
Abstract
Genomic imprinting is a remarkable phenomenon through which certain genes show monoallelic expression depending on their parent of origin. While imprinting may have evolved for viviparity and potentially as a mechanism to balance resource allocation in mammals, functional haploidy presents a clear risk to human health. Both epigenetic and genetic and aberrations at imprinted loci contribute to genomic imprinting disorders, such as Beckwith–Wiedemann, Silver–Russell, Prader–Willi and Angelman syndromes. Beyond these well-documented disorders, changes in the tissue-specific expression levels of imprinted genes may contribute far more widely to human disease. The expression of imprinted genes can be disrupted at the level of a single gene, at the level of an imprinted domain or through changes in imprinted gene networks. Importantly, imprinted genes can respond to prenatal adversity leading to persistent changes in gene expression. Consequently, in addition to identifying the functions of individual imprinted genes, it is important to understand the mechanisms through which imprints are established, maintained and erased, with erasure critical to ensure comprehensive erasure of epimutations in the germline. We review the critical aspects of genomic imprinting and imprinted human diseases as a paradigm for future studies on epigenetics of human development and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Genomic imprinting
- Imprinted domains
- Imprinted gene networks
- Imprinting disorders
- Germline
- DNA methylation
1 Introduction
One of the remarkable discoveries of the twentieth century was the finding that some genes in mammals do not obey the rules of Mendelian inheritance. Accordingly, “imprinted” genes classically exhibit monoallelic expression in a parent-of-origin dependent manner [1,2,3]. Some imprinted genes are monoallelically expressed in all expressing cell types, while others show partial, tissue-specific, and/or temporal imprinting. Their most critical and fundamentally important characteristic is that parent-of-origin expression is dependent on passage through the parental germline [1]. Functional differences between the parental genomes were first experimentally demonstrated using pronuclear transfer experiments to artificially generate diploid mouse embryos whose genomes were exclusive of either maternal or paternal origin [4,5,6,7,8,9]. Development of these monoparental embryos was initially relatively normal. However, embryos consisting of two maternally-derived genomes became progressively growth-restricted and died around embryonic day (E) 10, with particularly limited development of extra-embryonic tissue. Conversely, embryos with two paternal copies were both developmentally delayed and growth restricted, dying around E8.5 with an abundance of extra-embryonic tissue. Similarly, monoparental embryonic stem cells in chimeras with wild-type cells allocate to essentially reciprocal regions in chimeric embryos [10, 11], highlighting the functional differences and requirement for both parental genomes. Studies of uniparental disomic (UPD) embryos delineated parent-of-origin functional differences to specific chromosomal regions [12], with these regions found to contain domains of maternally and paternally expressed genes [13,14,15].
The discovery of imprinted genes, along with their phylogenetic distribution with an increasing knowledge of their functions, has led to considerable speculation concerning the evolution of genomic imprinting with two prevalent explanations being genetic conflict over maternal resources [16, 17] and maternal-offspring adaptation [18]. These, and many other theories that attempt to ascribe the importance of genomic imprinting, account for the intimate and complex relationship between the mammalian mother and her offspring with increasing investment toward viviparity dependent on an elaborate placenta and considerable postnatal nurturing [19] (Fig. 8.1). An important aspect of imprinting is the control of gene dosage by epigenetic marks inherited from the parental germline. Epigenetic marks under some circumstances can undergo modifications in response to environmental cues such as low protein diet in pregnancy [20], with a potential to affect fetal growth and other phenotypic consequences later in life. The consequent flexibility conferred by the epigenetic mechanism may be advantageous for supporting adaptation to different environments, but may also be responsible for diseases. Understanding the mechanisms that establish, maintain, and erase imprinting is therefore important for our understanding of some human diseases.

Genomic imprinting and the increasing investment in viviparity in mammals. Within vertebrate lineages, genomic imprinting has only been observed in marsupials and, more prominently, in eutherians with prolonged fetal development in utero before birth. Mya—million years ago
2 Establishment of Canonical Imprinting
Canonical imprinting resulting in monoallelic gene expression depends on the establishment of heritable DNA methylation marks in either the maternal or paternal germline, and these differentially methylated regions (gDMRs) are propagated in post-zygotic cells [1, 21, 22] (Fig. 8.2). In mice de novo DNA methylation at gDMRs is initiated in the male germline before birth [23,24,25] and detectable as DNA methylation more broadly at intergenic sequences and transposons [26]. For the two paternally imprinted loci, H19-DMR and IG-DMR, changes in histone modifications and high transcriptional read-through precede DNA methylation [27] by the de novo DNA methyltransferase DNMT3A and a DNMT-like protein DNMT3L; the latter lacks enzymatic activity [28,29,30,31]. De novo paternal methylation of the rodent-specific Rasgrf1 gDMR requires components of the PIWI-interacting RNA pathway which normally silences transposable elements in the male germline [32] and a second de novo DNA methyltransferase, DNMT3B [24]. For the H19-DMR and IG-DMR, these sequences acquire DNA methylation as part of a global mechanism methylating the mature sperm genome to an average level of ~90% [26]. A recent study showed recruitment of the DNMT3A/3L complex extensively to the genome of male germ cells by histone H3 lysine 36 dimethylation (H3K36me2) marks broadly deposited by the histone methyltransferase NSD1 [33].

The imprinting cycle of establishment, maintenance, spreading and erasure. DNA methylation imprints are initially established in the male and female germlines. After fertilization, imprints evade the waves of genome-wide reprogramming to become germline differentially methylated regions (gDMRs). These gDMRs act as imprinting centers from which the imprints “spread” to neighboring genes, establishing domains of maternally and paternally expressed genes. These gDMRs are maintained for the lifetime of the animal along with the monoallelic expression of a subset of imprinted genes. For many genes, monoallelic gene expression does not persist and/or can be tissue-specific
De novo DNA methylation in the female germline occurs after birth, during the growth phase of oogenesis [34]. Between birth and adulthood, the average genomic DNA methylation level goes from ~2% in non-growing oocytes, to nearly 40% in fully grown, germinal vesicle oocytes [26, 35]. DNA methylation is preceded by the loss of histone H3 lysine 4 di/trimethylation (H3K4me2/3), and gain of trimethylation at lysine 36 (H3K36me3) [36]. These domains with the H3K36me3 marks are established over transcribed gene bodies by the histone methyltransferase SETD2, which associates with the elongating RNA polymerase II [37, 38]. The recruitment of DNMT3A/3L complex follows to the H3K36me3-marked regions to catalyze de novo DNA methylation [28,29,30,31, 34, 39,40,41], resulting in a characteristic oocyte DNA methylome with a strong correlation between DNA methylation and the levels of transcription [26, 42]. All maternal gDMRs discovered to date are CpG-rich promoter elements [43], and each of them is covered by a transcript initiating from nearby oocyte-specific promoters [42]. Transcription through gDMRs has been shown experimentally to be functionally required for de novo DNA methylation at 6 maternal germline DMRs: Nespas/Gnasxl and 1A, KvDMR1, PWS-IC, Zac1 igDMR, and Peg3DMR [42, 44,45,46,47,48]. The generality of this mechanism for all maternal gDMRs is furthermore supported by the loss of all maternal imprints in oocytes deficient for DNMT3A, DNMT3L, or SETD2 [29, 35, 37].
3 Maintenance
After fertilization, both parental genomes undergo extensive passive (maternal) and active (maternal and paternal) DNA demethylation [49]. However, DNA methylation at gDMRs survives this reprogramming, which is not seen for the rest of gametically methylated regions. The protection of DNA methylation at gDMRs is, in part, mediated by the zinc-finger proteins ZFP57 and ZFP445, which bind to a methylated TGCCGC consensus motif within gDMRs [50,51,52,53,54]. These KRAB zinc finger proteins recruit TRIM28 (also known as KAP1) and the histone methyltransferase SETDB1. Consequently, the recruitment of this protein complex at the methylated gDMRs introduces H3K9me3 marks in the region, which promotes preferential recruitment of the DNMT1 DNA methylation maintenance machinery. DNA methylation at gDMRs is subsequently propagated for the lifetime of the individual through the repeated action of the maintenance DNA methyltransferase DNMT1 [55,56,57] in conjunction with UHRF1 (also known as NP95) [58].
4 Spreading
gDMRs operate as imprinting centers (ICs; also known as imprinting control regions) to establish domains of maternally and paternally expressed genes [13,14,15]. In mice, there are estimated to be 21 gDMRs originating during oogenesis, while three of them are inherited from sperm. These epigenetic marks regulate the allelic expression of around 100 non-coding transcripts and protein-coding genes [15, 59, 60]. There is strong evidence that approximately fifteen of these domains are conserved in humans, while some other are unique to humans, notably with uniparental expression of genes restricted to the placenta [61]. While not all gDMRs have been functionally demonstrated to act as imprinting centers in mice, there are six that have been studied through targeted deletion and have been found to regulate imprinting of the domains within which they reside: Airn/Igf2r, Igf2/H19, Snrpn, Kcnq1ot1/Cdkn1c, Dlk1/Gtl2, and Gnas cluster [62,63,64,65,66,67]. Both spreading and maintenance of imprinted expression must be carried out in cis because the two alleles exist in the same nuclear environment. Some domains contain long non-coding RNAs (lncRNAs) [68] with the expression of the unmethylated parental allele. Some of these lncRNAs have been shown to have a role in the imprinting of a domain containing several imprinted loci, while others lncRNAs appear not to play such a direct epigenetic role [68,69,70,71,72,73,74,75,76,77].
5 Erasure
Erasure of imprints is prerequisite in each germline cycle in preparation for the establishment of new epigenetic marks according to the sex of the embryo. The erasure of DNA methylation imprints occurs in the precursors of the germline, the primordial germ cells (PGCs). Genome-wide analyses of DNA methylation in mouse PGCs showed that not all genomic regions follow the same demethylation kinetics and that near-complete erasure of the genome occurs in two distinct waves. In phase I of reprogramming, from E8.5 to E10, the genome of migrating PGCs is gradually demethylated by passive dilution of 5mC marks [25, 78]. Specific genomic sequences—called late-demethylating—are largely protected from this first wave of passive demethylation. These include germline-specific genes, X-linked CpG islands (CGIs) in female embryos, specific families of repetitive elements, and imprinted gDMRs [25]. The beginning of phase II of demethylation coincides with the colonization of the genital ridges by PGCs [79]. It is characterized by a rapid conversion of 5mC to 5-hydroxymethylcytosine (5hmC) by the ten-eleven translocation dioxygenase TET1, followed by a passive loss of this epigenetic mark, which is not maintained by DNMT1 [80,81,82,83]. From E10.5 to E13.5, the methylation levels of imprints fall from 70% to ~10% on average, while the genome reaches its lowest level of global DNA methylation known, at close to only 3% [25]. A strikingly similar dynamic of demethylation has also been observed in human PGCs, suggesting that epigenetic reprogramming of the PGC genome is a conserved process, but the temporal sequence and possibly the mechanism might differ from the observations in mice [84]. Failure to erase imprints in gonocytes leads to abnormal expression of imprinted genes and embryonic lethality in the next generation because of the direct inheritance of abnormal grand-parental imprints [81, 85].
So far, TET1 is the only factor known to be implicated in phase II reprogramming in gonocytes in mice. A careful analysis of the progeny from Tet1-null mice revealed a broad spectrum of embryonic and postnatal phenotypes, suggesting abnormal expression of imprinted genes [85]. The analysis of DNA methylation patterns at imprinted gDMRs revealed that loss of TET1 in one of the parents is associated with abnormal biallelic DNA methylation at gDMRs in the offspring [85, 86]. These important results established the essential function of TET1 in imprint erasure since they are consistent with a failure to erase grand-parental imprints, which are then abnormally passed on to the progeny: perdurance of grand-paternal DNA methylation marks would be manifested when inherited maternally from Tet1-null females, and the converse for grand-maternal marks. The precise timing and mechanism of imprints erasure in the human germline merits further investigation.
6 Portrait of an Imprinted Domain: Mouse Distal Chromosome 7
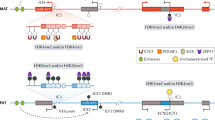
The imprinted region on mouse distal chromosome 7/human chromosome 11p15 has been the focus of considerable study in both humans and mice due to links with the imprinting disorders Beckwith–Wiedemann Syndrome (OMIM 130650; BWS) and Silver-Russell Syndrome (OMIM 180860; SRS), which both show parent-of-origin features. Evidence for the presence of imprinted genes at this chromosomal location came from studies on UPD7 mice which suggested the presence of more than one imprinted gene [87, 88]. Maternal UPD of the distal chromosome 7 region resulted in significant impairment of fetal and placental growth, with fetal death by embryonic day (E) 17.5 [88]. Paternal UPD of the same region resulted in delayed embryonic development with lethality by E10.5, alongside significant developmental abnormalities in the placenta [88,89,90]. We now know that this region contains two adjacent but mechanistically distinct imprinted domains (Fig. 8.3A). Moreover, these domains best exemplify what is known about the mechanisms that establish domain-wide imprinting.

