
Abstract
Xeroderma pigmentosum (XP) is an autosomal recessive disorder characterized by hyperpigmentation and extreme skin sensitivity to ultraviolet (UV) light as a result of a defective nucleotide excision repair (NER) system. It frequently leads to various cutaneous carcinomas, particularly affecting the sun-exposed areas. The first skin cancer could appear before the age of 10 years. Ocular and neurological deficits soon follow. Avoiding exposure to the sun, wearing protective clothing and applying sunscreens are the mainstay of prevention, along with oral isotretinoin and topical 5-fluorouracil. Genetic counselling should be undertaken to avoid consanguine marriages.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
A condition with conspicuous findings of parched pigmented skin was first introduced in 1874 by the Hungarian dermatologist Moritz Kohn Kaposi in a textbook published with his father-in-law, Ferdinand Ritter von Hebra, at that time the department chair of dermatology at Vienna University [1]. The disease was later labelled as “xeroderma pigmentosum” to connote the dry pigmented skin seen in these patients, but perhaps a misnomer as cranio-ocular abnormalities also surface with high frequency, along with various carcinomas. XP can be referred to as Kaposi dermatosis, not to be confused with Kaposi sarcoma seen in immunosuppressed individuals. The heterogeneity in XP genes that span several chromosomes makes the condition markedly complicated to diagnose and treat. Therapeutic options are being created to counter the defective DNA repair pathway. This chapter discusses the clinical features and pathophysiology of xeroderma pigmentosum.
Clinical Characteristics
The prevalence of xeroderma pigmentosum (XP) is the highest in Japan at 1 in 20,000 compared to 1 in a million in the United States [2]. Certain regions in the world carry a higher prevalence, such as Japan, North African countries and the Middle East, where consanguinity might be higher. Cases of XP occur in all races without sexual bias. The disease is usually detected within the first 2 years of life. The skin, the eye and the nervous system could be involved simultaneously or at different stages of the disease. The triad of cognitive deficit, short stature and hypogonadism in children with XP is referred to as De Sanctis-Cacchione syndrome [3].
Cutaneous Involvement
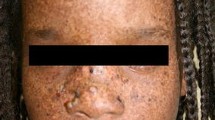
The skin is normal at birth, but the first stages of abnormalities begin at 6 months. Minimal UV exposure leads to profound sunburn reaction with blistering or persistent erythema in half of the individuals. In all children, numerous hyperpigmented patchy freckling (solar lentigines) on the face appears, a sign of unrepaired DNA damage. Without aggressive sunblock usage and UV light avoidance, the skin progressively becomes parched (xerosis), pigmented like salt-and-pepper (Fig. 31.1) and atrophic (poikiloderma) [4]. Telangiectatic skin lesions are also noted. Premalignant keratoses soon develop and convert into basal cell carcinoma, squamous cell carcinoma or melanoma by the age of 8 years. Children with XP have a 10,000-fold increased risk of developing cutaneous cancers than the age-matched population. Skin tumours tend to be multiple, and as many as 100 mixed tumours have been reported. This may result in disfigurement in severely affected subjects.

Hyper- and hypopigmented skin resembling salt-and-pepper appearance. (From Mareddy et al. 2013, with kind permission [4])
Ocular Involvement
Ocular problems arise in nearly 80% of children with XP. Photophobia often exists along with conjunctivitis, vascular pterygia and fibrovascular pannus of the cornea. Symblepharon (a partial or complete adhesion of the palpebral conjunctiva of the eyelid to the bulbar conjunctiva of the eyeball) with ulceration and atrophy of the eyelids results in ectropion, entropion or complete loss of the lids in severe cases. Keratitis, corneal opacification and tumours (squamous cell carcinoma, basal cell carcinoma, sebaceous cell carcinoma, papilloma, fibrosarcoma, melanoma and epithelioma) of the conjunctiva and corneo-conjunctival junction are the major source of ophthalmological morbidity. Patients with XP are at 2000-fold higher risk of developing cancer of the eyes. The ocular features appear as commonly as the cutaneous abnormalities, but may be more severe in black individuals. The retina is shielded by the anterior structures and, therefore, is usually not involved.
Neurological Involvement
Neurological involvement is often part of the phenotypic spectrum, and the symptoms could be quite protean. The presence of progressive neurological deficit and the age of onset correlate with the degree of defect in the DNA repair. Early onset of symptoms in infancy has been observed, or it could be delayed until the second decade. Neurological symptoms (in 20–30% of the cases) are progressive and may result in severe disability. The majority exhibit cognitive impairment, having a median intelligence quotient score of 45. Spasticity, ataxia and acquired microcephaly also exist.
