
Abstract
A decade of genomic analyses of acute lymphoblastic leukemia (ALL), the commonest childhood cancer, have transformed our understanding of the genetic basis of B-progenitor ALL. These studies have identified multiple new subtypes of B-ALL and have shown that many of these new subtypes are driven by diverse genetic alterations in addition to those previously characterized by aneuploidy or a single fusion oncoprotein. These include subtypes driven by multiple fusions converging on a single gene, phenocopies of fusions, and sequence mutations. Large-scale RNA sequencing has shown convergence of genetic alterations on distinct transcriptional signatures characteristic of most subtypes of ALL and has shown the potential for this modality to identify the causal aneuploidy, rearrangements and sequence mutations that define most subtypes. Genomic analyses have also identified coding and non-coding germline variants that predispose to familial and sporadic ALL, the constellations of genetic changes that define each subtype, and the relationship between genetic variegation, clonal diversity and relapse in ALL. Many of these genetic alterations are of clinical importance as they refine diagnosis and risk stratification or serve as therapeutic targets in ALL.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Acute lymphoblastic leukemia
- ALL
- B-ALL
- DUX4
- ETV6-RUNX1
- Hyperdiploid
- Hypodiploid
- KMT2A
- TCF3
- IKZF1
- MEF2D
- NUTM1
- PAX5
- Ph-like
- ZNF384
- Relapse
Introduction
Acute lymphoblastic leukemia (ALL) is a neoplasm of B- or T-lineage lymphoid progenitors, with B-cell precursor ALL (BCP-ALL) representing the more common lineage of disease in both children and adults. BCP-ALL comprises over 20 subtypes characterized by constellations of genetic alterations, including aneuploidy, chromosomal rearrangements, DNA copy number alterations, and sequence mutations and, typically, distinct gene expression profiles [1,2,3]. As described in this review, the subtypes of B-ALL show variability in the nature of the initiating lesion (e.g., single or multiple chromosomal rearrangements, sequence mutations, or aneuploidy), secondary genetic alterations, and outcome. The prevalence and prognosis of each subtype is age dependent. Moreover, there is growing appreciation of the role of germline coding and non-coding variants in predisposing to ALL, both in familial and sporadic cases, and, in some instances, predisposing to specific subtypes of ALL, a striking example being germline TP53 alterations and low hypodiploid ALL [4]. In the majority of subtypes of B-ALL, secondary genomic alterations are important events required for leukemogenesis, and also influence the risk of relapse [5, 6] (Fig. 1.1). Indeed, it is now recognized that in the majority of cases of B-ALL, the disease is usually polyclonal at the time of diagnosis, and when relapse occurs, there is substantial genomic evolution with clonal rise and fall and mutational extinction, convergence, and emergence [7,8,9]. Herein, we review the genomic landscape of BCP-ALL, including discussion of the role of germline predisposition and the genetics of clonal evolution and relapse. This review will emphasize illustrative examples of recently defined subtypes of ALL and highlight potential avenues for diagnostic implementation and therapeutic targeting of relapsed ALL with an emphasis on newly described entities and targets during the past decade.

Schema of the temporal pathogenesis of BCP-ALL
Historic Aspects of Genetic and Genomic Classification of B-ALL
For many years, genetic classification of B-ALL was performed by cytogenetic karyotyping and complementary targeted fluorescence in situ hybridization (FISH) and molecular assays for specific chromosomal rearrangements and fusions [10]. These identified aneuploid B-ALL with high hyperdiploidy and hypodiploidy; chimeric fusions including ETV6-RUNX1, BCR-ABL1, and TCF3-PBX1; and rearrangement of KMT2A (MLL) in approximately two thirds of childhood ALL. Due to the low prevalence of high hyperdiploidy and ETV6-RUNX1 in older individuals, over 50% of adult cases were unclassified [11]. This, coupled with the observation that many of these alterations were insufficient for leukemogenesis in experimental models, and the ability to detect several alterations at birth in cord blood or blood spots [12] years prior to the onset of leukemia, indicated that many cases of ALL had unidentified drivers and that collaborating genetic alterations are required for leukemogenesis in many cases.
