
Abstract
In recent years, molecular tools and techniques became popular to analyse and characterize the microorganisms involved in environmental clean-up. Further, high throughput genome sequencing methods and advanced protein-based technologies have completely revolutionized the era. The present chapter discusses some of the genomic and proteomic technologies that can be explored for analysing microbial diversity and community structure in a polluted ecosystem. However, continuous efforts are needed to increase their sensitivity, reproducibility and wide sample coverage along with the cost-effectivity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

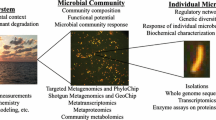

Keywords
18.1 Introduction
Since the earliest identification of living beings or microbes, researchers have been developing systematic categorization methods in the field of evolution and phylogeny. This gets more problematic in the context of bacteria, the most common kind of microbe. Bacteria reproduce asexually, which explains the traditional concept of species as a collection of organisms capable of interbreeding and procreating fertile offspring is not universally applicable. Additionally, bacteria’s tiny size contributes to their restricted variety of morphological features. Bacteria show a broad variety of biochemical variation in terms of cell structure and metabolism, and although this provides some background knowledge about their taxonomy, it is far from comprehensive. With the advent of molecular biology, a new revolution has occurred, and this new revolution has made major contributions to bacterial taxonomy and systematics, as well as to other fields of biological taxonomy.
In the 1970s, Carl Woese proposed a classification system based on the molecular comparisons of evolutionarily conserved ribosomal genes and segregated two domains, Bacteria and Archaea as they are different from the Eukaryotes (contains all the higher forms of the organism) (Woese 1987). Now the ribosomal based classification system is widely accepted by the microbiologist across the world. Still the bacterial systematic is still evolving and also the standardized concept of bacterial species constituents (Berg et al. 2020). However, molecular-based systematics has given a strong outline for designing classification schemes.
Despite the lack of a clear logical and precise definition of species, traditional techniques continue to be used in a wide variety of fields or sectors. However, modern molecular methods (genomics and proteomics) provide superior characterisation than traditional techniques. They rapidly produce multidirectional information on both microbial communities and their taxonomic relationships.
In most cases, bacteria can only be identified using restricted approaches that rely on genetic techniques that use microorganism genetic profiling and phenotypic techniques that use metabolic characteristics and chemical composition of the organism to identify the microbes in question. The benefits of genotypic methods above phenotypic techniques are not influenced by the physiological condition, medium composition, or growth phase of the organisms. The phenotypic techniques, on the other hand, revealed the functional features of organisms, such as metabolic processes, that are required for their survival, growth, and development.
18.2 Genomic Methods
These methods are based on the analysis of the genome that is represented by the haploid set of genes or chromosomes within an organism. It may be classified as structural genomics and functional genomics (Wang et al. 2020; Raghu et al. 2021; Soni et al. 2021). Structural genotyping involves the gene location, sequence, and physical characterization; while, functional genotyping involves gene regulation and protein expression (Soni et al. 2016; Suyal et al. 2019a). Moreover, the combinations of various “meta-” and “-omics” technologies have made it beneficial to humankind especially in environmental, medical, industrial, and agricultural fields (Suyal et al. 2015a, b, 2019c).
