
Abstract
Studies in epigenetic epidemiology have reported increasing numbers of epigenetic biomarkers associated with a wide range of exposures and outcomes. Due to cost and technical difficulties, these markers are usually derived from complex tissues that are composed of many different cell-types. This cell-type heterogeneity prevents the identification of cell-type specific epigenetic alterations, posing significant challenges to the interpretation and understanding of these markers. Consequently, there is a strong need to develop cost-effective computational solutions to tackle the cell-type heterogeneity problem. Here, I discuss some recently proposed cell-type deconvolution algorithms aimed at estimating cell-type fractions and identifying cell-type specific differential DNA methylation changes. I describe their successful application to epigenome studies. We also discuss their main limitations, providing general guidelines for their successful implementation and for correctly interpretating results derived from them.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Over the last two decades, we have seen a rapid increase in the number of studies reporting associations of epigenetic marks, in particular DNA methylation (DNAm), with epidemiological and disease risk factors, as well as with disease itself [1,2,3,4,5,6,7,8,9,10]. Many of these associations have been derived by measuring DNA methylation in the tissue-of-origin, for example, in precursor cancer lesions [8, 11, 12], cancer-tissue [13] or post-mortem brain [14, 15]). However, by far most associations have been derived from easily accessible “surrogate” tissues like blood [1, 16], often under the assumption that DNAm changes in such tissues can be informative of disease or disease risk [17,18,19,20]. An ever-increasing number of epigenetic biomarker studies are also measuring DNAm of cell-free DNA fragments (cfDNAm) in serum, which offers great potential for noninvasive early detection of a wide range of diseases, including type-1 diabetes, multiple sclerosis, ischemic brain damage, pancreatitis, and cancer [21,22,23,24,25,26]. In most cases, however, the biological interpretation of the measured epigenetic alterations remains challenging [27]. One obvious reason for this is that, unlike gene or protein expression, an epigenetic change may not necessarily be functional. As a concrete example, age-associated DNAm alterations have been widely reported [6, 7, 28,29,30,31], yet a significant proportion of these changes do not appear to be functional or may only act to stabilize gene expression [32]. Another reason, which is still often overlooked, is cell-type heterogeneity [33]. This refers to the fact that most epigenome studies deriving biomarkers or molecular classifications of disease, do so by measuring DNAm in a complex tissue comprised of many different cell-types, each with its own characteristic DNAm profile. This is true not only for tissues like lung, liver, or blood, but also when measuring cfDNAm in serum, as most of the cfDNA derives from lymphocytes [21, 26]. Thus, by only measuring an average DNAm profile over many underlying cell-types, it is difficult to ascertain if DNAm changes associated with an exposure or outcome of interest is the result of DNAm changes in individual cell-types, and if so, in which cell-types, or whether the DNAm change is merely the result of underlying changes in cell-type proportions (Fig. 4.1).

Broad classes of DMCs. Epigenome studies performed in complex tissues (by definition these are composed of many different cell-types) only measure an average DNAm profile, and thus can only detect differentially methylated cytosines (DMCs) without knowledge of the underlying sources driving these DNAm changes. DMCs may result from shifts in cell-type proportions between cases and controls, or may result from DNAm changes in one or more cell-types (CT). All types of DMCs may be informative of say diagnosis or prognosis, but detection of cell-type specific DMCs is important to improve our understanding of the molecular pathways involved in disease development
Is it important, from a biological, clinical, or epidemiological perspective, to determine the source or nature of a DNAm change? In general, the answer to this question is yes: knowing in which cell-type (or cell-types) a DNAm change may be occurring in is critically important in order to understand how putative functional consequences of the DNAm changes may affect cell-function and disease development. For instance, in the context of asthma, an EWAS for immunoglobulin-E concentrations in blood revealed associations that were later validated in isolated eosinophils, highlighting molecular pathways in a relevant cell-type that mediate allergic inflammation [34]. Another example is that of HAND2, a transcription factor that mediates the tumor-suppressive effects of progesterone in the endometrium: here promoter hypermethylation and silencing of HAND2 is observed in endometrial fibroblasts from precursor cancer lesions, which results in increased FGF paracrine signaling with endometrial epithelial cells, sensitizing these to oncogenic estrogen [35]. Another scenario where cell-type specificity may play an important role is in the context of mQTLs, i.e., SNP-CpG pairs where the methylation of the CpG is associated with genotype, with recent studies indicating that up to 20–30% of mQTLs derived in blood may be cell-type specific [36, 37]. Such cell-type-specific mQTLs could be informative of molecular pathways that contribute to disease predisposition in a cell-type-specific manner. Another example is aging, where the promoter of a given cell-specific transcription factor may become gradually hypermethylated with age, which could lead to irreversible silencing of the TF and to a skewed or impaired differentiation [38]. The same DNAm change occurring in a cell-type where the TF is already switched off, say by a repressive H3K27me3 mark, would not have a functional consequence.
One solution to the cell-type heterogeneity problem would be to perform the epigenome study in purified cell-types, but this remains labor intensive, costly and has mostly only been carried out in immune cells [36, 39,40,41], or to validate findings at a few selected loci [34]. Another potential solution is to generate DNAm data at the resolution of single cells [42,43,44]. However, this also remains costly, is unscalable to large numbers of individuals, and only generates very sparse data that cannot be used for building DNAm reference profiles for individual cell-types [44, 45]. Thus, there has been an ongoing attempt to address the cell-type heterogeneity problem computationally, by devising statistical algorithms (i) that can infer cell-type fractions and DMCs [46,47,48], (ii) that can infer latent components of variation and DMCs [49,50,51], and (iii) that can infer cell-type specific differential DNAm, i.e., differentially methylated cell-types (DMCTs) [52, 53] [54, 55], all in the context of epigenome studies performed in complex tissues.
The computational complexity posed by the cell-type heterogeneity problem can be quite substantial: assuming a study is performed in whole blood (a tissue with seven main blood cell subtypes: neutrophils, eosinophils, monocytes, natural-killer, B-cells, CD4+, and CD8+ T-cells), and that one finds a DMC associated with a factor of interest, there could be at least 27–1 = 127 different combinatorial differential methylation (DM) patterns among the seven cell-types that could give rise to the observed DM at the whole tissue level. For instance, at one extreme, a DMC could be present in all cell-types of the tissue, while at another, it may only be present in one of the cell-types (Fig. 4.1). The above estimate does not even consider the potential combinatorial possibilities in terms of the directionality of DNAm change, which could involve increased (i.e., hyper) or decreased (i.e., hypo) methylation in different cell-types. Nor does the above estimate include scenarios where the DMC is driven purely by a change in cell-type composition. A well-known example of the latter is the increased myeloid to lymphocyte ratio that is observed in blood as a function of age [7, 56], cancer [57] or Rheumatoid Arthritis (RA) [58]. Shifts in cell-type composition are of course critically important in the context of cfDNAm in serum, since it is these shifts (e.g., increased circulating tumor DNA burden) that are informative of disease [26]. In other easily accessible tissues such as saliva or buccal swabs, the number of cell-types will be even bigger than in blood, because these tissues contain squamous epithelial cells besides immune cells [59,60,61]. The complexity only increases even further when we start to consider solid tissues, which in addition to immune cells, may contain different types of epithelial, endothelial, and fibroblast cells. The full repertoire of cell-types within human tissues and organs is only now been elucidated thanks to major international efforts such as the Human Cell Atlas [62, 63]. For instance, some studies have estimated over 50 different cell-types in a tissue like lung [64, 65]. It should be noted though that given an observed magnitude of DNAm change, that this may impose substantial constraints on the allowed cell-type-specific DM patterns. For instance, if we observe a close to 100% change in DNAm between cases and controls, then this can only be realized if the change is happening unidirectionally in all the major cell-types of the issue. In general, it should be clear that the complexity of calling cell-type-specific DM can be at least 100-fold higher compared to calling DM [66].
Given this complexity, and given the inevitable limitations on the sample size of epigenome studies, it is understandable that statistical algorithms alone may not be able to fully address the above challenge. Nevertheless, as we shall see, statistical and computational methodology can help towards partial solutions or to solving the challenge in simpler scenarios, which can still be very informative and useful for disease diagnosis and early detection, for hypothesis generation or for devising validation experiments in purified cell-types. For instance, one way to simplify the problem is to consider a small number of “coarse” or “representative” cell-types, or only restrict to the main dominant cell-types within a tissue.
In the next sections, we shall describe some of the computational and statistical methods that have been proposed to estimate cell-type fractions and to detect cell-type-specific DNAm changes, as well as a number of applications where it has led to important novel insights.
2 General Considerations
In that follows, we shall refer to a cell-type specific differentially methylated cytosine as a differentially methylated cell-type, abbreviating this with “DMCT.” Given a DNAm matrix defined over cytosines and samples, and given a factor of interest (e.g., an exposure, disease-status), the inference of DMCTs generally proceeds in two steps:
-
1.