The imprinted domains on mouse distal chromosome 7. (A) The imprinted regions on mouse distal chromosome 7 showing the organisation and direction of imprinting of genes within the IC1 and IC2 domains. (B) IC1 domain imprinting involves differential conformation of the paternal and maternal alleles dependent on methylation-sensitive binding of CTCF. (C) IC2 domain imprinting requires the transcription of the long, non-coding RNA Kcnq1ot1 from the paternal allele, which is thought to attract the chromatin complexes PRC1 and PRC2 that add silencing histone marks along the domain, and may involve physical tethering to nucleoporins
7 The IC1 Imprinted Domain
In mice, the IC1 domain spans over 100 kilobases and contains the paternally expressed protein-coding genes Igf2 [87, 91] and Ins2 [92], and the maternally expressed non-coding RNA H19 [93]. Contained within H19 is a functionally important imprinted microRNA, miR-675 [94]. The gDMR within the IC1 domain, called H19-DMR, is located approximately 2 kb upstream of the H19 promoter. This region acquires DNA methylation exclusively in the male germline, resulting in paternal-allele-specific DNA methylation in offspring [95]. Other regions of differential DNA methylation in the IC1 domain include the promoter of H19 on the silenced paternal allele, three other DMRs within the body of the Igf2 gene also on the paternal allele which, in the case of Igf2, is the expressed allele. These sites in sperm are unmethylated but they acquire methylation after implantation and are referred to as somatic DMRs (sDMR) [96]. Maternal inheritance of targeted deletions of H19-DMR results in biallelic Igf2 and Ins2 expression alongside loss-of-expression of H19, even when the H19 promoter is left intact [63, 97]. The physical arrangement of paternally and maternally expressed genes at this locus and the observation of similar expression patterns for Igf2 and H19 in the developing embryos led to the idea that these genes compete for the same enhancers [97]. H19-DMR contains binding sites for zinc finger protein CCCTC-binding factor (CTCF) that is conserved amongst mammals [98,99,100]. Binding of CTCF to H19-DMR only occurs when this region is unmethylated [98,99,100]. Accordingly, the binding of CTCF is thought to organize the domain into a specific chromatin conformation [101,102,103,104] which permits expression of only H19 from the maternal allele [102, 105, 106]. In the absence of CTCF binding, a different conformation is adopted, resulting in the expression of Igf2 and Ins2 but not of H19 (Fig. 8.3B). Similar mechanism involving a structural conformation dependent on methylation-sensitive CTCF binding regulates imprinted expression of the paternally methylated Dlk1-Dio3 domain [107].
8 The IC2 Imprinted Domain
The IC2 domain spans approximately 800 kb and contains a number of protein-coding genes with preferential expression of the maternally inherited allele, including Phlda2 [108], Cdkn1c [109] and Ascl2 [110], and the paternally-expressed lncRNA, Kcnq1ot1 (aka Lit1) [65] (Fig. 8.3A). The gDMR within the IC2 domain is known as KvDMR1 (Kv-differentially methylated region 1) [65, 111, 112]. KvDMR1 is methylated on the maternal allele and contains the promoter for Kcnq1ot1 [111]. Paternal inheritance of a targeted deletion of this region in mice results in biallelic expression of the normally maternal-expressed genes upstream and downstream of the gDMR, and the loss of repressive histone modifications that normally coat the paternal allele [65, 75, 113,114,115]. Unlike in the IC1 domain, transcription of the lncRNA Kcnq1ot1 rather than the DMR itself, is required to establish and maintain paternal silencing of the IC2 domain [75, 76]. All the protein-coding IC2 domain genes require DNA methylation in cis on the maternal KvDMR1 allele for their maternal expression [28,29,30, 116,117,118,119]. One of the consequences of Kcnq1ot1 expression is the acquisition of a somatic DNA methylation mark on the silent paternal allele of Cdkn1c [120]. In mice deficient for the HELLS helicase (aka LSH), this somatic DMR is not appropriately established resulting in the biallelic expression of Cdkn1c [121]. Furthermore, the maintenance of this somatic DMR by DNMT1 is required for the continued silencing of the paternal allele of Cdkn1c during development [120, 122, 123]. This additional layer of epigenetic gene silencing suggests the importance of sustaining imprinting of Cdkn1c into adulthood.
The loss of domain-wide imprinting at the IC2 domain mirrors the earlier findings for the imprinted locus spanning the maternally-expressed Igf2r gene, whereby truncation of the lncRNA Airn resulted in biallelic expression of Igf2r in the embryo and placenta, and of the downstream genes Slc22a2 and Slc22a3, normally imprinted and maternally expressed in the placenta [69]. Various models have been proposed to explain how the expression of paternal lncRNAs mediates domain-wide imprinting. The presence of lncRNAs overlapping with and oppositely transcribed to protein-coding genes, as observed for Kcnq1ot1/Kcnq1 and Airn/Igf2r, suggested the possibility of transcriptional interference [124], supported by the finding that Airn transcription through the Igf2r promoter was necessary to silence Igf2r [73]. However, while this could account for imprinted genes with promoters overlapping with lncRNAs, for the majority of loci there are imprinted genes that lie upstream of the lncRNA and ones that are too far downstream to be overlapping with the transcription of the lncRNA itself. An alternative idea was that lncRNAs function by disrupting the promoter-enhancer interactions required for active gene transcription, although there are no enhancers for Slc22a2 or Slc22a2 within the Airn transcribed region [125], and enhancers for Cdkn1c lie outside the region spanned by Kcnq1ot1 [126]. LncRNAs may instead prevent the expression of nearby genes in cis by forming RNA “clouds” that coat the domain and recruit inactivating histone-modifying complexes [114, 127,128,129] (Fig. 8.3C). LncRNAs such as Kcnq1ot1 may additionally function by physically localizing and tethering the paternal allele to the nuclear periphery [127, 130, 131]. There is some evidence that tethering involves complexing with nucleoporins which are the main components of nuclear pore complexes embedded in the nuclear membrane [132]. While posttranscriptional knockdown of Kcnq1ot1 by RNAi does not impact imprinted gene expression in stem cells [133], conditional deletion of KvDMR1/IC2 in the early mouse embryo results in loss of both paternal gene silencing and acquisition of somatic DNA methylation [134], which suggests differences in the epigenetic mechanisms involved in short and longer-term silencing of the IC2 domain.
9 Non-canonical Imprinting
Recent genome-wide next-generation sequencing screens have identified genes exhibiting parent-of-origin behavior that lie outside imprinted domains and are not clearly associated with gDMRs [22, 135, 136]. These are termed non-canonical imprinted genes and their imprinted expression is mostly transient, generally restricted to the early embryo and extra-embryonic lineages. These genes rely on allelic histone modification, particularly histone H3 lysine 27 trimethylation (H3K27me3), following direct inheritance from the oocyte [137,138,139]. These observations revealed that inheritance of DNA methylation is not the only epigenetic mark for the direct inheritance from the mature gametes to regulate the expression of imprinted genes during development. The functional relevance of non-canonical imprinting in humans merits further investigation [140, 141].
10 Evolution of Genomic Imprinting
The evolution of genomic imprinting remains a fascinating subject for discussion since imprinting leads to monoallelic expression of specific genes with critical roles in development. Why would evolution favor a system that is vulnerable to recessive mutations [16, 17], and why is this specific phenomenon observed in mammals and flowering plants [142]. Clues may come from the observation that genomic imprinting leading to parent-of-origin-specific expression in not universal to all mammals and has not been reported in egg-laying mammalian species, such as monotremes, platypus and echidnas [143]. The emergence of imprinting in mammals predates the bifurcation between metatherians (marsupials) and eutherians (placentals), with some imprinted genes being shared amongst them. This suggests a link to the emergence of viviparity and fetal development in utero, leading to new energetic demands on the pregnant female [144] (Fig. 8.1). A few “classical” imprinted genes appear to be well conserved in mammals, but several species-specific differences have also been observed.
With respect to mechanisms, two different mechanisms that might have contributed to the emergence of species-specific imprinted genes, both related to transcription-coupled de novo DNA methylation in oocytes, have been described. The first mechanism implicates the generation of a new gene via retrotransposition within the intron of a host gene. Since this host gene is expressed in growing oocytes, the inserted retrogene acquires a maternal gDMR and becomes imprinted and paternally expressed in the progeny. Four different examples have been documented, one of which is rodent-specific (Zrsr1/U2af1-rs1), while the other three are more evolutionarily ancient, and also imprinted in human (Mcts2, Nap1l5, Inpp5f_v2) [145,146,147,148]. Although the retrotransposition event might have triggered the formation of a new imprinted gene, it is also possible that the necessary signals were already present at the ancestral host gene, although these are not imprinted themselves.
A second mechanism is based on the insertion of a new oocyte-specific promoter next to a host gene promoter. Here, the oocyte promoter is provided by a long terminal repeat (LTR) retrotransposon. Specific families of LTR elements evade silencing mechanisms operating in oocytes and act as promoters for diverse transcripts in growing oocytes. Since LTR elements are highly polymorphic and different families of elements have colonized different lineages during evolution, they have been shown to be responsible for species-specific differences in the DNA methylome of oocytes, which is guided by transcription [149]. Notably, specific LTR elements drive oocyte transcription through a number of species-specific gDMR. Indeed, a comparison of mouse and human maternal gDMRs has identified 4 mouse-specific and 17 human-specific examples of paternally-expressed imprinted genes acquiring DNA methylation in oocytes as a consequence of a nearby LTR promoter [61]. These results suggest that novel imprinted genes can be generated during evolution at least in part via the insertion of an active LTR promoter through retrotransposition. The targeted deletions of the upstream LTR at the imprinted genes Slc38a4 and Impact lead to loss of imprinting and biallelic expression, providing functional support for this model [61].
Once established, further elaboration of canonical imprinted genes and arrangements in domains can be envisaged. Regulated by gDMRs, three essential requirements must be met for functional imprinting to evolve: (i) acquisition of a DNA methylation imprint in one germ line; (ii) maintenance of the gDMR through the preimplantation phase of epigenetic erasure; and (iii) prevention of acquisition of DNA methylation on the unmethylated allele during postimplantation waves of de novo methylation [150].
Endogenous retroviruses have also been implicated in the regulation of allelic expression at non-canonical imprinted genes, which are all paternally expressed and show imprinted expression only in preimplantation embryos and extra-embryonic lineages. More importantly, imprinting in this case is independent of gametic DNA methylation and relies instead on direct inheritance of repressive H3K27me3 histone marks from the oocyte [137], but these imprints are transient and do not persist after implantation. The expression of paternally inherited non-canonical imprinted genes in the extra-embryonic ectoderm showed that the maternal allele acquires DNA methylation as a somatic DMR, via DNMT3A/3B activity [138, 139]. So the germline imprint at those genes with a histone mark (H3K27me3) is eventually replaced by a DNA methylation mark for maintenance of imprinted expression in the extra-embryonic tissues. Furthermore, this epigenetic mechanism preferentially targets endogenous retroviruses, specifically ERVK LTRs [138]. These repetitive elements, which can act as promoters or enhancers in extra-embryonic tissues, appear to guide this unusual form of tissue-specific imprinting by protecting the paternal allele from DNA methylation-mediated silencing, which is the default pathway in embryonic lineages. Since endogenous retroviruses are implicated in non-canonical imprinting, we can expect this mechanism to regulate mostly species-specific imprinted genes.
11 The Function of Imprinted Genes in Mouse Development
Several imprinted genes function in a dosage-sensitive manner to regulate fetal growth and placental development early in life, and to influence both metabolic and behavioral processes later in life including those relating to mothering, as has been extensively and elegantly reviewed [22, 151,152,153,154,155,156]. As with imprinting mechanisms, the function of imprinted gene located within the mouse distal chromosome 7 epitomize the function of imprinted genes more generally.
12 IC1 Domain Genes
Igf2 was one of the first imprinted genes to be identified in mice [87, 91], along with H19 [93] and Igf2r [157]. Igf2 encodes a fetal growth factor structurally related to insulin that normally signals to promote cell proliferation, growth, differentiation, and survival via IGF1R. IGF2 also binds to IGF2R, which sequesters and degrades excess IGF2 [158]. The finding that IGF2R is a maternally-expressed imprinted gene [157] was instrumental in supporting the idea that genomic imprinting evolved in response to a parental “tug-of-war” imposed by the development of mammalian offspring in utero [16, 17]. While deletion studies were important for understanding the normal function of IGF2, manipulating the dosage of Igf2 can provide greater insight into the function of imprinting. In mice, loss of imprinting (LOI) of Igf2 results from disruption of H19-DMR. Expression of both Igf2 and Ins2 occurs at approximately twice the normal level in this model with concomitant loss of H19, resulting in increased birth weight of between 8 and 30% together with placental overgrowth [97, 159]. Fetal growth is supported by the placenta which functions both in the transport of nutrients and in securing the availability of nutrients through the action of placental hormones on the mother [160]. IGF2 positively regulates the development of the region of the placenta involved in nutrient transport [161, 162]. Together, these experiments identify Igf2 as a paternally-expressed imprinted gene that normally functions to promote fetal growth and instruct changes in the placenta required to enhance nutrient transport.
H19 was the first noncoding transcript to be identified as an imprinted gene, and one of the most abundant polyadenylated RNAs in the developing mouse embryo [93, 163]. The precise function of H19 has been challenging to study because of the mechanistic and reciprocal link with Igf2 imprinting. Accordingly, a deletion to facilitate the loss of H19 resulted in upregulation of Igf2 alongside an overgrowth phenotype despite not disrupting the gDMR [159]. Mice lacking the H19 transcript appear to develop normally [164] but there is evidence that H19 functions as a tumor suppressor with the loss of H19 associated with faster progression in mouse tumorigenesis models [165]. H19 function may, in part, be mediated by the microRNA mir-675 which is processed from the first exon of the H19 transcript in both mice and humans [94, 166, 167]. In mice, loss-of-function of Ins2 alone does not lead to developmental or metabolic dysfunction [168, 169] but when combined with ablation of the related Ins1 gene, pups are born small and die shortly thereafter from neonatal diabetes [168].
13 IC2 Domain Genes
Loss of imprinting of the IC2 domain, in which all paternally-silenced IC2 domain genes become biallelically expressed, results in both fetal and placental growth restriction [65]. Within this domain, the function of Cdkn1c, Phlda2, and Ascl2 has been studied most intently. Cdkn1c encodes a cyclin-dependent kinase inhibitor, which normally suppresses proliferation and promotes differentiation in a wide range of tissues. Ablation of Cdkn1c in mice results in fetal overgrowth, placentomegaly with expansion of placental endocrine lineages, abdominal wall defects, cleft palate, renal dysplasia, adrenal cytomegaly, maternal preeclampsia, and premature birth [170,171,172,173,174,175]. Transgenic mice expressing Cdkn1c at twice the normal level are growth restricted identifying Cdkn1c as the major regulator of embryonic growth within the IC2 domain [176]. These experiments in mice identified Cdkn1c as a maternally-expressed imprinted gene that normally functions to restrain fetal growth and placental development. Imprinting of Cdkn1c (reduced paternal expression) would therefore be predicted to enhance fetal and placental growth, and potentially support longer gestation.