Other neurological abnormalities include choreoathetoid movements, seizures, polyneuropathy with segmental demyelination, sensorineural deafness and supranuclear ophthalmoplegia. The neurological problems may overshadow the cutaneous manifestations in some patients with XP. Rarely, in some patients, dysphagia or vocal cord paralysis may develop during a respiratory infection [5].
Pathogenesis
Early histological findings include hyperkeratosis and increased melanin in the basal cell layer. Atrophic or elongated rete ridges could be observed, along with chronic inflammatory processes in the upper dermis. Later, the skin develops features indistinguishable from actinic keratosis, having atypical and elastic architecture.
UV light naturally depletes Langerhans cells in the epidermis. In normal circumstances, UV radiation induces cross-link photoproducts (dimerization) between thymine nucleotides that require excision and insertion. This repair process, known as nucleotide excision repair (NER), is deficient in XP [8]. Two overlapping pathways for NER have been proposed: the rapid transcription-coupled (TC) repair directed at the transcribed strand and a slower global genome (GG) repair [9].
There are nine specific XP repair genes, eight of which constitute the NER pathway. They include ERCC1 and ERCC2 (XP-D) on the long arm of chromosome 19, ERCC3 (XP-G) on chromosome 2, ERCC4 (XP-F) on 16p, ERCC5 (XP-B) on 13q, XP-A on 9q, DDB2 on 11p, POLH (XP-V) on 6p and XP-C on 3p. The ninth gene bypasses unrepaired damage. The seven complementary groups A through G are defective in both pathways for NER [10]. Complementation refers to different molecular abnormalities in XP that when combined could reverse the DNA repair defect [11, 12]. These entities vary significantly in frequency and in populations. For example, XP-A accounts for up to 40% of all cases in Japan. In the United States, XP-C and XP-D complementation represent 30% and 20% of all XP cases, respectively, whereas XP-A is rare. The XP-G carries a severe form; on the flipside, XP-F is mild.
Neurological problems are seen more commonly in groups XP-A and XP-D [13]. Up to 50% of XP-D patients may show neurological deterioration. The presence of neurological abnormalities correlates with the degree of NER repair defect; patients with greatest impairment of DNA repair are more prone to developing neurodegeneration. Pathologic studies have shown neuronal loss without other histological hallmarks. Diffuse axonal loss with secondary demyelination has been determined in patients with clinical evidence of polyneuropathy [14].
Differential Diagnosis
The differential diagnosis is broad and includes cortical basal ganglionic degeneration, malignant astrocytoma, Hallervorden-Spatz disease, inherited metabolic disorders, multiple sclerosis, multiple system atrophy, olivopontocerebellar atrophy, thyroid disease, Bloom syndrome, LEOPARD syndrome, Osler-Weber-Rendu syndrome, Werner syndrome (progeria) and Hartnup disease. Other conditions also host mutations in the NER pathway, but with different symptoms, such as Cockayne syndrome (see Chap. 30), cerebro-oculo-facio-skeletal syndrome (COFS) and trichothiodystrophy.
Diagnosis
The diagnosis of XP is made clinically based on skin, eye and neurological manifestations. Cranial imaging studies typically reveal ventriculomegaly, along with cortical and brainstem atrophy in most patients, although the white matter is usually preserved. Nerve conduction velocities are reduced and may show axonal (or mixed) polyneuropathy. Audiometry usually reveals early high-tone hearing loss. Laboratory abnormalities are absent in XP patients. Genetic testing is not readily available, which involves cell-fusion techniques, followed by DNA repair analysis or gene sequencing. Amniocentesis or chorionic villi sampling is possible in the third trimester via unscheduled DNA synthesis or the alkaline comet assay [15].
Therapy
Patients with XP need to be monitored every 3–4 months for various dermatological and ophthalmological conditions. Consultation with a geneticist may help to differentiate XP from other related conditions, such as Cockayne syndrome and progeria. Patients must avoid the sun at all times, even fluorescent lights that emit radiation below 320 nm. If sun exposure cannot be entirely avoided, patients should wear full clothing, use sunscreen with SPF 50 or greater and wear dark glasses. A UV light meter can be used at home, in vehicles and classrooms to measure the amount, and window filters applied. Some families reverse their day/night cycle to eliminate sun exposure entirely. Methylcellulose eye drops or soft contact lenses have been used to keep the cornea moist and to protect against mechanical trauma in individuals with deformed eyelids.