The advent of microarray profiling of gene expression and DNA content (array-based comparative genomic hybridization, array-CGH, and single-nucleotide polymorphism (SNP) arrays) demonstrated that known subtypes of B-ALL exhibited relatively distinct gene expression profiles and could identify cases and subgroups that lacked a known driver [13,14,15]. SNP arrays identified multiple recurring DNA copy number alterations (CNA), particularly alterations in transcriptional regulators of lymphoid development (PAX5, IKZF1, EBF1), providing valuable insights into the nature of co-alterations in B-ALL [16, 17]. These approaches were largely incapable of robustly identifying subtype-defining new alterations, in part due to the limited ability to identify chromosomal rearrangements and chimeric fusion oncoproteins.
Transcriptome sequencing (RNA-seq) has been the most powerful single experimental approach in enabling a near-complete understanding of the molecular classification of B-ALL and the genomic drivers responsible. Although not able to fully identify all sequence and structural alterations, RNA-seq provides a wealth of data regarding gene expression, gene rearrangement, chromosomal aneuploidy, and mutations. The combination of all four data types has proven necessary in classifying B-ALL. The first advance in subtyping of B-ALL using RNA-seq was the identification of Ph-like (BCR-ABL1-like) ALL, a subtype first recognized using microarray gene expression profiling [18, 19], but requiring RNA-seq to resolve the remarkable diversity in genetic alterations, particularly chromosomal rearrangements resulting in enhancer hijacking and chimeric fusion oncoprotein formation, characteristic of this subtype of ALL [20, 21].
In the last 5 years, multiple groups from the USA, Europe, Japan, and China have generated or used B-ALL RNA-seq data to identify new targets of recurring rearrangement (e.g., DUX4, MEF2D, and ZNF384) associated with distinct gene expression profiles [22,23,24,25,26,27,28,29] and the presence of cases with alterations that phenocopy additional canonical B-ALL drivers, e.g., ETV6-RUNX1-like ALL [27]. Several of these subtypes have diverse rearrangements involving a single gene, some of which are cryptic and eluding classification by conventional cytogenetic analysis. Several large-scale B-ALL RNA-seq generation/aggregation studies encompassing up to almost 2000 samples enabled additional observations: additional, less prevalent subtypes driven by chromosomal rearrangements (e.g., rearrangement of NUTM1 and BCL2MYC/BCL6), identification of subtypes driven by initiating sequence mutations rather than chromosomal rearrangements (e.g., PAX5 P80R and IKZF1 N159Y), and subtypes with relatively distinct gene expression but diverse alterations targeting a single gene (PAX5alt, with fusions, sequence mutations, and intragenic amplification of this DNA-binding transcription factor) [5, 6, 26, 29] (Fig. 1.2).

t-SNE plot of gene expression data showing major B-cell precursor acute lymphoblastic leukemia (BCP-ALL) subtypes based on gene expression profiling of 1988 cases [5]. Each dot represents an individual case
By extending these studies across the age spectrum, these data have been particularly valuable in defining the genetic basis of B-ALL in older individuals, which is more parsimonious in the repertoire of subtypes, and commonly driven by alterations that are now recognized as inherently high risk: BCR-ABL1, Ph-like, low hypodiploid, and KMT2A- rearranged ALL, providing a partial explanation for the historically poor outcomes of B-ALL in adults [30] (Fig. 1.3).

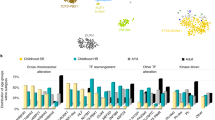
Prevalence of each major subtype in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) across age or risk. AYA, adolescent and young adult; SR, standard risk; HR, high risk. (a) Distribution of key groups of ALL according to age. (b) Cumulative prevalence of ALL subtypes by age. Outcome of selected subtypes for (c) high-risk childhood B-ALL and (d) adult ALL. (Data taken from Gu et al. [5])
Collectively, these studies have enabled classification of over 90% of childhood and adult ALL cases (Table 1.1). A minority of cases remain unclassified, but their driver alterations will likely be identified by the application of WGS that can identify non-coding mutations and rearrangements that deregulate genes without generating a chimeric RNA molecule and thus are not detected by RNA-seq alone.