Phlda2 encodes a PH domain-only protein normally maternally expressed in the mouse and human placenta where expression is highest [108, 177]. Phlda2 functions to limit expansion of a major placental endocrine lineage in mice [178,179,180,181]. Precisely regulated expression of Phlda2 is necessary for normal fetal growth with both loss- and gain-in-expression resulting in growth restriction [179, 181, 182]. Just two-fold expression of Phlda2 driven by a transgene resulted in >10% reduction in birthweight with relative sparing of the head, neonatal hypoglycemia followed by catch-up growth [182]. Rather than playing a direct role in regulating fetal growth, fetal growth restriction occurs as a consequence of the placental endocrine insufficiency induced by the reduction in placental endocrine cells. In addition to regulating placental development and fetal growth, Phlda2 plays a critically important role in instructing maternal caregiving behavior through controlling the production of placental hormones that act on the mother. Wild-type dams carrying and caring for offspring with different doses of Phlda2, behave atypically toward their offspring [183]. Dams carrying and caring for Phlda2 knockout offspring with an expanded placental endocrine compartment engage in more pup-focused behaviors whereas dams carrying and caring for Phlda2 transgenic offspring with a reduced placental endocrine compartment neglect their pups and prioritize nest building [183]. A direct role for imprinted genes in regulating maternal behavior has previously been highlighted by studies on the paternally expressed genes Peg1 and Peg3 but in both these examples, the dam was genetically altered [184, 185]. In the case of the Phlda2 studies it is the genetically altered offspring that influence their mother’s behavior before birth, consistent with the function of placental hormones in programming the maternal brain in pregnancy. High-quality maternal care is important for the offspring’s later life behavior, and both transgenic Phlda2 offspring and their wild-type siblings sharing the same abnormal environment were found to exhibit anxiety-like symptoms, mild depression, atypical social behavior, and reduced cognitive abilities as adults [186]. Together, these studies illustrate the far-reaching roles of imprinted genes both within and across generations.
Ascl2 encodes a bHLH transcription factor imprinted in the mouse placenta but not the fetus [110]. Ascl2 plays a critically important role in the placenta supporting the development of the endocrine lineages with full ablation resulting in embryonic lethality at E9.5 [110, 187, 188]. More modest reductions in the expression level of Ascl2 are also associated with placental defects alongside fetal growth restriction [189, 190]. Conversely, overexpression of Ascl2 results in reduced development of several placental endocrine lineages and fetal growth restriction [191]. These experiments indicate that a single dose of Ascl2 in the placenta is required for the proper allocation of progenitors to the different placental endocrine lineages required to support optimal fetal growth.
Cdkn1c, Ascl2, and Phlda2, through limiting the developing of the placental endocrine lineages, all function to limit the production of placental hormones. Consequently, alterations in their dosage can impact both fetal growth and maternal adaptations to pregnancy which in human populations may manifest as classic complications of pregnancy [192].
14 Imprinted Genes Influencing Adult Physiology and Behavior
In addition to these fundamentally important roles during gestation, imprinted genes also function to influence behavioral [22] and metabolic processes [193] critically important for the survival of mammals. Briefly, both Igf2 and Cdkn1c are expressed in the developing and adult nervous system and both genes are important for adult neurogenesis, with IGF2 functioning both as a paracrine and autocrine factor [194] while Cdkn1c is required for neural stem cell quiescence and differentiation [195]. Cdkn1c is also required for the maturation of midbrain dopamine neuronal cells [196]. Consistent with important roles during brain development and into adulthood, manipulating the expression of these genes results in alterations in behavior. Increased dosage of Cdkn1c has been linked to altered reward behaviors and social functions in mice [197,198,199] and Igf2 has been shown to be important for memory consolidation and cognitive function [200]. There is evidence for expression of the normally silenced allele of these two genes in the adult brain with the maternal expression of Igf2 in the choroid plexus and leptomeninges [201], functionally important for neurogenesis [194], and paternal expression of Cdkn1c in the brain important for neocortical development [202]. In addition to the nervous system, both Igf2 and Cdkn1c are expressed during development in tissues with important metabolic functions. For example, Igf2 and Cdkn1c are both relatively highly expressed in the developing pituitary [203] where Cdkn1c regulates progenitor proliferation but not differentiation [204]. Igf2 null mice possess more brown adipose tissue just prior to their birth [205] while Cdkn1c is both required for the proper differentiation of intrascapular brown adipose and stimulates “browning” within white adipose depots in mice [206]. Igf2 functions in different lineages to regulate both the size and function of the pancreas [207] while Cdkn1c regulates cell cycle exit of progenitors during the early stages of pancreas formation [208]. These multi-fold, multi-tissue roles mean that precisely regulated expression of Igf2 and Cdkn1c is key not only for early development but also for later life metabolism and behavior.
15 Variations in Genomic Imprinting and Human Disease
15.1 Genomic Imprinting Disorders
A number of classic congenital disorders are associated with alterations in imprinted domains, involving both genetic and epigenetic abnormalities in patients (Table 8.1) [209, 210]. These disorders classically exhibit parent-of-origin transmission, where transmission is possible, with clinical features commonly affecting growth, metabolism, and behavior consistent with the identified functions of many imprinted genes.
16 Beckwith–Wiedemann Syndrome (BWS; OMIM #130650)
One of the most common imprinting disorders is BWS, which is associated with a variety of genetic and/or epigenetic alterations localized to human chromosome 11p15.5 [210] (Fig. 8.4). BWS is estimated to occur in one in every 15,000 births and primarily involves overgrowth features [211]. BWS is usually diagnosed based on the presence characteristics including macrosomia (birth weight >97th percentile), macroglossia (unusually large tongue), neonatal hypoglycemia, ear creases or pits and abdominal wall defects, hemihypertrophy (one side of the body or a part of one side of the body is larger than the other), visceromegaly, nervus flammeus (port-wine stain), cleft palate, cardiac abnormalities, advanced bone age, enlarged placenta and abnormalities in placental vasculature. There is a high incidence of premature birth for BWS infants, sometimes in combination with polyhydramnios (excessive amniotic fluid) and gestational hypertension [212], with some BWS mother’s suffering the potentially life-threatening disorder of preeclampsia with HELLP (hemolysis, elevated liver enzymes, and low platelet count) [213]. Infants are at increased risk of developing congenital/childhood tumors such as Wilms’ tumor, adrenocortical carcinoma, hepatoblastoma, and neuroblastoma, although this risk decreases with age. Five to ten percent of BWS patients exhibit gain of methylation at H19-DMR (IC1), predicted from mouse studies to result in overexpression of IGF2, while 40–50% of patients have loss of DNA methylation at KvDMR1 (aka IC2) predicted to result in loss of expression of all the IC2 domain protein-coding genes. Although several BWS patients may sporadically lose DNA methylation at IC2 as an epimutation, recent evidence points to cis-acting genetic causes in some cases. Since IC2 is located in an intron of the KCNQ1 gene, implicated in long QT syndrome-1 [111], de novo establishment of the maternal DNA methylation imprint at IC2 is predicted to be guided by transcription through the region during oocyte growth, possibly from a KCNQ1 transcript initiating at its canonical start site. Such a model is in fact supported by studies of mouse mutants carrying the insertion of a transcriptional termination sequence adjacent to IC2, which prevents extension of a Kcnq1 transcript across IC2 [46]. That such a mechanism is conserved in humans is supported by rare BWS patients, also affected by long QT syndrome, in which transcription from the KCNQ1 promoter through IC2 is perturbed by maternally inherited translocations, promoter deletions, splice variants, or duplications within the KCNQ1 locus [214,215,216,217]. Patients with familial BWS carry inactivating germ line mutations in the coding sequence of the maternally inherited CDKN1C allele [218, 219], further highlighting the pathological contribution of CDKN1C in BWS. Functional studies in mice have been highly informative for our understanding of the gene changes underlying BWS [220]. While overgrowth in BWS could result from either too much IGF2 or too little CDKN1C, mice that overexpress IGF2 do not exhibit defining features of BWS such as cleft palate and abdominal wall defects which are observed in response to loss of murine Cdkn1c [170, 171]. It seems likely that either or both alterations have potential to contribute to BWS.

Contribution of imprinted genes to both rare and highly common human diseases exemplified by human chromosome 11p15. Genetic and epigenetic disruptions to imprinted domains are associated with rare imprinting disorders while variations in the expression levels of individual genes or IGNs may underlie highly common conditions such as low birth weight, type 2 diabetes, obesity, neurodevelopmental and behavioral disorders
17 Silver-Russell Syndrome (SRS; OMIM #180860)
SRS is a very rare imprinting disorder with approximately 70% of patients having alterations affecting human chromosome 7 or 11, with 30% of unknown origin [210] (Fig. 8.4). SRS is diagnosed with an approximate frequency of 1 in 300,000 but may be far more common. SRS is defined by fetal growth restriction and failure to thrive postnatally and some or all of the following: normal head circumference, triangular-shaped face with a large protruding forehead, clinodactyly, undergrowth of one side of the body (hemihypotrophy), fasting hypoglycaemia, night sweats and excessive thinness into adulthood. An international consensus statement summarizing recommendations for clinical diagnosis, investigation, and management was published in 2017 [221]. As with BWS, the cognitive and behavioral characteristics of SRS are less well established but there are reports of specific learning difficulties [222,223,224,225], hyperactivity [226], attention deficits [224], autistic regression [227], and eating difficulties [228,229,230,231]. Maternal uniparental disomy of chromosome 7 is present in 5–10% of SRS patients (mUPD7; two maternal copies). Within the duplicated region there are two imprinted domains one of which contains the maternally-expressed GRB10 gene which has been implicated in both growth and behavior in mice [232, 233] and one of which spans MEST (aka PEG1) which has similar growth regulatory properties in mutant mice [184]. The larger proportion of SRS cases (30–60%) have hypomethylation of H19-DMR predicted, from mouse studies, to be associated with loss of expression of IGF2. A few SRS patients carry maternal microduplications of the 11p15 IC2 domain with the minimally duplicated region spanning CDKN1C, KCNQ1, PHLDA2, and SLC22A18 [234,235,236]. These patients are predicted to have twice the normal level of CDKN1C expression. Rarely, patients diagnosed with SRS possess mutations within CDKN1C thought to increase the stability of the protein [237,238,239]. Maternally inherited dominant missense mutations primarily within the PCNA-binding domain of CDKN1C have been reported in IMAGe syndrome (Intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies; # 614732) [240, 241] (Fig. 8.4). IMAGe syndrome is a very rare condition combining intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies and is clinically distinguished from SRS by the presence of adrenal insufficiency. As with BWS, mouse studies provide support for too little IGF2 or too much CDKN1C in SRS cases linked to chromosome 11p15. However, mice with loss-of-expression of Igf2 do not exhibit the more defining features of SRS such as lack of body fat and altered behaviors, which are seen in mouse models with gain-in expression of Cdkn1c [197,198,199, 206, 242].
18 Multilocus Imprinting Disorders (MLID)
A number of patients with BWS have epigenetic alterations affecting additional imprinted regions with the first suggestion coming from the finding that some patients diagnosed with transient neonatal diabetes mellitus (TNDM) also have hypomethylation at KvDMR1 [243]. It is now clear that a significant proportion of patients with a diagnosed imprinting disorder have an MLID, whereby more than one imprinted domain is impacted [244, 245]. In BWS 20–50% of patients with loss of KvDMR1 (IC2) methylation have an MLID, and MLID have been reported in approximately 15% of SRS cases [246]. One widely held explanation for simultaneous epigenetic defects at multiple ICs is a failure to maintain DNA methylation at these sites during the genome-wide reprogramming that occurs shortly after fertilization. Evidence for such a mechanism comes from studies on TNDM patients with DNA methylation defects at other imprinted loci, many of whom carry recessive mutations in ZFP57 [247], one of the proteins that maintain allelic DNA methylation and H3K9me3 at imprinted DMRs in mouse preimplantation embryos [52, 53]. Although mutations in ZFP57 have not, as yet, been reported in BWS or SRS patients with MLID [248, 249], a case of BWS with MLID has been described in the progeny of a mother homozygous for mutation in NLRP2 [250]. Interestingly, mutations in the related NLRP5 gene, coding for a component of the oocyte subcortical complex, are associated with MLID [251]. Given the multiple epigenetic modifiers involved in establishing and maintaining genomic imprinting, it is likely that other epigenetic regulators (such as ZNF445, DNMT1, UHRF1, SETDB1, or TRIM28) could be found to be involved in the DNA maintenance defects characteristic of MLID.
19 Imprinted Genes and More Common Complications of Pregnancy
Imprinting disorders are relatively rare (Table 8.1) but low birth weight (LBW), defined by The World Health Organisation as birth weight <2500 g at any gestational age (United Nations Children’s Fund and World Health Organisation 2004), is one of the most common complications of pregnancy, affecting up to 19% of all births in the developing world and between 5 and 7.5% of births in developed countries [252]. Several genes within the 11p15.5 IC1/IC2 domain are known to play an important role in determining birth weight in mice, either by intrinsically regulating fetal growth potential or through regulating placental development to extrinsically impact fetal growth (Fig. 8.4). Alterations in the expression of IC1/IC2 domain genes have been reported in association with fetal growth disorders in human pregnancy. Placental expression of IGF2 generally positively correlates with birthweight consistent with studies in mice [91, 253]. For example, elevated placental IGF2 has been reported in large for gestational age infants [254, 255] and lower placental IGF2 in small for gestational age infants [256, 257]. Conversely, placental expression of CDKN1C generally negatively correlates with birthweight [234, 235, 256, 258,259,260], consistent with the finding that birth weight decreases with increasing levels of Cdkn1c in mice [176, 206]. IGF2 and CDKN1C are not the only fetal growth restriction genes located on human chromosome 11p15. In mice elevated Phlda2 drives late fetal growth restriction resulting in LBW followed by catch-up growth [182]. Abnormally elevated placental PHLDA2 is a highly common alteration linked to LBW [261] with a prevalence that may be as high as 25% in confirmed cases of fetal growth restriction [262]. Increased placental expression of both PHLDA2 and CDKN1C has been observed in small-for-gestational-age (SGA) infants, highlighting the potential for domain-wide loss of imprinting [256].
