response to mechanical stress.
More recently, caveolae were found to modulate mechanical tension at the tissue level. Teo et al. [110] demonstrated that caveolae modulate tissue tension within epithelial monolayers. Here, caveolae were found to be at the apex of a novel signalling pathway, where intact caveolae suppress the availability of phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) at the PM. By inhibiting caveolar biogenesis, PtdIns(4,5)P2 levels are enhanced, directly increasing cortical recruitment of the formin-like protein, FMNL2. In turn, enhanced FMNL2 increased the pool and organisation of cortical F-actin, resulting in elevated tension within the tissue. A significant ramification of this dysregulation was perturbation of oncogenic cell extrusion, resulting in the formation of large cell masses when a minority of oncogenic cells were incorporated into an otherwise healthy epithelial monolayer.
Intriguingly, recent studies show that the mechanosensitivity of caveolae and the Hippo-YAP/TAZ signalling pathway influence each other. The Hippo pathway, a major regulator or cell proliferation, migration and survival [134], is sensitive to mechanical stimuli such as tension [135], cell density [136] and stiffness of the extracellular matrix (ECM) [17], all of which affects the localisation and activity of YAP/TAZ. In 2018, Moreno-Vincente et al. [137] revealed that the mechanoresponsiveness of YAP to ECM stiffness was positively modulated by Cav-1, whose transcription, as well as that of cavin-1, is also dependent on the presence of YAP/TAZ [138]. Furthermore, caveolae are protective against mechanical stresses which arise from haemodynamic force [116, 139], a process which activates YAP/TAZ signalling [140, 141]. It has been suggested that interplay between caveolae and the Hippo pathway has significance in the pathogenesis of diseases such as atherosclerosis and vascular malformations, both of which result from dysfunctional endothelial shear stress sensing [142].
Conclusion
The past decade has witnessed exciting, rapid progress in understanding how cell-cell junctions contribute to the mechanobiology of epithelia. We now appreciate that epithelia are mechanically active tissues, which themselves generate forces and respond to forces generated extrinsically, and such changes in force can indicate events that challenge tissue integrity or homeostasis. In closing, we would identify a number of directions that we think warrant attention in the future. First is integrating molecular mechanisms into systems. We have begun to identify the molecular mechanisms that allow epithelia to detect changes in force and elicit compensatory homeostatic responses. Many more molecular details will inevitably be revealed in the next few years. As this molecular picture grows in richness and specificity, it will be important to consider how they may be integrated by feedback into functional networks rather than individual, linear pathways. For example, the contractile protein non-muscle Myosin II, which is activated by RhoA signalling, can also participate in mechanochemical feedback to support RhoA [143, 144]. Insights from statistical physics and mathematical modelling provide valuable resources to tackle such systems analysis. Second, can the homeostatic mechanisms of epithelial mechanotransduction be disrupted in disease? Clues include the observation that inflammatory cytokines such as TNF-α can increase mechanical tension at cell-cell junctions, including TJ [145]. Also, depletion of caveolae, which has been implicated in cancer, increases epithelial tension to compromise the elimination of transformed cells [110]. Therefore, elucidating the mechanobiology of junctions may provide many new insights into the biology and pathobiology of epithelia.
References
Charras, G. and Yap, A. S. (2018) ‘Tensile Forces and Mechanotransduction at Cell–Cell Junctions’, Current Biology, 28(8), pp. R445–R457. https://doi.org/10.1016/j.cub.2018.02.003.
Farge, E. (2003) ‘Mechanical Induction of Twist in the Drosophila Foregut/Stomodeal Primordium’, Current Biology, 13(16), pp. 1365–1377. https://doi.org/10.1016/S0960-9822(03)00576-1.
Hashimoto, Y. et al. (2019) ‘Mechanical Force Induces Phosphorylation-Mediated Signaling that Underlies Tissue Response and Robustness in Xenopus Embryos’, Cell Systems, 8(3), pp. 226-241.e7. https://doi.org/10.1016/j.cels.2019.01.006.
Eisenhoffer, G. T. et al. (2012) ‘Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia’, Nature, 484(7395), pp. 546–549. https://doi.org/10.1038/nature10999.
Marinari, E. et al. (2012) ‘Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding.’, Nature, 484(7395), pp. 542–5. https://doi.org/10.1038/nature10984.
Streichan, S. J. et al. (2014) ‘Spatial constraints control cell proliferation in tissues’, Proceedings of the National Academy of Sciences, 111(15), pp. 5586–5591. https://doi.org/10.1073/pnas.1323016111.
