
Abstract
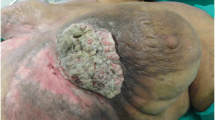
A 16-year-old boy presented to our hospital because of a huge mass on the face and a large ulcer on the scalp. Physical examination revealed a giant necrotic, ulcerating mass involving the two third upper part of the face (Fig. 23.1) and an ulcer sized 10 × 7 cm on the cranial vertex of the scalp (Fig. 23.2). The mass covered almost the two eyes and half of the nose with multiple bleedings and pus-discharging. This mass had been gradually progressing for the last 7 years. There were also diffuse hypo-hyperpigmented atrophic lesions all over the body (Fig. 23.3). His skin was unnaturally dry and rough. Telengiectases were observed over some lesions. These lesions have increased over the skin since the age of three. There was no palpable lymph node. He had a significant alteration of general condition with a BMI (body mass index) of 13.6 kg/m2. He has a family history of consanguinity, but no other person in the family has similar presentation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
A 16-year-old boy presented to our hospital because of a huge mass on the face and a large ulcer on the scalp. Physical examination revealed a giant necrotic, ulcerating mass involving the two third upper part of the face (Fig. 23.1) and an ulcer sized 10 × 7 cm on the cranial vertex of the scalp (Fig. 23.2). The mass covered almost the two eyes and half of the nose with multiple bleedings and pus-discharging. This mass had been gradually progressing for the last 7 years. There were also diffuse hypo-hyperpigmented atrophic lesions all over the body (Fig. 23.3). His skin was unnaturally dry and rough. Telangiectasias! were observed over some lesions. These lesions have increased over the skin since the age of three. There was no palpable lymph node. He had a significant alteration of general condition with a BMI (body mass index) of 13.6 kg/m2. He has a family history of consanguinity, but no other person in the family has similar presentation.

Giant mass involving the two third upper part of the face, covering almost the two eyes and half of the nose

Large ulcer on the cranial vertex of the scalp

Diffuse hypo-hyperpigmented atrophic lesions all over the body
Based on the case description and photographs, what is your diagnosis?
-
1.
Carcinoma in ichthyosis
-
2.
Squamous cell carcinoma in xeroderma pigmentosum
-
3.
Porphyria
-
4.
Cutaneous lymphoma
Diagnosis
Giant squamous cell carcinoma in xeroderma pigmentosum.
Discussion
Xeroderma pigmentosum (XP) is an autosomal recessive genetic disorder that appears in early childhood, in which the ability to repair damage to DNA caused by ultraviolet (UV) light is deficient [1]. It is associated with extreme sensitivity to sunlight resulting in sunburn, pigment changes in the skin with increase in the frequency of skin cancers. In children with XP, there is a 10000-fold increased risk of skin cancer under 20 years of age [2]. The mean age for skin cancer is 8 years compared to 60 years in the healthy individuals [3].
Clinically, XP patients develop skin sensitivity to sunlight in early years (as early as 6 months) of childhood with sunburn, actinic keratosis, xerosis, poikiloderma, ocular abnormalities, and malignant neoplasms of the skin, especially in sun-exposed areas [1]. The most common neoplasms are squamous cell carcinoma and basal cell carcinoma. The central nervous system (CNS) is often affected in XP patients. The CNS does not have direct UV radiation exposure making the mechanism of disease unclear, but unrepaired oxidative damage has been proposed as a possible cause [4]. Neurodegeneration occurs in an estimated 24% [2], including loss of intellectual functioning, deterioration of neurologic status, impaired hearing, abnormal speech, areflexia, ataxia, peripheral neuropathy, and loss of the ability to walk and talk.
The laboratory diagnosis of XP can be established with studies that include cellular hypersensitivity to UV radiation and chromosomal breakage studies and gene sequencing to identify the specific gene. Prenatal diagnosis is possible by amniocentesis or chorionic villi sampling [5]. Unfortunately, all these investigations are not available in our center and were, therefore, not undertaken for this patient. Clinical presentation provided the clue to the diagnosis.
In term of treatment, there is no cure for xeroderma pigmentosum. Investigative therapies using gene therapy and antioxidants to reduce oxidative damage may result in future treatment options [6]. Current management of XP patients mostly focuses on prevention of skin cancers by sunscreen use, protective clothing (long sleeves and pants, shirts with collars, tightly woven fabrics, wide-brimmed hat), restriction of outdoor activities to night-time. Frequent consultation and examination by dermatology and ophthalmology is recommended every 6 months to monitor for skin and ocular damage. Decreasing UV radiation exposure may not decrease neurodegenerations. Patients often require Vitamin D supplementation to compensate for sun avoidance [7]. Systemic treatment with retinoids has showed some benefits in reducing the number of skin cancers [8]. Surgical excision is the most common modality used for the treatment of XP-related neoplasms. However, lesions at some sites may not be amenable to adequate surgical intervention. In addition, significant esthetic deformities may result from multiple excisions. Therefore, early diagnosis and management are crucial in this disease. In our case, his poor socioeconomic background precludes these early interventions. The parents of this patient did not seek medical advice for him and he was brought late to hospital in spite of his early presentation at the age of 3-year-olds.
Key Points
-
Early diagnosis is very important for the prognosis of XP patients
-
Current management of XP patients focuses on prevention and treatment of skin cancers
References
Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis. 2011;6:70.
Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48(3):168–76.
Emir S, Hacısalihoğlu Ş, Özyörük D, et al. Squamous cell carcinoma associated with xeroderma pigmentosum: an unusual presentation with a tremendously huge mass over the face and paraneoplastic hypercalcemia-hyperleukocytosis. Turk J Pediatr. 2017;59(6):711.
Anttinen A, Koulu L, Nikoskelainen E, et al. Neurological symptoms and natural course of xeroderma pigmentosum. Brain J Neurol. 2008;131(Pt 8):1979–89.
Kleijer WJ, van der Sterre MLT, Garritsen VH, et al. Prenatal diagnosis of xeroderma pigmentosum and trichothiodystrophy in 76 pregnancies at risk. Prenat Diagn. 2007;27(12):1133–7.
Dupuy A, và Sarasin A. DNA damage and gene therapy of xeroderma pigmentosum, a human DNA repair-deficient disease. Mutat Res. 2015;776:2–8.
Masaki T, Wang Y, DiGiovanna JJ, et al. High frequency of PTEN mutations in nevi and melanomas from xeroderma pigmentosum patients. Pigment Cell Melanoma Res. 2014;27(3):454–64.
DiGiovanna JJ. Retinoid chemoprevention in patients at high risk for skin cancer. Med Pediatr Oncol. 2001;36(5):564–7.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Doanh, L.H., Van Thuong, N., Tirant, M. (2022). Giant Squamous Cell Carcinoma in Xeroderma Pigmentosum. In: Satolli, F., Tirant, M., Wollina, U., Lotti, T.M. (eds) Clinical Cases in Pediatric Skin Cancers. Clinical Cases in Dermatology. Springer, Cham. https://doi.org/10.1007/978-3-030-93666-2_23
Download citation
DOI: https://doi.org/10.1007/978-3-030-93666-2_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-93665-5
Online ISBN: 978-3-030-93666-2
eBook Packages: MedicineMedicine (R0)