For the identification, genotypic methods are classified into two distinctive categories: (1) pattern- or fingerprint-based techniques and (2) sequence-based techniques. Pattern-based methods make use of a systematic process that generates a sequence of fragments from the chromosomal DNA of the organism being studied. The fragments are then segregated based on the size which generates a profile (or fingerprint) unique to that organism and its close relatives. Then, using the information gathered, researchers can create a database of the particular organism (fingerprint), which can then be used as a reference for the test organisms to compare against (Emerson et al. 2008). If two profiles of different organisms’ match, they can be viewed as a close relative of each other, particularly at the level of strain or species. On the other hand, sequence-based techniques usually depend on the specific stretch of DNA or chromosome but do not always like the specific gene. In general, the approach is similar to the genotyping method: a specific sequence of DNA database created and then the test organism’s sequences compared with it. The degree of homology or similarity or matched sequences between the compared organisms is an estimate of how closely linked the organisms are among the compared organisms. Several computer-based algorithms have been created to build the phylogenetic tree in which we can compare the multiple sequences of different organisms to one another at a time. Thus, by making use of sequence comparisons of ribosomal RNA (rRNA) gene, archaea and bacteria can be easily distinguished as separate branches or having different relationships among the microorganisms (Raina et al. 2019). Both the techniques discussed above have merits and demerits. Conventionally, for the establishment of phylogenetic relationship among the bacteria at phylum, order, family, genus level, 16S rRNA gene sequence was analysed whereas for the establishment of relatedness at the species level or genus level the fingerprinting-based methods are good but less dependable above those levels (Vandamme et al. 1996). Fingerprinting and sequence-based methods combined with phenotypic characters is called polyphasic technique, is the standard approach nowadays to describe a new species or genus (Carro and Nouioui 2017).
18.3 Specific Genotyping Methodologies
The current techniques for characterisation may make use of a variety of fingerprinting or sequence-based methods, which may be employed either individually or in combination. These methods are continuously evolving and improving in terms of accuracy. Some of the most frequently used methods are listed below.
18.3.1 Fingerprinting-Based Methodologies
Among the genotypic methods, fingerprinting techniques are the most widely used presently. Techniques like Amplified fragment length polymorphism (AFLP), repetitive element PCR (rep-PCR), and random amplification of polymorphic DNA, utilize PCR for amplification of desired short DNA fragments by using specific primer sets (Sharma et al. 2020). These methods use the advantages of polymorphism in the DNA of the concerned organism which might be formed from the evolutionary process. A unique set of primer is used for more than one organism in the multiplex PCR; based on the molecular weight of amplicon (size) these sets can be separated through electrophoresis. They enable the fast identification of many microorganisms from a single sample combination (Settanni and Corsetti 2007).
Riboprinting utilizes sensitive probes instead of PCR to detect the difference in gene sequence or pattern between species and strain (Bruce 1996). It is one of several molecular methods that generates comparative data which is independent of the complexity of the morphology of the organisms. Diversi Lab system for rep-PCR (http://biomerieux-usa.com/diversilab) (Dou et al. 2015) and DuPont’s Ribo-Printer system (www2.dupont.com/Qualicon/en_US/) (Shintani 2013) have been exclusively developed commercial products bacterial identification. All the techniques discussed here are already mentioned in many kinds of literature as identification methods. These applications include source tracing, authentication of bacterial isolates for archiving reasons, taxonomy and systematics, as well as the identification of microbial population patterns, among other things.
18.3.2 Sequence-Based Methodologies
As the housekeeping genes are conserved and present universally in all the cells, the primer can be designed for the amplification of similar genes across the multiple genera. Multilocus sequencing (MLS) is a promising method developed to identify microbiological species. The principle is almost similar to 16S rRNA gene sequencing, but fragments of multiple “housekeeping” genes are sequenced. Later the combined sequences are put into one long sequence which is then compared with other sequences.
Since designing universal sets of primers is not possible, designing specific sets of primers for families or orders is a good concept. Two multilocus sequencing approaches used are multilocus sequence typing (MLST) and multilocus sequence analysis (MLSA). In MLST, a set of primers are used according to 6–10 genetic loci which allow the PCR amplification (amplicon size 400–600 bp). The concatenated sequences are then compared with the existing sequence database for the same organism. The result exhibits a very strong identification of a particular strain and showed a very close evolutionary relationship (Huebner et al. 2021). Among other things, this method may be used to monitor the spread of a disease and to demonstrate their usefulness in epidemiological research (Pérez-Losada et al. 2011). On the other hand, MLSA involves sequencing of multiple fragments of conserved protein-encoding genes, but with the more ad-hoc approach for gene selection for comparative analysis as it uses identification using a small subset of genes or loci (Glaeser and Kämpfer 2015). It identifies the organisms and finds relationships of species within genera of families in detail. One of the major limitations of this approach is the lack of standardization and central databases. Recently, several studies conducted with MLSA showed that instead of using a single common gene, different sets of genes were used for the identification of various bacterial phyla (Glaeser and Kämpfer 2015; Palmer et al. 2018). Hence comparative analysis is impossible with this technique.
