
Abstract
Mitochondrial diseases affect both children and adults with a wide range of clinical presentations ranging from mild chronic progressive external ophthalmoparesis to severe, life-threatening multisystem diseases. When patients are admitted to an ICU, they do not necessarily have a pre-existing diagnosis of mitochondrial disease. Establishing a diagnosis of mitochondrial disease is important because it can have therapeutic and prognostic implications. Patients will often have respiratory weakness, cardiomyopathy or arrhythmia, gastrointestinal problems that lead to malnutrition and pseudo-obstruction, bone marrow failure or diabetes. In addition, mitochondrial dysfunction will further increase vulnerability of many other body systems when confronted with acute illness. Cardiac decompensation due to cardiomyopathy or arrhythmias, and acute cerebral events like seizures or stroke-like episodes (seizure-mediated) are well-recognized and are the leading causes of death in patients with mitochondrial disease. Renal failure, acute liver failure, respiratory failure, encephalopathy with central respiratory depression, lactic acidaemia, and endocrine disturbances like diabetes, hypo- and hyperthyroidism, adrenal insufficiency or hypoparathyroidism, may all further contribute to the significant morbidity and increased mortality in hospitalized patients with mitochondrial disease. This chapter summarizes the clinical presentation, genetic cause and management of mitochondrial diseases leading to intensive care unit (ICU) admission.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mitochondrial encephalomyopathy
- Stroke-like episodes
- Lactic acidosis
- Respiratory failure
- Gastrointestinal pseudo-obstruction
- Metabolic decompensation
Introduction
Mitochondria are organelles that play a key role in many different cellular processes. They are best known for their role in energy production through respiration but are also involved in maintenance of the redox balance, amino acid and lipid metabolism, calcium homeostasis, temperature regulation, apoptosis and much more.
Mitochondrial energy production occurs through oxidative phosphorylation, whereby electrons travel through four large complexes located at the inner mitochondrial membrane. Electrons enter the electron transport chain (ETC) through complex I or II and are donated to oxygen at complex IV. Complexes I, III and IV use the energy derived from these electrons to pump protons (H+) from the matrix into the intermembrane space. This results in a proton gradient, used by the ATP synthase complex (complex V) to generate ATP from ADP.
Although defects in many mitochondrial processes may be involved in human disease, mitochondrial diseases are defined as genetic disorders that impair oxidative phosphorylation and mitochondrial ATP synthesis. Together, they affect about 1 in 5000 adults and children [1], and may lead to a range of well-defined, but overlapping, clinical syndromes (Table 13.1). However, the clinical diagnosis of a mitochondrial disease is often challenging. Many of the presenting symptoms are vague (e.g. chronic fatigue or abdominal discomfort), some of the typical complaints or signs are common in the general population (e.g. migraine or diabetes mellitus), and the range of phenotypes associated with mitochondrial disease is continuously expanding.
Diagnostic Work-Up in Suspected Mitochondrial Disease at the ICU
When a patient presents with one of the classical mitochondrial syndromes (see Table 13.1.), a clinical diagnosis can often be made and will then guide downstream targeted genetic analysis. However, most patients do not present with a clinically recognizable syndrome, and diagnosis is often difficult and delayed, both in children and adults [3]. Nevertheless, some symptoms, and in particular the co-occurrence of these symptoms in a patient or their family might raise suspicion of an underlying mitochondrial disease. For this purpose, mitochondrial disease criteria (MDC) have previously been established, based on scoring the presence of specific symptoms that are typically seen in patients [4] (Table 13.2). Traditionally, these criteria were used to decide on the need for invasive investigations like muscle biopsy. With the advent of next-generation sequencing, the diagnostic approach has changed significantly, but the same diagnostic criteria have recently been validated in a clinical context where both children and adults were diagnosed by whole exome or mtDNA sequencing [5]. A diagnostic flowchart including these criteria and current next-generation sequencing approaches is shown in Fig. 13.1 (adapted from [3]).

Diagnostic flowchart for suspected mitochondrial disease (adapted from Schon et al. 2020 [3])
Neither the MDC nor the flowchart are routinely used in the everyday clinic, and they have not been validated in the emergency or intensive care context as far as we are aware. However, we believe that their value remains in raising clinical suspicion and guiding subsequent diagnostic work-up.
Genetics of Mitochondrial Disease
The proportion of genetic diagnosis in mitochondrial disease has significantly improved over recent years [3]. However, some patients do not yet receive a molecular genetic diagnosis, and clinical suspicion of mitochondrial disease is further supported only by biochemical or histological studies. Thanks to novel genotyping and phenotyping technologies, we expect this proportion to continue to decrease in the near future.
Currently (in 2020), over 300 nuclear genes [3, 6] are known to cause mitochondrial disease, with autosomal recessive, autosomal dominant and X-linked inheritance patterns, affecting both children and adults. However, in adult patients, mitochondrial disease is mainly caused by mutations in the mitochondrial genome (mtDNA) and then shows exclusive maternal inheritance. The mitochondrial genome is a 16.6 kb circular double-stranded DNA molecule that is located in the mitochondrial matrix and contains 37 intron-less genes: 13 genes encode essential peptides of 4 out of the 5 respiratory chain complexes (complex I, III, IV and V); the remaining genes are 12 transfer RNA (tRNA) and 2 ribosomal RNA (rRNA) genes required for local protein synthesis. Because every nucleated cell contains many copies of mtDNA, cells can hold a mixture of different mitochondrial genomes, which is called heteroplasmy. Much of the mtDNA variation is neutral and does not cause disease, but when a deleterious variant arises, pathogenicity is determined in part, by the nature of the variant and the level of heteroplasmy required to manifest a biochemical defect or clinical phenotype. Heteroplasmy levels can differ between cells and tissues of a single individual, and it is this variability that likely contributes, in part, to the phenotypic heterogeneity encountered in mitochondrial disease.
MtDNA defects are not always inherited through the germline but can also arise during development and aging. This may lead to point mutations, large-scale deletions and progressive mtDNA depletion, usually as a consequence of primary defects in nuclear genes involved in maintenance and replication of the mtDNA. When these secondary mtDNA defects reach a high level in individual cells, they can cause a similar clinical phenotype to that seen for primary mtDNA disorders by directly affecting mitochondrial function.
General Approach to Critical Illness in Patients with Mitochondrial Disease
Preventing Acute Illness in Mitochondrial Disease
Because no curative treatment is available for mitochondrial diseases, a major aspect of their management is supportive therapies and the prevention of acute illness. Emergencies in mitochondrial disease are often related to deterioration of chronic conditions or to environmental changes that alter energy demands, like febrile infections, surgery and anaesthesia, dietary changes, prolonged fasting or specific medications (such as valproic acid [7]).
It is conceivable that stressors may induce a more catabolic state, whereby cells need to rewire metabolic pathways and increase utilization of intracellular stores of amino acids, fatty acids, nucleotides and carbohydrates. In the context of chronic mitochondrial dysfunction, this metabolic flexibility is often impaired and intracellular reserves are exhausted, resulting in increased production of toxic metabolites like reactive oxygen species that damage the cellular and extracellular environment. Together, this may lead to exacerbation of pre-existing conditions and even generalized multisystemic decompensation, particularly in children.
