
Abstract
The presence of a large number of genes in the gene expression profiles imposes a computational challenge for cancer classification. To deal with the high-dimensional feature space, in this paper, we present a 3-step feature selection framework, RRO (Relevancy-Redundancy-Optimization). In the first step, RRO identifies top-ranked class-relevant genes utilizing the analysis of variance (ANOVA) and F-test. In the second step, class correlated but redundant genes are removed by employing the Kendall rank correlation coefficient (Kendall’s \(\tau \)). Finally, we utilize a metaheuristic optimization algorithm, binary whale optimization algorithm (BWOA), with the support vector machine (SVM) classifier to select an optimal gene subset. The comparisons with thirteen state-of-the-art methods in ten gene expression datasets show that RRO yields better or comparable accuracy.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others
1 Introduction
The microarray gene expression profiles contain a large number of genes with a limited number of samples. Feature selection plays a crucial role in cancer classification as it allows researchers to identify the most substantial genes that provide insight into the mechanisms responsible for cancer. Furthermore, it decreases the computational costs associated with the classification by excluding non-informative and redundant genes. Moreover, it yields a simpler classification model, which refers to more interpretable results.
Researchers mainly utilized three approaches for gene selection: filter, wrapper, and hybrid approaches. The filter-based methods estimate the relevance scores of every feature by computing statistical measures and then remove the statistically non-significant features. The wrapper methods examine various combinations of gene subsets and determine the best subset by integrating them into a classifier. Although they are usually more accurate than the filter-based methods, they require extensive computational resources and are more prone to over-fitting. The hybrid methods usually apply one or multiple filter-based methods in the first step to reduce the feature space. Afterward, a wrapper-based method is employed to select the optimal feature subset.
The proposed methodology, RRO, starts with distinguishing the top-class discriminative genes using the filter-based ANOVA method. The Kendall rank-order correlation coefficient (Kendall’s \(\tau \)) is employed to eliminate class-correlated but redundant genes. Then, we apply the binary whale optimization algorithm (BWOA) with the SVM classifier to identify an optimized feature subset. Finally, we feed the selected gene subset to the SVM classifier to determine cancer types. We compare RRO with thirteen state-of-the-art methods on ten gene expressions datasets considering both best and average accuracy. The results demonstrate that RRO yields comparable or better accuracy than most of the existing methods.
1.1 Objective and Contributions
The main objective of this study is to demonstrate the efficacy of the combination of several feature selection methods (i.e., ANOVA, Kendall, and BWOA) and the SVM classifier for gene selection and cancer classification. The main contributions of this paper can be summarized as follows:
-
We show that the combinations of ANOVA and Kendall’s \(\tau \) can effectively remove irrelevant and redundant genes in gene expression datasets.
-
We demonstrate that the SVM classifier with the BWOA feature selection algorithm can effectively identify cancer subtypes (or presence).
2 Related Work
A number of univariate feature selection methods such as Chi-square [21], Information Gain (IG) [13, 19], Laplacian score (L-score) [37] and Fisher score (F-score) [11] have been employed for gene selection in literature. Besides, researchers also studied multivariate filter methods such as Minimal Redundancy Maximal Relevance (mRMR) [28], Random Subspace Method (RSM) [18], and Relevance Redundancy Feature Selection (RRFS) [12] which considers the dependencies among features.
Due to the high computational cost associated with wrapper-based feature selection methods, they are often combined with a filter-based method. The hybrid methodology initially employs a filter-based method to exclude non-informative genes to reduce search space. Mundra et al. [25] hybridized two of the most popular feature selection approaches, namely SVM-RFE and mRMR. Shreem et al. [34] proposed RM-GA approach that combines ReliefF, mRMR, and genetic algorithm (GA). In [9], the authors introduced a hybrid method named CFS-TGA, which utilizes correlation-based feature selection (CFS), Taguchi-Genetic Algorithm (TGA), and K-NN classifier. In [5], the authors proposed a feature selection algorithm, mRMR, and combined it with the Artificial Bee Colony (ABC) algorithm to select informative genes. The performance of the mRMR-ABC algorithm was evaluated on six binary and multiclass gene expression microarray datasets. Lee and Leu [20] presented Genetic Algorithm Dynamic Parameter (GADP) that produces every possible subset of genes and ranks the genes using their occurrence frequency. Yassi and Moattar [38] proposed a feature selection approach for microarray data that fuses both ranking and wrapper-based methods.