18.4 The Genomic Future
Whole-genome comparisons have proven to be more accurate and precise than DNA-DNA hybridization and has gained more popularity over the phenotypic traits concept for bacterial classification and identification. At the moment, the notion of “species” is defined by digital whole genome comparisons utilising average nucleotide identities (ANIs) or genome-to-genome distance computations (GGDCs). Since the advent of whole genome sequencing, phylogenomics has made significant contributions to the field of contemporary taxonomy (Lalucat et al. 2020). Complete genome comparisons identify a species at the genomic level based on 95% average nucleotide identity between two related strains (Olm et al. 2020). The advanced technology of next-generation sequencing has provided more rapid, economical, and easily available sequence-based methods for the identification and classification of bacteria at all levels. Another promising approach for the identification and characterization of microbes at the community level is Microarray. It works by probing several genes on a substrate (example: glass, silicon, nylon, etc.) and further hybridizing with DNA or RNA samples (Solieri et al. 2013). For rapid detection of hybridized samples with probes, fluorescent reporter molecules are used as markers on the microarray. In addition, use of microarray is of great importance for medical purposes such as disease diagnosis and pathogen identification (Herrera-Rodriguez et al. 2013). Some modifications such as phylochips are used to identify specific or various groups of bacteria directly from the environment samples and geochips for the identification of microbes responsible for biogeochemical processes (Liu et al. 2021).
18.5 Proteomics Technologies in Bacterial Identification and Characterization
Genotypic and phenotypic methods are not enough to understand the physiological and functional activities of an organism at the protein level. Proteomics a new approach is a rapid way to explore biomolecules and understand their activity. It is based on mass spectrometry and provides an integrative study of genotypic and proteomic data with all the vital information (Suyal et al. 2018, 2019b). Several of the most widely used technologies include electrospray ionisation mass spectrometry (ESI-MS), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS), one- or two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and surface-enhanced laser desorption/ionization (SELDI) mass spectrometry, etc.
18.5.1 Mass Spectrometry-Based Bacterial Characterization and Identification
Thomson has invented mass spectrometry to determine the mass to charge ratio of electrons in the late nineteenth century. The method is used to identify, quantify, and deduce the structure of a wide range of molecules (Baghel et al. 2017). The twentieth century saw an expansion of technology and its applications to chemical characterization, physical measurement, and biological identification.
Some soft ionization methods in mass spectrometry such as ESI-MS and MALDI-TOF-MS have made it easier to analyze larger molecules. It allows direct use of samples in their native form for interrogation (Fenn et al. 1989). MS has shown better outcomes than traditional approaches: in resolving the time constraint and generating protein profiles. Applications of these abovementioned techniques for identification and characterization are described below:
18.5.1.1 Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry
The mass spectral method gives: detailed overview of whole bacterial cells, spectrum patterns over a broad mass range, and identification and characterization are done by comparing with reference data. Initially, application of MALDI-TOF-MS in rapid identification of whole bacteria was shown by Holland et al. (1996) after which various strains such as Mycobacteria sp. (Pignone et al. 2006), Staphylococcus sp. (Edwards-Jones et al. 2000), and extremophilic bacteria and archaea (Krader and Emerson 2004) have been analyzed using the same.