Gjorevski, N. and Nelson, C. M. (2012) ‘Mapping of mechanical strains and stresses around quiescent engineered three-dimensional epithelial tissues.’, Biophysical journal, 103(1), pp. 152–62. https://doi.org/10.1016/j.bpj.2012.05.048.
Choi, H.-J. et al. (2012) ‘E-catenin is an autoinhibited molecule that coactivates vinculin’, Proceedings of the National Academy of Sciences, 109(22), pp. 8576–8581. https://doi.org/10.1073/pnas.1203906109.
Haas, A. J. et al. (2020) ‘Interplay between Extracellular Matrix Stiffness and JAM-A Regulates Mechanical Load on ZO-1 and Tight Junction Assembly.’, Cell reports, 32(3), p. 107924. https://doi.org/10.1016/j.celrep.2020.107924.
Huveneers, S. et al. (2012) ‘Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling.’, The Journal of cell biology, 196(5), pp. 641–52. https://doi.org/10.1083/jcb.201108120.
Pokutta, S. et al. (2002) ‘Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin.’, The Journal of biological chemistry, 277(21), pp. 18868–74. https://doi.org/10.1074/jbc.M201463200.
Spadaro, D. et al. (2017) ‘Tension-Dependent Stretching Activates ZO-1 to Control the Junctional Localization of Its Interactors.’, Current biology: CB, 27(24), pp. 3783-3795.e8. https://doi.org/10.1016/j.cub.2017.11.014.
Yao, M. et al. (2014) ‘Force-dependent conformational switch of α-catenin controls vinculin binding.’, Nature communications, 5, p. 4525. https://doi.org/10.1038/ncomms5525.
Yonemura, S. et al. (2010) ‘alpha-Catenin as a tension transducer that induces adherens junction development.’, Nature cell biology, 12(6), pp. 533–42. https://doi.org/10.1038/ncb2055.
Acharya, B. R. et al. (2017) ‘Mammalian Diaphanous 1 Mediates a Pathway for E-cadherin to Stabilize Epithelial Barriers through Junctional Contractility’, Cell Reports, 18(12), pp. 2854–2867. https://doi.org/10.1016/j.celrep.2017.02.078.
Pan, Y. et al. (2016) ‘Differential growth triggers mechanical feedback that elevates Hippo signaling’, Proceedings of the National Academy of Sciences, 113(45), pp. E6974–E6983. https://doi.org/10.1073/pnas.1615012113.
Dupont, S. et al. (2011) ‘Role of YAP/TAZ in mechanotransduction.’, Nature, 474(7350), pp. 179–83. https://doi.org/10.1038/nature10137.
Irvine, K. D. and Harvey, K. F. (2015) ‘Control of organ growth by patterning and hippo signaling in Drosophila.’, Cold Spring Harbor perspectives in biology, 7(6). https://doi.org/10.1101/cshperspect.a019224.
Wu, Y., Kanchanawong, P. and Zaidel-Bar, R. (2015) ‘Actin-Delimited Adhesion-Independent Clustering of E-Cadherin Forms the Nanoscale Building Blocks of Adherens Junctions’, Developmental Cell, 32(2), pp. 139–154. https://doi.org/10.1016/j.devcel.2014.12.003.
Maitre, J.-L. et al. (2012) ‘Adhesion Functions in Cell Sorting by Mechanically Coupling the Cortices of Adhering Cells’, Science, 338(6104), pp. 253–256. https://doi.org/10.1126/science.1225399.
Takeichi, M. (1995) ‘Morphogenetic roles of classic cadherins.’, Current opinion in cell biology, 7(5), pp. 619–27. https://doi.org/10.1016/0955-0674(95)80102-2.
Foty, R. A. and Steinberg, M. S. (2005) ‘The differential adhesion hypothesis: a direct evaluation’, Developmental Biology, 278(1), pp. 255–263. https://doi.org/10.1016/j.ydbio.2004.11.012.
Berx, G. et al. (1995) ‘E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers.’, The EMBO Journal, 14(24), pp. 6107–6115. https://doi.org/10.1002/j.1460-2075.1995.tb00301.x.
Bogenrieder, T. and Herlyn, M. (2003) ‘Axis of evil: molecular mechanisms of cancer metastasis’, Oncogene, 22(42), pp. 6524–6536. https://doi.org/10.1038/sj.onc.1206757.
Li, C. I. (2003) ‘Trends in Incidence Rates of Invasive Lobular and Ductal Breast Carcinoma’, JAMA, 289(11), p. 1421. https://doi.org/10.1001/jama.289.11.1421.
Padmanaban, V. et al. (2019) ‘E-cadherin is required for metastasis in multiple models of breast cancer’, Nature, 573(7774), pp. 439–444. https://doi.org/10.1038/s41586-019-1526-3.