MIMAGA-Selection [22] is a hybrid feature selection algorithm that combines Mutual Information Maximization (MIM) and Adaptive Genetic Algorithm (AGA). CLA-ACO [31] is a hybrid approach that employs Cellular Learning Automata (CLA) and Ant Colony Optimization (ACO). CLA-ACO consists of three phases: filter-based Fisher criterion method, cellular learning automata, and ant colony optimization.
Shreem et al. [33] combined Symmetrical Uncertainty (SU) with the Harmony Search Algorithm (HSA) for gene selection. The authors first eliminated non-essential genes using the SU method. In the second stage, HSA was employed as a wrapper method to find the most informative genes. Two classifiers, IB1 and NB, were utilized to assess the performance of SU-HSA. The authors applied SU-HSA to 10 microarray datasets and achieved 100% accuracy in four of them.
Salem et al. [29] proposed an Information Gain (IG) and Standard Genetic Algorithm (SGA) based method, IG/SGA, for feature selection. IG was applied at the initial step for feature reduction. Then, a genetic algorithm was applied to select the optimal features, and finally, Genetic Programming (GP) classifier was employed. Their method was evaluated on seven cancer microarray datasets and achieved 100% accuracy in two datasets. A GA and Intelligent Dynamic Genetic Algorithm (IDGA) based method for gene selection was proposed in [10]. The authors applied Laplacian and Fisher scores independently in the first phase to select the top 500 genes. In the second phase, the reinforcement learning-based IDGA method was applied. Support Vector Machine (SVM), Naïve Bayes (NB), and K-Nearest Neighbour (KNN) were employed as classifiers on five microarray cancer datasets. It was observed that when combined with the IDGA, the Fisher score outperformed the Laplacian score on four datasets.
A hybrid framework that employs both extraction and wrapper gene selection methods was presented by Aziz et al. [8]; as an extraction method, the authors used ICA, while as a wrapper method, ABC was utilized. Their experimental results utilizing the NB classifier yielded better performance compared to other gene selection algorithms. A Gene Selection Programming (GSP) method was proposed in [2] to select relevant genes for cancer classification. The authors adopted the SVM with a linear kernel as a classifier for the GSP. A neighborhood entropy-based feature selection algorithm was introduced in [35]. At first, the neighborhood entropy-based uncertainty measures were utilized to assess the uncertainty and exclude the noise present in gene expression datasets. In the subsequent steps, the neighborhood credibility degree and coverage degree were applied. The authors employed a heuristic reduction algorithm to decrease the computational complexity and to improve the classification performance.
A hybrid method that employs Adaptive Elastic Net (AEN) with Conditional Mutual Information (CMI) was introduced in [36]. AEN-CMI obtained better performance compared to L1-SVM, Elastic Net, Adaptive Lasso, and classic Adaptive Elastic Net algorithms on two cancer microarray datasets. In [1], an unsupervised two-stage feature selection technique was presented. In the first stage, three filter-based methods techniques were applied. A genetic algorithm was utilized in the second stage. Finally, three ML classifiers, SVM, k-NN, and random forest (RF) were employed for classification.
Several variants of Particle Swarm Optimization (PSO) algorithm have been proposed in the literature for cancer classification [15, 16, 24, 30]. HPSO-LS [24] embedded a local search strategy within the PSO to reduce the feature space before applying PSO. The authors also used the Pearson correlation coefficient to identify correlation among features. As a classifier, the k-NN classifier was used. In another PSO-based work [16], k-NN with PSO was utilized for feature selection and classification. However, their work focused more on selecting the best k value for the k-NN classifier to achieve better accuracy. In [15], the correlation-based feature selection (CFS) algorithm and iBPSO were applied for identifying the best feature set, and then NB classifiers were employed for classification. In [30], ANOVA, Spearman rank-order correlation (SRC), and BPSO were employed for feature selection.