One of the most famous examples was during the first outbreak of methicillin-resistant S. aureus (MRSA) in European Hospitals (1960). The threat of spreading resistant S. aureus urgently required some rapid identification method. Edward-Jones et al. (2000) developed matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for the same purpose as well as for the differentiation of methicillin-sensitive Staphylococcus aureus (MSSA) from methicillin-resistant Staphylococcus aureus (MRSA). The procedure involves smear preparation of a single bacterial colony on slide followed by applying matrix which is then observed using MALDI-TOF-MS. The analysis shows distinct spectral peaks for MRSA and MSSA. Based on this technique, several other instruments such as Bruker Daltonics’ MALDI BioTyper equipped with bioinformatics tools were developed. It also serves the same function by targeting some ribosomal proteins and proteins found in high amounts (Mellmann et al. 2008).
18.5.1.2 Electrospray Ionization Mass Spectrometry
ESI-MS is a potential approach for the characterization and analysis of various cellular components in microbes. It is considered more accurate in protein identification than MALDI-TOF-MS (as it is based on only molecular weight). ESI-MS uses peptide fragmentation fingerprints to search in the database and identifies the specific protein. The fingerprint is obtained by tandem mass spectrometry, in which target protein can be fragmented for second mass analysis. Modified approach, developed by integrating PCR with ESI-MS introduced Ibis Biosciences (the T5000 Biosensor System) for identification and characterization of bacteria (Sampath et al. 2007). Few major advantages associated with ESI-MS are rapid and fast process (Banerjee and Mazumdar 2012) provides specific identification of target bacteria in mixed culture; high resolution; and identification of virulence factors (Ho and Reddy 2011).
18.5.1.3 Surface-Enhanced Laser Desorption/Ionization
SELDI is a relatively new technique that separates proteins based on their binding affinity to a chip surface. Chemically and biologically modified chips are used for mass spectrometric analysis of complex protein mixtures. SELDI-MS generates a unique spectra pattern for proteins in the mixture based on their mass-to-charge ratio (You et al. 2013). Furthermore, different proteins can be identified from these profiles by comparing the respective peak intensities (Lu et al. 2010). Lundquist et al. (2005) demonstrated that SELDI-TOF-MS is one of the potential methods to produce distinct and reproducible protein profiles for the identification and discrimination of different species. For example, it made it possible to identify and distinguish the most infectious subspecies of Francisella tularensis out of four, the only subspecies found in North America causing tularemia in humans. This technique is used in the identification and characterization of bacteria, exploring bacterial proteomes, pathogen detection (Ho and Reddy 2011; Ardito et al. 2016), virulence factor identification, biomarker, and protein profiling in oncology (Langbein et al. 2006; Liu 2011), etc.
Although mass spectrometry plays a great role in the identification and characterization of bacteria by generating spectral patterns various factors cause difficulty in the reproducibility of protein profiles. Factors associated are physiological state of cell (García-Flores et al. 2012), growth medium of the cell (Wieme et al. 2014), sample preparation, the difference in instrument quality, and matrix selection (Wunschel et al. 2005; Vats et al. 2016). Scientists resolved this issue by introducing standard techniques for MALDI-TOF-MS of whole cells (Strejcek et al. 2018).
18.5.2 Gel-Based Method
SDS-PAGE is a widely used method for differentiating bacteria based on their protein contents. It separates the entire protein complement based on their charge and molecular weight. The difference in mobility of charged molecules leads to different migration patterns of proteins. This unique pattern helps to differentiate and characterize the variety of bacterial strains. It is considered promising fractionation technique and provides good resolution for proteins based on sizes, isoelectric points, and hydrophobic behaviour (Carruthers et al. 2015). The drawback of this approach is that it is time-consuming and tedious.