In order to prevent acute deterioration, most patients with mitochondrial disease will require life-long surveillance, involving multiple medical and allied-health disciplines. This is usually organized with input from specialist centres, experienced in the diagnosis and management of mitochondrial disease. Routine follow-up includes regular screening for recognized complications. Surgery needs to be properly planned, and patients need to be educated about avoiding precipitating factors, in particular specific medications (see below) [7], medication compliance and prolonged periods of fasting. End-of-life care planning should be integrated into a framework for routine clinical practice for those with such life-limiting disorders. In addition, carrying an emergency care plan or a medical alert bracelet could be considered for some patients, which will help to guide decisions for clinicians in an acute care setting.
Management of Patients with Mitochondrial Disease in the Critical Care Setting
When patients are admitted to an ICU, they do not necessarily have a previous diagnosis of mitochondrial disease. Nevertheless, establishing a diagnosis of mitochondrial disease is important because the severity and chronicity of pre-existing problems, and the multi-system involvement will pose additional challenges that may require specialist expertise. Patients will often have respiratory insufficiency or dysphagia due to myopathy; they may have a cardiomyopathy or arrhythmia, suffer from gastrointestinal problems that lead to malnutrition and intestinal pseudo-obstruction, or have epilepsy, bone marrow failure or diabetes mellitus. In addition, mitochondrial dysfunction will further increase vulnerability of many other body systems when confronted with acute illness. Cardiac decompensation due to cardiomyopathy or arrhythmias, and acute cerebral events like seizures or stroke-like episodes (seizure-mediated) are well-recognized and are the leading causes of death in patients with mitochondrial disease [8]. Renal failure, acute liver failure, ventilator failure, encephalopathy with central respiratory depression, lactic acidaemia, and endocrine disturbances like diabetes, hypo- and hyperthyroidism, adrenal insufficiency or hypoparathyroidism, may all further contribute to the significant morbidity and threefold (for adults) or sixfold (for children) increased mortality in hospitalized patients with mitochondrial disease [9].
Once a diagnosis is made, there is little evidence to guide the specific management of patients with mitochondrial disease in the critical care setting [10]. Treatable metabolic conditions should of course be excluded (see Chap. 11), but in the absence of curative treatment for most mitochondrial diseases, recommendations are mainly based on expert-opinion guidelines.
Some general recommendations have been described in [11] and are copied here in Table 13.3. Advice on the specific management of more frequent acute complications of mitochondrial disease is described in the following chapters.
The Newcastle Mitochondrial Disease Clinical Guidelines provide expert guidance to health professionals about the management of various aspects of mitochondrial disease. They have been developed using consensus expert opinion sourced from the National Commissioning Group Service for Rare Mitochondrial Diseases for Adults and Children in Newcastle, UK, with associated experts from other hospitals. The guidelines are a very useful resource for any clinician involved in care for patients with mitochondrial disease and are available online at: https://www.newcastle-mitochondria.com/clinical-professional-homepage/clinical-publications/clinical-guidelines/
Severe, Infantile Mitochondrial Disease
Genetic Causes and Pathophysiology
Mitochondrial diseases often manifest in infants, and there are several characteristic tissue specific and multisystem presentations reported in this age which require admission to the ICU. These diseases are both clinically and genetically very heterogeneous. For example, in a cohort of 42 infants with suspected mitochondrial disease, the molecular cause involved >10 different nuclear mitochondrial disease genes, subunits of mitochondrial respiratory chain enzymes, assembly genes, genes involved in mitochondrial translation, mtDNA maintenance or other mitochondrial functions [12]. However, there are several non-mitochondrial diseases with similar clinical presentation [11, 13, 14].
There is growing evidence that rare single gene disorders present in the neonatal period, therefore there is a need for rapid and comprehensive genetic testing in ICUs to assist acute and long-term clinical decision making [15]. In specialized centres rapid (turnaround time 2–3 weeks) whole genome sequencing has been used to identify the molecular diagnosis in 21% of infants in a large cohort of 567 samples. This study showed that the clinical presentation was a poor predictor of the molecular diagnosis in 90% of cases, and the diagnosis affected the clinical management in more than 65% of patients (83% of neonates) including modification of treatments and care pathways or guiding palliative care decisions. Therefore, if biochemical or imaging studies do not highlight a specific diagnosis, WGS has the potential to be a first-line diagnostic tool in critically ill children in ICU [15].
Manifestations
There are a few phenotypes which need special attention, as treatment options may vary based on the exact molecular defect such as severe infantile muscle weakness (floppy baby), severe lactic acidosis with metabolic crisis, acute infantile liver failure (see under Liver failure in mitochondrial disease) and multi-organ failure in infants.
There are a number of non-mitochondrial neuromuscular (e.g. spinal muscular atrophy, congenital muscular dystrophies, congenital myopathies, congenital myasthenic syndromes) and metabolic (such as inborn errors of metabolism, congenital defects of glycosylation) diseases which can manifest in severely ill infants, therefore extensive investigation of these patients is important. Laboratory investigations include creatine kinase (CK), lactate (in serum and CSF), amino acids, organic acids, fatty acids, very long chain fatty acids, white cell enzymes and national new-born blood spot screening programmes and may identify some of the underlying causes. Neuroimaging may be also useful in the differential diagnosis, however rapid and unbiased genetic studies (trio exome/genome) in these infants are of utmost importance to achieve an early diagnosis enabling a timely treatment and guiding management decisions [15]. Although many of the severe infantile conditions are progressive and fatal, there are some rare treatable genetic forms [16] and some of these result in an infantile reversible disease presentation, albeit exceptionally rare [16, 17].
Management
Lactic acidaemia is frequent in infants with mitochondrial disease in ICU. Under stable clinical conditions, this process may remain well compensated and does not require specific therapy. However, especially in situations with altered energy demands, such as febrile infections or longer periods of fasting, children with mitochondrial disorders have a high risk of metabolic decompensation with exacerbation of hyperlactaemia and severe metabolic acidosis. Unfortunately, there are no controlled studies assessing the treatment of this critical condition, and clinical outcome is often unfavourable. There are some expert-based suggestions to treat lactic acidosis including dietary recommendations, buffering strategies and specific drug therapy using sodium bicarbonate, sodium citrate, THAM (Tris-hydroxymethyl-amino-methane) buffering or less frequently dichloroacetate [18]. These strategies can stabilize the metabolic crisis enabling time to achieve the genetic diagnosis, which will guide further treatment of these conditions.
Further options to support OXPHOS in critically ill infants are supplementation of respiratory chain cofactors, which may result in significant improvement in some conditions [16]. This includes thiamine, biotin, riboflavin, coenzyme Q10 or carnitine or ketogenic diet in defects of pyruvate dehydrogenase deficiency [18]. However, ketogenic diet is contraindicated in patients with known fatty acid oxidation disorders and pyruvate carboxylase deficiency [18].