3 Proposed Methodology
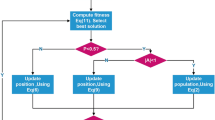
The RRO starts with normalizing the gene expression values. Afterward, the feature selection process consisting of three phases, relevance analysis (ANOVA), redundancy analysis (Kendall’s \(\tau \)), and feature set optimization (BWOA-SVM) are employed. Finally, the SVM classifier is applied for classification. The flow diagram of the proposed methodology is shown in Fig. 1.
3.1 Feature Scaling
The value of each feature (i.e., gene expression value) is scaled to fit within a range between 0 and 1. The following equation is used to normalize the d’th value (i.e., value for the d’th instance) of feature i in set X,
where, \(X_i.max\) and \({X_i.min}\) are the maximum and minimum gene expression values of gene i acorss all the instances, respectively.

Flowchart of the proposed methodology
3.2 Phase 1 Feature Selection: Relevance Analysis
In phase 1, ANOVA, a statistical method, is applied to reduce the feature search space by identifying top class-correlated genes. ANOVA F-test determines the variance between and within the groups, calculates F-value, and utilizes it to identify informative genes. This step selects a set of top class-correlated genes. The number of genes in the set is either 200 genes or 5% of the genes present in the dataset, whichever is minimum. The selected top class-correlated genes are then forwarded to the redundancy measure step.
3.3 Phase 2 Feature Selection: Redundancy Analysis
As univariate filter-based feature selection methods do not consider the interaction among features, they may select class-correlated but redundant genes. In phase 2, we employ Kendall’s \(\tau \) coefficient to remove redundant genes identified in phase 1. The Kendall rank correlation coefficient (often called Kendall’s \(\tau \) coefficient) is a non-parametric measure of the correspondence between two rankings. A value close to +1 indicates strong agreement, a value around –1 indicates strong disagreement, and a 0 value indicates no correlation.
Let \((x_1,y_1)\),...,\((x_n,y_n)\) be a set of observations of the joint random variables X and Y such that all the values of \(x_i\) and \(y_i\) are unique (assume their is no tie). Any pair of observations \((x_i,y_i)\) and \((x_j,y_j)\) , where i < j, are said to be concordant if the sort order of \((x_i,x_j)\) and \((y_i,y_j)\) agrees; otherwise they are said to be discordant. The Kendall \(\tau \) coefficient is defined as:
The redundancy threshold value is selected as 0.5 to identify the redundant genes. When two genes show a Kendall’s \(\tau \) correlation coefficient above the redundancy threshold, the lower class-relevant gene is removed.
3.4 Phase 3 Feature Selection: Meta-heuristic Optimization
Phase 3 of the feature selection step leverages binary whale optimization algorithm (BWOA) and SVM classifier. Whale Optimization Algorithm (WOA) [23] is a nature-inspired optimization algorithm that imitates the social behavior of humpback whales. The WOA optimization algorithm assumes that the present best candidate solution is the target prey or is close to the optimum solution. The other search agents (i.e., whale) gradually change their positions towards the current best solution (i.e., best whale). The following equations describe this behavior of i’th whale:
where t indicates the current iteration, A and C are coefficient values, \(r_1\) and \(r_2\) are random vectors in the range of [0, 1]; a decreases linearly through the iterations from 2 to 0. \(X_i\) is the position vector of the i’th whale. \({X_b}\) is the position vector of the current optimal solution (best search agent position).

The steps of BWOA
The two phases of bubble-net attacking behaviour of whales are the exploitation and exploration phases.
Exploitation Phase
Shrinking Encircling Mechanism: This behavior is achieved by decreasing the value of a. The fluctuation range of A decreases as a is reduced from 2 to 0 gradually over the course of iterations. By setting random values for A in [–1, 1], the updated position of each agent can be defined anywhere in between the position of the current best agent and the original position of the agent.
Spiral-Based Position Update: In this step, for each whale i, the distance to current best solution is computed. To imitate the helix-shaped movement of the humpback whale, a spiral equation is used, which is shown below-
where D = \(|X_b(t) - X_i(t)|\) indicates the distance of the prey (i.e., best solution) to i’th whale, b is the logarithmic spiral shape constant, l is a random number in [–1, 1].
The Exploration Phase
The exploration phase searches randomly instead of moving towards the current best solution. The position of a search agent is updated respect to a randomly chosen search agent instead of using the best search agent found so far. This strategy is employed when \(|A| > 1\), it emphasizes exploration and allow the WOA algorithm to perform a global search. The mathematical model is described as follows:
where, \(X_{rand}\) is the position vector of a randomly chosen search agent.