Premalignant lesions such as actinic keratoses may be treated with topical 5-fluorouracil or by freezing with liquid nitrogen. Cutaneous neoplasms are treated in the same manner as in individuals who do not have XP. This involves electro-desiccation and curettage, surgical excision or chemosurgery. Oral isotretinoin at 2 mg/kg/d is rendered only to those with multiple skin cancers. It may produce hepatotoxicity and dose-related irreversible calcification of ligaments and tendons [16]. The combination of 5% imiquimod cream and oral acitretin in a few cases of XP-related cutaneous neoplasms has shown some promise anecdotally [17, 18].
Neurological care is mostly supportive. Seizures can be treated like other complex partial seizures. Spasticity is usually mild. If it interferes with mobility, baclofen or botulinum neurotoxin could be beneficial.
New approaches are being investigated and utilized to engineer mega-nucleases, zinc-finger nucleases or TALE nucleases to accurately generate a double-strand break at a specific locus and to insert an exogenous DNA repair matrix [19].
Prognosis
Fewer than 40% of patients survive beyond the age of 20 years. Individuals with milder disease may survive beyond middle ages. Neoplasms are usually the chief cause of death. Many patients become bedridden and incontinent. Some develop significant cachexia in the terminal stages despite adequate caloric intake.
Bibliography
Hebra F, Kaposi M. On diseases of the skin including exanthemata. New Sydenham Soc. 1874;61:252–8.
Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum: cutaneous, ocular and neurological abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50.
De Sanctis C. Cacchione: a L’idiozia xerodermica. Riv Sper Freniatr. 1932;56:269–92.
Mareddy S, Reddy J, Babu S, Balan P. Xeroderma pigmentosum: man deprived of his right to light. Sci World J. 2013;2013:534752.
Ohto T, Iwasaki N, Okubo H, et al. Life-threatening vocal cord paralysis in a patient with group a xeroderma pigmentosum. Pediatr Neurol. 2004;30:222–4.
Benhamou S, Sarasin A. ERCC2/XPD gene polymorphisms and lung cancer: a HuGE review. Am J Epidemiol. 2005;161:1–14.
Hu Z, Wei Q, Wang X, Shen H. DNA repair gene XPD polymorphism and lung cancer risk: a meta-analysis. Lung Cancer. 2004;46:1–10.
Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5:a012609.
Park CJ, Choi BS. The protein shuffle. Sequential interactions among components of the human nucleotide excision repair pathway. FEBS J. 2006;273:1600–8.
Lehmann J, Schubert S, Schafer A, et al. An unusual mutation in the XPG gene leads to an internal in-frame deletion and a XP/CS complex phenotype. Br J Dermatol. 2014;171:903–5.
DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–96.
Black JO. Xeroderma pigmentosum. Head Neck Pathol. 2016;10:139–44.
Lai JP, Liu YC, Alimchandani M, et al. The influence of DNA repair on neurological degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D). Acta Neuropathol Commun. 2013;1:4.
Rapin I, Lindenbaum Y, Dickson DW, et al. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–9.
Alapetite C, Benoit A, Moustacchi E, Sarasin A. The comet assay as a repair test for prenatal diagnosis of Xeroderma pigmentosum and trichothiodystrophy. J Invest Dermatol. 1997;108:154–9.
Roseeuw D. The treatment of basal skin carcinomas in two sisters with xeroderma pigmentosum. Clin Exp Dermatol. 2003;28(Suppl 1):30–2.
Giannotti B, Vanzi L, Difonzo EM, Pimpinelli N. The treatment of basal cell carcinomas in a patient with xeroderma pigmentosum with a combination of imiquimod 5% cream and oral acitretin. Clin Exp Dermatol. 2003;28(Suppl 1):33–5.
Nagore E, Sevila A, Sanmartin O, et al. Excellent response of basal cell carcinomas and pigmentary changes in xeroderma pigmentosum to imiquimod 5% cream. Br J Dermatol. 2003;149:858–61.
Dupuy A, Valton J, Leduc S, et al. Targeted gene therapy of xeroderma pigmentosum cells using mega-nuclease and TALEN™. PLoS One. 2013;8:e78678.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Benjamin, R. (2022). Xeroderma Pigmentosum (Kaposi Dermatosis). In: Panteliadis, C.P., Benjamin, R., Hagel, C. (eds) Neurocutaneous Disorders. Springer, Cham. https://doi.org/10.1007/978-3-030-87893-1_31
Download citation
DOI: https://doi.org/10.1007/978-3-030-87893-1_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-87892-4
Online ISBN: 978-3-030-87893-1
eBook Packages: MedicineMedicine (R0)