First, we need to estimate the fractions for all cell-types in the tissue. Without knowledge of the proportions of each cell-type in a given tissue, it is not possible to infer in which cell-types putative DNAm changes are happening. Some cell-types in a tissue may be present in such low numbers that their estimation is not possible, and therefore in this step one aims to infer fractions for as many cell-types so as to account for at least 95% of the tissue composition. This is because the error-rate of estimating cell-type fractions generally is about 5% [48].
-
2.
Second, the estimated cell-type fractions are used as covariates in a linear model relating the observed DNAm profile of a cytosine to a factor/exposure of interest and other covariates representing potential confounding factors (e.g., batch effects). Importantly, the cell-type fractions enter the equations not only as ordinary covariates, but also as part of interaction terms with the factor/exposure of interest. Later we shall explain why interaction terms capture DMCTs.
While in general, only these two steps are required, in practice, it is strongly advised to include two intermediate analyses in order to better interpret the overall results. Once we have estimated cell-type fractions, it is important to establish if these cell-type fractions vary significantly in relation to our factor/exposure of interest. Indeed, if tissue composition changes, then the underlying shifts in cellular proportions could play an important causal role in disease development. A concrete example is the infiltration of CD8+ T-cells in triple-negative breast cancer, which is a well-known predictor of good outcome in this usually aggressive type of cancer [67]. Cell-type compositional changes could potentially also be used for disease risk prediction or diagnosis [21, 22, 68,69,70]. For instance, in the context of cfDNAm in serum, an increased proportion of DNA fragments reflecting tissue-specific cell-death, as e.g., it happens with pancreatic beta-cell death in type-1 diabetes or with exocrine cell-death in pancreatic cancer, could be used for early detection or monitoring of therapy response [26]. A shift in tissue composition can also reflect a systemic effect of disease, as for instance, with the observed increased myeloid to lymphoid ratio in the blood of cancer patients [57]. If such shifts occur before the conventional diagnosis of disease, they could be used for early detection or for quantifying disease risk.
The other intermediate step one is advised to perform is the inference of differentially methylated cytosines (DMCs). This is accomplished using the same linear model as for DMCT inference, but without the inclusion of interaction terms. In this model, cell-type fractions only enter as ordinary covariates alongside other potential confounders, aim being to identify DMCs that are not driven by changes in cell-type composition or by any of the other potential confounders. It is important to stress that once we have identified such a DMC, that this does not tell us anything about which cell-types the DNAm change is happening in, although there could be constraints on this depending on the observed effect size, as mentioned earlier. We shall see one concrete example of this later. Now we turn to the specific task of estimating cell-type fractions in complex tissues.
3 Estimating Cell-Type Fractions in Complex Tissues
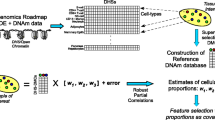
As mentioned earlier, this task is required before we can infer DMCTs. And the reliability of the inference of DMCTs in a given study hinges on our ability to accurately estimate the underlying cell-type fractions. Currently, the best way to estimate cell-type fractions in a given sample for which a genome-wide DNAm profile is available, is through the construction of a DNA methylation reference matrix (DMRM) [71]: the columns of this matrix represent the cell-types within the tissue, with the rows representing marker CpGs (or marker genes) whose DNAm levels vary substantially between cell-types. Here we shall discuss three different strategies that have been applied to build a DMRM (Fig. 4.2). The choice of strategy largely depends on the tissue-type being considered, which is why the ensuing discussion is structured based on tissue-type.

Strategies to building a DNAm reference matrix. Depicted are three strategies to building a DNAm reference matrix (DMRM). One approach (left branch) is based on FACS sorting cells and subsequently generating genome-wide DNAm profiles for these purified cell populations. Subsequently, one identifies DMCs between these purified cell populations to build the DMRM. Another strategy (middle) is to use existing DNAm profiles of relevant purified cell samples in the public domain to build the DMRM, once again by identifying DMCs between these purified samples. This strategy can be applied more broadly to solid tissues, but compromising cellular resolution. Another strategy (right) is to leverage the high-resolution nature of a tissue-specific scRNA-Seq atlas to build an mRNA expression reference matrix and to subsequently impute a corresponding tissue-specific DMRM. The cellular resolution of this strategy is high (only limited by the cell-types the scRNA-Seq assay can measure), but the quality of the imputation may not suffice to distinguish similar cell-types
3.1 Estimation of Cell-Type Fractions in Blood
We first consider the case of whole blood (WB), peripheral blood mononuclear cells (PMBCs) and cord blood (CB), because for these tissues, it is possible to generate genome-wide DNAm reference profiles for all major underlying cell-types. This can be accomplished by the use of well-known markers for blood cell subtypes, whose specificity is relatively high in order to be able to generate purified samples (e.g., CD19 for B-cells) through, e.g., FACS sorting. Thus, in the case of WB, it has been possible to generate genome-wide DNAm profiles for all seven major cell subtypes, which includes neutrophils, eosinophils, monocytes, B-cells, CD4+ T-cells, CD8+ T-cells, natural-killer cells, and B-cells [72]. PBMCs are generally devoid of granulocytes which includes neutrophils, eosinophils, and basophils. For CB, a tissue of increasing importance in epigenetic epidemiology [73,74,75,76,77], it has also been possible to build tailored DNAm reference matrices [78, 79], as indeed it is well recognized that both the composition and cell-type-specific DNAm profiles are different to those of whole blood. In all these cases, once genome-wide DNAm profiles for purified samples have been generated, it is then possible to build corresponding DMRMs.
It is important to briefly describe how the DMRM should be constructed and indeed why it is necessary to use this DMRM for estimating cell-type fractions. If we have a genome-wide DNAm profile for a WB/PBMC/CB sample, represented by a vector \( \overrightarrow{\beta} \) defined over a large number of CpGs (around 450k or 850k, depending on the Illumina beadarray version), then it is sensible to express it as a linear mixture of corresponding genome-wide DNAm profiles for the constituent cell-types, as in:
where k denotes the cell-type, K is the total number of different cell-types and fk is the proportion/fraction of cells of type k in the mixture. These fractions are obviously unknown, and we would like to infer them given the measured DNAm profile of the mixture and that of the constituent cell-types. Mathematically, the inference of these fractions is indeed possible, by formulating the above equation as a linear least squares multivariate regression problem, with one key difference, however, which is that the fractions must all be bounded between 0 and 1, and that their sum must add to 1 (or to a number less than 1 since in practice we cannot possibly know or have DNAm profiles for all underlying cell-types in a tissue). These additional constraints on the regression problem turn the ordinary least squares regression (LSR) into a constrained LSR or constrained projection (CP) problem, which can be solved using techniques in quadratic programming (QP) [46]. While it might be tempting to solve this CP problem using all available CpGs, this is not advisable because a large chunk of the genome will not differ between blood cell subtypes. In other words, uninformative CpGs that show little variability between blood cell subtypes generally do not contribute, and therefore it is sensible to exclude these from the inference as they are not needed. Computationally, if the CP problem can be solved over a small number of informative CpGs this will also speed up the estimation of cell-type fractions very substantially, which is an important consideration. Thus, once the DNAm profiles for the purified cell-types have been generated, the next step is to identify the informative CpGs. There are several strategies to accomplish this [46, 48, 78, 79], but in general, they involve a process of identifying cell-type specific DMCs, i.e., CpGs that are highly methylated (or unmethylated) in one cell-type, with correspondingly low (or high) DNAm values in all other cell-types. Ideally, one desires a reasonable number of cell-type specific DMCs for each cell-type, which ensures robustness to potential “dropouts” in independent datasets where the fractions are to be estimated. By dropout we here mean a probe that does not pass QC in the independent dataset and which therefore cannot be used in the inference procedure. Ideally, one would also want to ensure equal or approximately equal numbers of cell-type-specific markers for each cell-type, because can avoid biasing the inference to those cell-types with more markers. And thirdly, one would ideally only include cell-type-specific DMCs, where the difference in DNAm between the marker cell-type and all others is as large as possible, i.e., typically over 0.7 in the beta value [0,1] scale, i.e., at least a 70% methylation change. This number is motivated by the heavy use of Illumina DNAm beadarray technologies, where the two main modes (unmethylated and methylated peaks) are generally about 0.7 methylation units apart, with a value of 1 being the theoretical maximum. In practice, all three requirements above may not be met. For instance, for similar cell-types (e.g., CD4+ and CD8+ T-cells) it might be difficult to find many DMCs with over 70% DNAm difference between them, and indeed accurately estimating the relative fractions of highly similar cell-types still remains a challenge. Mathematically, the quality of a DNAm reference matrix can be expressed in terms of its conditioning number [79], which is an indicator of how robust the inference would be. Based on extensive experience estimating cell-type fractions, we advise on the following guidelines for selecting DMCs for a reference DNAm matrix. We note that these guidelines are not unique to DNAm but apply equally well to other data types (e.g., RNA-Seq):
-
1.