Outcome
Many of the severe infantile mitochondrial diseases are progressive and do not recover. However, in contrast to fatal infantile mitochondrial and neuromuscular diseases (such as severe congenital myopathies or muscular dystrophies), patients with reversible infantile mitochondrial myopathy (m.14674T>C in mt-tRNAGlu) or hepatopathy (TRMU mutations) have an excellent prognosis and spontaneously improve and recover after the 6–12 month of life, therefore these patients should receive all intensive care and life-sustaining measures. Mechanical ventilation, tube feeding, monitoring and treating other organ dysfunctions result in most cases in regression of clinical symptoms. Because some patients present with low levels of L-carnitine or mitochondrial cofactors (coenzyme Q10, thiamine, riboflavin), supplementing these factors may facilitate the clinical improvement of this condition. Low dietary cysteine intake has been suggested to compromize mitochondrial translation in infants carrying the homoplasmic m.14674T>C/G mutation, therefore supplementation with L-cysteine or N-acetyl-cysteine can be considered, especially in infants with clinical manifestations [19]. Furthermore, recent studies highlight the importance of nutrition in mitochondrial energy metabolism, therefore supplementation of critically ill infants with amino acids and nutrients may have a more important role than it has been suggested previously [20].
Case Vignette 1
A 3-months-old infant developed subacute severe muscle weakness, respiratory failure and feeding difficulties (floppy baby), requiring mechanical ventilation, tube feeding, and supportive treatment on ICU. Mitochondrial disease was suspected because of high lactate. Muscle biopsy showed numerous COX-negative RRFs and severe complex I and IV defects. Spontaneous improvement was observed at 6 months of age. Delayed motor milestones, some muscular hypotonia, but able to walk at 2.5 years of age. MtDNA analysis detected the homoplasmic m.14674T>C mutation, which was done by Sanger sequencing. Based on the clinical presentation and high lactate the clinicians wanted to screen directly for the genetic form leading to a reversible disease.
Learning Points
In infants with clinical signs of muscle weakness and high lactate, the reversible infantile form of mitochondrial disease has to be considered and tested as soon as possible. This has relevant implications about the prognosis and the ITU treatment of the patients.
Stroke-like Episodes and Epileptic Encephalopathy
Genetic Causes and Pathophysiology
Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes (MELAS) is one of the most severe syndromes in mitochondrial disease [21]. Stroke-like episodes, the acute presentation of MELAS syndrome, are clinically characterized by headache, nausea and vomiting, encephalopathy, focal-onset seizures with or without associated focal neurological deficits, with cortical and sub-cortical signal abnormalities that are usually cross-vascular territories identified on MRI brain. While stroke-like episodes were originally described in individuals under the age of 40, late-onset presentation is increasingly recognized [22]. The first genetic defect linked to MELAS was a heteroplasmic mitochondrial DNA mutation, m.3243A>G, which accounts for ~80% of the cases reported in the early literature [23]. Many other rarer mtDNA mutations such as m.3271T>C (MT-TL1) and m.13513G>A (MT-ND5) have also been linked to MELAS [24]. More recently, recessive mutations in the mitochondrial polymerase gamma (POLG) have emerged as another important cause of refractory/super-refractory seizures and stroke-like episodes [25,26,27].
Manifestations
The diagnosis of a stroke-like episode requires the combination of clinical assessment, cranial MRI and electroencephalogram (EEG). A mitochondrial stroke-like episode is a subacute event, driven by seizure activity in a patient with genetic mitochondrial disease [28]. It should not be confounded with a typical vascular stroke involving acute loss of function symptoms, and when this occurs in a patient with known mitochondrial disease, the presence of mitochondrial disease should not delay conventional evidence-based treatment for acute ischemic or haemorrhagic stroke. Instead, symptom onset is often subacute, may evolve over several days and is frequently accompanied by focal-onset seizures. Neurological symptoms are complex, may have positive (e.g. hallucinations or focal motor seizures) and negative (e.g. visual field defect, sensory deficit or motor weakness, apraxia and aphasia) signs, include neuropsychiatric disturbances, and are typically not limited to vascular territory.
These potentially treatable seizure-mediated episodes can present at any age and are typically associated with confluent cortical/subcortical MRI changes and EEG abnormalities. Potential triggers are often metabolic stressors including infection, gut dysmotility, dehydration, prolonged fasting or changes in anti-seizure medication. All patients with previous stroke-like episodes, as well as their carers, should be provided with an emergency personalized care plan which they can present to their local primary care and emergency services. An example care plan is shown in Fig. 13.2.

Example personalized care plan for patients with MELAS due to the m.3243A>G variant, as used at the Highly Specialized NHS Service for Rare Mitochondrial Disorders in Newcastle upon Tyne, UK. For POLG-related disease it also includes contraindication of Valproate
Management
The limitations in our understanding of disease mechanisms are reflected in the controversy surrounding the acute management of stroke-like episodes in clinical practice. There is no evidence that intravenous L-arginine works [29] and indeed it may delay instigation of appropriate therapies [30], although some clinicians do advocate its use in patients presenting with stroke-like episodes [31], based on the findings derived from a single open-label trial with a small number of patients [32] and other anecdotal case reports. There is consensus however that aggressive seizure management is too often still over-looked in patients presenting with stroke-like episodes, resulting in metabolic crisis and prolonged hospital admission [32,33,35].
A recent study used the modified Delphi process to harness the clinical expertise from a group of specialists in mitochondrial disease from five European countries to produce consensus guidance for acute management of stroke-like episodes and commonly associated complications [28].
Early anti-epileptic treatment with a benzodiazepine outside hospital, or an IV anti-epileptic drug (not valproate in the setting of known POLG mutations) should be the mainstay of treatment, as well as routine investigations aimed at treating potential triggers and preventing complications. Transferring patients to the intensive care unit is recommended in the following circumstances:
-
1.
Generalized, convulsive status epilepticus
-
2.
Intrusive, frequent focal motor seizures with breakthrough generalized seizures which fail to respond to IV AEDs (and titration of usual maintenance AEDs)
-
3.
Severe encephalopathy (with breakthrough focal motor or generalized seizures) with a high risk of aspiration
-
4.
Focal motor status epilepticus with retained consciousness failing to respond to benzodiazepine and two IV AEDs
Midazolam is often the first choice of general anaesthetic agent to treat refractory status epilepticus associated with stroke-like episodes. Anecdotal reports suggest that propofol, especially in the paediatric population, may lead to propofol infusion syndrome. However, propofol is not absolutely contraindicated in refractory status epilepticus associated with stroke-like episodes and oftentimes is required in combination with midazolam due to the refractory nature of the ictal activity. The decision to use propofol should be made on a case by case basis. Thiopentone sodium (high dose) remains an important drug in terminating refractory status epilepticus particularly in cases of POLG-related MELAS. However, recovery is slow from this anaesthesia and the need for intensive care is prolonged. Following general anaesthesia, continuous EEG monitoring of patients with stroke like episodes should be performed to ensure that no breakthrough seizures occur. If this is unavailable, EEG should be performed as soon as possible after induction of anaesthesia, and at regular intervals for the duration of anaesthesia.
Maintenance intravenous fluid should be administered for patients who are at risk of dehydration, especially in those presenting with vomiting due to intestinal pseudo-obstruction. Careful monitoring of fluid and electrolyte balance may be necessary in those patients with low body mass index, cardiomyopathy or chronic kidney disease as part of the multisystem mitochondrial disease.
Mild to moderate lactic acidosis (serum lactate level: 2.2–5.0 mmol/L with pH >7.30) often responds well to rehydration and does not require any buffering agent. Some patients with mitochondrial disease have baseline hyperlactaemia. Buffering agent such as sodium bicarbonate can be used with care in severe lactic acidosis (pH <7.1), but management of severe metabolic acidosis should be shared with the intensive care specialist or nephrologist. Dichloroacetate should not be used, as it has been shown to cause unacceptable levels of toxicity (neuropathy) that outweigh any potential benefits [36].