18.5.2.1 Two-Dimensional Gel Electrophoresis (2DE)
Combining SDS-PAGE with isoelectric focusing (IEF)—lead to the development of a new high-resolution technique named 2DE discovered by O’Farrell in 1975. It is capable of separating complicated protein mixtures in a single gel analysis. Two-dimensional gel electrophoresis begins with initial segregation on the basis of pH gradient associated with the isoelectric point of the proteins in the first dimension, followed by SDS-PAGE separation in the second dimension. Further staining of a gel with standard staining solutions for visualization of protein spots and analysis of protein gel patterns or 2DE maps (Soni et al. 2015; Kendrick et al. 2019). These patterns can be studied further and stored in reference databases for future use. It is often used for isolating and analyzing target protein from complex protein mixtures, and identification of unknown species by comparing differential expression 2DE maps, with a reference database. To obtain more efficient and complex proteome analysis 2DE is merged with mass spectrometry. Numerous reports demonstrated that this combined approach can be used to study the entire proteome or subproteome of a variety of species, including the exosporium of Bacillus anthracis spores (Redmond et al. 2004), Bacillus subtilis, Helicobacter pylori, E. coli, Pseudomonas aeruginosa, and Staphylococcus aureus (Hecker et al. 2003; Peng et al. 2005; Pieper et al. 2006). Databases with complete information of 2DE maps and mass spectra of known bacteria will provide rapid identification and efficient comparative study of unknown bacteria (Curreem et al. 2012). However, building such a database is generally a very tedious job.
18.6 Databases
To generate, archive, process and integrate large data sets of many samples with robust quality is a real challenge for both the (genomic and proteomic) approaches. There are a variety of databases and tools available which provide integrated data for the particular type of analysis. For genomic analysis databases based on 16SrRNA genes include green genes (DeSantis et al. 2006) and Ribosomal Database Project (Cole et al. 2009). On the other side, in-depth data analysis of proteomics has been carried out with tools like GlycoMod and databases such as Phospho Site (Gasteiger et al. 2003). Moreover, new algorithms have been developed that adapt to actual experimental phenomenon and parameters (Lees et al. 2016; San et al. 2020). Wilke et al. (2003) introduced the ProDB platform which provided enriched protein profile along with experimental set-up and parameters, like growth and culture conditions to check impact generation on mass-spectra profile.
18.7 Conclusion
The use of molecular technologies is at the core of the identification and characterisation of microorganisms. However, there are certain problems that need to be addressed, such as the functional knowledge of related instruments, their mobility, cost-effectiveness, and accessibility, among others. It will undoubtedly inspire students, researchers, and the scientific community to use a variety of technologies in order to achieve environmental sustainability.
References
Ardito F, Perrone D, Cocchi R, Lo Russo L, De Lillo A, Giannatempo G, Lo Muzio L (2016) Novel possibilities in the study of the salivary proteomic profile using SELDI-TOF/MS technology. Oncol Lett 11(3):1967–1972
Baghel US, Singh A, Singh D, Sinha M (2017) Application of mass spectroscopy in pharmaceutical and biomedical analysis. In: Spectroscopic analyses: developments and applications. IntechOpen, London, p 105
Banerjee S, Mazumdar S (2012) Electrospray ionization mass spectrometry: a technique to access the information beyond the molecular weight of the analyte. Int J Anal Chem 2012:282574
Berg G, Rybakova D, Fischer D, Cernava T, Vergès MCC, Charles T et al (2020) Microbiome definition re-visited: old concepts and new challenges. Microbiome 8(1):1–22