Heritable Susceptibility to Leukemia
Several lines of evidence support genetic predisposition for many subtypes of BCP-ALL, including (a) Down syndrome and other rare constitutional syndromes with increased risks for leukemia; (b) kindreds with familial BCP-ALL; (c) genome-wide association studies (GWAS) that have identified non-coding DNA polymorphisms which influence risk of BCP-ALL; and (d) a growing number of genes harboring germline non-silent variants presumed to confer risk of sporadic HM.
Children with constitutional syndromes such as Down syndrome, Noonan syndrome, neurofibromatosis type 1, ataxia-telangiectasia, Fanconi anemia, and other bone marrow failure syndromes (severe congenital neutropenia, dyskeratosis congenita, Shwachman-Diamond syndrome, and Diamond-Blackfan anemia) have an increased risk of leukemia. The spectrum of risk is syndrome specific. For example, Down syndrome is associated with a markedly increased risk of AML and B-ALL; Noonan syndrome and neurofibromatosis type 1 have increased risk of JMML (discussed later in this chapter), ataxia-telangiectasia increases T-ALL risk, and bone marrow failure syndromes primarily increase risk of AML [31,32,33,34].
Familial cancer syndromes such as Li-Fraumeni syndrome, constitutional mismatch repair deficiency syndrome, or DNA repair syndromes (Bloom, Werner, Nijmegen breakage) have increased incidence of malignancy, including ALL in a proportion of cases [35, 36]. Familial BCP-ALL is uncommon, but genomic analyses of such kindreds has been tremendously informative by identifying non-silent germline variants in transcription factor and tumor suppressor genes segregating with ALL that in many cases are also present as germline events in sporadic BCP-ALL. Key examples are TP53 germline mutations and low hypodiploid B-ALL, ETV6 variants and hyperdiploid and ETV6-RUNX1-like ALL [37], and PAX5 mutations and B-ALL with dicentric/isochromosome 9 [4, 38,39,40]. These susceptibility genes are targets of somatic mutation in ALL: ETV6 and PAX5 are rearranged, amplified, deleted, and mutated in B-ALL [5, 16]. Germline variants of IKZF1 predispose to a syndrome with immunodeficiency, autoimmunity, and sporadic/familial BCP-ALL [41, 42]; somatic IKZF1 alterations are enriched in BCR-ABL1, Ph-like, and DUX4-rearranged B-ALL [19, 23, 43].
Genome-wide association studies (GWAS) have identified at least 13 loci with primarily non-coding variants associated with BCP-ALL. The relative risk associated with these variants is modest compared with constitutional syndromes or familial leukemia. Risk variants are frequently at or near hematopoietic transcription factor or tumor suppressor gene loci, including ARID5B, BAK1, CDKN2A/CDKN2B, BMI1-PIP4K2A, CEBPE, ELK3, ERG, GATA3, IGF2BP1, IKZF1 IKZF3, USP7, and LHPP [36, 44, 45]. Several variants display ancestry and ALL subtype-specific associations, such as GATA3 with Hispanics and Ph-like B-ALL, ERG with African Americans and TCF3-PBX1 B-ALL, and USP7 with African Americans and T-ALL with TAL1 deregulation [46,47,48].
Genomic analyses have identified additional susceptibility variants in sporadic hyperdiploid B-ALL (NBN, ETV6, FLT3, SH2B3, and CREBBP), Down syndrome-associated B-ALL (IKZF1, NBN, RTEL1), and T-ALL (Fanconi-BRCA pathway mutations) [49,50,51].