Early consultation with the nutritional team is recommended during admission for stroke-like episodes. Gastroparesis and small bowel intestinal pseudo-obstruction with recurrent vomiting can be particularly dangerous, certainly in patients at increased risk of aspiration—such as those with encephalopathy, seizures, or bulbar dysfunction. Prompt recognition of pseudo-obstruction based on clinical symptoms and radiological findings is crucial, so that drainage of the stomach content can be achieved with the insertion of a wide-bore nasogastric tube. Concomitant constipation often occurs and should be treated appropriately. Swallowing should be assessed and regularly monitored.
Acute neuropsychiatric symptoms (hallucinations, confusion) can be managed with anti-psychotic medications, but advice from the psychiatric service should be sought (Table 13.4).
Outcome
The outcome of severe intractable stroke-like episodes can be fatal. The long-term consequence of recurrent stroke-like episodes is cognitive impairment due to neurodegeneration (Fig. 13.3).

Acute management workflow of seizures in mitochondrial stroke-like episodes, based on the “At a glance guidelines on the management of mitochondrial stroke-like episodes” used at the Highly Specialised NHS Service for Rare Mitochondrial Disorders in Newcastle upon Tyne, UK
Case Vignette 2
A 17-year-old woman presented to the emergency department with a 2-day history of a constant flashing light in her left hemi-visual field (also with eyes closed), blurred vision and worsening ataxia. She described experiencing intermittent blurred vision, a rainbow of coloured flashes with eyelid flickering lasting around 1 min and headache (described as her worst headache ever) for 2 weeks before attending the emergency department.
She had been diagnosed with recessive POLG-related mitochondrial disease characterized by ataxia, motor-sensory neuropathy and stable epilepsy at the age of 16 years. Her epilepsy was initially managed with topiramate which then changed to carbamazepine due to inefficacy, and levetiracetam was a subsequent add-on therapy. Gabapentin was started for neuropathic pain.
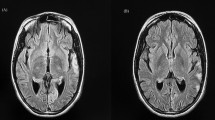
On admission, her GCS was 15 and left homonymous hemianopia was evident. Other positive neurological findings were mild, distal ankle weakness, areflexia and sensory loss below the knee level. Stroke-like episode and prolonged occipital seizures were suspected; carbamazepine and levetiracetam were increased to 400 mg BD and 1750 mg, respectively, and clobazam 10 mg BD was added. MRI head performed a week after the onset of positive visual phenomena identified T2-signal abnormalities involving the right medial occipital lobe with cytotoxic changes (Fig. 13.4a–d). Her EEG demonstrated an active ictal focus over the right posterior quadrant with occasional epileptic discharges within the contra-lateral parieto-occipital region. A decision for escalating to general anaesthesia treatment was made given the patient had developed refractory non-convulsive status epilepticus (occipital seizures) and unilateral visual field loss. Burst suppression pattern was achieved and maintained with the combined administration of midazolam (265 mg) and thiopentone (18,875 mg) over 3 days. She remained in a comatose state for 6 days after stopping thiopentone. Repeat MRI head (day 7) showed resolution of signal abnormalities in the right occipital lobe and corpus callosum (Fig. 13.4e–h). Her ICU stay was complicated by hospital-acquired pneumonia and clostridium difficile diarrhoea, which were treated with appropriate antibiotics. Extubation was successfully performed 10 days after ICU admission, and the patient was stepped down for rehabilitation on day 14. However, a left-sided visual field loss returned 5 days later, but without positive visual phenomena on this occasion. Her EEG only showed encephalopathic changes; repeat MRI head (day 22) showed a stroke-like lesion that had appeared in the same location as per the previous scan but without restricted diffusion (Fig. 13.4i–l).

Serial MRI scans from the patient described in case vignette 2. a, e and i = T2-weighted image; b, f and j + d, h and l = Diffusion-weighted image (DWI); c, g and k = apparent diffusion coefficient (ADC) image
Learning Points
-
1.
Non-convulsive seizures originating from the occipital lobe could be misdiagnosed as migrainous visual aura.
-
2.
Seizure management is challenging in POLG-related stroke-like episodes, and aggressive treatment is crucial to limit neuronal loss and development of significant disability.
Severe Cardiac Complications in Mitochondrial Disease (Cardiomyopathy, Arrhythmia)
Genetic Causes and Pathophysiology
The heart heavily relies on mitochondrial respiration to produce energy required for contraction and electric conduction and is therefore amongst the most vulnerable tissues when confronted with mitochondrial dysfunction. Estimates of the prevalence of cardiac manifestations in mitochondrial disease vary [36,37,39], but overall, around 50–60% of patients are reported to be affected by some form of cardiac dysfunction. The prevalence and type of involvement are highly dependent on the underlying mutation (Table 13.5).
Cardiac manifestations are independent of the severity of other symptoms, and onset is most often insidious with clinical symptoms usually emerging after a diagnosis of mitochondrial disease has been made. However, in rare instances, a diagnosis of mitochondrial disease is made in ICU, when acute cardiovascular involvement is the initial and/or prevailing manifestation.
Cardiac Manifestations
The most common manifestations of cardiac pathology in patients with mitochondrial disease are hypertrophic cardiomyopathy and conduction block often predicated by genotype. When a patient presents with these conditions to the emergency department and critical care unit, the absence of concomitant coronary artery disease, hypertension, valvular disease, or congenital heart disease might raise suspicion of an underlying metabolic or mitochondrial disorder. Nevertheless, many different cardiac manifestations have been reported in the context of mitochondrial disease, and presence of these certainly does not exclude this diagnosis.
Structural Defects
Hypertrophic cardiomyopathy (HCM) is the most common structural abnormality found in mitochondrial disease and can sometimes even be seen on prenatal ultrasound [40]. Patients often remain asymptomatic for a long time before chest pain, dyspnoea, palpitations or syncope develop. Diagnosis is supported by ECG and 2D-echocardiogram (and/or cardiac MRI).
Overall, HCM is most frequently associated with autosomal dominant mutations in genes encoding cardiac sarcomere proteins. Only a minority of cases are caused by pathogenic mitochondrial disease variants, but the signs in Table 13.2 (MDC score) should raise suspicion and initiate a diagnostic work-up for mitochondrial disease as in Fig. 13.1 (flowchart). Specific patterns on cardiac ultrasound or magnetic resonance imaging (MRI) may be preferentially associated with particular syndromes or genetic mutations and guide further diagnostic investigations. For example, concentric left ventricular hypertrophy with patchy non-ischemic (intra-mural) late gadolinium enhancement in multiple segments on cardiac MRI is thought to be more suggestive of m.3243A>G-associated cadiomyopathy [39]. However, these patterns are not diagnostic, and phenotypes are most likely further confounded by variability in heteroplasmy levels.
Arrhythmias
Arrhythmias caused by mitochondrial dysfunction may include various degrees of atrio-ventricular conduction block, sick sinus syndrome, left or right bundle branch blocks, intra-ventricular delays, Wolff-Parkinson-White tachyarrhythmia or left ventricular pre-excitation [41, 42].