Perform DM analysis between one cell-type (the “marker” cell-type) against all others (as one group) to identify DMCs using some sensible significance threshold (e.g., FDR < 0.05).
-
2.
For these DMCs, compare the mean DNAm level in the marker cell-type to the highest (or lowest) mean DNAm level from among all other cell-types, to then subselect DMCs that have an effect size (i.e., difference in mean DNAm) larger than +/−0.7.
-
3.
For each cell-type, rank these DMCs according to the absolute effect size and select a given top number of these. We recommend about 50 for each cell-type, if possible.
-
4.
If in step-2 or step-3 there are not enough DMCs for a given cell-type, then the corresponding effect size threshold could be relaxed (for instance, one could go as low as +/−0.3 or +/−0.4), or alternatively, one may select a smaller number of top-ranked DMCs in step-3. However, as mentioned, due to QC-issues in independent data, we do not recommend that the final number of cell-type specific DMCs per cell-type should be less than 10.
Once the cell-type-specific DMCs have been identified, the DMRM is then built by taking the average or median DNAm of these CpGs in each cell-type separately. We note that although theoretically, one should take the average, in practice, taking the median is also justified as it may provide a more robust estimator. For K cell-types, the DMRM would then ideally be a matrix with K*50 rows labeling the cell-type-specific DMCs, and K columns labeling the cell-types. With this DMRM in place, we would then estimate cell-type fractions by solving the following CP problem:
DMRMs for blood and cord blood are available from various Bioconductor R-packages, including minfi [80], EpiDISH [48] and FlowSorted.CordBlood.450 K [81].
In the case of tissues like blood, it is also possible to obtain cell-type fractions using magnetic flow cytometric techniques (e.g., MACS), which thus allows objective testing of the DNAm-based estimates. This assumes of course that the sample taken for the DNAm-assay and the sample used to obtain MACS-based estimates are taken at the same timepoint, as blood cell fractions are known to vary with time [82,83,84]. In general, the agreement of estimated cell-type fractions obtained using a DMRM with those derived with MACS is very good (Fig. 4.3).

Agreement between DNAm and flow cytometry-based cell fraction estimates in whole blood. For each of the six major blood cell subtypes, a scatterplot of the flow cytometric cell-type fraction estimate (y-axis) vs. the DNAm-based estimate (x-axis). The dashed lines represent the best fit regression line. R2 and P-values are given. Dataset consists of 162 whole blood samples from healthy individuals

Identification of DMCTs. (a) Overall strategy to infer DMCTs involves a step where we infer cell-type fractions for each sample, followed by fitting a linear model with interaction terms between phenotype and cell-type fractions to infer a map over CpGs and cell-types indicating which CpGs are altered in which cell-types. (b–d) Three examples of CpGs that define DMCTs in one cell-type (CT1) (b), all cell-types (CT1–3) (c) and two cell-types where the direction of DNAm change is different (CT1–2) (d). The scatterplots display the adjusted DNAm beta value against the corresponding cell-type fraction, with red datapoints indicating “cases,” blue datapoints indicating “controls.” Hyper = hypermethylated in cases, Hypo = hypomethylated in cases
3.2 Estimation of Cell-Type Fractions in Saliva and Buccal Swabs
Like blood, saliva and buccal tissue can be obtained fairly easily and cheaply, allowing noninvasive measurement of DNA methylation in a tissue that contains squamous epithelial cells in addition to immune cells [59,60,61]. The reason for treating saliva and buccal swabs separately from blood and solid tissue-types is that it is relatively straightforward to build DMRMs for these two tissue-types. Since they contain immune cells, the same DNAm profiles of immune cell subtypes in blood can be used for building the DMRM for saliva/buccal swabs. The only additional requirement is to generate a DNAm reference profile for squamous epithelial cells. However, since effectively most epithelial cells in saliva/buccal swabs are squamous in origin, and there is therefore no need to distinguish different epithelial cell-types from each other, a DNAm reference profile for any epithelial cell-type may suffice. For instance, ENCODE [85] and the NIH Epigenomics roadmap [86, 87] have generated genome-wide DNAm profiles for many different normal epithelial cell lines and primary epithelial cells. By using different types of epithelial samples one can thus hone in on the generic DNAm patterns of any epithelial sample, including the squamous epithelial cells from the oral cavity. Thus, for these tissues and given genome-wide DNAm profiles for epithelial and immune cell subtypes, one can apply the same DMRM-construction strategy outlined above, i.e., by first deriving cell-type-specific DMCs, from which the DMRM is then built. Estimation of cell-type fractions then proceeds exactly as described before for the case of blood.
A slight variation to the above procedure is obtained by recognizing that cell-types within a tissue exhibit markedly different levels of similarity, depending on their shared developmental trajectories. Thus, one expects substantially more DMCs between epithelial and immune cells than between CD4+ and CD8+ T-cells. In recognition of this, an algorithm called HEpiDISH (Hierarchical Epigenetic Dissection of Intra-Sample Heterogeneity) [59] uses a hierarchical 2-step strategy with two DNAm reference matrices, one aimed at obtaining a total epithelial and total immune cell fraction, and a second one to obtain fractions for all immune cell subtypes. Because we will revisit HEpiDISH in the context of solid tissue-types, we refer details to the next subsection.
3.3 Estimation of Cell-Type Fractions in Solid Tissues
The most challenging scenario is to estimate cell-type fractions in a solid tissue-type. This is because the number of cell-types is obviously much higher than in blood (there will be different kinds of fibroblasts, endothelial, and epithelial cell subtypes, besides immune cells), but also because this number is still generally unknown and generating DNAm profiles for even just a few of these cell-types is technically challenging. This means that for most cell-types in a solid tissue, there are no available DNAm reference profiles. This is despite efforts from the Epigenomics Roadmap [86] to generate a number of these (e.g., kidney podocytes, breast myoepithelial, breast luminal cells). Given these major challenges, how does one then aim to estimate cell-type fractions in a solid tissue like breast or lung where the number of cell-types will be substantially higher than 10 and for which reference DNAm profiles may not be available for specific cell-types?
In principle, one may be tempted to apply reference-free cell-type deconvolution algorithms [49, 50, 88], which aim to infer latent (i.e., hidden/unknown) sources of variation in the data, some of which may correspond to variations in cell-type fractions. However, reference-free algorithms cannot yield direct cell-type fraction estimates and are therefore limited as far as DMCT inference is concerned, their application being primarily to infer DMCs not driven by changes in cell-type composition [49]. Given that biotechnology will eventually advance to a level that makes single-cell DNA methylomics more reliable, affordable, and scalable, thus allowing routine generation of DNAm reference matrices, it is sensible to focus on reference-based approaches as we are doing here.
So far, two different reference-based strategies have been developed. One approach is based on the HEpiDISH algorithm [59], mentioned earlier. This uses a two DMRM strategy, whereby in the first step, the algorithm uses one DMRM to estimate a total epithelial, a total fibroblast and a total immune cell fraction. While solid tissues like lung or breast contain other cell-types (e.g., endothelial cells), the lack of sufficient reference DNAm profiles for purified endothelial cells (to allow for training and validation) means that these may be challenging to include. In the case of breast, adipocytes form a major component in addition to epithelial, fibroblast, and immune cells, and for adipocytes, reference DNAm profiles are available in the public domain to allow construction and independent validation of a 4 cell-type DNAm reference matrix, defined over a generic epithelial, fibroblast, fat, and immune cell [59]. In the second optional step, HEpiDISH then estimates fractions for the immune cell subtypes. This is accomplished with a second DMRM defined over CpGs that discriminate the different immune cell subtypes from each other. Importantly, for each of these CpGs, it is also required that their baseline DNAm level, i.e., the level of DNAm seen across most of the immune cell subtypes, is similar to that of the epithelial, fibroblast, and fat cells, to ensure that variations in these other cell-type fractions would not bias the relative fractions of immune cell subtypes [59]. Thus, this second DMRM is distinct from the one used when inferring cell-type fractions in WB.