3.5 Binary Whale Optimization Algorithm (BWOA)
For feature selection, we use a modified version of WOA, called binary WOA (BWOA) [14], that finds the candidate solution with a binary value (i.e., not-selected (0) or selected (1)) of individual genes. We use the S-shaped sigmoid function in each dimension to map the continuous-valued velocity given by Eq. 2 to the range [0, 1], as shown in Eq. 10.
The feature (i.e., gene) states in the solutions are changed based on the Eq. 11. For example, the state of the d’th gene in solution i at time t is determined by,
where \(\rho _{id}\) is a random number with uniform distribution within the range of [0.0, 1.0).
3.6 BWOA for Gene Selection
The BWOA optimization process involves several steps, as shown in Fig. 2. The BWOA algorithm initially starts with a set of random solutions. Each solution represents an d-dimensional feature (i.e., gene) set, where \(g_i\) = 1 means the gene i is selected as a feature, while 0 value means it is not selected (an example provided in Fig. 3). At each iteration, candidate solutions update their positions towards either the best solution obtained so far or a randomly chosen solution. The parameter a is decreased from 2 to 0 to support exploration and exploitation. A random solution is chosen when \(|A|\ge \) 1, while the current best solution is selected when \(|A| < 1\) for updating the position of the candidate solutions. Depending on the value of p, BWOA can switch between either a circular movement or spiral. The BWOA algorithm terminates when the maximum number of iteration is reached. We use 300 as a maximum limit for the iterations.

Representation of a candidate solution of BWOA with a gene set with n number of genes (\(d_1,d_2, ....., d_n\))
Optimization Goal
In RRO, the optimization goal of the BWOA is set to identify a gene subset that provides the highest accuracy for cancer classification. The optimization functions of BWOA only consider accuracy to find the best solution; minimizing the number of genes is not considered in the optimization step, as irrelevant and redundant genes are discarded in the earlier phases.
Training and Testing Data Splitting
The SVM classifier is used to assess the accuracy of a candidate solution. We use 90% data for training and the remaining 10% data are used to asses the accuracy of the candidate feature set. The data split is performed randomly and in a stratified fashion. We use the BWOA implementation of [17].
3.7 Classification
In the classification stage, the SVM classifier is utilized for determining the types (or presence) of cancer leveraging the informative genes identified in the feature selection stages. The default parameters settings of scikit-learn [27] implementation of the SVM classifier (with class-balanced weights) are used. The results are reported based on the 10-fold cross-validation.
4 Datasets and Baselines
4.1 Gene Expression Datasets
We report the performance of RRO on ten gene expression benchmark datasetsFootnote 1\(^{,}\)Footnote 2, shown in Table 1. The datasets represent gene expression profiles of different types of cancers, such as Colon, Central Nerve System (CNS), DLBCL, Leukemia, Lung, Prostate, SRBCT, Ovarian, Leukemia-3 and Lymphoma shown in Table 1.
The Colon dataset is a binary dataset consisting of gene expression profiles of 40 cancerous and 22 normal colon tissues. The CNS dataset comprises 61 examples from two classes. The DLBCL dataset consists of 77 gene expression samples from two classes, DLBCL and Follicular Lymphoma (FL) morphology. In the Leukemia dataset, 25 samples belong to AML and 47 samples belong to ALL. Lung cancer consists of 181 samples, 31 from Malignant Pleural Mesothelioma (MPM) and 150 from Adenocarcinoma (ADCA). The Prostate and SRBCT datasets contain 102 and 63 samples, respectively. The four classes of the SRBCT dataset are neuroblastoma (NB), rhabdomyosarcoma (RMS), non-Hodgkin lymphoma (NHL), and the Ewing family of tumors (EWS). The three classes of the Lymphoma-3 dataset are DLBCL, FL, and CLL. The Ovarian dataset represents cancer and normal class.