Diagnosis of arrhythmias is usually based on history and 12-lead resting or longer-term ECG. Brady-arrhythmias can be asymptomatic, but with persistent or severe abnormalities, symptoms are common and include easy fatigability, reduced exercise capacity, dizziness, dyspnoea, presyncope and syncope. Tachycardia will usually produce symptoms and may result in palpitations, fatigue, light-headedness, chest pain, dyspnoea and less commonly syncope.
Conduction blocks are most frequently seen in association with heteroplasmic large-scale deletions of mtDNA, leading to CPEO and Kearns-Sayre Syndrome (KSS). Additional symptoms of KSS are a pigmentary retinopathy, often accompanied by ataxia, deafness and dementia. Severe cardiac manifestations of KSS include complete heart block and sudden death; therefore cardiac monitoring and considering defibrillator or pacemaker implantation in at-risk patients are of primordial importance in the follow-up of these patients. Patients with m.8344A>G [43] or m.3243A>G mutations [37] in contrast have a higher incidence of tachy arrhythmias such as Wolff-Parkinson-White Syndrome.
Management
All patients with a suspected or confirmed diagnosis of mtDNA-related mitochondrial disease and/or those considered susceptible to cardiac involvement should undergo repeated assessment to ensure early detection and treatment of cardiac pathology [11]. Guidelines suggest blood pressure measurement, ECG and cardiac ultrasound at the initial visit, and afterwards 1- to 2-yearly follow-up for the first 5 years. Further screening then depends on findings and symptoms but could be extended to once every 3 years if investigations are normal. Frequent (every 3–6 months) Holter monitoring should be considered in those patients with high risk of conduction abnormalities, in particular single deletions leading to CPEO and Kearns-Sayre Syndrome. Asymptomatic carriers should be screened as well, as even low levels of heteroplasmy may lead to cardiac abnormalities, but the intervals can be tailored to individual mutations and symptoms.
When acute cardiac pathology is the primary manifestation, and patients have not had a diagnosis as yet, more advanced imaging modalities, for example through cardiac MRI or MRS, may aid in excluding other possible causes of apparent ventricular hypertrophy and can guide further testing and genetic analysis [44]. Endomyocardial biopsy may aid in the differential diagnosis of various forms of hypertrophic cardiomyopathy, but in the current era of genomics, the risks often outweigh the possible benefits.
Good guidelines exist for management of hypertrophic cardiomyopathy, including from the European Society of Cardiology (ESC) [45], and these are applicable to patients with mitochondrial disease also. Cardiac arrhythmias should be investigated and treated accordingly, and ablation in Wolff-Parkinson-White syndrome and pacemakers or implantable defibrillators should be considered where appropriate. When patients are at high risk of completed heart block (e.g. KSS), current guidelines suggest to follow the same approach as for other patients without genetic conditions [46, 47].
Heart transplantation as a treatment for intractable heart failure caused by mitochondrial hypertrophic or dilated cardiomyopathy may be considered too. Several case reports [47,48,50] and one larger series of 11 patients [51] have been published describing successful heart transplants in adult patients with mitochondrial disease. In children, heart transplantation has a similarly favourable outcome [51,52,54], and overall survival seems comparable to children without mitochondrial disease. Nevertheless, mitochondrial disease does confer a greater risk of complications and longer hospital stay both in children and adults [54].
Prognosis
In adults with mitochondrial disease, sudden death is sometimes reported in the family history of patients [38, 55]. Nevertheless, life-threatening cardiovascular complications are relatively rare and when properly treated, the prognosis of cardiac involvement is overall good. In one series of 32 adult patients with mitochondrial disease, 5-year cardiovascular event free survival was 67%. Four out of 32 patients received invasive treatment (heart transplant, pacemaker or ablation) and none died of cardiovascular disease [38]. In children, cardiovascular involvement is less frequent than in adults, but when cardiomyopathy is present, it is a significant contributor to mortality. It increased mortality risk almost three times (up to 71%) in hospitalized children with mitochondrial disease [56].
Case Vignette 3
A 32-year-old man with a 6-year history of sensorineural hearing loss presented to the emergency department with intermittent vague chest pain for 3 weeks. Troponin I was slightly raised (94 ng/L; ref. 0–56) with lateral T-wave inversion and infero-lateral ST-depression on ECG. He was hyperglycaemic and HbA1c was 89 mmol/mol (ref. <48 mmol/mol). Serum lactate was increased (3.1 mmol/L; ref. 0.6–1.4) and it was noted that 6 months previously, on a routine blood test, this also was raised to 3.3 mmol/L. Troponin I remained stable between 69 and 94 ng/L over the next 3 days. Echocardiogram and CT coronary angiogram were normal, but CT chest showed mild thickening of the left ventricular myocardium. Two weeks later, he experienced palpitations from sleep and was admitted the same day after a pre-syncopal episode. Holter monitoring was repeatedly normal. Clinical examination only suggested mild bilateral ptosis that was not reported by the patient.
His grandmother and maternal uncle were known to have hearing problems, and his mother was previously diagnosed with type II diabetes. Mitochondrial disease was suspected, and he was referred for outpatient cardiac MRI and genetic testing. Cardiac MRI confirmed minor concentric left ventricular hypertrophy up to 12 mm with normal ventricle size and systolic function, and he was found to carry the common m.3243A>G mutation at 34% heteroplasmy in blood. A diagnosis of maternal inherited diabetes and deafness (MIDD) was made. Ophthalmology screening showed bilateral macula pattern-like dystrophy without visual symptoms. Ramipril 2.5 mg daily, Aspirin 75 mg daily and subcutaneous insulin were started, and further follow-up with yearly ECG, 24-h ECG and echocardiogram have been stable.
Learning Points
Acute or subacute heart disease can be a presenting feature in mitochondrial disease. Attention for other signs and symptoms in patients and teir families, even when they are very common in the population (like diabetes, migraine or hearing problems), may point to an underlying genetic mitochondrial disease. Long-term screening and standard treatment of cardiac disease should then be initiated.
Liver Failure in Mitochondrial Disease
Genetic Causes and Pathophysiology
Liver failure is a life-threatening critical illness, often leading to admission to intensive care. Symptoms include jaundice, encephalopathy, bleeding, fatigue and lactic acidosis. Although the cause of liver failure is often unknown, inherited disorders of mitochondrial oxidative phosphorylation may be responsible, especially in infancy and young children <2 years of age [57]. In general, mitochondrial liver disease often occurs with extra-hepatic involvement [58]. However, isolated hepatic failure may also be related to mitochondrial dysfunction caused by defects of mitochondrial DNA (mtDNA) maintenance such as mtDNA deletion (Pearson syndrome) and depletion [58]. Genetic forms of mtDNA depletion and liver failure can be associated with autosomal recessive mutations in DGUOK [59], MPV17 [60], TWNK [61], SUCLG1 [62] or POLG (Alpers-Huttenlocher syndrome) [57, 63]. Patients with mutations in POLG are at high risk for developing valproate induced liver failure. Low mtDNA copy number has also been detected in patients (often infants) with acute liver failure without a specific mitochondrial genetic diagnosis [57, 64], and these patients may have a good prognosis and do not develop other symptoms, suggesting that low liver mtDNA copy number may be a secondary phenomenon caused by various liver pathologies in infants [57].