Prenatal Origin of Leukemia
Several observations indicate that a proportion of childhood leukemia cases are initiated before birth [52,53,54]. Chromosomal translocations such as ETV6-RUNX1 may be detected at birth in blood spots and cord blood, years before the clinical onset of leukemia, providing support for a multistep process of leukemogenesis. This is supported by genomic analyses of monozygotic, monochorionic twins concordant for leukemia, showing genetic identity of initiating lesions and discordance for secondary genetic alterations, indicating inter-twin, intrauterine transmission of leukemia [53, 55]. Evidence for in utero origin is strongest for KMT2A-rearranged and ETV6-RUNX1 ALL. Anecdotal evidence supports in utero origin for other subtypes of B-ALL, including hyperdiploid and ZNF384-rearranged leukemia [56].
Aneuploid BCP-ALL : Hyperdiploidy, Hypodiploidy, and Intrachromosomal Amplification of Chromosome 21
High hyperdiploidy (51–67 chromosomes) comprises approximately 30% of pediatric BCP-ALL and is associated with a favorable prognosis (Table 1.1) [57]. High hyperdiploidy is characterized by a nonrandom gain of chromosomes, typically +X, +4, +6, +10, +14, +17, +18, and +21 [57]. In particular, combined gain of chromosome 4, 10, and 17 is associated with favorable prognosis [58]. Alterations involving the Ras pathway (KRAS, NRAS, FLT3, PTPN11) and epigenetic modifiers (CREBBP, WHSC1) are frequent genetic events, with deletions leading to enhancer hijacking and deregulation of FLT3 particularly common in high hyperdiploid ALL [57, 59]. These secondary genomic alterations and the gene expression profiles of high hyperdiploid and the near-haploid subset of hypodiploid ALL are similar, suggesting a common origin [60]. Low hyperdiploid cases (47–50 chromosomes) harbor a diverse range of chromosomal changes and alterations rather than representing a genetically distinct subtype of ALL.
Hypodiploid ALL comprises three subtypes, two of which have an unfavorable prognosis: near-haploid ALL (24–31 chromosomes) and low hypodiploid ALL (32–39 chromosomes) [61,62,63]. Notably, chromosome 21 is never lost in hypodiploid ALL nor in other forms of ALL, suggesting an essential role in tumor cell fitness [4]. High hypodiploid ALL (40–44 chromosomes) is genetically heterogeneous, is not a distinct subtype of B-ALL, and does not share the unfavorable outcome of the other two groups. Accurate identification of low/near-haploid ALL is important in view of the poor prognosis and inherited genetic basis of low hypodiploid ALL in children [4]. Duplication of the aneuploid genome, or masked hypodiploidy, is common and may be mistaken for high hyperdiploidy [64]. These entities can be distinguished by the patterns of chromosomal gain and loss of heterozygosity observed on cytogenetic or SNP array analysis: masked hypodiploidy typically has diploid and tetraploid chromosomes, whereas hyperdiploidy has a mixture of triploid and some tetraploid (e.g., 21, X); masked hypodiploid cases typically have LOH of the duplicated chromosomes. Flow cytometric analysis of DNA index frequently demonstrates peaks for both non-duplicated and masked clones in hypodiploid ALL, even if cytogenetic analysis demonstrates an apparently predominant masked clone.
Near-haploid ALL presents at a younger age and commonly exhibits alterations activating the Ras pathway (particularly NF1) and inactivating mutations/deletions of IKZF3 (AIOLOS) [4]. Low hypodiploid ALL is rare but increases with age. Frequent secondary alterations include IKZF2 (HELIOS), RB1, and CDKN2A/CDKN2B. The mechanistic differences between the IKAROS gene family members in leukemogenesis (IKZF1 in kinase-driven and DUX4-rearranged leukemia and IKZF2/3 in hypodiploid ALL) remain to be determined. Importantly, almost all cases of low hypodiploid ALL in children and adults have biallelic alterations of TP53 due to mutation (or less commonly focal deletion) and aneuploidy of the second chromosome [4]. In approximately half of pediatric cases (but not adult), the TP53 mutations are germline, indicating that low hypodiploid ALL is a manifestation of Li-Fraumeni syndrome [4, 65]. Although still associated with an unfavorable prognosis, minimal residual disease (MRD) risk-stratified therapy has improved the outcome of hypodiploid ALL [66]. Hypodiploid ALL cells are sensitive to BCL2 inhibition, and BCL2 inhibitors are being evaluated in prospective clinical trials of newly diagnosed and relapsed/refractory ALL [67].