4.2 Performance Metrics
The precision, recall, macro F1 score, accuracy, and the number of genes used are provided to demonstrate the efficacy of the RRO. The recall (\(R_c\)), precision (\(P_c\)), and F1 score (\(F1_c\)) of a class c is calculated as follows:
The \(TP_c\), \(FP_c\), \(FN_c\) of a class c is defined as follows:
-
\(TP_c\) = Both prediction and true label refer to the same class c
-
\(FP_c\) = Prediction indicates class c, while true label is different (i.e., not class c)
-
\(FN_c\) = Prediction indicates any of the other classes (i.e., non c-class) while true class is c
Finally, the macro F1 score is calculated by taking the average of F1 scores of all the classes.
Besides, the accuracy of RRO is reported in various gene expression datasets to compare it with state-of-the-art methods. The accuracy is defined as follows:
Besides, we investigate the number of genes different methodologies use for classification. The number of genes utilized is a critical indicator of the efficacy of various methods since the fewer number of genes usually makes the results more interpretable and less prone to over-fitting.
4.3 Baseline Methods
The efficacy of RRO is shown by comparing it with thirteen state-of-the-art methods. The summary of each method is provided in Table 2. The comparative performances of RRO and the state-of-the-art methods are shown in terms of accuracy and the number of genes utilized.
5 Results and Discussion
Table 3 shows the precision, recall, F1 score, and accuracy of RRO. Among the 10 datasets, RRO obtains a perfect F1 score of 1.0 in 5 datasets (DLBCL, Leukemia, SRBCT, Lymphoma, and Ovarian). In two datasets, Lung and Prostate, it achieves almost perfect F1 scores (0.994 and 0.990).
Table 4 shows the comparison results of RRO with thirteen state-of-the-art methods. Both accuracy (Acc.) and the number of genes (#G) used by various methods are provided (except [1, 26], which did not report the number of genes utilized). Note that existing works used distinct sets of evaluation datasets in their experiments; thus, they may not have experimental results for all the datasets used in this study (indicated by ‘-’).
The comparisons with the state-of-the-art methods demonstrate that RRO provides better accuracy than most of the existing methodologies. We notice, in several datasets, such as Lung, Ovarian, and SRBCT datasets, RRO achieves the perfect (i.e.,100%) accuracy using less than seven genes. In three other datasets, it also attains 100% accuracy. In other datasets such as Colon, Leukemia-3, and Prostate, it is noticed that RRO shows better performance compared to most of the existing methods; Although, yields a bit less accurate results than [4, 15] in Colon and [29] in Prostate datasets, respectively. When the average accuracy is considered, RRO yields better results than other methods (which reported average accuracy) in CNS and Prostate datasets (shown by non-integer values for the number of genes).
The running time could be a limiting factor of the applicability of evolutionary algorithms for feature selection; especially, with the presence of thousands of features in the dataset. RRO utilizes two filter-based approaches in early steps to reduce the feature search space, thus, keeps the running time feasible.
The efficacy of RRO is shown on datasets of distinct characteristics. We use 2-class, 3-class, and 4-class gene expression datasets (Table 1). Besides, the datasets used in the experiment have distinct class distribution ratios. The results reveal that RRO yields fairly good performances across different types of datasets.
The comparison results reveal that the efficiency of various methods depends on the dataset, the number of genes utilized, and the parameter settings. It is noticed that none of the methods (including the proposed RRO) is constantly better than others.
6 Summary and Conclusions
In this study, a hybrid feature selection framework RRO is proposed for cancer classification. RRO utilizes ANOVA for selecting the initial set of class-discriminative genes. In the subsequent step, redundant genes are eliminated using Kendal’s \(\tau \) correlation. Finally, BWOA and SVM are employed to identify an optimized feature set for classification. It is observed that among the ten gene expression datasets, RRO reaches a perfect F1 score of 1.0 in five datasets. Besides, RRO realizes 100% classification accuracy in six datasets using a varied number of genes. The comparisons with the thirteen state-of-the-art methods on ten microarray datasets demonstrate that RRO exhibits better or comparable performance in terms of classification accuracy and the number of genes utilized. The results demonstrate that RRO is a highly effective approach for feature selection in the microarray dataset. The comparative analysis also reveals that the performances of various feature selection and classification methods are not consistent across datasets. The future work will involve investigating the performance of RRO in other types of gene expression datasets, such as the RNA-Seq dataset.