Bruce J (1996) Automated system rapidly identifies and characterizes microorganisms in food. Food Technol (Chicago) 50(1):77–81
Carro L, Nouioui I (2017) Taxonomy and systematics of plant probiotic bacteria in the genomic era. AIMS Microbiol 3(3):383
Carruthers NJ, Parker GC, Gratsch T, Caruso JA, Stemmer PM (2015) Protein mobility shifts contribute to gel electrophoresis liquid chromatography analysis. J Biomol Tech 26(3):103
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37(suppl_1):D141–D145
Curreem SO, Watt RM, Lau SK, Woo PC (2012) Two-dimensional gel electrophoresis in bacterial proteomics. Protein Cell 3(5):346–363
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Dou H, Xu Y, Li T (2015) Application of the DiversiLab system for tracing the source of the mixed infections caused by Cryptococcus neoformans var. grubii from a patient with systemic lupus erythematosus. Mycoses 58(3):149–159
Edwards-Jones V, Claydon MA, Evason DJ, Walker J, Fox AJ, Gordon DB (2000) Rapid discrimination between methicillin-sensitive and methicillin-resistant Staphylococcus aureus by intact cell mass spectrometry. J Med Microbiol 49(3):295–300
Emerson D, Agulto L, Liu H, Liu L (2008) Identifying and characterizing bacteria in an era of genomics and proteomics. Bioscience 58(10):925–936
Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM (1989) Electrospray ionization for mass spectrometry of large biomolecules. Science 246(4926):64–71
García-Flores M, Juárez-Colunga S, Montero-Vargas JM, López-Arciniega JAI, Chagolla A, Tiessen A, Winkler R (2012) Evaluating the physiological state of maize (Zea mays L.) plants by direct-injection electrospray mass spectrometry (DIESI-MS). Mol BioSyst 8(6):1658–1660
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31(13):3784–3788
Glaeser SP, Kämpfer P (2015) Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Syst Appl Microbiol 38(4):237–245
Hecker M, Engelmann S, Cordwell SJ (2003) Proteomics of Staphylococcus aureus—current state and future challenges. J Chromatogr B 787(1):179–195
Herrera-Rodriguez SE, Elizondo-Quiroga D, Alvarez-Maya I (2013) Infectious diseases detection by microarray: an overview of clinical relevant infections. J Biomed Sci Eng 2013
Ho YP, Reddy PM (2011) Advances in mass spectrometry for the identification of pathogens. Mass Spectrom Rev 30(6):1203–1224
Holland RD, Wilkes JG, Rafii F, Sutherland JB, Persons CC, Voorhees KJ, Lay JO Jr (1996) Rapid identification of intact whole bacteria based on spectral patterns using matrix-assisted laser desorption/ionization with time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 10(10):1227–1232
Huebner R, Mugabi R, Hetesy G, Fox L, De Vliegher S, De Visscher A et al (2021) Characterization of genetic diversity and population structure within Staphylococcus chromogenes by multilocus sequence typing. PLoS One 16(3):e0243688
Kendrick N, Darie CC, Hoelter M, Powers G, Johansen J (2019) 2D SDS PAGE in combination with Western blotting and mass spectrometry is a robust method for protein analysis with many applications. In: Advancements of mass spectrometry in biomedical research. Springer, Cham, pp 563–574
Krader P, Emerson D (2004) Identification of archaea and some extremophilic bacteria using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. Extremophiles 8(4):259–268
Lalucat J, Mulet M, Gomila M, García-Valdés E (2020) Genomics in bacterial taxonomy: impact on the genus Pseudomonas. Genes 11(2):139
Langbein S, Lehmann J, Harder A, Steidler A, Michel MS, Alken P, Badawi JK (2006) Protein profiling of bladder cancer using the 2D-PAGE and SELDI-TOF-MS technique. Technol Cancer Res Treat 5(1):67–71
Lees JA, Vehkala M, Välimäki N, Harris SR, Chewapreecha C, Croucher NJ et al (2016) Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat Commun 7(1):1–8