Mège, R. M. and Ishiyama, N. (2017) ‘Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions.’, Cold Spring Harbor perspectives in biology, 9(5). https://doi.org/10.1101/cshperspect.a028738.
Knudsen, K. A. et al. (1995) ‘Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin.’, The Journal of cell biology, 130(1), pp. 67–77. https://doi.org/10.1083/jcb.130.1.67.
Nieset, J. E. et al. (1997) ‘Characterization of the interactions of alpha-catenin with alpha-actinin and beta-catenin/plakoglobin.’, Journal of cell science, 110 (Pt 8), pp. 1013–22. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9152027.
Abe, K. and Takeichi, M. (2008) ‘EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt.’, Proceedings of the National Academy of Sciences of the United States of America, 105(1), pp. 13–9. https://doi.org/10.1073/pnas.0710504105.
Marie, H. et al. (2003) ‘The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin.’, The Journal of biological chemistry, 278(2), pp. 1220–8. https://doi.org/10.1074/jbc.M205391200.
Sarpal, R. et al. (2019) ‘Role of α-Catenin and its mechanosensing properties in regulating Hippo/YAP-dependent tissue growth.’, PLoS genetics, 15(11), p. e1008454. https://doi.org/10.1371/journal.pgen.1008454.
Bays, J. L. et al. (2014) ‘Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions.’, The Journal of cell biology, 205(2), pp. 251–63. https://doi.org/10.1083/jcb.201309092.
Bertocchi, C. et al. (2017) ‘Nanoscale architecture of cadherin-based cell adhesions.’, Nature cell biology, 19(1), pp. 28–37. https://doi.org/10.1038/ncb3456.
Hazan, R. B. et al. (1997) ‘Vinculin is associated with the E-cadherin adhesion complex.’, The Journal of biological chemistry, 272(51), pp. 32448–53. https://doi.org/10.1074/jbc.272.51.32448.
Weiss, E. E. et al. (1998) ‘Vinculin is part of the cadherin-catenin junctional complex: complex formation between alpha-catenin and vinculin.’, The Journal of cell biology, 141(3), pp. 755–64. https://doi.org/10.1083/jcb.141.3.755.
Leerberg, J. M. et al. (2014) ‘Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens.’, Current biology : CB, 24(15), pp. 1689–99. https://doi.org/10.1016/j.cub.2014.06.028.
le Duc, Q. et al. (2010) ‘Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner.’, The Journal of cell biology, 189(7), pp. 1107–15. https://doi.org/10.1083/jcb.201001149.
Maddugoda, M. P. et al. (2007) ‘Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell–cell contacts in mammalian epithelial cells’, Journal of Cell Biology, 178(3), pp. 529–540. https://doi.org/10.1083/jcb.200612042.
Chuan, P., Spudich, J. A. and Dunn, A. R. (2011) ‘Robust mechanosensing and tension generation by myosin VI.’, Journal of molecular biology, 405(1), pp. 105–12. https://doi.org/10.1016/j.jmb.2010.10.010.
Acharya, B. R. et al. (2018) ‘A Mechanosensitive RhoA Pathway that Protects Epithelia against Acute Tensile Stress.’, Developmental cell, 47(4), pp. 439-452.e6. https://doi.org/10.1016/j.devcel.2018.09.016.
Aijaz, S., Balda, M. S. and Matter, K. (2006) ‘Tight junctions: molecular architecture and function.’, International review of cytology, 248, pp. 261–98. https://doi.org/10.1016/S0074-7696(06)48005-0.
Zihni, C. et al. (2016) ‘Tight junctions: from simple barriers to multifunctional molecular gates.’ Nature reviews. Molecular cell biology, 17(9), pp. 564–80. https://doi.org/10.1038/nrm.2016.80.
González-Mariscal, L. et al. (2003) ‘Tight junction proteins.’, Progress in biophysics and molecular biology, 81(1), pp. 1–44. https://doi.org/10.1016/s0079-6107(02)00037-8.
Bazzoni, G. and Dejana, E. (2004) ‘Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis.’, Physiological reviews, 84(3), pp. 869–901. https://doi.org/10.1152/physrev.00035.2003.
Garcia, M. A., Nelson, W. J. and Chavez, N. (2018) ‘Cell-Cell Junctions Organize Structural and Signaling Networks.’, Cold Spring Harbor perspectives in biology, 10(4). https://doi.org/10.1101/cshperspect.a029181.
Cartagena-Rivera, A. X. et al. (2017) ‘Apical surface supracellular mechanical properties in polarized epithelium using noninvasive acoustic force spectroscopy.’, Nature communications, 8(1), p. 1030. https://doi.org/10.1038/s41467-017-01145-8.