Another strategy is based on the EpiSCORE algorithm [89, 90]. EpiSCORE leverages the high-resolution nature of a tissue-specific scRNA-Seq atlas to first construct a mRNA expression reference matrix for all major cell-types in the tissue. This assumes that the scRNA-Seq assay has captured the most important cell-types in the tissue, which is not always the case: for instance, in the case of breast, fat cells are abundant but they are large cells that are often missed by current scRNA-Seq protocols [91]. Given the mRNA expression reference matrix, EpiSCORE then imputes a corresponding DMRM, defined over the promoters of a subset of the expression marker genes and the same number of cell-types. Of note, this imputation only works for 20–30% of the marker genes in the expression reference matrix, because for most marker genes there is no strong anti-correlative pattern between promoter DNAm and gene expression. The smaller number of “imputable” marker genes, for which there is such a strong anti-correlation, are identified using matched RNA-Seq and whole-genome bisulfite sequencing (WGBS) data from resources like the NIH Epigenomics Roadmap [86] and ENCODE [92, 93]. Unlike HEpiDISH, EpiSCORE can then yield cell-type fractions for all cell-types in the tissue in one step, by applying an analogous multivariate linear model as the one described earlier for blood.
3.4 Estimation of Cell-Type Fractions from cfDNAm in Serum
For completeness, we also describe briefly methods for estimating cell-type fractions from cfDNAm in serum, framing this in the context of cancer where such methods offer particular promise. Broadly speaking, the underlying strategy has been to build analogs of DMRMs by identifying informative CpGs (i.e., DMCs) from comparisons between DNAm measured in serum or blood from healthy individuals and DNAm measured in solid cancer-types. Since the cfDNA in plasma from healthy individuals stems predominantly from lymphocytes, such differential DNAm analyses naturally adjust for the immune cell contamination that is inevitable present in solid cancers [79, 94], thus identifying DNAm changes that are unique to the non-immune cells (e.g., epithelial, fibroblast cells) present in cancer-tissue. An algorithm called Cancer Locator [95] then builds a DMRM defined over a healthy and multiple cancer-type states, subsequently applying this DMRM in a beta-mixture modeling maximum likelihood framework to infer both tumor fraction and tumor-type. An important element of this approach has been the explicit modeling of variance in addition to the mean when building the DMRM [95]. Another strategy has been to perform targeted bisulfite sequencing of informative regions in case/control cohorts, using Machine Learning (ML) methods on a training set to build predictors of cancer-type, which are subsequently validated in independent plasma samples [21, 22]. Here, the ML method returns a probability measure that an individual has a particular cancer-type, which can be interpreted roughly as a relative measure of the corresponding tumor cell burden in the serum sample. Overall, these strategies have shown that sensitive detection, i.e., with sensitivities and specificities close to 90% or higher, is possible for a wide range of common tumor types (e.g., lung, liver, esophageal cancer) [21, 95], in some cases even up to 4 years before conventional diagnosis [22]. In this regard, it is worth noting that the cell-type specificity and meta-stability of DNAm offer significant advantages over other molecular data types in distinguishing the tissue-of-origin, as demonstrated by many studies (see e.g [96,97,98,99].). However, challenges remain in that informative features are not yet selected optimally. For instance, given that DNAm changes are widely altered and shared between cancer-types [100, 101], or that they could reflect alterations in other cellular compartments (e.g., fibroblasts or endothelial cells), it is plausible that these confounders could limit performance when validating in independent cohorts. Thus, cell-type deconvolution methods for solid tissues as described earlier should be the ideal starting point in which to identify an optimal set of informative DMCs that are unique to the cells of origin of each cancer-type.
4 Inferring Cell-Type-Specific Differential DNA Methylation (DMCTs)
Once we have inferred the cell-type fractions, we are now in a position to infer DMCTs. Here we shall describe one proposed DMCT-calling algorithm, the CellDMC algorithm [52]. Another very similar algorithm that subsumes CellDMC is TOAST [54]. Assuming for the time being that a CpG is altered in only one cell-type of the tissue, CellDMC/TOAST is based on the intuitive notion that the difference in DNAm between case/control status (for convenience we here assume a binary factor of interest, but similar arguments apply to a factor of interest that is continuously-valued, e.g., age or smoking exposure), would be bigger in those samples where the altered cell-type is more abundant. At the other extreme, i.e., in samples where the altered cell-type is only present in small numbers or not present at all, the difference in DNAm will correspondingly be much smaller. Extending this to more general scenarios, it is therefore plausible that by studying the patterns of DNAm change as a function of cell-type fractions, one can identify DMCTs (Fig. 3.4 to Fig. 4.4).
Statistically, the dependence of DNAm change with cell-type fractions can be captured using linear interaction terms, in this case, interactions between the factor of interest and cell-type fractions. From a modeling perspective, these interaction terms also emerge naturally: for a given cytosine c in a sample s, the methylation value βcs will be given by the formula:
where \( {\hat{f}}_{ks} \) are the estimated cell-type fractions and where βcks denotes the DNAm value in cell-type k in sample s. It is natural to assume that this value follows a linear model in relation to the factor of interest (denoted here by y),
which means that the expression above becomes
where εcs is a Gaussian error term. The second term in brackets is the linear interaction term between cell-type fraction and our factor of interest. If there are covariates or confounders that affect DNAm independently of cell-type (e.g., batch effects), and which we represent as Wq, it is easy to show that the above formula becomes
We note that here the cell-type fractions have already been estimated and that their sum must add to 1, which means that implicit in the equation above, there is an intercept term, and a linear term in y (without interactions). The regression coefficients can be solved under a linear LSR, which returns t-statistics and P-values for the interaction terms, i.e., for assessing if there are DMCTs or not. It is important to note that the above model is a marginal conditional model, i.e., one aims to determine if a CpG is altered in a given cell-type conditioned on all other cell-types. This is to be contrasted with the marginal (unconditional) model for cell-type k
where we estimate an interaction between the factor and the given cell-type fraction, ignoring
all other potential interaction terms. Incidentally, the model for inferring ordinary DMCs, would be
Later we present a power calculation to indicate that the marginal conditional model can display in certain scenarios limited sensitivity to detect DMCTs that occur in all cell-types. Thus, our guideline is the following 2-step strategy:
-
1.
First, we infer ordinary DMCs adjusting for cell-type fractions, i.e., by adding cell-type fractions as covariates in the linear model. This ensures that DMCs are not driven by changes in cell-type proportion, while also keeping high sensitivity to detect most alterations, albeit without knowledge of which specific cell-types are altered.
-
2.
Second, we run the marginal conditional model to identify DMCTs. Any DMCs called in step-2, which are not found to be DMCTs, most likely indicate CpGs that are altered in all cell-types.
5 Applications
We next describe a number of concrete examples where the CellDMC algorithm has been applied to real epigenome datasets, not only validating the algorithm, but also demonstrating how novel insights can be gained by inferring DMCTs as opposed to just DMCs.
5.1 Identification of Smoking-Associated DMCTs in Buccal Swabs
A compelling way to validate the CellDMC algorithm is by performing an EWAS in relation to smoking exposure in buccal swabs. To understand why, we first note that buccal swabs contain on average about 50% immune cells and 50% squamous epithelial cells [59], i.e., two main cell-types. Second, many smoking-EWAS performed in blood have shown that there is a highly reproducible signature of smoking-associated DNAm changes. For instance, a recent meta-analysis identified a gold-standard set of 62 smoking-associated CpGs, the great majority of which undergo hypomethylation in the blood of smokers [2]. This includes a well-known CpG mapping to the repressor of the aryl-hydrocarbon receptor (AHRR). A more recent study encompassing a much larger number of samples [102] further validated these 62 smoking-DMCs, while also identifying a larger number of alterations. Thus, it is reasonable to posit that in a smoking-EWAS performed in buccal swabs, that we should be able to retrieve this 62 CpG smoking hypomethylation signature in the immune cell compartment of the tissue. The result of applying CellDMC to an EWAS of 790 buccal swabs [18] is shown in Fig. 4.5a, which demonstrates that the algorithm indeed predicts most of the 62 CpGs to be hypomethylated in the immune cells from smokers. Interestingly, the algorithm also predicts that these alterations are not happening in the squamous epithelial compartment of buccal swabs, suggesting that these DNAm alterations do not occur in the cells of origin of smoking-related diseases like squamous cell carcinoma of the lung or head and neck. Instead, it has been shown that the DMCTs occurring in the epithelial compartment may be more relevant in mediating the risk of smoking in these cancer-types [18, 59, 103, 104].