Intrachromosomal amplification of chromosome 21 (iAMP21) is more common in older children and is characterized by gain of three or more extra copies of a region of chromosome 21 including RUNX1 generated by breakage-fusion-bridge cycles and chromothripsis [68,69,70,71]. The germline Robertsonian translocation rob(15;21) or a germline ring chromosome 21 is associated with a markedly elevated risk of iAMP21 [72]. Patients with iAMP21 usually lack other key cytogenetic alterations, although it is observed as a secondary event in ETV6-RUNX1 and BCR-ABL1 ALL in a minority of cases. Historically associated with an unfavorable outcome, intensive therapy improves prognosis [73, 74]. The key driver gene(s) located on chromosome 21 resulting in requirement for this chromosome in ALL, and mediating leukemogenesis in iAMP21 ALL, remains to be determined.
ETV6-RUNX1 and ETV6-RUNX1-Like ALL
The t(12:21)(p13:q22) translocation encodes ETV6-RUNX1, the most common fusion in BCP-ALL (20–25% in children) that is associated with a favorable prognosis [5, 75]. This translocation is frequently cryptic on cytogenetic analysis, and leukemic cells have a distinct immunophenotype (CD27 positive and CD44 low/negative) [76]. The ETV6-RUNX1 fusion may be identified in umbilical cord blood and, thus, is considered to arise in utero as a leukemia-initiating alteration [75]. However, ETV6-RUNX1 itself is insufficient to induce overt leukemia and requires the prolonged latency with additional genetic events including deletion of the non-rearranged ETV6 allele, focal deletion of PAX5, and mutation of WHSC1 [2, 16, 17, 75, 77]. This is supported by heterogeneity in the subclonal composition of ETV6-RUNX1 ALL [75, 78, 79].
ETV6-RUNX1-like ALL exhibits a similar GEP and immunophenotype to ETV6-RUNX1 ALL despite the lack of ETV6-RUNX1 fusion [5, 6, 27, 76]. ETV6-RUNX1-like ALL is also most common in children and has relatively favorable outcome [27, 76]. This subtype includes several alternate rearrangements in ETV6 (e.g., ETV6-ELMO1), IKZF1 (e.g., IKZF1-ETV6), TCF3 (e.g., TCF3-FLI1), and FUS-ERG as well as copy number alterations in ETV6, IKZF1, and ARPP21, suggesting that alteration of multiple ETS and other transcription factors are converging on the same mechanism of transformation (although not ERG, which is distinct in the DUX4-rearranged ALL) [5, 27, 76].
TCF3-PBX1 and TCF3-HLF BCP-ALL
The t(1;19)(q23;p13) translocation encoding TCF3-PBX1 fusion is present in 5–6% of pediatric BCP-ALL and is associated with a pre-B in transition (cytoplasmic immunoglobulin heavy chain positive) immunophenotype [80]. Previously considered high risk due to higher central nervous system involvement and relapse [15, 81, 82], TCF3-PBX1 ALL is classified as favorable or intermediate risk with current treatment regimens [83]. Conditional activation of TCF3-PBX1 in B-cell progenitors results in enhanced self-renewal and eventual development of leukemia with PAX5 deletion and activation of JAK-STAT or Ras signaling pathways [84]. Importantly, TCF3-PBX1 ALL exhibits sensitivity to dasatinib and ponatinib, but not imatinib, which occurs as a result of inhibition of pre-BCR signaling by SRC kinases. Due to compensatory upregulation of ROR1 expression, combination with ROR1 inhibition may enhance the sensitivity of dasatinib [85].