Severe infantile liver failure can also be caused by dysfunction of mitochondrial transcription and translation [65]. Pathogenic mutations in TFAM cause infantile-onset progressive fatal liver failure, while intrauterine growth restriction and severe hepatopathy was reported in patients with mutations in translation elongation factors GFM1 and TSFM [65].
In contrast to these progressive and often fatal conditions, a unique reversible infantile hepatopathy has been shown in association with mutations in the mitochondrial tRNA modifying factor TRMU [66, 67]. Considering that acute infantile liver failure may be reversible is of utmost importance in treating patients with liver failure in ICU. These patients need rapid diagnostic testing and consideration of active therapeutic interventions including liver transplantation [51].
Rare cases of liver dysfunction in single respiratory chain complex deficiencies were reported due to mutations in SCO1 (complex IV assembly factor) and BCS1L (complex III assembly factor) [58]. Biallelic mutations in NBAS have been identified as a new molecular cause of acute recurrent liver failure and dysmorphic signs with onset in infancy [68].
Manifestations
Mitochondrial liver failure often presents in early infancy [57]. Liver biopsy often shows cirrhosis, micro- and macrovesicular steatosis and cholestasis and abnormal mitochondrial morphology on electron microscopy as well as low mtDNA copy number. Liver crises are triggered by febrile infections; they become less frequent with age but are not restricted to childhood. Complete recovery is typical for TRMU mutations, but acute crises can be fatal in other genetic forms, such as POLG mutations (see above). Furthermore, respiratory chain abnormalities were frequently detected in liver samples of patients with severe liver failure requiring transplantation due to various forms of non-mitochondrial liver diseases [64].
Management
Treatment of liver failure in NBAS deficiency is symptomatic, antipyretic therapy and induction of anabolism including glucose and parenteral lipids effectively ameliorate the course of liver crises [69]. Patients with biallelic TRMU mutations usually recover spontaneously after 1–2 years of age [67]. Active intensive care support of these patients is of utmost importance, as they have a good prognosis, although long-term follow up data are not available in many cases.
Liver transplantation can be lifesaving in severe cases [61, 70]. Liver transplantation has been performed in patients with liver failure due to mutations in DGUOK or POLG [59, 60, 71], but may be considered in isolated or predominant severe liver presentations of other forms of mitochondrial diseases [72, 73]. Liver transplantation has been effective in stabilizing symptoms and nearly normalizing thymidine levels in patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) [72]. These patients do not have acute liver failure, but the donor liver produces wild-type TYMP enzyme, and even a small increase in enzyme activity enables elimination of toxic thymidine nucleotides in MNGIE. Therefore, liver transplantation may have an improved safety profile over hematopoietic stem cell transplant in this condition [72].
Outcome
A significant proportion of patients benefit from liver transplantation with long-term survival and a stabilization of neurological features despite initial neurological abnormalities [60, 71]. Decisions to list a patient with mitochondrial disease for liver transplant should be taken in the light of other comorbidities and the natural history of the specific mitochondrial disease [51].
Case Vignette 4
Acute liver failure and muscle weakness in a 4-months-old child. Diagnostic work-up revealed a female infant with skin icterus, enlarged liver, body weight was under the third percentile. Laboratory investigations showed lactic acidosis (12.4 mmol/L, normal<2.2 mmol/L) with pH 7.3, BE-13 mmol/L, bicarbonate 14 mmol/L and hypoglycaemia (12 mg/dL, normal 60–100 mg/dL), elevated liver enzymes (AST 1310 U/L, normal<80 U/L; ALT 1364 U/L, normal <65 U/L; LDH 709 U/L, normal 158–353 U/L), elevated total bilirubin (6.3 mg/dL, normal 0.2–1.0 mg/dL) with elevated direct bilirubin (4.8 mg/dL, normal <0.2 mg/dL) and normal indirect bilirubin (0.5 mg/dL) as well as an impaired liver synthesis (PTT 160 s, normal 25–60 s; TPZ 17%, normal range 70–120%, AT III 17%, normal range 80–123%) and thrombocytopenia (60/nL, normal 200–480/nL). On the intensive care unit symptomatic therapy was started immediately, comprising invasive ventilation, volume substitution, glucose infusion, substitution with sodium bicarbonate, platelets and erythrocytes and fresh frozen plasma. Antibiotic and antiviral therapy (Cefotaxim, Ampicillin, acyclovir) were initiated. Muscle and liver biopsy showed combined complex I and IV deficiency and normal mtDNA copy number. WES detected compound heterozygous TRMU mutations. The child recovered at 12 months of age after careful management of liver failure and doing well at 10 years of age.
Learning Points
Severe infantile liver failure can be reversible , therefore diagnosing TRMU mutations is of utmost importance, as these patients have a very good prognosis.
Acute Gastrointestinal Manifestations in Mitochondrial Disease
Causes and Manifestations
Gastrointestinal involvement in mitochondrial disease is common, affecting more than 50% of patients and their presentation may vary considerably between syndromes and individuals. Although well recognized in patients with TYMP mutations causing Mitochondrial NeuroGastro-Intestinal Encephalopathy (MNGIE), gastrointestinal symptoms are often underappreciated in other patients with mitochondrial disease. Common features include chronic constipation or diarrhoea, poor appetite with weight loss, nausea, recurrent vomiting, dysphagia, and sphincter dysfunction causing reflux or rarely achalasia. Many of these symptoms could be attributed to smooth muscle myopathy related to mitochondrial dysfunction, but they may also be a sign of underlying neuropathy, central nervous system problems, or even mood disturbance [74]. Chronic gastrointestinal symptoms can lead to acute complications such as aspiration pneumonia due to dysphagia, or electrolyte disturbances associated with chronic diarrhoea and vomiting. In addition, they may complicate care for patients with mitochondrial disease hospitalized for other conditions, in particular in patients with MELAS and stroke-like episodes.
One of the most severe manifestations of gastrointestinal dysmotility is chronic intestinal pseudo-obstruction (IPO), which is particularly common in patients with MELAS. IPO is characterized by more than 6 months of recurrent episodes of bowel obstruction in absence of a mechanical cause. Symptoms include abdominal distension and pain, nausea and vomiting with evidence of a dilated bowel on clinical examination or radiography (abdominal X-ray, ultrasound or CT scan). In a retrospective study from a tertiary care centre, up to 19% of patients with IPO were found to have an underlying mitochondrial disorder [75], and these patients were more likely to have severe nutritional deficiency requiring parenteral nutrition and multisystem complications leading to premature death [74, 75]. Importantly, acute exacerbations of IPO may accompany or precipitate stroke-like episodes in patients with MELAS [74].
Management of Acute Pseudo-Obstruction
(Sub-)Acute intestinal pseudo-obstruction should be managed aggressively after exclusion of a mechanical obstruction (e.g. volvulus, adhesion, tumour or impacted stool). Patients should be kept nil by mouth until the pseudo-obstruction is resolved, while maintaining adequate IV hydration with dextrose (see Table 13.3). For patients who present with predominantly gastroparesis and small bowel involvement, prompt gastric drainage and decompression is crucial to circumvent severe aspiration. Parenteral nutrition should be considered, in particular when the episode is long-lasting and patients are already underweight. Water-soluble enemas and flatus tubes may help to relieve distal small bowel and large bowel pseudo-obstruction. Medications that may have precipitated the acute deterioration should be withdrawn and accompanying urinary retention treated accordingly. Surgical resection should only be a last resort, in case of true obstruction or complications requiring surgery like perforation, after consultation with specialized gastroenterologists and clinicians with experience in treating patients with mitochondrial disease. Certainly in patients with MELAS, close monitoring for other multisystem manifestations and neurologic decline is required.