Examples of DMCTs. (a) Scatterplot of t-statistics of association between smoking and DNA methylation (~480,000 CpGs) in immune cells (y-axis,IC) vs. the corresponding statistics in the epithelial cells (x-axis, Epi), as predicted by CellDMC in an EWAS of buccal swabs (n = 790 samples). CpGs belonging to a known 62 CpG hypomethylation smoking signature are highlighted in red. (b) Patterns of cell-type-specific differential DNAm for HAND2 (promoter & first exon region) in endometrial cancer, as predicted by CellDMC. Boxplot on the lower right corner is the differential DNAm pattern for whole tissue, i.e., averaged over all cell-types. (c) Distribution of DMCTs in an application of CellDMC to lung squamous cell carcinoma (LSCC) (TCGA dataset). Scatterplots at the bottom illustrate two DMCT examples: an epithelial and an endothelial-specific hypermethylated CpG. N = normal, C = cancer. (d) Scatterplots summarizing results of the GSEA of epithelial and endothelial-specific DMCTs. y-axis labels the statistical significance (−log10[P-value]), x-axis labels the log2[OddsRatio]. P-value and OR computed from a Fisher-test
5.2 HAND2 Hypermethylation in Endometrial Cancer
Another insightful application is to endometrial cancer. Endometrial tissue is composed mainly of epithelial, fibroblast, and immune cells. As mentioned in an earlier section, the promoter of HAND2, a transcription factor that mediates the tumor-suppressive effects of progesterone, is hypermethylated in the endometrial fibroblasts of precursor lesions, as well as in endometrial cancer itself, events that are associated with silencing of HAND2 [35]. HAND2’s promoter also undergoes hypermethylation in blood as a function of age [32]. Thus, application of CellDMC to say the TCGA endometrial cancer study (403 cancers +46 normal-adjacent) [105] should reveal that HAND2 is hypermethylated in fibroblasts and possibly also in the immune cells present within the endometrial tissue, because the average age of the 403 endometrial cancers is higher than that of the 46 normal-adjacent normals. Moreover, the average DNAm change between endometrial cancer and normal-adjacent samples is relatively large at around 0.6, a strong indication that HAND2 undergoes promoter hypermethylation in all main cell-types, including the epithelial compartment. Thus, it is not surprising that when applied to the TCGA dataset, at the resolution of three cell-types (epithelial, fibroblast, and immune cell) and using the corresponding DNAm reference matrix from HEpiDISH (see Fig. 4.2), that CellDMC predicts HAND2 promoter hypermethylation in all three cell-types (Fig. 4.5b). We note that in this instance, the marginal conditional model did have the sensitivity to detect HAND2 as changing in all three cell-types, despite being a scenario where an ordinary DMC-model is more appropriate. That the algorithm correctly predicts a DNAm change in the fibroblasts is critical, as it is the silencing of HAND2 in the fibroblasts that leads to an altered paracrine signaling with the epithelial cells, sensitizing these to oncogenic estrogen [35].
5.3 An Endothelial-to-Mesenchymal (EndoMT) Transformation Signature in Lung Cancer
An example of how novel insight can be gained through application of a tool like CellDMC is illustrated in the case of lung squamous cell carcinoma (LSCC). Here, the EpiSCORE algorithm (Fig. 4.2) was used to build a lung-specific DNAm reference matrix defined over epithelial, fibroblast, endothelial, and immune cells. While one could also consider different immune cell subtypes, we can refrain from doing so if our focus is on alterations occurring in the other cell-types. By simplifying the problem to four main cell-types also yields more power to detect changes in specific cellular compartments. The distribution of inferred DMCTs is displayed in Fig. 4.5c, together with a few examples, and a Gene Set Enrichment Analysis (GSEA) on the DMCTs appearing in the specific cellular compartments, which reveals patterns that are strongly consistent. For instance, the enrichment of bivalent and PRC2 marked domains among epithelial-specific hypermethylated DMCTs is a well-known universal cancer signature (Fig. 4.5d) [89]. Likewise, the concomitant enrichment of SMAD2/SMAD3 binding targets among endothelial-specific hypermethylated DMCTs, and of terms related to mesenchyme, migration and invasion among endothelial-specific hypomethylated DMCTs, is noteworthy given the role of SMAD2/SMAD3 in maintaining vascular integrity (Fig. 4.5d). Therefore, CellDMC makes the prediction that the observed hypermethylation in lung cancer endothelial cells is associated with disruption of SMAD2/SMAD3 binding, compromising vascular integrity and associated with an endothelial-to-mesenchymal (EndoMT) transformation. Such an EndoMT has been reported previously [106,107,108], but as revealed by CellDMC, epigenetic alterations could be critical in promoting such a transformation.
5.4 Myeloid and Lymphoid Specific Smoking-Associated DMCTs
As a final example, we revisit the case of smoking in blood. As mentioned earlier, smoking is associated with a highly reproducible DNAm signature in whole blood [2, 102]. However, to date, no large-scale EWAS in specific blood cell subtypes has been performed in relation to detecting smoking-associated DMCTs. Hence it is unknown whether the observed smoking signature is present in myeloid, lymphoid, or in both cell lineages together. Only smaller scale studies have converged on identifying a small number of CpGs that appear to be altered specifically in myeloid or lymphoid cells [109]. Specifically, Su et al. reported 7 CpGs that exhibit lineage-specific smoking-associated DNAm changes, with 5 of these being specific to myeloid cells, and 2 being specific to lymphocytes [109]. A recent study applied CellDMC to a number of independent EWAS with smoking information (2 in whole blood and 1 in buccal swabs), and at the resolution of 2 cell-types (myeloid and lymphoid), revealing good consistency with Su et al. [110]. A meta-analysis over a large number of EWAS in whole blood further revealed myeloid-specific hypermethylation and hypomethylation signatures, with the latter being strongly enriched for DNase Hypersensitive Sites (DHS) as defined in inflammatory macrophages, and with the former one only being enriched for DHSs as defined in acute myeloid leukemia (AML) [110]. In contrast, the same meta-analysis did not reveal an extended lymphocyte-specific smoking signature, suggesting that smoking imparts its effect on DNAm patterns in blood mostly via alterations in myeloid cells. This is a significant observation given that smoking is a moderate risk factor for AML but not for lymphocytic leukemias [111].
In summary, all the above examples illustrate successful applications of an algorithm such as CellDMC in identifying cell-type-specific DMCs, in some cases validating known DNAm signatures, and in other cases generating novel concrete hypotheses for further exploration and testing.
6 Limitations
Despite the successful applications described above, it is important to also emphasize the limitations associated with identifying DMCTs. We illustrate some of these limitations in the context of a power simulation on realistic data, focusing on the last example considered in the previous section, namely identifying smoking-associated DM in two cell lineages of blood (myeloid vs. lymphoid) [110]. This is a scenario where one cell-lineage (myeloid cells) accounts for most of the cells in the tissue (i.e., approximately 60–70% of cells in whole blood derive from the myeloid lineage), with variations in the myeloid fraction between individuals also accounting for most of the variation in DNAm. Details of the actual simulation model can be found in You et al. [110]. Very briefly, in this in-silico model, one simulates three classes of DMCTs, and subsequently assesses the sensitivity or power of the algorithm (in this case CellDMC) to detect these alterations. One class of DMCTs is specific to the lymphoid lineage, another class is specific to the myeloid lineage, and the third class of DMCTs is present in both lineages, and therefore this class is non-specific. The sensitivity to detect each category of DMCT in a whole blood EWAS is shown in Fig. 4.6, for a range of different effect sizes and two different sample sizes.

Power calculation. Plots of the sensivity (y-axis) to detect each class of DMCT vs effect size (x-axis), and for two different sample sizes (n = 200 and n = 600). We note that n is the total sample size and number of cases and controls is assumed similar. The effect sizes are realistic and corresponding to average DNAm changes in individual cell-types that range from 0.05 to just over 0.6. In the last plot, we display the sensitivity to detect the non-specific DMCTs in each lineage separately
Thus, for appropriately powered studies detecting lymphoid or myeloid-specific DMCTs is in general not a problem. For instance, for an epigenome study with 200 samples (100 cases and 100 controls), the sensitivity to detect myeloid-specific DMCTs is around 80% for an effect size of 2, which roughly corresponds to about a 10–20% average absolute DNAm change in myeloid cells. The corresponding sensitivity for lymphoid-specific DMCTs is just over 60%, i.e., lower than for myeloid-specific DMCTs, owing to the lower proportion and variance of lymphoid fractions in blood, but still reasonably high. For larger effect sizes, i.e., for average DNAm differences larger than say +/− 0.3, sensitivities are higher than 80 or 90% for both lineage-specific DMCTs. However, for non-specific DMCTs, the power calculation indicates markedly reduced sensitivity to detect the change in the lymphoid compartment. Thus, larger sample sizes would be needed to detect non-specific DMCTs as being non-specific. As far as the FPR and FDR are concerned, these are generally quite low, with the corresponding specificity and precision being close to 100% for all three classes of DMCTs (see You et al. [110]).
The above simple power calculation clearly illustrates the potential limitations associated with inferring DMCTs in real epigenome studies. As one wishes to infer DMCTs in the context of ever-increasing numbers of cell-types, one main limitation is on the sensitivity to detect non-specific DMCTs, and thus great caution needs to be exercised when interpreting the specificity of inferred DMCTs. Another major limitation is the potentially low variance in the fraction displayed by a given cell-type, as noted recently [112].