A variant of the t(1;19) translocation, t(17;19)(q22;p13), encodes the TCF3-HLF fusion, a rare subtype of ALL associated with an extremely poor prognosis [86, 87]. TCF3-PBX1 and TCF3-HLF ALL have distinct gene expression profiles and mutational landscapes [88]. TCF3-HLF ALL exhibited stem cell and myeloid features with enrichment of PAX5 deletions and alterations of Ras pathway genes [88]. The TCF-HLF fusion may act as a pioneer transcription factor, recruiting EP300 to activate MYC, with vulnerability to EP300 inhibition [89]. TCF3-HLF leukemic cells are sensitive to the BCL2 inhibitor venetoclax (ABT-199), representing a potential targeted therapeutic approach [88].
KMT2A-Rearranged ALL
KMT2A (MLL) on chromosome 11q23 is rearranged to more than 80 different partner genes, and these rearrangements describe a distinct subtype of leukemia with variable immunophenotype spanning ALL, AML, and mixed phenotype leukemia with both lymphoid and myeloid features and poor outcome [90]. KMT2A-rearranged BCP-ALL is typically of the pro-B phenotype, lacking CD10 expression, with co-expression of myeloid markers. Approximately 80% of KMT2A-rearranged ALL is observed in infants, in whom KMT2A rearrangement is acquired in utero. There is also a second peak in prevalence in adults, and more than 75% of cases are fused to AFF1. KMT2A-rearranged leukemia may also follow exposure to topoisomerase II inhibitors, with similar breakpoints to infant leukemia suggesting a common mechanism of leukemogenesis [91]. In infant ALL, the most commonly perturbed pathways include PI3K and Ras pathways [92,93,94]. KMT2A rearrangement results in assembly of a large multi-protein complex that results in aberrant transcriptional and epigenetic dysregulation via H3K79 methylation and recruitment of the H3K79 methyltransferase DOT1L, which interacts with multiple KMT2A rearrangement partners [95,96,97]. Multiple therapeutic approaches are being pursued, including inhibition of DOT1L, bromodomain, Menin, and the polycomb repressive complex [90, 97,98,99].
Kinase-Driven BCP-ALL : BCR-ABL1 ALL and Ph-like ALL
The derivative chromosome 22, Philadelphia chromosome (Ph), arises from the reciprocal t(9;22)(q34;q11) translocation and encodes BCR-ABL1 [7, 41, 100]. Although BCR-ABL1 ALL is associated with poor prognosis, the addition of tyrosine kinase inhibitors (TKIs) to the conventional chemotherapy has improved outcome in children and adults [101,102,103,104]. In contrast to BCR-ABL1-positive chronic myeloid leukemia at chronic phase, BCR-ABL1 ALL is characterized by a high frequency of secondary genetic alterations, particularly of the lymphoid transcription factor gene IKZF1 and CDKN2A/B encoding the INK4/ARF cell cycle regulators [43, 105], and IKZF1 alterations are associated with unfavorable outcome irrespective of TKI exposure [102, 105]. Moreover, mutations in the kinase domain of ABL1 (most frequently T315I) induce TKI resistance and are more commonly observed in patients treated with TKI monotherapy or in adults treated with less intensive chemotherapy and less common in children treated with intensive chemotherapy [106]. Current treatment approaches to mitigate the poor outcome of BCR-ABL1 ALL include frontline treatment with the third-generation TKI ponatinib with chemotherapy [101]. The adverse effect of IKZF1 mutations in the pathogenesis of BCR-ABL1 ALL is in part due to loss of IKZF1 repression of stemness and cell-cell adhesion [107, 108]. This may be reversed by rexinoids (via agonism of rexinoid X receptor alpha, which induces expression of wild-type IKZF1) and focal adhesion kinase inhibitors (which inhibit downstream integrin signaling pathways) [108, 109].
Before consensus guidelines for MRD assessment in BCR-ABL1 ALL were provided [110], several approaches have been tested for MRD monitoring (genome or transcriptome BCR-ABL1 and Ig/TCR rearrangements) [111]. Importantly, some patients showed discrepancy of MRD results as assessed by measurement of Ig/TCR and BCR-ABL1 transcript levels, due to the presence of the BCR-ABL1 fusion in progenitors in addition to the blast population [111]. This BCR-ABL1-positive clonal hematopoiesis is suggestive of a CML-like disease exhibiting lymphoid blast crisis.