References
Al-Obeidat, F., Tubaishat, A., Shah, B., Halim, Z., et al.: Gene encoder: a feature selection technique through unsupervised deep learning-based clustering for large gene expression data. Neural Comput. Appl. 788, 1–23 (2020)
Alanni, R., Hou, J., Azzawi, H., Xiang, Y.: A novel gene selection algorithm for cancer classification using microarray datasets. BMC Med. Genomics 12(1), 10 (2019)
Almugren, N., Alshamlan, H.: FF-SVM: new firefly-based gene selection algorithm for microarray cancer classification. In: 2019 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), pp. 1–6. IEEE (2019)
Alomari, O.A., Khader, A.T., Al-Betar, M.A., Abualigah, L.M.: Gene selection for cancer classification by combining minimum redundancy maximum relevancy and bat-inspired algorithm. Int. J. Data Min. Bioinform. 19(1), 32–51 (2017)
Alshamlan, H., Badr, G., Alohali, Y.: MRMR-ABC: a hybrid gene selection algorithm for cancer classification using microarray gene expression profiling. Biomed Res. Int. 2015 (2015)
Alshamlan, H.M., Badr, G.H., Alohali, Y.A.: Genetic bee colony (GBC) algorithm: a new gene selection method for microarray cancer classification. Comput. Biol. Chem. 56, 49–60 (2015)
Alshamlan, H.M., Badr, G.H., Alohali, Y.A.: ABC-SVM: artificial bee colony and SVM method for microarray gene selection and multi class cancer classification. Int. J. Mach. Learn. Comput. 6(3), 184 (2016)
Aziz, R., Verma, C., Srivastava, N.: A novel approach for dimension reduction of microarray. Comput. Biol. Chem. 71, 161–169 (2017)
Chuang, L.Y., Yang, C.H., Wu, K.C., Yang, C.H.: A hybrid feature selection method for DNA microarray data. Comput. Biol. Med. 41(4), 228–237 (2011)
Dashtban, M., Balafar, M.: Gene selection for microarray cancer classification using a new evolutionary method employing artificial intelligence concepts. Genomics 109(2), 91–107 (2017)
Dudoit, S., Fridlyand, J., Speed, T.P.: Comparison of discrimination methods for the classification of tumors using gene expression data. J. Am. Stat. Assoc. 97(457), 77–87 (2002)
Ferreira, A.J., Figueiredo, M.A.: An unsupervised approach to feature discretization and selection. Pattern Recogn. 45(9), 3048–3060 (2012)
Gao, L., Ye, M., Lu, X., Huang, D.: Hybrid method based on information gain and support vector machine for gene selection in cancer classification. Genomics Proteomics Bioinform 15(6), 389–395 (2017)
Hussien, A.G., Hassanien, A.E., Houssein, E.H., Bhattacharyya, S., Amin, M.: S-shaped binary whale optimization algorithm for feature selection. In: Bhattacharyya, S., Mukherjee, A., Bhaumik, H., Das, S., Yoshida, K. (eds.) Recent Trends in Signal and Image Processing. AISC, vol. 727, pp. 79–87. Springer, Singapore (2019). https://doi.org/10.1007/978-981-10-8863-6_9
Jain, I., Jain, V.K., Jain, R.: Correlation feature selection based improved-binary particle swarm optimization for gene selection and cancer classification. Appl. Soft Comput. 62, 203–215 (2018)
Kar, S., Sharma, K.D., Maitra, M.: Gene selection from microarray gene expression data for classification of cancer subgroups employing PSO and adaptive k-nearest neighborhood technique. Expert Syst. Appl. 42(1), 612–627 (2015)
Khurma, R.A., Aljarah, I., Sharieh, A., Mirjalili, S.: EvoloPy-FS: an open-source nature-inspired optimization framework in python for feature selection. In: Mirjalili, S., Faris, H., Aljarah, I. (eds.) Evolutionary Machine Learning Techniques. AIS, pp. 131–173. Springer, Singapore (2020). https://doi.org/10.1007/978-981-32-9990-0_8
Lai, C., Reinders, M.J., Wessels, L.: Random subspace method for multivariate feature selection. Pattern Recogn. Lett. 27(10), 1067–1076 (2006)
Lai, C.M., Yeh, W.C., Chang, C.Y.: Gene selection using information gain and improved simplified swarm optimization. Neurocomputing 218, 331–338 (2016)