7 Useful Software
Here we give a brief overview of some of the important software that has been developed in the cell-type heterogeneity field. Most of this software is freely available as R-code from the websites as specified in the publications. For estimating cell-type fractions in whole blood or peripheral blood mononuclear cells, we can recommend the minfi Bioconductor package [80] which uses Houseman’s constrained projection algorithm [46] as well as the original DMRM for blood used when validating the Houseman algorithm. Another option is the EpiDISH Bioconductor package [48], which offers robust partial correlations (RPCs) and Support Vector Regression as an alternative to constrained projection for estimating cell-type fractions. For those unfamiliar with R, the EpiDISH package has an associated webserver [113] and also incorporates its own DMRM for blood. Another important option for blood is the IDOL algorithm [78] and associated DMRM [114]. For cord blood, there are dedicated DMRMs available from the FlowSorted.CordBlood.450 K Bioconductor R-package [81] and from [115, 129]. For saliva, there is also a dedicated DMRM, and which has been specially designed for epidemiological studies in children [116]. For saliva and buccal swabs, one can also use a general three cell-type DNAm reference matrix defined for generic epithelial, fibroblast, and immune cells [59, 117], and which is also available within the EpiDISH package. For complex solid tissues, one can use the HEpiDISH algorithm [59] and associated 2-layer DMRMs for estimating cell-type fractions for epithelial, stromal, and all immune cell subtypes, and which is also part of the EpiDISH package. Another option for inferring epithelial and stromal fractions in complex tissues is MethylCIBERSORT [118]. The EpiSCORE algorithm can be used to build DMRMs for tissue-types for which there is a matching scRNA-Seq atlas, and DMRMs for lung and breast tissue have been provided [89]. An alternative statistical strategy for estimating cell-type fractions called dtangle that improves upon the constraint projection and the other frameworks has also been proposed [119]. A method called MethylResolver has recently been proposed to evaluate the quality of an inferred cell-type decomposition [120], which is an important step missing from all other software. Algorithms for identifying cell-type-specific DNA methylation changes include TOAST [54], CellDMC [52], HIRE [55], Omicwas [121] and TCA [122]. Omicwas, which implements a non-linear ridge regression framework is noteworthy for also dealing with the co-linearity problem in DMRMs of high cellular resolution. Reference-free and semi-reference-free methods remain useful for the purpose of identifying DMCs not confounded by cell-type heterogeneity [49,50,51, 123,124,125], but are generally speaking limited for the tasks of cell-type fraction estimation and inference of cell-type-specific DNAm changes.
8 Outlook
It is likely that over the next years, technologies for single-cell methylomics [126] will improve to allow the construction of DNAm reference profiles for most cell-types within a tissue. This would provide an alternative means to build a DMRM, which is more tailored to the tissue of interest, thus overcoming some of the limitations of existing strategies. However, in the foreseeable future, generating single-cell methylomics for large numbers of tissues (and individuals) will remain impractical. This means that for epigenome studies aiming to identify cell-type-specific biomarkers, and which will require genome-wide profiling of many 100s to 1000s of individuals, it will still be necessary to use cell-type deconvolution methods. Thus, single-cell methylomics may help towards the construction of tissue-specific DMRMs, and this in turn may improve the inference of cell-type fractions and DMCTs from large-scale epigenome studies.
Another important area of future development will be applications in personalized epigenetic epidemiology and medicine. One key area that is likely to grow even further is noninvasive detection of disease via measurement of cfDNAm in plasma. Cell-type deconvolution methods as well as more conventional Machine Learning methods need to be improved further, specially in relation to how informative features are selected. Currently, algorithms are also aimed at identifying biomarkers (cell-type specific or not) that display average differences in DNAm associated with an exposure or outcome. However, averaging over individuals also obscures in which particular individuals a DNAm change is happening. In the context of DMCTs, thus one would wish to infer in which cell-types and individual a particular DNAm change is happening in. Thus, given a DNAm data matrix and estimates of cell-type fractions, one would ideally want to infer an array of DNAm values, defined over CpGs, cell-types, and individuals. Mathematically, this is known as a tensor-object [37] and attempts to infer such a tensor in the context of EWAS have been made [122], albeit not yet successfully so [127]. Of note, such tensor-inference methods are also likely to overfit and to be computationally very intensive if applied in a genome-wide context, as the number of parameters to estimate is substantially higher.
An important biological question for the future is the relative fractions of non-specific vs. specific DMCTs. This is likely to depend on the tissue as well as phenotype. For instance, in the case of SNPs, the proportion of mQTLs that are shared between blood cell-types is likely to be large (approx. 70–80%) [36, 37]. Thus, in the case of DNAm variation associated with genetic variants, this variation appears to be largely cell-type independent, which however still allows for downstream functional effects to display cell-type specificity. In aging too, a recent study has shown that most age-associated DNAm changes appear to be independent of tissue and cell-type [128]. EWAS for body-mass index have also revealed shared DNAm changes between blood and fat cells, but also differences [3]. In the case of smoking in blood, there is evidence for both non-specific as well as specific DMCTs, whereas in buccal swabs the effects appear to be much more cell-type specific [18]. Other conditions such as type-1 diabetes [39], asthma [34], Rheumatoid Arthritis [41] or cancer [89] appear to be associated with larger numbers of cell-type-specific DMCTs. Understanding why specific factors may affect DNAm in a cell-type independent manner and why others do not will be an important question for the epigenetics community that is also highly relevant for epigenetic epidemiological studies.
To conclude, this chapter provides a brief overview of the key concepts and tools needed to tackle the challenge posed by cell-type heterogeneity in the context of DNA methylation studies. We recommend that future epigenetic epidemiological studies make an attempt to tackle this challenge, using the software tools described herein, mainly as hypothesis generation tools, to be followed up with functional studies or validations in purified cell populations.
Abbreviations
- DMC:
-
differentially methylated cytosine
- DMCT:
-
differentially methylated cell-type
- DNAm:
-
DNA methylation
- EWAS:
-
Epigenome-Wide Association Study
- FDR:
-
False Discovery Rate
- FPR:
-
False Positive Rate
- LSR:
-
least squares regression
- mQTL:
-
methylation quantitative trait loci
- PR C2:
-
Polycomb-Repressive-Complex-2
- scRNA-Seq:
-
single-cell RNA-Seq
- SE:
-
Sensitivity
References
Petronis A (2010) Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465:721–727
Gao X, Jia M, Zhang Y, Breitling LP, Brenner H (2015) DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics 7:113
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, Tsai PC, Ried JS, Zhang W, Yang Y et al (2017) Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541:81–86
van Otterdijk SD, Binder AM, Michels KB (2017) Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy. Epigenetics 12:875–885
Non AL, Binder AM, Kubzansky LD, Michels KB (2014) Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics 9:964–972
Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R et al (2009) Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 5:e1000602
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP et al (2010) Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20:440–446
Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, Widschwendter M (2012) Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med 4:24
Vandiver AR, Irizarry RA, Hansen KD, Garza LA, Runarsson A, Li X, Chien AL, Wang TS, Leung SG, Kang S, Feinberg AP (2015) Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol 16:80
Laird PW (2003) The power and the promise of DNA methylation markers. Nat Rev Cancer 3:253–266
Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD et al (2014) Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A 111:15538–15543
Issa JP (2014) Aging and epigenetic drift: a vicious cycle. J Clin Invest 124:24–29
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45:1113–1120
Montano C, Taub MA, Jaffe A, Briem E, Feinberg JI, Trygvadottir R, Idrizi A, Runarsson A, Berndsen B, Gur RC et al (2016) Association of DNA methylation differences with schizophrenia in an epigenome-wide association study. JAMA Psychiatry 73:506–514
Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, Kam-Thong T, Xi HS, Quan J, Chen Q et al (2018) Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci 21:1117–1125
Rakyan VK, Down TA, Balding DJ, Beck S (2011) Epigenome-wide association studies for common human diseases. Nat Rev Genet 12:529–541