In patients with MNGIE, several treatments have been proposed, including allogeneic hematopoietic stem cell transplantation [76], carrier erythrocyte entrapped thymidine phosphorylase therapy [77] or liver [72] transplantation. However, the outcome of these invasive therapies is worse in patients with significant comorbidity and more advanced disease: pre-transplant liver disease or a history of IPO are negative predictors of outcome and overall survival [76].
Safe Use of Medications in Patients with Mitochondrial Disease
Traditionally, patients with mitochondrial disease are given a long list of medications that should be avoided if at all possible. Although this cautionary approach has certainly helped to raise awareness of the risk for medication- or stressor-induced metabolic decompensation, for example in the perioperative period, there is very little evidence that most of these medications actually cause any harm to these patients.
Recently, a mitochondrial expert panel followed a Delphi method to establish consensus guidelines for the safe use of medications in patients with mitochondrial disease [7]. Their main conclusions are summarized in Table 13.6 [7]. The key recommendations regarding use of medications in patients with mitochondrial disease are the following:
-
1.
Valproate should be avoided in patients with POLG-related mitochondrial disease.
-
2.
Prolonged use of specific drugs (see Table 13.6) should be avoided if good alternative treatment options are available.
-
3.
The usual standards of good practice prevail when prescribing any drug, irrespective of the drug’s mitochondrial toxicity potential or profile (e.g. corticosteroids may cause muscle toxicity or spironolactone may cause metabolic acidosis, independent of an underlying mitochondrial disease or not).
Self Assessment Questions
-
1.
Mitochondrial diseases frequently present with a clinically recognizable syndrome.
-
(a)
True.
-
(b)
False (*)
-
(a)
-
2.
Which of the following examination findings is most characteristic for mitochondrial myopathy?
-
(a)
Delayed tonic pupillary reaction on light stimuli.
-
(b)
External ophthalmoplegia. (*)
-
(c)
Muscle weakness fluctuating within 5 min.
-
(d)
Painful cramps with fasciculations.
-
(a)
-
3.
Which of the following symptoms should raise a suspicion of a mitochondrial disease if there is no other explanation?
-
(a)
Burning feet.
-
(b)
Kinesiogenic dystonia.
-
(c)
Paroxysmal vertigo.
-
(d)
Sensorineural hearing loss. (*)
-
(a)
-
4.
Abnormalities in which type of DNA cause a mitochondrial disease?
-
(a)
Only in mitochondrial DNA.
-
(b)
Only in nuclear DNA.
-
(c)
Both in mitochondrial and nuclear DNA. (*)
-
(a)
-
5.
What is the most common inheritance pattern in adult-onset mitochondrial disease?
-
(a)
Autosomal dominant.
-
(b)
Autosomal recessive.
-
(c)
X-linked.
-
(d)
Maternally-inherited. (*)
-
(a)
-
6.
What is the most common inheritance pattern in paediatric mitochondrial disease?
-
(a)
Autosomal dominant.
-
(b)
Autosomal recessive. (*)
-
(c)
X-linked.
-
(d)
Maternally-inherited.
-
(a)
-
7.
A patient with a mitochondrial cytopathy should avoid fasting for the prevention of an acute deterioration.
-
(a)
True. (*)
-
(b)
False.
-
(a)
-
8.
Which of the following problems may be seen in the context of a mitochondrial disease?
-
(a)
Cardiac arrhythmias.
-
(b)
Heart failure.
-
(c)
Seizures.
-
(d)
Stroke-like episodes.
-
(e)
All of the above. (*)
-
(a)
-
9.
Which of the following are generally recommended for nutrition of a mitochondrial patient in the ICU?
-
(a)
Avoidance of lipids.
-
(b)
Dextrose containing IV fluids. (*)
-
(c)
L-arginine infusion
-
(a)
-
10.
All severe infantile mitochondrial diseases are progressive and do not recover.
-
(a)
True.
-
(b)
False. (*)
-
(a)
-
11.
Which of the following anti-epileptic drugs should be avoided in a patient with a mitochondrial cytopathy?
-
(a)
Carbamazepine.
-
(b)
Lamotrigine.
-
(c)
Levetiracetam.
-
(d)
Valproate. (*)
-
(a)
-
12.
Which is the first choice general anaesthetic agent to treat a refractory status epilepticus in a patient with mitochondrial cytopathy?
-
(a)
Midazolam. (*)
-
(b)
Thiopentone.
-
(c)
Ketamine.
-
(d)
Propofol.
-
(a)
-
13.
Which is the best way to treat a mild lactic acidosis in a patient with mitochondrial cytopathy?
-
(a)
Acetozolamide.
-
(b)
Glucose.
-
(c)
Rehydration. (*)
-
(d)
Sodium bicarbonate.
-
(a)
-
14.
Which antibiotic agent should be used with caution in patients with predisposing mtDNA mutations for ototoxicity.
-
(a)
Aminoglycosides. (*)
-
(b)
Cephalosporins.
-
(c)
Macrolides.
-
(d)
Penicillin.
-
(a)
-
15.
Life-threatening cardiovascular complications are common in all forms of mitochondrial disease.
-
(a)
True.
-
(b)
False. (*)
-
(a)
References
Gorman GS, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015.
Chinnery PF, Keogh MJ. Clinical mitochondrial medicine. Cambridge University Press; 2018.
Schon K, Ratnaike T, van den Ameele J, Horvath R, Chinnery PF. Mitochondrial diseases: a diagnostic revolution. Trends Genet. 2020.
Morava E, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006.
Witters P, et al. Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genet Med. 2018.
Stenton SL, Prokisch H. Genetics of mitochondrial diseases: identifying mutations to help diagnosis. EBioMedicine. 2020.
De Vries MC, et al. Safety of drug use in patients with a primary mitochondrial disease: an international Delphi-based consensus. J Inherit Metab Dis. 2020.
Barends M, et al. Causes of death in adults with mitochondrial disease. JIMD Rep. 2016.
McCormack SE, et al. Hospitalizations for mitochondrial disease across the lifespan in the U.S. Mol Genet Metab. 2017.
Howard RS, et al. Management of mitochondrial disease on an intensive care unit. QJM. 1995.
Parikh S, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017.
Calvo SE, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012.
Pyle A, et al. Respiratory chain deficiency in nonmitochondrial disease. Neurol Genet. 2015.
Parikh S, et al. Diagnosis of possible’ mitochondrial disease: an existential crisis. J Med Genet. 2019.
C.E., F., et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 2019.
Distelmaier F, Haack TB, Wortmann SB, Mayr JA, Prokisch H. Treatable mitochondrial diseases: cofactor metabolism and beyond. Brain. 2017.
Boczonadi V, Bansagi B, Horvath R. Reversible infantile mitochondrial diseases. J Inherit Metab Dis. 2015.
Danhauser K, et al. Treatment options for lactic acidosis and metabolic crisis in children with mitochondrial disease. J Inherit Metab Dis. 2015.