Liu C (2011) The application of SELDI-TOF-MS in clinical diagnosis of cancers. J Biomed Biotechnol 2011
Liu D, He X, Chater CC, Perez-Moreno J, Yu F (2021) Microbiome community structure and functional gene partitioning in different micro-niches within a sporocarp-forming fungus. Front Microbiol 12:629352
Lu HB, Zhou JH, Ma YY, Lu HL, Tang YL, Zhang QY, Zhao CH (2010) Five serum proteins identified using SELDI-TOF-MS as potential biomarkers of gastric cancer. Jpn J Clin Oncol 40(4):336–342
Lundquist M, Caspersen MB, Wikström P, Forsman M (2005) Discrimination of Francisella tularensis subspecies using surface enhanced laser desorption ionization mass spectrometry and multivariate data analysis. FEMS Microbiol Lett 243(1):303–310
Mellmann A, Cloud J, Maier T, Keckevoet U, Ramminger I, Iwen P et al (2008) Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J Clin Microbiol 46(6):1946–1954
O’Farrell PH (1975) High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250(10):4007–4021
Olm MR, Crits-Christoph A, Diamond S, Lavy A, Matheus Carnevali PB, Banfield JF (2020) Consistent metagenome-derived metrics verify and delineate bacterial species boundaries. Msystems 5(1):e00731–e00719
Palmer M, Steenkamp ET, Coetzee M, Blom J, Venter SN (2018) Genome-based characterization of biological processes that differentiate closely related bacteria. Front Microbiol 9:113
Peng X, Xu C, Ren H, Lin X, Wu L, Wang S (2005) Proteomic analysis of the sarcosine-insoluble outer membrane fraction of Pseudomonas aeruginosa responding to ampicilin, kanamycin, and tetracycline resistance. J Proteome Res 4(6):2257–2265
Pérez-Losada M, Porter ML, Viscidi RP, Crandall KA (2011) Multilocus sequence typing of pathogens. In: Genetics and evolution of infectious disease. Elsevier, London, pp 503–521
Pieper R, Gatlin-Bunai CL, Mongodin EF, Parmar PP, Huang ST, Clark DJ et al (2006) Comparative proteomic analysis of Staphylococcus aureus strains with differences in resistance to the cell wall-targeting antibiotic vancomycin. Proteomics 6(15):4246–4258
Pignone M, Greth KM, Cooper J, Emerson D, Tang J (2006) Identification of mycobacteria by matrix-assisted laser desorption ionization-time-of-flight mass spectrometry. J Clin Microbiol 44(6):1963–1970
Raghu S, Kumar S, Suyal DC, Sahu B, Kumar V, Soni R (2021) Molecular tools to explore rhizosphere microbiome. In: Nath M, Bhatt D, Bhargava P, Chaudhary DK (eds) Microbial metatranscriptomics belowground. Singapore, Springer Nature, pp 37–57. https://doi.org/10.1007/978-981-15-9758-9_2
Raina V, Nayak T, Ray L, Kumari K, Suar M (2019) A polyphasic taxonomic approach for designation and description of novel microbial species. In: Microbial diversity in the genomic era. Academic Press, London, pp 137–152
Redmond C, Baillie LW, Hibbs S, Moir AJ, Moir A (2004) Identification of proteins in the exosporium of Bacillus anthracis. Microbiology 150(2):355–363
Sampath R, Hall TA, Massire C, Li F, Blyn LB, Eshoo MW et al (2007) Rapid identification of emerging infectious agents using PCR and electrospray ionization mass spectrometry. Ann N Y Acad Sci 1102(1):109
San JE, Baichoo S, Kanzi A, Moosa Y, Lessells R, Fonseca V et al (2020) Current affairs of microbial genome-wide association studies: approaches, bottlenecks and analytical pitfalls. Front Microbiol 10:3119
Settanni L, Corsetti A (2007) The use of multiplex PCR to detect and differentiate food-and beverage-associated microorganisms: a review. J Microbiol Methods 69(1):1–22
Sharma A, Lee S, Park YS (2020) Molecular typing tools for identifying and characterizing lactic acid bacteria: a review. Food Sci Biotechnol 29(10):1301–1318
Shintani H (2013) Rapid assay of airborne microorganisms and bioburden using several procedures. Pharm Anal Acta 4(6):1–12
Solieri L, Dakal TC, Giudici P (2013) Next-generation sequencing and its potential impact on food microbial genomics. Ann Microbiol 63(1):21–37