In addition to regulating fetal growth, imprinted genes within the IC1/IC2 domain are critically important for placental development in mice, involved in both nutrient transport and the regulation of placental hormone production. Consequently, aberrant expression of imprinted genes in the fetally-derived placenta may contribute to disorders impacting the mother during pregnancy. For example, loss of function of Cdkn1c in the offspring has been linked to preeclampsia-like symptoms in the mouse dams [174], consistent with the finding of HELLP syndrome in human mothers of BWS infants with maternally inherited CDKN1C mutations [213]. Similarly in mice increased expression of fetal/placental Igf2 mediated by disrupted IC1 domain imprinting impacts maternal glucose management with potential relevance to gestational diabetes [263]. Changes in the expression of Phlda2 in the mouse placenta have been linked to alterations in the behavior of mothers toward their newborns [154] with potential relevance to pregnancy-related mood disorders [264].
20 Imprinted Gene Networks
In addition to the domain-wide regulation of allelic expression, some imprinted genes have been observed to interact within imprinted gene networks (IGNs) with important roles in development [265]. A number of imprinted genes located in different domains possess a strikingly similar pattern of expression suggesting interactions, with Cdkn1c and Plagl1 (aka Zac1) providing a key early example [243]. The interaction between imprinted genes in networks was further highlighted through the bioinformatic analysis of genes co-expressed with Plagl1 [266]. Disruption of these networks can result from changes in the expression of individual imprinted genes as shown for H19 [267] or disruption of epigenetic regulators, as shown for Trim28 [268]. To add further complexity, mutations disrupting networks can have more than one phenotypic outcome as a consequence of IGNs adjusting stochastically. Bimodal phenotypes have been reported for a number of mouse models in which individual imprinted genes have been genetically modified, further exemplified by studies on Trim28 mutants, where haploinsufficiency disrupted IGNs in mice manifesting an obese phenotype but not those of normal weight [268]. Evidence for similar stratification of IGNs in human populations was also reported in this study. Disrupted expression of IGNs may therefore contribute to the considerable clinical overlap noted for several imprinting disorders [210]. Disruptions to IGNs may contribute to a wide range of common human diseases (Fig. 8.4).
21 Imprinted Genes and Early Life Adversity
Imprinted genes may respond to external exposures at the individual, domain-wide, or IGN level contributing to human disease. Early life adversity is a major threat to human health because of the harmful outcomes for offspring in the short and longer term, a phenomenon described as fetal programming or developmental origins of health and disease [269, 270]. The clear role of imprinted genes in regulating fetal growth and postnatal phenotypes along with the predicted flexibility of epigenetic marks involved in the establishment, spreading, and maintenance of their expression has led to many researchers asking whether imprinted genes respond to adversity. Such studies are dependent on the demonstration of loss of monoallelic expression mediated by changes in epigenetics marks which can be challenging to demonstrate. Exposure to a maternal low-protein diet drives persistent loss-of-imprinting of at least one gene within the IC2 domain [20]. In this study, Cdkn1c luciferase reporter mice were used in which gene expression is visualized as a bioluminescent signal in living mice. Exposure to low-protein diet from conception resulted in loss of normal paternal silencing alongside loss of DNA methylation at the somatic Cdkn1c-sDMR but without impacting methylation of the IC2 domain gDMR, consistent with previous studies reporting that DNA methylation at gDMRs is relatively resistant to dietary adversities [271, 272]. Expression and methylation changes occurred before birth and persisted into adulthood even under conditions of a normal diet. Critically, supplementation with the methyl donor folate in pregnancy prevented loss of silencing. While this study provides definitive experimental evidence that imprinted genes can respond epigenetically to a dietary exposure, it remains to be determined how many genes are responsive, under what adversity conditions and to what extent the mechanisms underpinning responsiveness are conserved. Given that different mechanisms lead to the establishment and maintenance of imprinting, it seems likely that multiple pathways could disrupt the epigenetic regulation of imprinted loci. Outcomes of individual adversities may, however, be similar due to disruptions in IGNs.
22 Conclusion
Imprinted genes are associated with rare imprinting disorders such as BWS and SRS, but animal models suggest that imprinted genes have a greater potential to contribute more widely to human diseases including low birth weight, and chronic health conditions such as obesity, type 2 diabetes, and mental health disorders (Fig. 8.4). Imprinted genes may also, at least in part, be associated with the well-established relationship between early life adversity and human disease. A major challenge is how to translate functional outcomes from genetically modified mice to humans. Many studies have focused on comparing DNA methylation levels at gDMRs as a proxy for imprinted gene expression levels. However, the majority of imprinted genes are not directly spanned by gDMRs, and multistep epigenetic processes are required to establish and maintain their monoallelic expression. Developments in genome-wide approaches, single-cell technologies, combined with mathematical modeling are required to fully establish the extent to which variation in the expression of imprinted genes might contribute to wide-ranging human disease.
Abbreviations
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- Airn :
-
Antisense Of Igf2R non-protein coding RNA
- Ascl2 :
-
Achaete-scute family bHLH transcription factor 2
- BWS:
-
Beckwith–Wiedemann Syndrome
- Cdkn1c :
-
Cyclin-dependent kinase inhibitor 1c
- CpG:
-
Dinucleotide CG
- CTCF:
-
CCCTC-binding factor
- Dio3 :
-
Iodothyronine Deiodinase 3
- Dlk1 :
-
Delta Like Non-Canonical Notch Ligand 1
- DNMT1:
-
DNA methyltransferase 1
- DNMT3A:
-
DNA methyltransferase 3A
- DNMT3B:
-
DNA methyltransferase 3B
- DNMT3L:
-
DNA methyltransferase 3L
- (E):
-
Embryonic
- ERVK:
-
Endogenous retrovirus-K
- gDMR:
-
Germline differentially methylated region
- Gnas :
-
GNAS (guanine nucleotide binding protein, alpha stimulating) complex locus
- Grb10 :
-
Growth factor receptor-bound protein 10
- H19 :
-
H19 gene
- H3K27me3:
-
Histone H3 lysine 27 trimethylation
- H3K36me2/3:
-
Histone H3 di/trimethylated at lysine 36
- H3K4me2/3:
-
Histone H3 di/trimethylated at lysine 4
- HELL2:
-
Helicase, lymphoid specific
- HELLP:
-
Haemolysis, elevated liver enzymes and low platelet count
- IC:
-
Imprinting centre
- Igf2 :
-
Insulin-like growth factor 2
- Igf2r :
-
Insulin-like growth factor 2 receptor
- IGN:
-
Imprinted gene network
- IMAGe:
-
Intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies
- Inpp5f-v2 :
-
Inositol polyphosphate-5-phosphatase F variant 2
- Ins2 :
-
Insulin 2
- Kcnq1ot1 :
-
Kcnq1 opposite strand/antisense transcript 1 (non-protein coding) gene
- KvDMR:
-
DMR in the Kcnq1ot1 locus
- LBW:
-
Low birth weight
- LTR:
-
Long terminal repeat
- Mcts2 :
-
Malignant T cell amplified sequence
- Meg3 (aka Gtl2):
-
Maternally expressed 3
- Mest (aka Peg1):
-
Mesoderm-specific transcript
- MLID:
-
Multilocus imprinting disorders
- Nap1l5 :
-
Nucleosome assembly protein 1-like 5
- NLRP5:
-
NLR family pyrin domain containing 5
- NSD1:
-
Nuclear receptor binding SET domain protein 1
- Peg1 :
-
Paternally expressed gene-1
- Peg3 :
-
Paternally expressed gene-3
- PGCs:
-
Primordial germ cells
- Phlda2 :
-
Pleckstrin homology-like domain, family A, member 2 gene
- PIWI:
-
P-element induced Wimpy testis
- Plag1 (aka Zac1):
-
Pleiomorphic adenoma gene-like 1
- Rasgrf1 :
-
Ras protein-specific guanine nucleotide releasing factor 1
- sDMR:
-
Somatic differentially methylated region
- SETD2:
-
SET domain containing 2, histone lysine methyltransferase
- SETDB1:
-
SET domain bifurcated histone lysine methyltransferase 1
- Slc22a18 :
-
Solute carrier family 22, member 18
- Slc22a2 :
-
Solute carrier family 22 member 2
- Slc22a3 :
-
Solute carrier family 22 member 3
- Slc38a4 :
-
Solute carrier family 38 member 4
- Snrpn :
-
Small nuclear ribonucleoprotein polypeptide N
- SRS:
-
Silver–Russell Syndrome
- TAD:
-
Topologically associating domain
- Tet:
-
Ten-eleven translocation protein
- TNDM:
-
Transient neonatal diabetes mellitus
- TRIM28:
-
Tripartite motif containing 28
- UHRF1:
-
Ubiquitin like with PHD and ring finger domains 1
- UPD:
-
Uniparental disomy
- ZFP:
-
Zinc-finger protein
- Zrsr1 (aka U2af1-rs1):
-
Zinc finger (CCCH type), RNA binding motif and serine/arginine rich 1
References
Surani MA (1998) Imprinting and the initiation of gene silencing in the germ line. Cell 93(3):309–312
Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev Genet 2(1):21–32. https://doi.org/10.1038/35047554
Ferguson-Smith AC, Surani MA (2001) Imprinting and the epigenetic asymmetry between parental genomes. Science 293(5532):1086–1089
Surani MA, Barton SC (1983) Development of gynogenetic eggs in the mouse: implications for parthenogenetic embryos. Science 222(4627):1034–1036. https://doi.org/10.1126/science.6648518
McGrath J, Solter D (1983) Nuclear transplantation in the mouse embryo by microsurgery and cell fusion. Science 220(4603):1300–1302. https://doi.org/10.1126/science.6857250
McGrath J, Solter D (1984) Completion of mouse embryogenesis requires both the materal and paternal genomes. Cell 37:179–183
Surani MA, Barton SC, Norris ML (1984) Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308(5959):548–550
Barton SC, Surani MA, Norris ML (1984) Role of paternal and maternal genomes in mouse development. Nature 311:374–376
Solter D, Aronson J, Gilbert SF, McGrath J (1985) Nuclear transfer in mouse embryos: activation of the embryonic genome. Cold Spring Harb Symp Quant Biol 50:45–50
Surani MA, Barton SC, Norris ML (1987) Influence of parental chromosomes on spatial specificity in androgenetic-parthenogenetic chimaeras in the mouse. Nature 326:395–397
Thomson JA, Solter D (1988) The developmental fate of androgenetic, parthenogenetic and gynogenetic cells in chimeric gastrulating mouse embryos. Genes Dev 2:1344–1351
Cattanach BM, Kirk M (1985) Differential activity of maternally and paternally derived chromosome regions in mice. Nature 315(6019):496–498
Ferguson-Smith AC (2011) Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 12(8):565–575. https://doi.org/10.1038/nrg3032
Barlow DP, Bartolomei MS (2014) Genomic imprinting in mammals. Cold Spring Harb Perspect Biol 6(2). https://doi.org/10.1101/cshperspect.a018382
Ferguson-Smith AC, Bourc’his D (2018) The discovery and importance of genomic imprinting. Elife 7. https://doi.org/10.7554/eLife.42368
Moore T, Haig D (1991) Genomic imprinting in mammalian development: a parental tug-of-war. TIG 7(2):45–49
Haig D, Graham C (1991) Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell 64(6):1045–1046
Wolf JB, Hager R (2006) A maternal-offspring coadaptation theory for the evolution of genomic imprinting. PLoS Biol 4(12):e380. https://doi.org/10.1371/journal.pbio.0040380
Strahan R (1983) Complete book of Australian mammals. Angus and Robertson
Van de Pette M, Abbas A, Feytout A, McNamara G, Bruno L, To WK, Dimond A, Sardini A, Webster Z, McGinty J, Paul EJ, Ungless MA, French PMW, Withers DJ, Uren A, Ferguson-Smith AC, Merkenschlager M, John RM, Fisher AG (2017) Visualizing changes in cdkn1c expression links early-life adversity to imprint mis-regulation in adults. Cell Rep 18(5):1090–1099. https://doi.org/10.1016/j.celrep.2017.01.010
SanMiguel JM, Bartolomei MS (2018) DNA methylation dynamics of genomic imprinting in mouse development. Biol Reprod 99(1):252–262. https://doi.org/10.1093/biolre/ioy036
Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC, Erice Imprinting G (2019) Genomic imprinting and physiological processes in mammals. Cell 176(5):952–965. https://doi.org/10.1016/j.cell.2019.01.043
Li JY, Lees-Murdock DJ, Xu GL, Walsh CP (2004) Timing of establishment of paternal methylation imprints in the mouse. Genomics 84(6):952–960. https://doi.org/10.1016/j.ygeno.2004.08.012
Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, Okano M, Li E, Nozaki M, Sasaki H (2007) Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 16(19):2272–2280. https://doi.org/10.1093/hmg/ddm179
Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W (2012) The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell 48(6):849–862. https://doi.org/10.1016/j.molcel.2012.11.001
Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, Suzuki Y, Kono T (2012) Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8(1):e1002440. https://doi.org/10.1371/journal.pgen.1002440
Henckel A, Chebli K, Kota SK, Arnaud P, Feil R (2012) Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J 31(3):606–615. https://doi.org/10.1038/emboj.2011.425
Hata K, Okano M, Lei H, Li E (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129(8):1983–1993
Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294(5551):2536–2539. https://doi.org/10.1126/science.1065848
Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429(6994):900–903. https://doi.org/10.1038/nature02633
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99(3):247–257
Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, Gotoh K, Hiura H, Arima T, Fujiyama A, Sado T, Shibata T, Nakano T, Lin H, Ichiyanagi K, Soloway PD, Sasaki H (2011) Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 332(6031):848–852. https://doi.org/10.1126/science.1203919
Shirane K, Miura F, Ito T, Lorincz MC (2020) NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat Genet 52(10):1088–1098. https://doi.org/10.1038/s41588-020-0689-z
Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G (2011) Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43(8):811–814. https://doi.org/10.1038/ng.864
Shirane K, Toh H, Kobayashi H, Miura F, Chiba H, Ito T, Kono T, Sasaki H (2013) Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet 9(4):e1003439. https://doi.org/10.1371/journal.pgen.1003439
Stewart KR, Veselovska L, Kim J, Huang J, Saadeh H, Tomizawa S, Smallwood SA, Chen T, Kelsey G (2015) Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev 29(23):2449–2462. https://doi.org/10.1101/gad.271353.115
Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W, Du Z, Huang C, Ma J, Zheng H, Li Y, Liu C, Walker CL, Jonasch E, Lefebvre L, Wu M, Lorincz MC, Li W, Li L, Xie W (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51(5):844–856. https://doi.org/10.1038/s41588-019-0398-7
Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, Zhang QH, Chen SJ, Huang QH, Chen Z (2005) Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem 280(42):35261–35271. https://doi.org/10.1074/jbc.M504012200
Chedin F, Lieber MR, Hsieh CL (2002) The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A 99(26):16916–16921. https://doi.org/10.1073/pnas.262443999
Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S (2004) DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem 279(26):27816–27823. https://doi.org/10.1074/jbc.M400181200
Kaneda M, Hirasawa R, Chiba H, Okano M, Li E, Sasaki H (2010) Genetic evidence for Dnmt3a-dependent imprinting during oocyte growth obtained by conditional knockout with Zp3-Cre and complete exclusion of Dnmt3b by chimera formation. Genes Cells. https://doi.org/10.1111/j.1365-2443.2009.01374.x
Veselovska L, Smallwood SA, Saadeh H, Stewart KR, Krueger F, Maupetit-Mehouas S, Arnaud P, Tomizawa S, Andrews S, Kelsey G (2015) Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol 16(1):209. https://doi.org/10.1186/s13059-015-0769-z
Bourc’his D, Bestor TH (2006) Origins of extreme sexual dimorphism in genomic imprinting. Cytogenet Genome Res 113(1–4):36–40. https://doi.org/10.1159/000090813
Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G (2009) Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev 23(1):105–117. https://doi.org/10.1101/gad.495809
Smith EY, Futtner CR, Chamberlain SJ, Johnstone KA, Resnick JL (2011) Transcription is required to establish maternal imprinting at the Prader-Willi syndrome and Angelman syndrome locus. PLoS Genet 7(12):e1002422. https://doi.org/10.1371/journal.pgen.1002422
Singh VB, Sribenja S, Wilson KE, Attwood KM, Hillman JC, Pathak S, Higgins MJ (2017) Blocked transcription through KvDMR1 results in absence of methylation and gene silencing resembling Beckwith-Wiedemann syndrome. Development 144(10):1820–1830. https://doi.org/10.1242/dev.145136
Bretz CL, Kim J (2017) Transcription-driven DNA methylation setting on the mouse Peg3 locus. Epigenetics 12(11):945–952. https://doi.org/10.1080/15592294.2017.1377869
Joh K, Matsuhisa F, Kitajima S, Nishioka K, Higashimoto K, Yatsuki H, Kono T, Koseki H, Soejima H (2018) Growing oocyte-specific transcription-dependent de novo DNA methylation at the imprinted Zrsr1-DMR. Epigenetics Chromatin 11(1):28. https://doi.org/10.1186/s13072-018-0200-6
Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W, Li W, Zhou Q, Aluru N, Tang F, He C, Huang X, Liu J (2014) Programming and inheritance of parental DNA methylomes in mammals. Cell 157(4):979–991. https://doi.org/10.1016/j.cell.2014.04.017
Takahashi N, Coluccio A, Thorball CW, Planet E, Shi H, Offner S, Turelli P, Imbeault M, Ferguson-Smith AC, Trono D (2019) ZNF445 is a primary regulator of genomic imprinting. Genes Dev 33(1–2):49–54. https://doi.org/10.1101/gad.320069.118
Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB (2012) Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335(6075):1499–1502. https://doi.org/10.1126/science.1216154
Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC (2008) A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15(4):547–557. https://doi.org/10.1016/j.devcel.2008.08.014
Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, Trono D (2011) In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell 44(3):361–372. https://doi.org/10.1016/j.molcel.2011.08.032
Shi H, Strogantsev R, Takahashi N, Kazachenka A, Lorincz MC, Hemberger M, Ferguson-Smith AC (2019) ZFP57 regulation of transposable elements and gene expression within and beyond imprinted domains. Epigenetics Chromatin 12(1):49. https://doi.org/10.1186/s13072-019-0295-4
Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR (2001) Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104(6):829–838. https://doi.org/10.1016/s0092-8674(01)00280-x
Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H (2008) Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev 22(12):1607–1616. https://doi.org/10.1101/gad.1667008
Weaver JR, Sarkisian G, Krapp C, Mager J, Mann MR, Bartolomei MS (2010) Domain-specific response of imprinted genes to reduced DNMT1. Mol Cell Biol 30(16):3916–3928. https://doi.org/10.1128/MCB.01278-09
Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450(7171):908–912. https://doi.org/10.1038/nature06397
Hanna CW, Kelsey G (2014) The specification of imprints in mammals. Heredity 113(2):176–183. https://doi.org/10.1038/hdy.2014.54
White CR, MacDonald WA, Mann MR (2016) Conservation of DNA methylation programming between mouse and human gametes and preimplantation embryos. Biol Reprod 95(3):61. https://doi.org/10.1095/biolreprod.116.140319
Bogutz AB, Brind’Amour J, Kobayashi H, Jensen KN, Nakabayashi K, Imai H, Lorincz MC, Lefebvre L (2019) Evolution of imprinting via lineage-specific insertion of retroviral promoters. Nat Commun 10(1):5674. https://doi.org/10.1038/s41467-019-13662-9
Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP (1997) Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 389(6652):745–749
Thorvaldsen JL, Duran KL, Bartolomei MS (1998) Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev 12(23):3693–3702
Bielinska B, Blaydes SM, Buiting K, Yang T, Krajewska-Walasek M, Horsthemke B, Brannan CI (2000) De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat Genet 25(1):74–78. https://doi.org/10.1038/75629
Fitzpatrick GV, Soloway PD, Higgins MJ (2002) Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 32(3):426–431
Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC (2003) Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 35(1):97–102
Williamson CM, Turner MD, Ball ST, Nottingham WT, Glenister P, Fray M, Tymowska-Lalanne Z, Plagge A, Powles-Glover N, Kelsey G, Maconochie M, Peters J (2006) Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat Genet 38(3):350–355
MacDonald WA, Mann MRW (2020) Long noncoding RNA functionality in imprinted domain regulation. PLoS Genet 16(8):e1008930. https://doi.org/10.1371/journal.pgen.1008930
Sleutels F, Zwart R, Barlow DP (2002) The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415(6873):810–813. https://doi.org/10.1038/415810a
Santoro F, Mayer D, Klement RM, Warczok KE, Stukalov A, Barlow DP, Pauler FM (2013) Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development 140(6):1184–1195. https://doi.org/10.1242/dev.088849
Kota SK, Lleres D, Bouschet T, Hirasawa R, Marchand A, Begon-Pescia C, Sanli I, Arnaud P, Journot L, Girardot M, Feil R (2014) ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1-Dio3 domain. Dev Cell 31(1):19–33. https://doi.org/10.1016/j.devcel.2014.08.009
Tibbit CJ, Williamson CM, Mehta S, Ball ST, Chotalia M, Nottingham WT, Eaton SA, Quwailid MM, Teboul L, Kelsey G, Peters J (2015) Antisense activity across the nesp promoter is required for nespas-mediated silencing in the imprinted gnas cluster. Noncoding RNA 1(3):246–265. https://doi.org/10.3390/ncrna1030246
Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, Aumayr K, Pasierbek P, Barlow DP (2012) Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338(6113):1469–1472. https://doi.org/10.1126/science.1228110
Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL (2013) Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 9(12):e1004039. https://doi.org/10.1371/journal.pgen.1004039
Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM (2006) Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 20(10):1268–1282
Shin JY, Fitzpatrick GV, Higgins MJ (2008) Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J 27(1):168–178
Keshavarz M, Tautz D (2021) The imprinted lncRNA Peg13 regulates sexual preference and the sex-specific brain transcriptome in mice. Proc Natl Acad Sci U S A 118(10). https://doi.org/10.1073/pnas.2022172118
Kobayashi H, Sakurai T, Miura F, Imai M, Mochiduki K, Yanagisawa E, Sakashita A, Wakai T, Suzuki Y, Ito T, Matsui Y, Kono T (2013) High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res 23(4):616–627. https://doi.org/10.1101/gr.148023.112
Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA (2002) Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 117(1–2):15–23
Yamaguchi S, Hong K, Liu R, Inoue A, Shen L, Zhang K, Zhang Y (2013) Dynamics of 5-methylcytosine and 5-hydroxymethylcytosine during germ cell reprogramming. Cell Res 23(3):329–339. https://doi.org/10.1038/cr.2013.22
Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, Faull KF, Lyko F, Jaenisch R (2013) Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell 24(3):310–323. https://doi.org/10.1016/j.devcel.2012.12.015
Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA (2013) Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339(6118):448–452. https://doi.org/10.1126/science.1229277
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324(5929):930–935. https://doi.org/10.1126/science.1170116
Gkountela S, Zhang KX, Shafiq TA, Liao WW, Hargan-Calvopina J, Chen PY, Clark AT (2015) DNA demethylation dynamics in the human prenatal germline. Cell 161(6):1425–1436. https://doi.org/10.1016/j.cell.2015.05.012
Yamaguchi S, Shen L, Liu Y, Sendler D, Zhang Y (2013) Role of Tet1 in erasure of genomic imprinting. Nature 504(7480):460–464. https://doi.org/10.1038/nature12805
SanMiguel JM, Abramowitz LK, Bartolomei MS (2018) Imprinted gene dysregulation in a Tet1 null mouse model is stochastic and variable in the germline and offspring. Development 145(7). https://doi.org/10.1242/dev.160622
Ferguson-Smith AC, Cattanach BM, Barton SC, Beechey CV, Surani MA (1991) Molecular and embryological investigations of parental imprinting on mouse chromosome 7. Nature 351:667–670
Searle AG, Beechey CV (1990) Genome imprinting phenomena on mouse chromosome 7. Genet Res (Cambridge) 56:237–244
McLaughlin KJ, Solter D, Mann J (1997) Developmental consequences of two paternal copies of imprinted chromosome region distal 7 in mice. J Cell Physiol 173(2):242–246. https://doi.org/10.1002/(SICI)1097-4652(199711)173:2<242::AID-JCP29>3.0.CO;2-G
McLaughlin KJ, Szabò P, Haegel H, Mann JR (1996) Mouse embryos with paternal duplication of an imprinted chromosome 7 region die around midgestation and lack placental spongiotrophoblast. Development 122:265–270
DeChiara TM, Robertson EJ, Efstratiadis A (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64(4):849–859
Deltour L, Montagutelli X, Guenet J-L, Jami J, Paldi A (1995) Tissue and developmental stage-specific imprinting of the mouse proinsulin gene, Ins-2. Dev Biol 168:686–688
Bartolomei MS, Zemei S, Tilghman SM (1991) Parental imprinting of the mouse H19 gene. Nature 351:153–155
Cai X, Cullen BR (2007) The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA (New York, NY) 13(3):313–316. https://doi.org/10.1261/rna.351707
Ferguson-Smith AC, Sasaki H, Cattanach BM, Surani MA (1993) Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 362(6422):751–755. https://doi.org/10.1038/362751a0