Choi, W. et al. (2016) ‘Remodeling the zonula adherens in response to tension and the role of afadin in this response.’, The Journal of cell biology, 213(2), pp. 243–60. https://doi.org/10.1083/jcb.201506115.
Fanning, A. S., Van Itallie, C. M. and Anderson, J. M. (2012) ‘Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia.’, Molecular biology of the cell, 23(4), pp. 577–90. https://doi.org/10.1091/mbc.E11-09-0791.
Hatte, G., Prigent, C. and Tassan, J.-P. (2018) ‘Tight junctions negatively regulate mechanical forces applied to adherens junctions in vertebrate epithelial tissue.’, Journal of cell science, 131(3). https://doi.org/10.1242/jcs.208736.
Tornavaca, O. et al. (2015) ‘ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation.’, The Journal of cell biology, 208(6), pp. 821–38. https://doi.org/10.1083/jcb.201404140.
Itoh, M. et al. (1997) ‘Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments.’, The Journal of cell biology, 138(1), pp. 181–92. https://doi.org/10.1083/jcb.138.1.181.
Maiers, J. L. et al. (2013) ‘ZO-1 recruitment to α-catenin—a novel mechanism for coupling the assembly of tight junctions to adherens junctions.’, Journal of cell science, 126(Pt 17), pp. 3904–15. https://doi.org/10.1242/jcs.126565.
Zemljic-Harpf, A. E. et al. (2014) ‘Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes.’, Journal of cell science, 127(Pt 5), pp. 1104–16. https://doi.org/10.1242/jcs.143743.
Etournay, R. et al. (2007) ‘Shroom2, a myosin-VIIa- and actin-binding protein, directly interacts with ZO-1 at tight junctions.’, Journal of cell science, 120(Pt 16), pp. 2838–50. https://doi.org/10.1242/jcs.002568.
Katsube, T. et al. (1998) ‘Cortactin associates with the cell-cell junction protein ZO-1 in both Drosophila and mouse.’, The Journal of biological chemistry, 273(45), pp. 29672–7. https://doi.org/10.1074/jbc.273.45.29672.
Itoh, M. et al. (2012) ‘Rho GTP exchange factor ARHGEF11 regulates the integrity of epithelial junctions by connecting ZO-1 and RhoA-Myosin II signaling’, Proceedings of the National Academy of Sciences, 109(25), pp. 9905–9910. https://doi.org/10.1073/pnas.1115063109.
Otani, T. et al. (2006) ‘Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells.’, The Journal of cell biology, 175(1), pp. 135–46. https://doi.org/10.1083/jcb.200605012.
Aijaz, S. et al. (2005) ‘Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition.’, Developmental cell, 8(5), pp. 777–86. https://doi.org/10.1016/j.devcel.2005.03.003.
Guillemot, L. et al. (2014) ‘MgcRacGAP interacts with cingulin and paracingulin to regulate Rac1 activation and development of the tight junction barrier during epithelial junction assembly.’, Molecular biology of the cell, 25(13), pp. 1995–2005. https://doi.org/10.1091/mbc.E13-11-0680.
Terry, S. J. et al. (2011) ‘Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis.’, Nature cell biology, 13(2), pp. 159–66. https://doi.org/10.1038/ncb2156.
Hoffman, B. D. and Yap, A. S. (2015) ‘Towards a Dynamic Understanding of Cadherin-Based Mechanobiology.’, Trends in cell biology, 25(12), pp. 803–814. https://doi.org/10.1016/j.tcb.2015.09.008.
Yap, A. S., Duszyc, K. and Viasnoff, V. (2018) ‘Mechanosensing and Mechanotransduction at Cell-Cell Junctions.’, Cold Spring Harbor perspectives in biology, 10(8). https://doi.org/10.1101/cshperspect.a028761.
Fanning, A. S. et al. (1998) ‘The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton.’, The Journal of biological chemistry, 273(45), pp. 29745–53. https://doi.org/10.1074/jbc.273.45.29745.
Fanning, A. S., Ma, T. Y. and Anderson, J. M. (2002) ‘Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1.’, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 16(13), pp. 1835–7. https://doi.org/10.1096/fj.02-0121fje.
Lye, M. F. et al. (2010) ‘Insights into regulated ligand binding sites from the structure of ZO-1 Src homology 3-guanylate kinase module.’, The Journal of biological chemistry, 285(18), pp. 13907–17. https://doi.org/10.1074/jbc.M109.093674.
Sakakibara, S. et al. (2018) ‘Requirement of the F-actin-binding activity of l-afadin for enhancing the formation of adherens and tight junctions’, Genes to Cells, 23(3), pp. 185–199. https://doi.org/10.1111/gtc.12566.