Boczonadi V, et al. Altered 2-thiouridylation impairs mitochondrial translation in reversible infantile respiratory chain deficiency. Hum Mol Genet. 2013.
Hathazi D, et al. Metabolic shift underlies recovery in reversible infantile respiratory chain deficiency. EMBO J. 2020;39(23):e105364.
Goto YI, Nonaka I, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990.
Hirano M, et al. MELAS: An original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992.
Yu-Ichi G, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes(MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992.
Shanske S, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008.
Deschauer M, et al. MELAS associated with mutations in the POLG1 gene. Neurology. 2007.
Tzoulis C, et al. Localized cerebral energy failure in DNA polymerase gamma-associated encephalopathy syndromes. Brain. 2010.
Engelsen BA, et al. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain. 2008.
Ng YS, et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019.
Stefanetti RJ, Ng YS, Errington L, Blain AP, McFarland R, Gorman GS. L-arginine in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes: a systematic review. Neurology. 2022;98(23):e2318–28.
Kitamura M, et al. L-arginine intervention at hyper-acute phase protects the prolonged MRI abnormality in MELAS. J Neurol. 2016.
Koenig MK, et al. Recommendations for the management of strokelike episodes in patients with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes. JAMA Neurol. 2016.
Koga Y, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005.
Whittaker RG, et al. Epilepsy in adults with mitochondrial disease: a cohort study. Ann Neurol. 2015.
Kaufman KR, Zuber N, Rueda-Lara MA, Tobia A. MELAS with recurrent complex partial seizures, nonconvulsive status epilepticus, psychosis, and behavioral disturbances: case analysis with literature review. Epilepsy Behav. 2010.
Lee HN, et al. Epilepsy characteristics and clinical outcome in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Pediatr Neurol. 2016.
Kaufmann P, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006.
Anan R, et al. Cardiac involvement in mitochondrial diseases: a study on 17 patients with documented mitochondrial DNA defects. Circulation. 1995.
Limongelli G, et al. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010.
Florian A, et al. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J Cardiovasc Magn Reson. 2015.
García-Díaz L, Coserria F, Antiñolo G. Hypertrophic cardiomyopathy due to mitochondrial disease: prenatal diagnosis, management, and outcome. Case Rep Obstet Gynecol. 2013.
Kabunga P, et al. Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int J Cardiol. 2015.
Wahbi K, et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur Heart J. 2015.
Wahbi K, et al. Cardiac involvement is frequent in patients with the m.8344A>G mutation of mitochondrial DNA. Neurology. 2010.
Partington SL, Givertz MM, Gupta S, Kwong RY. Cardiac magnetic resonance aids in the diagnosis of mitochondrial cardiomyopathy. Circulation. 2011.
Elliott PM, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014.
Brignole M, et al. 213 ESC guidelines on cardiac pacing and cardiac resynchronization therapy. Eur Heart J. 2013.
Brugada J, et al. 2019 ESC guidelines for the management of patients with supraventricular tachycardia the task force for the management of patients with supraventricular tachycardia of the European Society of Cardiology (ESC). Eur Heart J. 2019.
Homan DJ, et al. Heart transplantation for a patient with Kearns-Sayre syndrome and end-stage heart failure. Congest Heart Fail. 2011.
Tranchant C, et al. Cardiac transplantation in an incomplete Kearns-Sayre syndrome with mitochondrial DNA deletion. Neuromuscul Disord. 1993.
Bhati RS, Sheridan BC, Mill MR, Selzman CH. Heart transplantation for progressive cardiomyopathy as a manifestation of MELAS syndrome. J Heart Lung Transpl. 2005.
Parikh S, et al. Solid organ transplantation in primary mitochondrial disease: proceed with caution. Mol Genet Metab. 2016.
Santorelli FM, et al. Hypertrophic cardiomyopathy and mtDNA depletion. Successful treatment with heart transplantation. Neuromuscul Disord. 2002.
Golden AS, Law YM, Shurtleff H, Warner M, Saneto RP. Mitochondrial electron transport chain deficiency, cardiomyopathy, and long-term cardiac transplant outcome. Pediatr Transplant. 2012.
Weiner JG, et al. Heart transplantation in children with mitochondrial disease. J Pediatr. 2020;217:46–51.e4.
Ng YS, et al. Sudden adult death syndrome in m.3243A>G-related mitochondrial disease: an unrecognized clinical entity in young, asymptomatic adults. Eur Heart J. 2016.
Holmgren D, et al. Cardiomyopathy in children with mitochondrial disease: clinical course and cardiological findings. Eur Heart J. 2003.
McKiernan P, et al. Incidence of primary mitochondrial disease in children younger than 2 years presenting with acute liver failure. J Pediatr Gastroenterol Nutr. 2016.
Rahman S. Gastrointestinal and hepatic manifestations of mitochondrial disorders. J Inherit Metab Dis. 2013.
Dimmock DP, et al. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency. Liver Transpl. 2008.
Hynynen J, et al. Acute liver failure after valproate exposure in patients with POLG1 mutations and the prognosis after liver transplantation. Liver Transpl. 2014.
Fellman V, Kotarsky H. Mitochondrial hepatopathies in the newborn period. Semin Fetal Neonatal Med. 2011.
Van Hove JLK, et al. Succinyl-coa ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res. 2010.
Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004.
Lane M, et al. Mitochondrial dysfunction in liver failure requiring transplantation. J Inherit Metab Dis. 2016.
Boczonadi V, Ricci G, Horvath R. Mitochondrial DNA transcription and translation: clinical syndromes. Essays Biochem. 2018.
Zeharia A, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009.
Schara U, et al. Acute liver failure with subsequent cirrhosis as the primary manifestation of TRMU mutations. J Inherit Metab Dis. 2011.
Carli D, et al. NBAS pathogenic variants: defining the associated clinical and facial phenotype and genotype–phenotype correlations. Hum Mutat. 2019.
Staufner C, et al. Recurrent acute liver failure due to NBAS deficiency: phenotypic spectrum, disease mechanisms, and therapeutic concepts. J Inherit Metab Dis. 2016.
Chinnery PF, DiMauro S. Mitochondrial hepatopathies. J Hepatol. 2005.
Grabhorn E, et al. Long-term outcomes after liver transplantation for deoxyguanosine kinase deficiency: a single-center experience and a review of the literature. Liver Transpl. 2014.
Kripps KA, et al. Successful liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Mol Genet Metab. 2020.
Parikh S, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015.
Ng YS, et al. Pseudo-obstruction, stroke, and mitochondrial dysfunction: a lethal combination. Ann Neurol. 2016.
Amiot A, et al. Frequency of mitochondrial defects in patients with chronic intestinal pseudo-obstruction. Gastroenterology. 2009.
Halter JP, et al. Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain. 2015.
Levene M, et al. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. J Clin Med. 2019.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
van den Ameele, J., Ng, Y.S., Gorman, G.S., Horvath, R. (2022). Intensive Care Implications in Primary Mitochondrial Disease. In: Damian, M., de Visser, M. (eds) Emergencies in Neuromuscular Disorders. Springer, Cham. https://doi.org/10.1007/978-3-030-91932-0_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-91932-0_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91931-3
Online ISBN: 978-3-030-91932-0
eBook Packages: MedicineMedicine (R0)