John RM, Lefebvre L (2011) Developmental regulation of somatic imprints. Differentiation 81(5):270–280. https://doi.org/10.1016/j.diff.2011.01.007
Leighton PA, Saam JR, Ingram RS, Stewart CL, Tilghman SM (1995) An enhancer deletion affects both H19 and Igf2 expression. Genes Dev 9(17):2079–2089
Bell AC, Felsenfeld G (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405(6785):482–485. https://doi.org/10.1038/35013100
Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405(6785):486–489. https://doi.org/10.1038/35013106
Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, Ohlsson R, Lobanenkov VV (2000) Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 10(14):853–856. https://doi.org/10.1016/s0960-9822(00)00597-2
Engel N, Raval AK, Thorvaldsen JL, Bartolomei SM (2008) Three-dimensional conformation at the H19/Igf2 locus supports a model of enhancer tracking. Hum Mol Genet 17(19):3021–3029. https://doi.org/10.1093/hmg/ddn200
Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, Ohlsson R (2006) CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A 103(28):10684–10689. https://doi.org/10.1073/pnas.0600326103
Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S, Hou A, Vu TH, Hoffman AR (2008) CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol Cell Biol 28(20):6473–6482. https://doi.org/10.1128/MCB.00204-08
Qiu X, Vu TH, Lu Q, Ling JQ, Li T, Hou A, Wang SK, Chen HL, Hu JF, Hoffman AR (2008) A complex deoxyribonucleic acid looping configuration associated with the silencing of the maternal Igf2 allele. Mol Endocrinol 22(6):1476–1488. https://doi.org/10.1210/me.2007-0474
Murrell A, Heeson S, Reik W (2004) Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet 36(8):889–893. https://doi.org/10.1038/ng1402
Court F, Baniol M, Hagege H, Petit JS, Lelay-Taha MN, Carbonell F, Weber M, Cathala G, Forne T (2011) Long-range chromatin interactions at the mouse Igf2/H19 locus reveal a novel paternally expressed long non-coding RNA. Nucleic Acids Res 39(14):5893–5906. https://doi.org/10.1093/nar/gkr209
Lleres D, Moindrot B, Pathak R, Piras V, Matelot M, Pignard B, Marchand A, Poncelet M, Perrin A, Tellier V, Feil R, Noordermeer D (2019) CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol 20(1):272. https://doi.org/10.1186/s13059-019-1896-8
Qian N, Frank D, O’Keefe D, Dao D, Zhao L, Yuan L, Wang Q, Keating M, Walsh C, Tycko B (1997) The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum Mol Genet 6(12):2021–2029
Hatada I, Mukai T (1995) Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse. Nat Genet 11(2):204–206
Guillemot F, Caspary T, Tilghman SM, Copeland NG, Gilbert DJ, Jenkins NA, Anderson DJ, Joyner AL, Rossant J, Nagy A (1995) Genomic imprinting of Mash2, a mouse gene required for trophoblast development. Nat Genet 9:235–242
Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, Nicholls RD, Weksberg R, Driscoll DJ, Maher ER, Shows TB, Higgins MJ (1999) A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A 96(14):8064–8069. https://doi.org/10.1073/pnas.96.14.8064
Engemann S, Strodicke M, Paulsen M, Franck O, Reinhardt R, Lane N, Reik W, Walter J (2000) Sequence and functional comparison in the beckwith-wiedemann region: implications for a novel imprinting centre and extended imprinting. Hum Mol Genet 9(18):2691–2706
Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W (2004) Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 36(12):1291–1295
Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, Peters AH (2008) Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell 15(5):668–679
Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R (2004) Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36(12):1296–1300
Kato Y, Iii WM, Hilton K, Barton SC, Tsunoda Y, Surani MA (1999) Developmental potential of mouse primordial germ cells. Development 126(9):1823–1832
Tada T, Tada M, Hilton K, Barton SC, Sado T, Takagi N, Surani MA (1998) Epigenotype switching of imprintable loci in embryonic germ cells. Dev Genes Evol 207(8):551–561
Kono T, Obata Y, Yoshimzu T, Nakahara T, Carroll J (1996) Epigenetic modifications during oocyte growth correlates with extended parthenogenetic development in the mouse. Nat Genet 13:91–94
Obata Y, Kaneko-Ishino T, Koide T, Takai Y, Ueda T, Domeki I, Shiroishi T, Ishino F, Kono T (1998) Disruption of primary imprinting during oocyte growth leads to the modified expression of imprinted genes during embryogenesis. Development 125(8):1553–1560
Bhogal B, Arnaudo A, Dymkowski A, Best A, Davis TL (2004) Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics 84(6):961–970
Fan T, Hagan JP, Kozlov SV, Stewart CL, Muegge K (2005) Lsh controls silencing of the imprinted Cdkn1c gene. Development 132(4):635–644
Caspary T, Cleary MA, Baker CC, Guan XJ, Tilghman SM (1998) Multiple mechanisms regulate imprinting of the mouse distal chromosome 7 gene cluster. Mol Cell Biol 18(6):3466–3474
Wood MD, Hiura H, Tunster S, Arima T, Shin JY, Higgins M, John RM (2010) Autonomous silencing of the imprinted Cdkn1c gene in stem cells. Epigenetics 5(3)
Pauler FM, Koerner MV, Barlow DP (2007) Silencing by imprinted noncoding RNAs: is transcription the answer? Trends Genet 23(6):284–292. https://doi.org/10.1016/j.tig.2007.03.018
Andergassen D, Muckenhuber M, Bammer PC, Kulinski TM, Theussl HC, Shimizu T, Penninger JM, Pauler FM, Hudson QJ (2019) The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genet 15(7):e1008268. https://doi.org/10.1371/journal.pgen.1008268
John RM, Ainscough JF, Barton SC, Surani MA (2001) Distant cis-elements regulate imprinted expression of the mouse p57 (Kip2) (Cdkn1c) gene: implications for the human disorder, Beckwith—Wiedemann syndrome. Hum Mol Genet 10(15):1601–1609
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32(2):232–246. https://doi.org/10.1016/j.molcel.2008.08.022
Redrup L, Branco MR, Perdeaux ER, Krueger C, Lewis A, Santos F, Nagano T, Cobb BS, Fraser P, Reik W (2009) The long noncoding RNA Kcnq1ot1 organises a lineage-specific nuclear domain for epigenetic gene silencing. Development 136(4):525–530
Schertzer MD, Braceros KCA, Starmer J, Cherney RE, Lee DM, Salazar G, Justice M, Bischoff SR, Cowley DO, Ariel P, Zylka MJ, Dowen JM, Magnuson T, Calabrese JM (2019) lncRNA-induced spread of polycomb controlled by genome architecture, RNA abundance, and CpG Island DNA. Mol Cell 75(3):523–537. https://doi.org/10.1016/j.molcel.2019.05.028
Mohammad F, Pandey RR, Nagano T, Chakalova L, Mondal T, Fraser P, Kanduri C (2008) Kcnq1ot1/Lit1 noncoding RNA mediates transcriptional silencing by targeting to the perinucleolar region. Mol Cell Biol 28(11):3713–3728
Fedoriw AM, Calabrese JM, Mu W, Yee D, Magnuson T (2012) Differentiation-driven nucleolar association of the mouse imprinted Kcnq1 locus. G3 (Bethesda) 2(12):1521–1528. https://doi.org/10.1534/g3.112.004226
Sachani SS, Landschoot LS, Zhang L, White CR, MacDonald WA, Golding MC, Mann MRW (2018) Nucleoporin 107, 62 and 153 mediate Kcnq1ot1 imprinted domain regulation in extraembryonic endoderm stem cells. Nat Commun 9(1):2795. https://doi.org/10.1038/s41467-018-05208-2
Golding MC, Magri LS, Zhang L, Lalone SA, Higgins MJ, Mann MR (2011) Depletion of Kcnq1ot1 non-coding RNA does not affect imprinting maintenance in stem cells. Development 138(17):3667–3678. https://doi.org/10.1242/dev.057778
Mohammad F, Pandey GK, Mondal T, Enroth S, Redrup L, Gyllensten U, Kanduri C (2012) Long noncoding RNA-mediated maintenance of DNA methylation and transcriptional gene silencing. Development 139(15):2792–2803. https://doi.org/10.1242/dev.079566
DeVeale B, van der Kooy D, Babak T (2012) Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet 8(3):e1002600. https://doi.org/10.1371/journal.pgen.1002600
Kelsey G, Bartolomei MS (2012) Imprinted genes … and the number is? PLoS Genet 8(3):e1002601. https://doi.org/10.1371/journal.pgen.1002601
Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y (2017) Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547(7664):419–424. https://doi.org/10.1038/nature23262
Hanna CW, Perez-Palacios R, Gahurova L, Schubert M, Krueger F, Biggins L, Andrews S, Colome-Tatche M, Bourc’his D, Dean W, Kelsey G (2019) Endogenous retroviral insertions drive non-canonical imprinting in extra-embryonic tissues. Genome Biol 20(1):225. https://doi.org/10.1186/s13059-019-1833-x
Chen Z, Yin Q, Inoue A, Zhang C, Zhang Y (2019) Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Sci Adv 5(12):eaay7246. https://doi.org/10.1126/sciadv.aay7246
Zhang W, Chen Z, Yin Q, Zhang D, Racowsky C, Zhang Y (2019) Maternal-biased H3K27me3 correlates with paternal-specific gene expression in the human morula. Genes Dev 33(7–8):382–387. https://doi.org/10.1101/gad.323105.118
Xia W, Xu J, Yu G, Yao G, Xu K, Ma X, Zhang N, Liu B, Li T, Lin Z, Chen X, Li L, Wang Q, Shi D, Shi S, Zhang Y, Song W, Jin H, Hu L, Bu Z, Wang Y, Na J, Xie W, Sun YP (2019) Resetting histone modifications during human parental-to-zygotic transition. Science 365(6451):353–360. https://doi.org/10.1126/science.aaw5118
Gehring M (2013) Genomic imprinting: insights from plants. Annu Rev Genet 47:187–208. https://doi.org/10.1146/annurev-genet-110711-155527
Renfree MB, Hore TA, Shaw G, Graves JA, Pask AJ (2009) Evolution of genomic imprinting: insights from marsupials and monotremes. Annu Rev Genomics Hum Genet 10:241–262. https://doi.org/10.1146/annurev-genom-082908-150026
Renfree MB, Ager EI, Shaw G, Pask AJ (2008) Genomic imprinting in marsupial placentation. Reproduction 136(5):523–531
Nabetani A, Hatada I, Morisaki H, Oshimura M, Mukai T (1997) Mouse U2af1-rs1 is a neomorphic imprinted gene. Mol Cell Biol 17(2):789–798
Choi JD, Underkoffler LA, Wood AJ, Collins JN, Williams PT, Golden JA, Schuster EF Jr, Loomes KM, Oakey RJ (2005) A novel variant of Inpp5f is imprinted in brain, and its expression is correlated with differential methylation of an internal CpG island. Mol Cell Biol 25(13):5514–5522. https://doi.org/10.1128/MCB.25.13.5514-5522.2005
Wood AJ, Roberts RG, Monk D, Moore GE, Schulz R, Oakey RJ (2007) A screen for retrotransposed imprinted genes reveals an association between X chromosome homology and maternal germ-line methylation. PLoS Genet 3(2):e20. https://doi.org/10.1371/journal.pgen.0030020
McCole RB, Loughran NB, Chahal M, Fernandes LP, Roberts RG, Fraternali F, O’Connell MJ, Oakey RJ (2011) A case-by-case evolutionary analysis of four imprinted retrogenes. Evolution 65(5):1413–1427. https://doi.org/10.1111/j.1558-5646.2010.01213.x
Brind’Amour J, Kobayashi H, Richard Albert J, Shirane K, Sakashita A, Kamio A, Bogutz A, Koike T, Karimi MM, Lefebvre L, Kono T, Lorincz MC (2018) LTR retrotransposons transcribed in oocytes drive species-specific and heritable changes in DNA methylation. Nat Commun 9(1):3331. https://doi.org/10.1038/s41467-018-05841-x
Proudhon C, Duffie R, Ajjan S, Cowley M, Iranzo J, Carbajosa G, Saadeh H, Holland ML, Oakey RJ, Rakyan VK, Schulz R, Bourc’his D (2012) Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol Cell 47(6):909–920. https://doi.org/10.1016/j.molcel.2012.07.010
Smith FM, Garfield AS, Ward A (2006) Regulation of growth and metabolism by imprinted genes. Cytogenet Genome Res 113(1–4):279–291
Peters J (2014) The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet 15(8):517–530. https://doi.org/10.1038/nrg3766
Cleaton MA, Edwards CA, Ferguson-Smith AC (2014) Phenotypic outcomes of imprinted gene models in mice: elucidation of pre- and postnatal functions of imprinted genes. Annu Rev Genomics Hum Genet 15:93–126. https://doi.org/10.1146/annurev-genom-091212-153441
Creeth HDJ, McNamara GI, Isles AR, John RM (2019) Imprinted genes influencing the quality of maternal care. Front Neuroendocrinol 53:100732. https://doi.org/10.1016/j.yfrne.2018.12.003
Kaneko-Ishino T, Ishino F (2019) Evolution of viviparity in mammals: what genomic imprinting tells us about mammalian placental evolution. Reprod Fertil Dev 31(7):1219–1227. https://doi.org/10.1071/RD18127
Hanin G, Ferguson-Smith AC (2020) The evolution of genomic imprinting: Epigenetic control of mammary gland development and postnatal resource control. Wiley Interdiscip Rev Syst Biol Med 12(3):e1476. https://doi.org/10.1002/wsbm.1476
Barlow DP, Stoger R, Herrmann BG, Saito K, Schweifer N (1991) The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349(6304):84–87. https://doi.org/10.1038/349084a0
Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth R, Rutter WJ (1987) Insulin-like growth factor II receptor as a multifunctional binding protein. Nature 329:301–307
Ripoche M-A, Kress C, Poirier F, Dandolo L (1997) Deletion of the H19 transcription unit reveals the existence of a putative imprinting control element. Genes Dev 11:1596–1604
John R, Hemberger M (2012) A placenta for life. Reprod Biomed Online 25(1):5–11. https://doi.org/10.1016/j.rbmo.2012.03.018