Tanaka-Okamoto, M. et al. (2011) ‘Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia.’, Journal of cell science, 124(Pt 13), pp. 2231–40. https://doi.org/10.1242/jcs.081000.
Cordenonsi, M. et al. (1999) ‘Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin.’, The Journal of cell biology, 147(7), pp. 1569–82. https://doi.org/10.1083/jcb.147.7.1569.
D’Atri, F. and Citi, S. (2001) ‘Cingulin interacts with F-actin in vitro.’, FEBS letters, 507(1), pp. 21–4. https://doi.org/10.1016/s0014-5793(01)02936-2.
Scott, D. W., Tolbert, C. E. and Burridge, K. (2016) ‘Tension on JAM-A activates RhoA via GEF-H1 and p115 RhoGEF.’, Molecular biology of the cell, 27(9), pp. 1420–30. https://doi.org/10.1091/mbc.E15-12-0833.
Fanning, A. S. and Anderson, J. M. (2009) ‘Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions.’, Annals of the New York Academy of Sciences, 1165, pp. 113–20. https://doi.org/10.1111/j.1749-6632.2009.04440.x.
Beutel, O. et al. (2019) ‘Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions.’, Cell, 179(4), pp. 923-936.e11. https://doi.org/10.1016/j.cell.2019.10.011.
Schwayer, C. et al. (2019) ‘Mechanosensation of Tight Junctions Depends on ZO-1 Phase Separation and Flow.’, Cell, 179(4), pp. 937-952.e18. https://doi.org/10.1016/j.cell.2019.10.006.
Vasileva, E. et al. (2020) ‘Cingulin unfolds ZO-1 and organizes myosin-2B and γ-actin to mechanoregulate apical and tight junction membranes’, bioRxiv, p. 2020.05.14.095364. https://doi.org/10.1101/2020.05.14.095364.
Martinac, B. and Kloda, A. (2003) ‘Evolutionary origins of mechanosensitive ion channels’, Progress in Biophysics and Molecular Biology, 82(1–3), pp. 11–24. https://doi.org/10.1016/S0079-6107(03)00002-6.
Bae, C. et al. (2013) ‘Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1’, Proceedings of the National Academy of Sciences, 110(12), pp. E1162–E1168. https://doi.org/10.1073/pnas.1219777110.
Martinac, B. (2014) ‘The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity Biochimica et Biophysica Acta (BBA)-Biomembranes, 1838(2), pp. 682–691. https://doi.org/10.1016/j.bbamem.2013.07.015.
Kung, C. (2005) ‘A possible unifying principle for mechanosensation.’, Nature, 436(7051), pp. 647–54. https://doi.org/10.1038/nature03896.
Markin, V. S. and Martinac, B. (1991) ‘Mechanosensitive ion channels as reporters of bilayer expansion. A theoretical model.’, Biophysical journal, 60(5), pp. 1120–7. https://doi.org/10.1016/S0006-3495(91)82147-6.
Martinac, B., Adler, J. and Kung, C. (1990) ‘Mechanosensitive ion channels of E. coli activated by amphipaths.’, Nature, 348(6298), pp. 261–3. https://doi.org/10.1038/348261a0.
Perozo, E. et al. (2002) ‘Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating.’, Nature structural biology, 9(9), pp. 696–703. https://doi.org/10.1038/nsb827.
Teng, J. et al. (2015) ‘The force-from-lipid (FFL) principle of mechanosensitivity, at large and in elements.’, Pflugers Archiv: European journal of physiology, 467(1), pp. 27–37. https://doi.org/10.1007/s00424-014-1530-2.
Różycki, B. and Lipowsky, R. (2015) ‘Spontaneous curvature of bilayer membranes from molecular simulations: asymmetric lipid densities and asymmetric adsorption.’, The Journal of chemical physics, 142(5), p. 054101. https://doi.org/10.1063/1.4906149.
Bavi, O. et al. (2016) ‘Influence of Global and Local Membrane Curvature on Mechanosensitive Ion Channels: A Finite Element Approach.’, Membranes, 6(1). https://doi.org/10.3390/membranes6010014.
Chalfie, M. (2009) ‘Neurosensory mechanotransduction.’, Nature reviews. Molecular cell biology, 10(1), pp. 44–52. https://doi.org/10.1038/nrm2595.
Katta, S., Krieg, M. and Goodman, M. B. (2015) ‘Feeling force: physical and physiological principles enabling sensory mechanotransduction.’, Annual review of cell and developmental biology, 31, pp. 347–71. https://doi.org/10.1146/annurev-cellbio-100913-013426.