Lee, C.P., Leu, Y.: A novel hybrid feature selection method for microarray data analysis. Appl. Soft Comput. 11(1), 208–213 (2011)
Liu, H., Setiono, R.: Chi2: feature selection and discretization of numeric attributes. In: Proceedings of 7th IEEE International Conference on Tools with Artificial Intelligence, pp. 388–391. IEEE (1995)
Lu, H., Chen, J., Yan, K., Jin, Q., Xue, Y., Gao, Z.: A hybrid feature selection algorithm for gene expression data classification. Neurocomputing 256, 56–62 (2017)
Mirjalili, S., Lewis, A.: The whale optimization algorithm. Adv. Eng. Softw. 95, 51–67 (2016)
Moradi, P., Gholampour, M.: A hybrid particle swarm optimization for feature subset selection by integrating a novel local search strategy. Appl. Soft Comput. 43, 117–130 (2016)
Mundra, P.A., Rajapakse, J.C.: SVM-RFE with MRMR filter for gene selection. IEEE Trans. Nanobiosci. 9(1), 31–37 (2009)
Nguyen, T., Khosravi, A., Creighton, D., Nahavandi, S.: Hidden Markov models for cancer classification using gene expression profiles. Inf. Sci. 316, 293–307 (2015)
Pedregosa, F.: Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011)
Peng, H., Long, F., Ding, C.: Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 27(8), 1226–1238 (2005)
Salem, H., Attiya, G., El-Fishawy, N.: Classification of human cancer diseases by gene expression profiles. Appl. Soft Comput. 50, 124–134 (2017)
Sazzed, S.: ANOVA-SRC-BPSO: a hybrid filter and swarm optimization-based method for gene selection and cancer classification using gene expression profiles. In: Proceedings of the Canadian Conference on Artificial Intelligence (2021). https://caiac.pubpub.org/pub/hay53dvq, https://caiac.pubpub.org/pub/hay53dvq
Sharbaf, F.V., Mosafer, S., Moattar, M.H.: A hybrid gene selection approach for microarray data classification using cellular learning automata and ant colony optimization. Genomics 107(6), 231–238 (2016)
Shreem, S.S., Abdullah, S., Nazri, M.Z.A.: Hybridising harmony search with a Markov blanket for gene selection problems. Inf. Sci. 258, 108–121 (2014)
Shreem, S.S., Abdullah, S., Nazri, M.Z.A.: Hybrid feature selection algorithm using symmetrical uncertainty and a harmony search algorithm. Int. J. Syst. Sci. 47(6), 1312–1329 (2016)
Shreem, S.S., Abdullah, S., Nazri, M.Z.A., Alzaqebah, M.: Hybridizing RELIEFF, MRMR filters and GA wrapper approaches for gene selection. J. Theor. Appl. Inf. Technol. 46(2), 1034–1039 (2012)
Sun, L., Zhang, X., Qian, Y., Xu, J., Zhang, S.: Feature selection using neighborhood entropy-based uncertainty measures for gene expression data classification. Inf. Sci. 502, 18–41 (2019)
Wang, Y., Yang, X.G., Lu, Y.: Informative gene selection for microarray classification via adaptive elastic net with conditional mutual information. Appl. Math. Model. 71, 286–297 (2019)
Xiaofei, H., Deng, C., Partha, N.: Laplacian score for feature selection. In: Advances in Neural Information Processing Systems, pp. 507–514 (2005)
Yassi, M., Moattar, M.H.: Robust and stable feature selection by integrating ranking methods and wrapper technique in genetic data classification. Biochem. Biophys. Res. Commun. 446(4), 850–856 (2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this paper

Cite this paper
Sazzed, S. (2022). Feature Selection in Gene Expression Profile Employing Relevancy and Redundancy Measures and Binary Whale Optimization Algorithm (BWOA). In: Li, B., et al. Advanced Data Mining and Applications. ADMA 2022. Lecture Notes in Computer Science(), vol 13087. Springer, Cham. https://doi.org/10.1007/978-3-030-95405-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-95405-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-95404-8
Online ISBN: 978-3-030-95405-5
eBook Packages: Computer ScienceComputer Science (R0)