Coan PM, Fowden AL, Constancia M, Ferguson-Smith AC, Burton GJ, Sibley CP (2008) Disproportional effects of Igf2 knockout on placental morphology and diffusional exchange characteristics in the mouse. J Physiol 586(Pt 20):5023–5032. https://doi.org/10.1113/jphysiol.2008.157313
Esquiliano DR, Guo W, Liang L, Dikkes P, Lopez MF (2009) Placental glycogen stores are increased in mice with H19 null mutations but not in those with insulin or IGF type 1 receptor mutations. Placenta 30(8):693–699
Brannan CI, Dees EC, Ingram RS, Tilghman SM (1990) The product of the H19 gene may function as an RNA. Mol Cell Biol 10:28–36
Jones BK, Levorse JM, Tilghman SM (1998) Igf2 imprinting does not require its own DNA methylation or H19 RNA. Genes Dev 12(14):2200–2207. https://doi.org/10.1101/gad.12.14.2200
Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, Dandolo L (2008) The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A 105(34):12417–12422. https://doi.org/10.1073/pnas.0801540105
Mineno J, Okamoto S, Ando T, Sato M, Chono H, Izu H, Takayama M, Asada K, Mirochnitchenko O, Inouye M, Kato I (2006) The expression profile of microRNAs in mouse embryos. Nucleic Acids Res 34(6):1765–1771. https://doi.org/10.1093/nar/gkl096
Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W (2012) The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol 14(7):659–665. https://doi.org/10.1038/ncb2521
Duvillie B, Cordonnier N, Deltour L, Dandoy-Dron F, Itier JM, Monthioux E, Jami J, Joshi RL, Bucchini D (1997) Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci U S A 94(10):5137–5140
Leroux L, Desbois P, Lamotte L, Duvillie B, Cordonnier N, Jackerott M, Jami J, Bucchini D, Joshi RL (2001) Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 50(Suppl 1):S150–S153. https://doi.org/10.2337/diabetes.50.2007.s150
Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ (1997) Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 387(6629):151–158
Yan Y, Frisen J, Lee MH, Massague J, Barbacid M (1997) Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 11(8):973–983
Takahashi K, Kobayashi T, Kanayama N (2000) p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol Hum Reprod 6(11):1019–1025
Takahashi K, Nakayama K, Nakayama K (2000) Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J Biochem (Tokyo) 127(1):73–83
Kanayama N, Takahashi K, Matsuura T, Sugimura M, Kobayashi T, Moniwa N, Tomita M, Nakayama K (2002) Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol Hum Reprod 8(12):1129–1135
Tunster SJ, Van de Pette M, John RM (2011) Fetal overgrowth in the Cdkn1c mouse model of Beckwith-Wiedemann syndrome. Dis Model Mech 4(6):814–821. https://doi.org/10.1242/dmm.007328
Andrews SC, Wood MD, Tunster SJ, Barton SC, Surani MA, John RM (2007) Cdkn1c (p57Kip2) is the major regulator of embryonic growth within its imprinted domain on mouse distal chromosome 7. BMC Dev Biol 7:53. https://doi.org/10.1186/1471-213X-7-53
Frank D, Mendelsohn CL, Ciccone E, Svensson K, Ohlsson R, Tycko B (1999) A novel pleckstrin homology-related gene family defined by Ipl/Tssc3, TDAG51, and Tih1: tissue-specific expression, chromosomal location, and parental imprinting. Mamm Genome 10(12):1150–1159
Frank D, Fortino W, Clark L, Musalo R, Wang W, Saxena A, Li CM, Reik W, Ludwig T, Tycko B (2002) Placental overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad Sci U S A 99(11):7490–7495
Salas M, John R, Saxena A, Barton S, Frank D, Fitzpatrick G, Higgins MJ, Tycko B (2004) Placental growth retardation due to loss of imprinting of Phlda2. Mech Dev 121(10):1199–1210
Tunster SJ, Tycko B, John RM (2010) The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol Cell Biol 30(1):295–306. https://doi.org/10.1128/MCB.00662-09
Tunster SJ, Creeth HD, John RM (2016) The imprinted Phlda2 gene modulates a major endocrine compartment of the placenta to regulate placental demands for maternal resources. Dev Biol 409(1):251–260. https://doi.org/10.1016/j.ydbio.2015.10.015
Tunster SJ, Van De Pette M, John RM (2014) Isolating the role of elevated Phlda2 in asymmetric late fetal growth restriction in mice. Dis Model Mech 7(10):1185–1191. https://doi.org/10.1242/dmm.017079
Creeth HDJ, McNamara GI, Tunster SJ, Boque-Sastre R, Allen B, Sumption L, Eddy JB, Isles AR, John RM (2018) Maternal care boosted by paternal imprinting in mammals. PLoS Biol 16(7):e2006599. https://doi.org/10.1371/journal.pbio.2006599
Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA (1998) Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest [see comments]. Nat Genet 20(2):163–169
Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA (1999) Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284(5412):330–333
Harrison DJ, Creeth HDJ, Tyson HR, Boque-Sastre R, Hunter S, Dwyer DM, Isles AR, John RM (2021) Placental endocrine insufficiency programs anxiety, deficits in cognition and atypical social behaviour in offspring. Hum Mol Genet. https://doi.org/10.1093/hmg/ddab154
Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner AL (1994) Essential role of Mash-2 in extraembryonic development. Nature 371(6495):333–336
Tanaka M, Gertsenstein M, Rossant J, Nagy A (1997) Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev Biol 190(1):55–65
Oh-McGinnis R, Bogutz AB, Lefebvre L (2011) Partial loss of Ascl2 function affects all three layers of the mature placenta and causes intrauterine growth restriction. Dev Biol 351(2):277–286. https://doi.org/10.1016/j.ydbio.2011.01.008
Bogutz AB, Oh-McGinnis R, Jacob KJ, Ho-Lau R, Gu T, Gertsenstein M, Nagy A, Lefebvre L (2018) Transcription factor ASCL2 is required for development of the glycogen trophoblast cell lineage. PLoS Genet 14(8):e1007587. https://doi.org/10.1371/journal.pgen.1007587
Tunster SJ, McNamara GI, Creeth HD, John RM (2016) Increased dosage of the imprinted Ascl2 gene restrains two key endocrine lineages of the mouse Placenta. Dev Biol 418(1):55–65. https://doi.org/10.1016/j.ydbio.2016.08.014
John RM (2017) Imprinted genes and the regulation of placental endocrine function: Pregnancy and beyond. Placenta 56:86–90. https://doi.org/10.1016/j.placenta.2017.01.099
Millership SJ, Van de Pette M, Withers DJ (2019) Genomic imprinting and its effects on postnatal growth and adult metabolism. Cell Mol Life Sci 76(20):4009–4021. https://doi.org/10.1007/s00018-019-03197-z
Ferron SR, Radford EJ, Domingo-Muelas A, Kleine I, Ramme A, Gray D, Sandovici I, Constancia M, Ward A, Menheniott TR, Ferguson-Smith AC (2015) Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat Commun 6:8265. https://doi.org/10.1038/ncomms9265
Furutachi S, Matsumoto A, Nakayama KI, Gotoh Y (2013) p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. https://doi.org/10.1038/emboj.2013.50
Joseph B, Wallen-Mackenzie A, Benoit G, Murata T, Joodmardi E, Okret S, Perlmann T (2003) p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc Natl Acad Sci U S A 100(26):15619–15624
McNamara GI, Davis BA, Dwyer DM, John RM, Isles AR (2016) Behavioural abnormalities in a novel mouse model for Silver Russell Syndrome. Hum Mol Genet. https://doi.org/10.1093/hmg/ddw357
McNamara GI, John RM, Isles AR (2018) Territorial behavior and social stability in the mouse require correct expression of imprinted Cdkn1c. Front Behav Neurosci 12:28. https://doi.org/10.3389/fnbeh.2018.00028
McNamara GI, Davis BA, Browne M, Humby T, Dalley JW, Xia J, John RM, Isles AR (2018) Dopaminergic and behavioural changes in a loss-of-imprinting model of Cdkn1c. Genes Brain Behav 17(2):149–157. https://doi.org/10.1111/gbb.12422
Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM (2011) A critical role for IGF-II in memory consolidation and enhancement. Nature 469(7331):491–497. https://doi.org/10.1038/nature09667
Charalambous M, Menheniott TR, Bennett WR, Kelly SM, Dell G, Dandolo L, Ward A (2004) An enhancer element at the Igf2/H19 locus drives gene expression in both imprinted and non-imprinted tissues. Dev Biol 271(2):488–497. https://doi.org/10.1016/j.ydbio.2004.04.022
Imaizumi Y, Furutachi S, Watanabe T, Miya H, Kawaguchi D, Gotoh Y (2020) Role of the imprinted allele of the Cdkn1c gene in mouse neocortical development. Sci Rep 10(1):1884. https://doi.org/10.1038/s41598-020-58629-9
Scagliotti V, Costa Fernandes Esse R, Willis TL, Howard M, Carrus I, Lodge E, Andoniadou CL, Charalambous M (2021) Dynamic expression of imprinted genes in the developing and postnatal pituitary gland. Genes (Basel) 12(4). https://doi.org/10.3390/genes12040509
Bilodeau S, Roussel-Gervais A, Drouin J (2009) Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol 29(7):1895–1908. https://doi.org/10.1128/MCB.01885-08
Borensztein M, Viengchareun S, Montarras D, Journot L, Binart N, Lombes M, Dandolo L (2012) Double Myod and Igf2 inactivation promotes brown adipose tissue development by increasing Prdm16 expression. FASEB J 26(11):4584–4591. https://doi.org/10.1096/fj.12-208496
Van De Pette M, Tunster SJ, McNamara GI, Shelkovnikova T, Millership S, Benson L, Peirson S, Christian M, Vidal-Puig A, John RM (2016) Cdkn1c boosts the development of brown adipose tissue in a murine model of silver russell syndrome. PLoS Genet 12(3):e1005916. https://doi.org/10.1371/journal.pgen.1005916
Hammerle CM, Sandovici I, Brierley GV, Smith NM, Zimmer WE, Zvetkova I, Prosser HM, Sekita Y, Lam BYH, Ma M, Cooper WN, Vidal-Puig A, Ozanne SE, Medina-Gomez G, Constancia M (2020) Mesenchyme-derived IGF2 is a major paracrine regulator of pancreatic growth and function. PLoS Genet 16(10):e1009069. https://doi.org/10.1371/journal.pgen.1009069
Georgia S, Soliz R, Li M, Zhang P, Bhushan A (2006) p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol 298(1):22–31. https://doi.org/10.1016/j.ydbio.2006.05.036
Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tumer Z, Monk D, Mackay DJ, Gronskov K, Riccio A, Linglart A, Netchine I (2015) Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics 7:123. https://doi.org/10.1186/s13148-015-0143-8
Eggermann T, Davies JH, Tauber M, van den Akker E, Hokken-Koelega A, Johansson G, Netchine I (2021) Growth restriction and genomic imprinting-overlapping phenotypes support the concept of an imprinting network. Genes (Basel) 12(4). https://doi.org/10.3390/genes12040585
Weksberg R, Shuman C, Beckwith JB (2010) Beckwith-Wiedemann syndrome. Eur J Hum Genet 18(1):8–14. https://doi.org/10.1038/ejhg.2009.106
Wangler MF, Chang AS, Moley KH, Feinberg AP, Debaun MR (2005) Factors associated with preterm delivery in mothers of children with Beckwith-Wiedemann syndrome: a case cohort study from the BWS registry. Am J Med Genet A 134A(2):187–191. https://doi.org/10.1002/ajmg.a.30595
Romanelli V, Belinchon A, Campos-Barros A, Heath KE, Garcia-Minaur S, Martinez-Glez V, Palomo R, Mercado G, Gracia R, Lapunzina P (2009) CDKN1C mutations in HELLP/preeclamptic mothers of Beckwith-Wiedemann Syndrome (BWS) patients. Placenta 30(6):551–554. https://doi.org/10.1016/j.placenta.2009.03.013
Valente FM, Sparago A, Freschi A, Hill-Harfe K, Maas SM, Frints SGM, Alders M, Pignata L, Franzese M, Angelini C, Carli D, Mussa A, Gazzin A, Gabbarini F, Acurzio B, Ferrero GB, Bliek J, Williams CA, Riccio A, Cerrato F (2019) Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith-Wiedemann locus. Genet Med 21(8):1808–1820. https://doi.org/10.1038/s41436-018-0416-7
Beygo J, Burger J, Strom TM, Kaya S, Buiting K (2019) Disruption of KCNQ1 prevents methylation of the ICR2 and supports the hypothesis that its transcription is necessary for imprint establishment. Eur J Hum Genet 27(6):903–908. https://doi.org/10.1038/s41431-019-0365-x
Beygo J, Joksic I, Strom TM, Ludecke HJ, Kolarova J, Siebert R, Mikovic Z, Horsthemke B, Buiting K (2016) A maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial Beckwith-Wiedemann syndrome. Eur J Hum Genet 24(9):1280–1286. https://doi.org/10.1038/ejhg.2016.3
Essinger C, Karch S, Moog U, Fekete G, Lengyel A, Pinti E, Eggermann T, Begemann M (2020) Frequency of KCNQ1 variants causing loss of methylation of Imprinting Centre 2 in Beckwith-Wiedemann syndrome. Clin Epigenetics 12(1):63. https://doi.org/10.1186/s13148-020-00856-y
Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, Okada A, Ohishi S, Nabetani A, Morisaki H, Nakayama M, Niikawa N, Mukai T (1996) An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet 14(2):171–173. https://doi.org/10.1038/ng1096-171
O’Keefe D, Dao D, Zhao L, Sanderson R, Warburton D, Weiss L, Anyane-Yeboa K, Tycko B (1997) Coding mutations in p57KIP2 are present in some cases of Beckwith-Wiedemann syndrome but are rare or absent in Wilms tumors. Am J Hum Genet 61(2):295–303
Chang S, Bartolomei MS (2020) Modeling human epigenetic disorders in mice: Beckwith-Wiedemann Syndrome and Silver-Russell Syndrome. Dis Model Mech. https://doi.org/10.1242/dmm.044123
Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, Canton AP, Chrzanowska KH, Davies JH, Dias RP, Dubern B, Elbracht M, Giabicani E, Grimberg A, Gronskov K, Hokken-Koelega AC, Jorge AA, Kagami M, Linglart A, Maghnie M, Mohnike K, Monk D, Moore GE, Murray PG, Ogata T, Petit IO, Russo S, Said E, Toumba M, Tumer Z, Binder G, Eggermann T, Harbison MD, Temple IK, Mackay DJ, Netchine I (2017) Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev 13(2):105–124. https://doi.org/10.1038/nrendo.2016.138