Soni R, Suyal DC, Agrawal K, Yadav A, Souche Y, Goel R (2015) Differential proteomic analysis of Himalayan psychrotolerant diazotroph Pseudomonas palleroniana N26 Strain under low temperature diazotrophic conditions. CryoLetters 36(2):74–82
Soni R, Suyal DC, Sai S, Goel R (2016) Exploration of nifH gene through soil metagenomes of the western Indian Himalayas. 3 Biotech 6(1):25
Soni R, Suyal DC, Sahu B, Phulara SC (2021) Metagenomics: an approach to unravel the plant microbiome and its function. In: Verma A, Saini JK, Hesham AEL, Singh HB (eds) Phytomicrobiome interactions and sustainable agriculture. Wiley-Blackwell, Hoboken, pp 32–44
Strejcek M, Smrhova T, Junkova P, Uhlik O (2018) Whole-cell MALDI-TOF MS versus 16S rRNA gene analysis for identification and dereplication of recurrent bacterial isolates. Front Microbiol 9:1294
Suyal DC, Yadav A, Shouche Y, Goel R (2015a) Diversified diazotrophs associated with the rhizosphere of Western Indian Himalayan native red kidney beans (Phaseolus vulgaris L.). 3 Biotech 5(4):433–441
Suyal DC, Yadav A, Shouche Y, Goel R (2015b) Bacterial diversity and community structure of Western Indian Himalayan red kidney bean (Phaseolus vulgaris) rhizosphere as revealed by 16S rRNA gene sequences. Biologia 70(3):305–313
Suyal DC, Kumar S, Joshi D, Soni R, Goel R (2018) Quantitative proteomics of psychotrophic diazotroph in response to nitrogen deficiency and cold stress. J Proteome 187:235–242
Suyal DC, Joshi D, Debbarma P, Soni R, Das B, Goel R (2019a) Soil metagenomics: unculturable microbial diversity and its function. In: Mycorrhizosphere and Pedogenesis. Springer, Singapore, pp 355–362
Suyal DC, Joshi D, Kumar S, Soni R, Goel R (2019b) Differential protein profiling of soil diazotroph Rhodococcus qingshengii S10107 towards low-temperature and nitrogen deficiency. Sci Rep 9(1):1–9
Suyal DC, Kumar S, Joshi D, Yadav A, Shouche Y, Goel R (2019c) Comparative overview of red kidney bean (Phaseolus vulgaris) rhizospheric bacterial diversity in perspective of altitudinal variations. Biologia 74(10):1405–1413
Vandamme P, Pot B, Gillis M, De Vos P, Kersters K, Swings J (1996) Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol Rev 60(2):407–438
Vats P, Verma SM, Monif T (2016) Mechanistic evaluation of matrix effect on three different model of mass spectrometer by using a model drug. J Anal Bioanal Tech 7:314
Wang D, Li F, Cao S, Zhang K (2020) Genomic and functional genomics analyses of gluten proteins and prospect for simultaneous improvement of end-use and health-related traits in wheat. Theor Appl Genet 133(5):1521–1539
Wieme AD, Spitaels F, Aerts M, De Bruyne K, Van Landschoot A, Vandamme P (2014) Effects of growth medium on matrix-assisted laser desorption–ionization time of flight mass spectra: a case study of acetic acid bacteria. Appl Environ Microbiol 80(4):1528–1538
Wilke A, Rückert C, Bartels D, Dondrup M, Goesmann A, Hüser AT et al (2003) Bioinformatics support for high-throughput proteomics. J Biotechnol 106(2–3):147–156
Woese CR (1987) Bacterial evolution. Microbiol Rev 51(2):221–271
Wunschel SC, Jarman KH, Petersen CE, Valentine NB, Wahl KL, Schauki D et al (2005) Bacterial analysis by MALDI-TOF mass spectrometry: an inter-laboratory comparison. J Am Soc Mass Spectrom 16(4):456–462
You J, Willcox MD, Madigan MC, Wasinger V, Schiller B, Walsh BJ et al (2013) Tear fluid protein biomarkers. Adv Clin Chem 62:151–196
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gangola, S. et al. (2022). Advanced Molecular Technologies for Environmental Restoration and Sustainability. In: Suyal, D.C., Soni, R. (eds) Bioremediation of Environmental Pollutants. Springer, Cham. https://doi.org/10.1007/978-3-030-86169-8_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-86169-8_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86168-1
Online ISBN: 978-3-030-86169-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)