Ph-like or BCR-ABL1-like ALL exhibits a gene expression profile similar to BCR-ABL1 ALL despite the lack of the BCR-ABL1 fusion [18, 19]. The prevalence and outcome of Ph-like ALL are similar to those of BCR-ABL1 ALL, increasing in incidence with age and associated with elevated MRD levels and/or higher rates of treatment failure [20, 112,113,114,115,116,117,118], although the prevalence of Ph-like ALL is higher than BCR-ABL1 ALL in the adolescent and young adult (AYA) population [20, 117, 119, 120]. Similar to BCR-ABL1 ALL, IKZF1 alterations are common, which result in acquisition of stem cell-like features and poor responsiveness to TKI. The heterogeneous genetic alterations driving Ph-like ALL may be classified into four main groups (Table 1.2., Fig. 1.4): (1) alterations driving JAK-STAT signaling, including rearrangements and mutations/deletions of CRLF2, JAK2, EPOR, TYK2, IL7R, SH2B3, JAK1, JAK3, TYK2, and IL2RB; (2) fusions involving ABL-class genes (ABL1, ABL2, CSF1R, LYN, PDGFRA, PDGFRB); (3) mutations activating Ras signaling (NRAS, KRAS, PTPN11); and (4) less common fusions (FLT3, FGFR1, NTRK3) [2, 121, 122]. Of these, CRLF2 alterations are found in almost half of Ph-like ALL in adolescents, young adults, and older adults, as well as in half of ALL associated with Down syndrome ALL [123,124,125]. These alterations are rearrangements of CRLF2 to IGH or P2RY8 resulting in enhancer hijacking or promoter swapping, respectively, and aberrant expression of CRLF2 as part of a heterodimer with IL-7 receptor alpha. CRLF2-rearranged ALL commonly has concomitant alterations that facilitate JAK-STAT signaling pathway activation, including sequence mutations of Janus kinases (most commonly at R683 of the pseudokinase domain of JAK2), IL-7RA, and deletions of negative regulators of JAK-STAT signaling (SH2B3 and USP9X) [126, 127]. CRLF2 rearrangement is associated with Hispanic ancestry and a germline GATA3 non-coding variant [46, 128].

Cartoon depicting targets of genetic alteration and type of mutation in Ph-like ALL
Importantly, most kinase-activating alterations in Ph-like ALL can, theoretically, be targeted by FDA-approved TKIs: JAK-STAT signaling (JAK inhibition), ABL-class fusions (ABL inhibitor), and FLT3 and NTRK3 fusions (FLT3 and NTRK3 inhibitor) with emerging evidence of efficacy in human leukemia, although evidence for efficacy of TKIs, at least as monotherapy, is strongest for ABL1-class and ETV6-NTRK3 Ph-like ALL [20, 129,130,131,132,133]. In contrast JAK inhibitor monotherapy in preclinical and clinical studies of CRLF2-rearranged Ph-like ALL is less effective [134]. Combination of kinase inhibitors against multiple signaling shows synergistic effects in PDX models of CRLF2/JAK mutant (JAK and PI3K/mTOR inhibitors) and ABL/PDGFR mutant (dasatinib and PI3K/mTOR inhibitor) [135]. Several of these (ruxolitinib, imatinib, dasatinib, ponatinib) are being tested in frontline studies [120, 133, 136]. As kinase-activating lesions also drive signaling through additional signaling pathways (e.g., PI3K, MEK, etc.), it is likely that additional therapeutic approaches will be required for optimal therapeutic response. Additional therapeutic approaches include BCL2 inhibitors, which exhibit synergy with TKIs in preclinical models [137, 138], and chimeric antigen receptor T cells directed against CRLF2 [139].