Sukharev, S. and Corey, D. P. (2004) ‘Mechanosensitive Channels: Multiplicity of Families and Gating Paradigms’, Science Signaling, 2004(219), pp. re4. https://doi.org/10.1126/stke.2192004re4.
Wang, J. et al. (2020) ‘Tethering Piezo channels to the actin cytoskeleton for mechanogating via the E-cadherin-β-catenin mechanotransduction complex’, bioRxiv, p. 2020.05.12.092148. https://doi.org/10.1101/2020.05.12.092148.
Chubinskiy-Nadezhdin, V. I. et al. (2019) ‘Agonist-induced Piezo1 activation suppresses migration of transformed fibroblasts.’, Biochemical and biophysical research communications, 514(1), pp. 173–179. https://doi.org/10.1016/j.bbrc.2019.04.139.
Pardo-Pastor, C. et al. (2018) ‘Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses.’, Proceedings of the National Academy of Sciences of the United States of America, 115(8), pp. 1925–1930. https://doi.org/10.1073/pnas.1718177115.
Yao, M. et al. (2020) ‘Force-dependent Piezo1 recruitment to focal adhesions regulates adhesion maturation and turnover specifically in non-transformed cells’, bioRxiv, p. 2020.03.09.972307. https://doi.org/10.1101/2020.03.09.972307.
Ranade, S. S., Syeda, R. and Patapoutian, A. (2015) ‘Mechanically Activated Ion Channels.’, Neuron, 87(6), pp. 1162–1179. https://doi.org/10.1016/j.neuron.2015.08.032.
Coste, B. et al. (2010) ‘Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels’, Science, 330(6000), pp. 55–60. https://doi.org/10.1126/science.1193270.
Li, J. et al. (2014) ‘Piezo1 integration of vascular architecture with physiological force.’, Nature, 515(7526), pp. 279–282. https://doi.org/10.1038/nature13701.
Ge, J. et al. (2015) ‘Architecture of the mammalian mechanosensitive Piezo1 channel’, Nature, 527(7576), pp. 64–69. https://doi.org/10.1038/nature15247.
Guo, Y. R. and MacKinnon, R. (2017) ‘Structure-based membrane dome mechanism for Piezo mechanosensitivity.’, eLife, 6. https://doi.org/10.7554/eLife.33660.
Zhao, Q. et al. (2018) ‘Structure and mechanogating mechanism of the Piezo1 channel’, Nature, 554(7693), pp. 487–492. https://doi.org/10.1038/nature25743.
Saotome, K. et al. (2018) ‘Structure of the mechanically activated ion channel Piezo1.’, Nature, 554(7693), pp. 481–486. https://doi.org/10.1038/nature25453.
De Vecchis, D., Beech, D. J. and Kalli, A. C. (2021) ‘Molecular dynamics simulations of Piezo1 channel opening by increases in membrane tension.’, Biophysical journal, 120(8), pp. 1510–1521. https://doi.org/10.1016/j.bpj.2021.02.006.
Coste, B. et al. (2015) ‘Piezo1 ion channel pore properties are dictated by C-terminal region.’, Nature communications, 6, p. 7223. https://doi.org/10.1038/ncomms8223.
Gudipaty, S. A. et al. (2017) ‘Mechanical stretch triggers rapid epithelial cell division through Piezo1’, Nature, 543(7643), pp. 118–121. https://doi.org/10.1038/nature21407.
Palade, G. (1953) ‘Fine structures of blood capillaries’, Journal of Applies Physics, 24, p. 1424.
Yamada, E. (1955) ‘The fine structure of the gall bladder epithelium of the mouse’, The Journal of Biophysical and Biochemical Cytology, 1(5), pp. 445–458. https://doi.org/10.1083/jcb.1.5.445.
Lo, H. P. et al. (2015) ‘The caveolin-cavin system plays a conserved and critical role in mechanoprotection of skeletal muscle.’, The Journal of cell biology, 210(5), pp. 833–49. https://doi.org/10.1083/jcb.201501046.
Lu, Z. et al. (2003) ‘Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion.’, Cancer cell, 4(6), pp. 499–515. https://doi.org/10.1016/s1535-6108(03)00304-0.
Orlichenko, L. et al. (2009) ‘Caveolae mediate growth factor-induced disassembly of adherens junctions to support tumor cell dissociation.’, Molecular biology of the cell, 20(19), pp. 4140–52. https://doi.org/10.1091/mbc.e08-10-1043.
Palacios, F. et al. (2002) ‘ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly.’, Nature cell biology, 4(12), pp. 929–36. https://doi.org/10.1038/ncb881.