Jaffe AE, Kleinman JE (2016) Genetic and epigenetic analysis of schizophrenia in blood-a no-brainer? Genome Med 8:96
Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, Anjum S, Hardy R, Salvesen HB, Thirlwell C et al (2015) Correlation of smoking-associated DNA methylation changes in buccal cells with DNA methylation changes in epithelial cancer. JAMA Oncol 1:476–485
Levine ME, Lu AT, Chen BH, Hernandez DG, Singleton AB, Ferrucci L, Bandinelli S, Salfati E, Manson JE, Quach A et al (2016) Menopause accelerates biological aging. Proc Natl Acad Sci U S A 113:9327–9332
Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, Nelson HH, Karagas MR, Wiencke JK, Kelsey KT (2012) Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomark Prev 21:1293–1302
Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T et al (2018) Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563:579–583
Chen X, Gole J, Gore A, He Q, Lu M, Min J, Yuan Z, Yang X, Jiang Y, Zhang T et al (2020) Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun 11:3475
Widschwendter M, Zikan M, Wahl B, Lempiainen H, Paprotka T, Evans I, Jones A, Ghazali S, Reisel D, Eichner J et al (2017) The potential of circulating tumor DNA methylation analysis for the early detection and management of ovarian cancer. Genome Med 9:116
Liu L, Toung JM, Jassowicz AF, Vijayaraghavan R, Kang H, Zhang R, Kruglyak KM, Huang HJ, Hinoue T, Shen H et al (2018) Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann Oncol 29:1445–1453
Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K (2017) Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 49:635–642
Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgard B, Blennow K, Zetterberg H et al (2016) Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A 113:E1826–E1834
Lappalainen T, Greally JM (2017) Associating cellular epigenetic models with human phenotypes. Nat Rev Genet 18:441–451
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102:10604–10609
Ahuja N, Issa JP (2000) Aging, methylation and cancer. Histol Histopathol 15:835–842
Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP (1998) Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 58:5489–5494
Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP (2010) Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20:332–340
Yuan T, Jiao Y, de Jong S, Ophoff RA, Beck S, Teschendorff AE (2015) An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet 11:e1004996
Jaffe AE, Irizarry RA (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15:R31
Liang L, Willis-Owen SA, Laprise C, Wong KC, Davies GA, Hudson TJ, Binia A, Hopkin JM, Yang IV, Grundberg E et al (2015) An epigenome-wide association study of total serum immunoglobulin E concentration. Nature 520:670–674
Jones A, Teschendorff AE, Li Q, Hayward JD, Kannan A, Mould T, West J, Zikan M, Cibula D, Fiegl H et al (2013) Role of DNA methylation and epigenetic silencing of HAND2 in endometrial cancer development. PLoS Med 10:e1001551
Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martin D, Watt S, Yan Y, Kundu K, Ecker S et al (2016) Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167:1398–1414.e24
Teschendorff AE, Jing H, Paul DS, Virta J, Nordhausen K (2018) Tensorial blind source separation for improved analysis of multi-omic data. Genome Biol 19:76
Baylin SB, Ohm JE (2006) Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 6:107–116
Paul DS, Teschendorff AE, Dang MA, Lowe R, Hawa MI, Ecker S, Beyan H, Cunningham S, Fouts AR, Ramelius A et al (2016) Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat Commun 7:13555
Reynolds LM, Taylor JR, Ding J, Lohman K, Johnson C, Siscovick D, Burke G, Post W, Shea S, Jacobs DR Jr et al (2014) Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat Commun 5:5366
Julia A, Absher D, Lopez-Lasanta M, Palau N, Pluma A, Waite Jones L, Glossop JR, Farrell WE, Myers RM, Marsal S (2017) Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in B cells. Hum Mol Genet 26:2803–2811
Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, Krueger F, Smallwood S, Ponting CP, Voet T et al (2016) Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 13:229–232
Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G (2014) Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11:817–820
Lee DS, Luo C, Zhou J, Chandran S, Rivkin A, Bartlett A, Nery JR, Fitzpatrick C, O’Connor C, Dixon JR, Ecker JR (2019) Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat Methods 16:999–1006
Kelsey G, Stegle O, Reik W (2017) Single-cell epigenomics: recording the past and predicting the future. Science 358:69–75
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform 13:86
Accomando WP, Wiencke JK, Houseman EA, Butler RA, Zheng S, Nelson HH, Kelsey KT (2012) Decreased NK cells in patients with head and neck cancer determined in archival DNA. Clin Cancer Res 18:6147–6154
Teschendorff AE, Breeze CE, Zheng SC, Beck S (2017) A comparison of reference-based algorithms for correcting cell-type heterogeneity in epigenome-wide association studies. BMC Bioinform 18:105
Houseman EA, Molitor J, Marsit CJ (2014) Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 30:1431–1439
Lutsik P, Slawski M, Gasparoni G, Vedeneev N, Hein M, Walter J (2017) MeDeCom: discovery and quantification of latent components of heterogeneous methylomes. Genome Biol 18:55
Li Z, Wu H (2019) TOAST: improving reference-free cell composition estimation by cross-cell type differential analysis. Genome Biol 20:190
Zheng SC, Breeze CE, Beck S, Teschendorff AE (2018) Identification of differentially methylated cell types in epigenome-wide association studies. Nat Methods 15:1059–1066
Montano CM, Irizarry RA, Kaufmann WE, Talbot K, Gur RE, Feinberg AP, Taub MA (2013) Measuring cell-type specific differential methylation in human brain tissue. Genome Biol 14:R94
Li Z, Wu Z, Jin P, Wu H (2019) Dissecting differential signals in high-throughput data from complex tissues. Bioinformatics 35(20):3898–3905
Luo X, Yang C, Wei Y (2019) Detection of cell-type-specific risk-CpG sites in epigenome-wide association studies. Nat Commun 10:3113
Maegawa S, Gough SM, Watanabe-Okochi N, Lu Y, Zhang N, Castoro RJ, Estecio MR, Jelinek J, Liang S, Kitamura T et al (2014) Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res 24:580–591
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, Jones A, Lechner M, Beck S, Jacobs IJ, Widschwendter M (2009) An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS One 4:e8274
Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M et al (2013) Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 31:142–147
Zheng SC, Webster AP, Dong D, Feber A, Graham DG, Sullivan R, Jevons S, Lovat LB, Beck S, Widschwendter M, Teschendorff AE (2018) A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics 10:925–940
Theda C, Hwang SH, Czajko A, Loke YJ, Leong P, Craig JM (2018) Quantitation of the cellular content of saliva and buccal swab samples. Sci Rep 8:6944
Wong YT, Tayeb MA, Stone TC, Lovat LB, Teschendorff AE, Iwasiow R, Craig JM (2021) A comparison of epithelial cell content of oral samples estimated using cytology and DNA methylation. Epigenetics:1–8. https://doi.org/10.1080/15592294.2021.1950977
Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M et al (2017) The human cell atlas. elife 6:e27041
Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA (2017) The human cell atlas: from vision to reality. Nature 550:451–453
Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, Brouwer S, Gomes T, Hesse L, Jiang J et al (2019) A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med 25:1153–1163
Schiller HB, Montoro DT, Simon LM, Rawlins EL, Meyer KB, Strunz M, Vieira Braga FA, Timens W, Koppelman GH, Budinger GRS et al (2019) The human lung cell atlas: a high-resolution reference map of the human lung in health and disease. Am J Respir Cell Mol Biol 61:31–41
Bock C (2012) Analysing and interpreting DNA methylation data. Nat Rev Genet 13:705–719
Ali HR, Provenzano E, Dawson SJ, Blows FM, Liu B, Shah M, Earl HM, Poole CJ, Hiller L, Dunn JA et al (2014) Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann Oncol 25:1536–1543
Koestler DC, Usset J, Christensen BC, Marsit CJ, Karagas MR, Kelsey KT, Wiencke JK (2017) DNA methylation-derived neutrophil-to-lymphocyte ratio: an epigenetic tool to explore cancer inflammation and outcomes. Cancer Epidemiol Biomark Prev 26:328–338
Wiencke JK, Koestler DC, Salas LA, Wiemels JL, Roy RP, Hansen HM, Rice T, McCoy LS, Bracci PM, Molinaro AM et al (2017) Immunomethylomic approach to explore the blood neutrophil lymphocyte ratio (NLR) in glioma survival. Clin Epigenetics 9:10
Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, Chan WK, Ma ES, Chan SL, Cheng SH et al (2015) Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A 112:E5503–E5512
Zheng SC, Beck S, Jaffe AE, Koestler DC, Hansen KD, Houseman AE, Irizarry RA, Teschendorff AE (2017) Correcting for cell-type heterogeneity in epigenome-wide association studies: revisiting previous analyses. Nat Methods 14:216–217
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7:e41361
den Dekker HT, Burrows K, Felix JF, Salas LA, Nedeljkovic I, Yao J, Rifas-Shiman SL, Ruiz-Arenas C, Amin N, Bustamante M et al (2019) Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur Respir J 53:1801795
Merid SK, Novoloaca A, Sharp GC, Kupers LK, Kho AT, Roy R, Gao L, Annesi-Maesano I, Jain P, Plusquin M et al (2020) Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med 12:25
Salas LA, Baker ER, Nieuwenhuijsen MJ, Marsit CJ, Christensen BC, Karagas MR (2019) Maternal swimming pool exposure during pregnancy in relation to birth outcomes and cord blood DNA methylation among private well users. Environ Int 123:459–466