Lai KY, Skuse D, Stanhope R, Hindmarsh P (1994) Cognitive abilities associated with the Silver-Russell syndrome. Arch Dis Child 71(6):490–496
Noeker M, Wollmann HA (2004) Cognitive development in Silver-Russell syndrome: a sibling-controlled study. Dev Med Child Neurol 46(5):340–346
Plotts CA, Livermore CL (2007) Russell-Silver syndrome and nonverbal learning disability: a case study. Appl Neuropsychol 14(2):124–134. https://doi.org/10.1080/09084280701322684
Saal HM, Pagon RA, Pepin MG (1985) Reevaluation of Russell-Silver syndrome. J Pediatr 107(5):733–737
Russell A (1954) A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). Proc R Soc Med 47(12):1040–1044
Vardi O, Davidovitch M, Vinkler C, Michelson M, Lerman-Sagie T, Lev D (2012) Autistic regression in a child with Silver-Russell syndrome and maternal UPD 7. Eur J Paediatr Neurol 16(1):95–98. https://doi.org/10.1016/j.ejpn.2011.05.009
Azcona C, Stanhope R (2005) Hypoglycaemia and Russell-Silver syndrome. J Pediatr Endocrinol Metab 18(7):663–670
Blissett J, Harris G, Kirk J (2001) Feeding problems in Silver-Russell syndrome. Dev Med Child Neurol 43(1):39–44
Marsaud C, Rossignol S, Tounian P, Netchine I, Dubern B (2015) Prevalence and management of gastrointestinal manifestations in Silver-Russell syndrome. Arch Dis Child 100(4):353–358. https://doi.org/10.1136/archdischild-2013-305864
Netchine I, Rossignol S, Dufourg MN, Azzi S, Rousseau A, Perin L, Houang M, Steunou V, Esteva B, Thibaud N, Demay MC, Danton F, Petriczko E, Bertrand AM, Heinrichs C, Carel JC, Loeuille GA, Pinto G, Jacquemont ML, Gicquel C, Cabrol S, Le Bouc Y (2007) 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab 92(8):3148–3154. https://doi.org/10.1210/jc.2007-0354
Charalambous M, Smith FM, Bennett WR, Crew TE, Mackenzie F, Ward A (2003) Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proc Natl Acad Sci U S A 100(14):8292–8297
Garfield AS, Cowley M, Smith FM, Moorwood K, Stewart-Cox JE, Gilroy K, Baker S, Xia J, Dalley JW, Hurst LD, Wilkinson LS, Isles AR, Ward A (2011) Distinct physiological and behavioural functions for parental alleles of imprinted Grb10. Nature 469(7331):534–538. https://doi.org/10.1038/nature09651
Schonherr N, Meyer E, Roos A, Schmidt A, Wollmann HA, Eggermann T (2007) The centromeric 11p15 imprinting centre is also involved in Silver-Russell syndrome. J Med Genet 44(1):59–63. https://doi.org/10.1136/jmg.2006.044370
Bonaldi A, Mazzeu JF, Costa SS, Honjo RS, Bertola DR, Albano LM, Furquim IM, Kim CA, Vianna-Morgante AM (2011) Microduplication of the ICR2 domain at chromosome 11p15 and familial Silver-Russell syndrome. Am J Med Genet A 155A(10):2479–2483. https://doi.org/10.1002/ajmg.a.34023
Nakashima S, Kato F, Kosho T, Nagasaki K, Kikuchi T, Kagami M, Fukami M, Ogata T (2015) Silver-Russell syndrome without body asymmetry in three patients with duplications of maternally derived chromosome 11p15 involving CDKN1C. J Hum Genet 60(2):91–95. https://doi.org/10.1038/jhg.2014.100
Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, Le Jule M, Thibaud N, Faussat AM, Tauber M, Le Bouc Y, Netchine I (2013) CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet 50(12):823–830. https://doi.org/10.1136/jmedgenet-2013-101691
Kerns SL, Guevara-Aguirre J, Andrew S, Geng J, Guevara C, Guevara-Aguirre M, Guo M, Oddoux C, Shen Y, Zurita A, Rosenfeld RG, Ostrer H, Hwa V, Dauber A (2014) A novel variant in CDKN1C is associated with intrauterine growth restriction, short stature, and early-adulthood-onset diabetes. J Clin Endocrinol Metab 99(10):E2117–E2122. https://doi.org/10.1210/jc.2014-1949
Sabir AH, Ryan G, Mohammed Z, Kirk J, Kiely N, Thyagarajan M, Cole T (2019) Familial Russell-Silver syndrome like phenotype in the PCNA domain of the CDKN1C gene, a further case. Case Rep Genet 2019:1398250. https://doi.org/10.1155/2019/1398250
Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, Delot EC, Rodriguez-Fernandez IA, Braslavsky D, Bergada I, Dell’angelica EC, Nelson SF, Martinez-Agosto JA, Achermann JC, Vilain E (2012) Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. https://doi.org/10.1038/ng.2275
Cabrera-Salcedo C, Kumar P, Hwa V, Dauber A (2017) IMAGe and related undergrowth syndromes: the complex spectrum of gain-of-function CDKN1C mutations. Pediatr Endocrinol Rev 14(3):289–297. https://doi.org/10.17458/per.vol14.2017.SKHD.imageandrelatedundergrowth
Van de Pette M, Tunster SJ, John RM (2018) Loss of imprinting of Cdkn1c protects against age and diet-induced obesity. Int J Mol Sci 19(9). https://doi.org/10.3390/ijms19092734
Arima T, Kamikihara T, Hayashida T, Kato K, Inoue T, Shirayoshi Y, Oshimura M, Soejima H, Mukai T, Wake N (2005) ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res 33(8):2650–2660
Mackay DJ, Eggermann T, Buiting K, Garin I, Netchine I, Linglart A, de Nanclares GP (2015) Multilocus methylation defects in imprinting disorders. Biomol Concepts 6(1):47–57. https://doi.org/10.1515/bmc-2014-0037
Eggermann T, Heilsberg AK, Bens S, Siebert R, Beygo J, Buiting K, Begemann M, Soellner L (2014) Additional molecular findings in 11p15-associated imprinting disorders: an urgent need for multi-locus testing. J Mol Med (Berl) 92(7):769–777. https://doi.org/10.1007/s00109-014-1141-6
Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D, Monk D (2016) Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet 32(7):444–455. https://doi.org/10.1016/j.tig.2016.05.001
Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, Hahnemann JM, Kordonouri O, Masoud AF, Oestergaard E, Storr J, Ellard S, Hattersley AT, Robinson DO, Temple IK (2008) Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 40(8):949–951. https://doi.org/10.1038/ng.187
Boonen SE, Hahnemann JM, Mackay D, Tommerup N, Brondum-Nielsen K, Tumer Z, Gronskov K (2012) No evidence for pathogenic variants or maternal effect of ZFP57 as the cause of Beckwith-Wiedemann Syndrome. Eur J Hum Genet 20(1):119–121. https://doi.org/10.1038/ejhg.2011.140
Spengler S, Gogiel M, Schonherr N, Binder G, Eggermann T (2009) Screening for genomic variants in ZFP57 in Silver-Russell syndrome patients with 11p15 epimutations. Eur J Med Genet 52(6):415–416. https://doi.org/10.1016/j.ejmg.2009.07.005
Meyer E, Lim D, Pasha S, Tee LJ, Rahman F, Yates JR, Woods CG, Reik W, Maher ER (2009) Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet 5(3):e1000423. https://doi.org/10.1371/journal.pgen.1000423
Docherty LE, Rezwan FI, Poole RL, Turner CL, Kivuva E, Maher ER, Smithson SF, Hamilton-Shield JP, Patalan M, Gizewska M, Peregud-Pogorzelski J, Beygo J, Buiting K, Horsthemke B, Soellner L, Begemann M, Eggermann T, Baple E, Mansour S, Temple IK, Mackay DJ (2015) Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat Commun 6:8086. https://doi.org/10.1038/ncomms9086
Valero De Bernabe J, Soriano T, Albaladejo R, Juarranz M, Calle ME, Martinez D, Dominguez-Rojas V (2004) Risk factors for low birth weight: a review. Eur J Obstet Gynecol Reprod Biol 116(1):3–15. https://doi.org/10.1016/j.ejogrb.2004.03.007
DeChiara TM, Efstratiadis A, Robertson EJ (1990) A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345:78–80
Kappil MA, Green BB, Armstrong DA, Sharp AJ, Lambertini L, Marsit CJ, Chen J (2015) Placental expression profile of imprinted genes impacts birth weight. Epigenetics 10(9):842–849. https://doi.org/10.1080/15592294.2015.1073881
Su R, Wang C, Feng H, Lin L, Liu X, Wei Y, Yang H (2016) Alteration in expression and methylation of IGF2/H19 in placenta and umbilical cord blood are associated with macrosomia exposed to intrauterine hyperglycemia. PLoS One 11(2):e0148399. https://doi.org/10.1371/journal.pone.0148399
Xing Y, Liu H, Cui Y, Wang X, Tong X (2019) Abundances of placental imprinted genes CDKN1C, PHLDA2 and IGF-2 are related to low birth weight and early catch-up growth in full-term infants born small for gestational age. PLoS One 14(6):e0218278. https://doi.org/10.1371/journal.pone.0218278
Demetriou C, Abu-Amero S, Thomas AC, Ishida M, Aggarwal R, Al-Olabi L, Leon LJ, Stafford JL, Syngelaki A, Peebles D, Nicolaides KH, Regan L, Stanier P, Moore GE (2014) Paternally expressed, imprinted insulin-like growth factor-2 in chorionic villi correlates significantly with birth weight. PLoS One 9(1):e85454. https://doi.org/10.1371/journal.pone.0085454
Boonen SE, Freschi A, Christensen R, Valente FM, Lildballe DL, Perone L, Palumbo O, Carella M, Uldbjerg N, Sparago A, Riccio A, Cerrato F (2016) Two maternal duplications involving the CDKN1C gene are associated with contrasting growth phenotypes. Clin Epigenetics 8:69. https://doi.org/10.1186/s13148-016-0236-z
Xue Y, Shankar S, Cornell K, Dai Z, Wang CE, Rudd MK, Coffee B (2015) Paternal duplication of the 11p15 centromeric imprinting control region is associated with increased expression of CDKN1C in a child with Russell-Silver syndrome. Am J Med Genet A 167A(12):3229–3233. https://doi.org/10.1002/ajmg.a.37371
López-Abad M, Iglesias-Platas I, Monk D (2016) Epigenetic characterization of CDKN1C in placenta samples from non-syndromic intrauterine growth restriction. Front Genet 7:62. https://doi.org/10.3389/fgene.2016.00062
Jensen AB, Tunster SJ, John RM (2014) The significance of elevated placental PHLDA2 in human growth restricted pregnancies. Placenta 35(8):528–532. https://doi.org/10.1016/j.placenta.2014.04.018
McMinn J, Wei M, Schupf N, Cusmai J, Johnson EB, Smith AC, Weksberg R, Thaker HM, Tycko B (2006) Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 27(6–7):540–549
Petry CJ, Evans ML, Wingate DL, Ong KK, Reik W, Constancia M, Dunger DB (2010) Raised late pregnancy glucose concentrations in mice carrying pups with targeted disruption of H19delta13. Diabetes 59(1):282–286. https://doi.org/10.2337/db09-0757
Janssen AB, Capron LE, O’Donnell K, Tunster SJ, Ramchandani PG, Heazell AE, Glover V, John RM (2016) Maternal prenatal depression is associated with decreased placental expression of the imprinted gene PEG3. Psychol Med 46(14):2999–3011. https://doi.org/10.1017/S0033291716001598
Patten MM, Cowley M, Oakey RJ, Feil R (2016) Regulatory links between imprinted genes: evolutionary predictions and consequences. Proc Biol Sci 283(1824). https://doi.org/10.1098/rspb.2015.2760
Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, Pavlidis P, Journot L (2006) Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11(5):711–722. https://doi.org/10.1016/j.devcel.2006.09.003
Gabory A, Ripoche MA, Le Digarcher A, Watrin F, Ziyyat A, Forne T, Jammes H, Ainscough JF, Surani MA, Journot L, Dandolo L (2009) H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 136(20):3413–3421. https://doi.org/10.1242/dev.036061
Dalgaard K, Landgraf K, Heyne S, Lempradl A, Longinotto J, Gossens K, Ruf M, Orthofer M, Strogantsev R, Selvaraj M, Lu TT, Casas E, Teperino R, Surani MA, Zvetkova I, Rimmington D, Tung YC, Lam B, Larder R, Yeo GS, O’Rahilly S, Vavouri T, Whitelaw E, Penninger JM, Jenuwein T, Cheung CL, Ferguson-Smith AC, Coll AP, Korner A, Pospisilik JA (2016) Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell 164(3):353–364. https://doi.org/10.1016/j.cell.2015.12.025
Barker DJ (1990) The fetal and infant origins of adult disease. BMJ 301(6761):1111
Gluckman PDaH M (2006) Developmental origins of health and disease, 1st edn. Cambridge University Press, Cambridge
Ivanova E, Chen JH, Segonds-Pichon A, Ozanne SE, Kelsey G (2012) DNA methylation at differentially methylated regions of imprinted genes is resistant to developmental programming by maternal nutrition. Epigenetics 7(10):1200–1210. https://doi.org/10.4161/epi.22141
Radford EJ, Isganaitis E, Jimenez-Chillaron J, Schroeder J, Molla M, Andrews S, Didier N, Charalambous M, McEwen K, Marazzi G, Sassoon D, Patti ME, Ferguson-Smith AC (2012) An unbiased assessment of the role of imprinted genes in an intergenerational model of developmental programming. PLoS Genet 8(4):e1002605. https://doi.org/10.1371/journal.pgen.1002605
Acknowledgments
M.A.S. is supported by Wellcome Trust grant 209475/Z/17/Z, L.L. is supported by CIHR grant PJT-165992 and R.M.J is supported by BBSRC grant BB/V008684/1. The authors are grateful to the wider imprinting community—both those whose work is directly covered and the many studies which we have not been able to include due to space constraints. Help with Fig. 8.1 from F. M. John.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
John, R.M., Lefebvre, L., Surani, M.A. (2022). Genomic Imprinting: A Paradigm for Epigenetics of Human Diseases. In: Michels, K.B. (eds) Epigenetic Epidemiology. Springer, Cham. https://doi.org/10.1007/978-3-030-94475-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-94475-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94474-2
Online ISBN: 978-3-030-94475-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)