Volontè, D., Galbiati, F. and Lisanti, M. P. (1999) ‘Visualization of caveolin-1, a caveolar marker protein, in living cells using green fluorescent protein (GFP) chimeras’, FEBS Letters, 445(2–3), pp. 431–439. https://doi.org/10.1016/S0014-5793(99)00164-7.
Teo, J. L. et al. (2020) ‘Caveolae Control Contractile Tension for Epithelia to Eliminate Tumor Cells.’, Developmental cell, 54(1), pp. 75-91.e7. https://doi.org/10.1016/j.devcel.2020.05.002.
Janshoff, A. and Steinem, C. (2015) ‘Mechanics of lipid bilayers: What do we learn from pore-spanning membranes?’, Biochimica et Biophysica Acta (BBA) – Molecular Cell Research, 1853(11), pp. 2977–2983. https://doi.org/10.1016/j.bbamcr.2015.05.029.
Sinha, B. et al. (2011) ‘Cells respond to mechanical stress by rapid disassembly of caveolae.’, Cell, 144(3), pp. 402–13. https://doi.org/10.1016/j.cell.2010.12.031.
Seemann, E. et al. (2017) ‘Deciphering caveolar functions by syndapin III KO-mediated impairment of caveolar invagination’, eLife, 6. https://doi.org/10.7554/eLife.29854.
Dulhunty, A. F. and Franzini-Armstrong, C. (1975) ‘The relative contributions of the folds and caveolae to the surface membrane of frog skeletal muscle fibres at different sarcomere lengths.’, The Journal of physiology, 250(3), pp. 513–39. https://doi.org/10.1113/jphysiol.1975.sp011068.
Prescott, L and Brightman, M. W. (1976a) ‘The sarcolemma of Aplysia smooth muscle in freeze-fracture preparations.’, Tissue & cell, 8(2), pp. 241–58. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18631588.
Cheng, J. P. X. et al. (2015) ‘Caveolae protect endothelial cells from membrane rupture during increased cardiac output.’, The Journal of cell biology, 211(1), pp. 53–61. https://doi.org/10.1083/jcb.201504042.
Garcia, J. et al. (2017) ‘Sheath Cell Invasion and Trans-differentiation Repair Mechanical Damage Caused by Loss of Caveolae in the Zebrafish Notochord.’, Current biology : CB, 27(13), pp. 1982-1989.e3. https://doi.org/10.1016/j.cub.2017.05.035.
Lim, Y.-W. et al. (2017) ‘Caveolae Protect Notochord Cells against Catastrophic Mechanical Failure during Development’, Current Biology, 27(13), pp. 1968-1981.e7. https://doi.org/10.1016/j.cub.2017.05.067.
Golani, G. et al. (2019) ‘Membrane Curvature and Tension Control the Formation and Collapse of Caveolar Superstructures’, Developmental Cell, 48(4), pp. 523-538.e4. https://doi.org/10.1016/j.devcel.2018.12.005.
Pelkmans, L. and Zerial, M. (2005) ‘Kinase-regulated quantal assemblies and kiss-and-run recycling of caveolae.’, Nature, 436(7047), pp. 128–33. https://doi.org/10.1038/nature03866.
Echarri, A. et al. (2019) ‘An Abl-FBP17 mechanosensing system couples local plasma membrane curvature and stress fiber remodeling during mechanoadaptation.’, Nature communications, 10(1), p. 5828. https://doi.org/10.1038/s41467-019-13782-2.
Boyd, N. L. et al. (2003) ‘Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells’, American Journal of Physiology-Heart and Circulatory Physiology, 285(3), pp. H1113–H1122. https://doi.org/10.1152/ajpheart.00302.2003.
Rizzo, V. et al. (1998) ‘In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association.’, The Journal of biological chemistry, 273(52), pp. 34724–9. https://doi.org/10.1074/jbc.273.52.34724.
Trouet, D. et al. (1999) ‘Caveolin-1 modulates the activity of the volume-regulated chloride channel.’, The Journal of physiology 520 Pt 1, pp. 113–9. https://doi.org/10.1111/j.1469-7793.1999.t01-1-00113.x.
Morén, B. et al. (2012) ‘EHD2 regulates caveolar dynamics via ATP-driven targeting and oligomerization.’, Molecular biology of the cell, 23(7), pp. 1316–29. https://doi.org/10.1091/mbc.E11-09-0787.
Torrino, S. et al. (2018) ‘EHD2 is a mechanotransducer connecting caveolae dynamics with gene transcription’, Journal of Cell Biology, 217(12), pp. 4092–4105. https://doi.org/10.1083/jcb.201801122.
Kawamura, S., Miyamoto, S. and Brown, J. H. (2003) ‘Initiation and Transduction of Stretch-induced RhoA and Rac1 Activation through Caveolae’, Journal of Biological Chemistry, 278(33), pp. 31111–31117. https://doi.org/10.1074/jbc.M300725200.