Vehmeijer FOL, Kupers LK, Sharp GC, Salas LA, Lent S, Jima DD, Tindula G, Reese S, Qi C, Gruzieva O et al (2020) DNA methylation and body mass index from birth to adolescence: meta-analyses of epigenome-wide association studies. Genome Med 12:105
Yeung EH, Guan W, Zeng X, Salas LA, Mumford SL, de Prado BP, van Meel ER, Malmberg A, Sunyer J, Duijts L et al (2020) Cord blood DNA methylation reflects cord blood C-reactive protein levels but not maternal levels: a longitudinal study and meta-analysis. Clin Epigenetics 12:60
Koestler DC, Jones MJ, Usset J, Christensen BC, Butler RA, Kobor MS, Wiencke JK, Kelsey KT (2016) Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL). BMC Bioinform 17:120
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453–457
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369
Gervin K, Salas LA, Bakulski KM, van Zelm MC, Koestler DC, Wiencke JK, Duijts L, Moll HA, Kelsey KT, Kobor MS et al (2019) Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin Epigenetics 11:125
Oh G, Koncevicius K, Ebrahimi S, Carlucci M, Groot DE, Nair A, Zhang A, Krisciunas A, Oh ES, Labrie V et al (2019) Circadian oscillations of cytosine modification in humans contribute to epigenetic variability, aging, and complex disease. Genome Biol 20:2
Oh G, Ebrahimi S, Carlucci M, Zhang A, Nair A, Groot DE, Labrie V, Jia P, Oh ES, Jeremian RH et al (2018) Cytosine modifications exhibit circadian oscillations that are involved in epigenetic diversity and aging. Nat Commun 9:644
Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, Kobor MS (2012) Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A 109(Suppl 2):17253–17260
Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R et al (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR et al (2010) The NIH Roadmap Epigenomics mapping consortium. Nat Biotechnol 28:1045–1048
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330
Teschendorff AE, Relton CL (2018) Statistical and integrative system-level analysis of DNA methylation data. Nat Rev Genet 19:129–147
Teschendorff AE, Zhu T, Breeze CE, Beck S (2020) EPISCORE: cell type deconvolution of bulk tissue DNA methylomes from single-cell RNA-seq data. Genome Biol 21:221
Teschendorff AE (2020) EpiSCORE: cell-type deconvolution of bulk tissue DNA methylomes from single-cell RNA-seq data (version 0.9.1). Zenodo. https://doi.org/10.5281/zenodo.3893646
Tabula Muris C (2018) Overall c, logistical c, organ c, processing, library p, sequencing, computational data a, cell type a, writing g, et al: single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562:367–372
Gerstein M (2012) Genomics: ENCODE leads the way on big data. Nature 489:208
Consortium EP, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A et al (2020) Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583:699–710
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD et al (2015) The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21:938–945
Kang S, Li Q, Chen Q, Zhou Y, Park S, Lee G, Grimes B, Krysan K, Yu M, Wang W et al (2017) CancerLocator: non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol 18:53
Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V et al (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173:291–304.e6
Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, Samet Y, Maoz M, Druid H, Arner P et al (2018) Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 9:5068
Tang W, Wan S, Yang Z, Teschendorff AE, Zou Q (2018) Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 34:398–406
Moran S, Martinez-Cardus A, Sayols S, Musulen E, Balana C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC et al (2016) Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 17:1386–1395
Nejman D, Straussman R, Steinfeld I, Ruvolo M, Roberts D, Yakhini Z, Cedar H (2014) Molecular rules governing de novo methylation in cancer. Cancer Res 74:1475–1483
Chen Y, Breeze CE, Zhen S, Beck S, Teschendorff AE (2016) Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenetics Chromatin 9:10
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S et al (2016) Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet 9:436–447
Chen Y, Widschwendter M, Teschendorff AE (2017) Systems-epigenomics inference of transcription factor activity implicates aryl-hydrocarbon-receptor inactivation as a key event in lung cancer development. Genome Biol 18:236
Zheng SC, Widschwendter M, Teschendorff AE (2016) Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 8:705–719
Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R et al (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497:67–73
Platel V, Faure S, Corre I, Clere N (2019) Endothelial-to-mesenchymal transition (EndoMT): roles in tumorigenesis, metastatic extravasation and therapy resistance. J Oncol 2019:8361945
Tang YN, Ding WQ, Guo XJ, Yuan XW, Wang DM, Song JG (2015) Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat Commun 6:8230
Potenta S, Zeisberg E, Kalluri R (2008) The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 99:1375–1379
Su D, Wang X, Campbell MR, Porter DK, Pittman GS, Bennett BD, Wan M, Englert NA, Crowl CL, Gimple RN et al (2016) Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PLoS One 11:e0166486
You C, Wu S, Zheng SC, Zhu T, Jing H, Flagg K, Wang G, Jin L, Wang S, Teschendorff AE (2020) A cell-type deconvolution meta-analysis of whole blood EWAS reveals lineage-specific smoking-associated DNA methylation changes. Nat Commun 11:4779
Fircanis S, Merriam P, Khan N, Castillo JJ (2014) The relation between cigarette smoking and risk of acute myeloid leukemia: an updated meta-analysis of epidemiological studies. Am J Hematol 89:E125–E132
Meier R, Nissen E, Koestler DC (2021) Low variability in the underlying cellular landscape adversely affects the performance of interaction-based approaches for conducting cell-specific analyses of DNA methylation in bulk samples. Stat Appl Genet Mol Biol 20(3):73–84
Zheng SC, Breeze CE, Beck S, Dong D, Zhu T, Ma L, Ye W, Zhang G, Teschendorff AE (2019) EpiDISH web server: epigenetic dissection of intra-sample-heterogeneity with online GUI. Bioinformatics 36(6):1950–1951
Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, Christensen BC (2018) An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol 19:64
Lin X, Tan JYL, Teh AL, Lim IY, Liew SJ, MacIsaac JL, Chong YS, Gluckman PD, Kobor MS, Cheong CY, Karnani N (2018) Cell type-specific DNA methylation in neonatal cord tissue and cord blood: a 850K-reference panel and comparison of cell types. Epigenetics 13:941–958
Middleton LYM, Dou J, Fisher J, Heiss JA, Nguyen VK, Just AC, Faul J, Ware EB, Mitchell C, Colacino JA, Bakulski KM (2021) Saliva cell type DNA methylation reference panel for epidemiological studies in children. Epigenetics:1–17. https://doi.org/10.1080/15592294.2021.1890874
van Dongen J, Ehli EA, Jansen R, van Beijsterveldt CEM, Willemsen G, Hottenga JJ, Kallsen NA, Peyton SA, Breeze CE, Kluft C et al (2018) Genome-wide analysis of DNA methylation in buccal cells: a study of monozygotic twins and mQTLs. Epigenetics Chromatin 11:54
Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, King EV, Lechner M, Marafioti T, Quezada SA et al (2018) Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun 9:3220
Hunt GJ, Freytag S, Bahlo M, Gagnon-Bartsch JA (2019) Dtangle: accurate and robust cell type deconvolution. Bioinformatics 35:2093–2099
Arneson D, Yang X, Wang K (2020) MethylResolver-a method for deconvoluting bulk DNA methylation profiles into known and unknown cell contents. Commun Biol 3:422
Takeuchi F, Kato N (2021) Nonlinear ridge regression improves cell-type-specific differential expression analysis. BMC Bioinform 22:141
Rahmani E, Schweiger R, Rhead B, Criswell LA, Barcellos LF, Eskin E, Rosset S, Sankararaman S, Halperin E (2019) Cell-type-specific resolution epigenetics without the need for cell sorting or single-cell biology. Nat Commun 10:3417
Onuchic V, Hartmaier RJ, Boone DN, Samuels ML, Patel RY, White WM, Garovic VD, Oesterreich S, Roth ME, Lee AV, Milosavljevic A (2016) Epigenomic deconvolution of breast tumors reveals metabolic coupling between constituent cell types. Cell Rep 17:2075–2086
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28(6):882–883
Teschendorff AE, Zhuang J, Widschwendter M (2011) Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics 27:1496–1505
Karemaker ID, Vermeulen M (2018) Single-cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol 36:952–965
Jing H, Zheng SC, Breeze CE, Beck S, Teschendorff AE (2019) Calling differential DNA methylation at cell-type resolution: an objective status-quo. bioRxiv. https://doi.org/10.1101/822940
Zhu T, Zheng SC, Paul DS, Horvath S, Teschendorff AE (2018) Cell and tissue type independent age-associated DNA methylation changes are not rare but common. Aging (Albany NY) 10:3541–3557
Cardenas A, Allard C, Doyon M, Houseman EA, Bakulski KM, Perron P, Bouchard L, Hivert MF (2016) Validation of a DNA methylation reference panel for the estimation of nucleated cells types in cord blood. Epigenetics 11:773–779
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Teschendorff, A.E. (2022). Cell-Type Heterogeneity in DNA Methylation Studies: Statistical Methods and Guidelines. In: Michels, K.B. (eds) Epigenetic Epidemiology. Springer, Cham. https://doi.org/10.1007/978-3-030-94475-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-94475-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94474-2
Online ISBN: 978-3-030-94475-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)