Nomura, R. and Fujimoto, T. (1999) ‘Tyrosine-phosphorylated caveolin-1: immunolocalization and molecular characterization.’, Molecular biology of the cell, 10(4), pp. 975–86. https://doi.org/10.1091/mbc.10.4.975.
Radel, C. and Rizzo, V. (2005) ‘Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of Csk to mediate actin reorganization.’, American journal of physiology. Heart and circulatory physiology, 288(2), pp. H936-45. https://doi.org/10.1152/ajpheart.00519.2004.
Peng, F. et al. (2007) ‘RhoA activation in mesangial cells by mechanical strain depends on caveolae and caveolin-1 interaction.’, Journal of the American Society of Nephrology : JASN, 18(1), pp. 189–98. https://doi.org/10.1681/ASN.2006050498.
Grande-García, A. et al. (2007) ‘Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases’, Journal of Cell Biology, 177(4), pp. 683–694. https://doi.org/10.1083/jcb.200701006.
Hetmanski, J. H. R. et al. (2019) ‘Membrane Tension Orchestrates Rear Retraction in Matrix-Directed Cell Migration.’, Developmental cell, 51(4), pp. 460-475.e10. https://doi.org/10.1016/j.devcel.2019.09.006.
Ariotti, N. et al. (2014) ‘Caveolae regulate the nanoscale organization of the plasma membrane to remotely control Ras signaling’, Journal of Cell Biology, 204(5), pp. 777–792. https://doi.org/10.1083/jcb.201307055.
Boopathy, G. T. K. and Hong, W. (2019) ‘Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis’, Frontiers in Cell and Developmental Biology, 7. https://doi.org/10.3389/fcell.2019.00049.
Ibar, C. et al. (2018) ‘Tension-dependent regulation of mammalian Hippo signaling through LIMD1’, Journal of Cell Science, 131(5). https://doi.org/10.1242/jcs.214700.
Zhao, B. et al. (2007) ‘Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control.’, Genes & development, 21(21), pp. 2747–61. https://doi.org/10.1101/gad.1602907.
Moreno-Vicente, R. et al. (2018) ‘Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP.’, Cell reports, 25(6), pp. 1622-1635.e6. https://doi.org/10.1016/j.celrep.2018.10.024.
Rausch, V. et al. (2019) ‘The Hippo Pathway Regulates Caveolae Expression and Mediates Flow Response via Caveolae’, Current Biology, 29(2), pp. 242-255.e6. https://doi.org/10.1016/j.cub.2018.11.066.
Yu, J. (2006) ‘Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels’, Journal of Clinical Investigation, 116(5), pp. 1284–1291. https://doi.org/10.1172/JCI27100.
Lee, H. J. et al. (2017) ‘Fluid shear stress activates YAP1 to promote cancer cell motility’, Nature Communications, 8(1), p. 14122. https://doi.org/10.1038/ncomms14122.
Wang, K.-C. et al. (2016) ‘Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis’, Proceedings of the National Academy of Sciences, 113(41), pp. 11525–11530. https://doi.org/10.1073/pnas.1613121113.
Rausch, V. and Hansen, C. G. (2020) ‘The Hippo Pathway, YAP/TAZ, and the Plasma Membrane’, Trends in Cell Biology, 30(1), pp. 32–48. https://doi.org/10.1016/j.tcb.2019.10.005.
Munjal, A. et al. (2015) ‘A self-organized biomechanical network drives shape changes during tissue morphogenesis.’, Nature, 524(7565), pp. 351–5. https://doi.org/10.1038/nature14603.
Priya, R. et al. (2015) ‘Feedback regulation through myosin II confers robustness on RhoA signalling at E-cadherin junctions.’, Nature cell biology, 17(10), pp. 1282–93. https://doi.org/10.1038/ncb3239.
He, W.-Q. et al. (2020) ‘Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis.’, International journal of molecular sciences, 21(3). https://doi.org/10.3390/ijms21030993.
Acknowledgements
The authors were supported by grants and fellowships from the National Health and Medical Research Council of Australia (GNT1164462, 1136592 to ASY; APP1156489 to RGP; GNT1140090 to ASY and RGP) and the Australian Research Council (DP19010287 to ASY).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Brooks, J.W., Parton, R.G., Yap, A.S., Duszyc, K. (2022). Epithelial Mechanosensing at Cell-Cell Contacts and Tight Junctions. In: González-Mariscal, L. (eds) Tight Junctions. Springer, Cham. https://doi.org/10.1007/978-3-030-97204-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-97204-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-97203-5
Online ISBN: 978-3-030-97204-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)