DUX4-Rearranged ALL
Rearrangement and overexpression of the homeobox transcription factor gene DUX4 defines a distinct subgroup of BCP-ALL [5, 22, 23, 27]. This subtype also exhibits deregulation of the ETS family transcription factor ERG and comprises up to 5–10% of BCP-ALL with a slight peak in the AYA population. It has a distinct immunophenotype (CD2 and CD371 positive) and favorable outcome [140]. The pathogenesis of this form of leukemia is remarkable for the interrelated, sequential genetic events that deregulate two DNA-binding transcription factors characteristic of this disease (Fig. 1.5). Deregulation of DUX4 is induced by rearrangement to strong enhancer elements, most commonly the immunoglobulin heavy chain (IGH) enhancer, which results in expression of a C-terminal truncated DUX4 protein that is not normally expressed in B cells [22, 23]. This truncated isoform of DUX4 then binds to an intragenic region of ERG resulting in transcriptional deregulation and expression of multiple aberrant coding and non-coding ERG isoforms and deletion of ERG in up to 70% of DUX4-rearranged cases [23]. One isoform is ERGalt, a C-terminal fragment which retains the DNA-binding and transactivating domain of ERG, that exerts a dominant negative effect and is transforming [23]. The deletions of ERG are commonly polyclonal [141], supporting a model in which an initiating rearrangement of DUX4 results in gross transcriptional deregulation of ERG and primes the locus for RAG-mediated deletion. Loss of ERG activity, either through deletion and/or expression of ERGalt, cooperates with DUX4 deregulation in leukemogenesis [23, 141]. DUX4 rearrangement is associated with a favorable outcome in children and adults, even with IKZF1 deletion [142]. As clonal ERG deletions are not present in all DUX4-rearranged cases, the use of ERG deletion as a surrogate for this subtype, as is used in the definition of IKZFplus [143], is suboptimal and should be avoided. Accurate identification of this favorable subtype of ALL requires identification of DUX4 rearrangement (either directly or through identification of elevated DUX4 expression) [23]. In this regard , detection of strong CD371 cell surface expression by flow cytometry might serve as a promising surrogate marker for this subtype [140].

Schema of the sequence of transcription factor alterations driving leukemogenesis in DUX4-rearranged ALL: rearrangement of DUX4 to strong enhancers results in deregulation of DUX4 expression with truncation of the C-terminus. This shortened form of DUX4 binds to intron 6 of ERG, resulting in gross transcriptional deregulation and expression of multiple coding, non-coding, and enhancer RNA species, including a C-terminal isoform initiated by a novel first exon, ERGalt. This aberrancy also permits deletion of ERG as a secondary event
MEF2D-Rearranged ALL
Rearrangement of MEF2D is associated with older age of onset and relatively inferior outcome due to early relapse [24, 26, 144, 145]. MEF2D-rearranged ALL is characterized by an aberrant immunophenotype (low or absent expression of CD10, high expression of CD38, and cytoplasmic μ-chain), mature B-ALL-like morphology, and distinct expression profiles. The N-terminal of MEF2D is fused to several partner genes, retaining its DNA-binding domain [24, 144, 145]. High expression of MEF2D fusion protein is induced by evasion from miRNA-mediated degradation [146] and results in transcriptional activation of MEF2D targets [24]. Dysregulated MEF2D targets include overexpression of HDAC9, which confers therapeutic sensitivity to HDAC inhibitors such as panobinostat [24].
ZNF384-Rearranged ALL
ZNF384 rearrangement defines a distinct subtype of leukemia that can be diagnosed as BCP-ALL or B/myeloid mixed phenotype acute leukemia (MPAL) [147]. ZNF384 is rearranged as the C-terminal partner to multiple genes, including the histone acetyltransferases and transcriptional regulators EP300 and CREBBP, SWI/SNF proteins SMARCA2 and ARID1B, and others (TAF15, EWSR1, TCF3, NIPBL, and CLTC) [5, 6, 22, 24,25,26, 29, 147,148,149,150,151,152,153,154]. The most common are EP300-ZNF384 (particularly in BCP-ALL) and TCF3-ZNF384 (in both BCP-ALL and B/myeloid MPAL). In BCP-ALL, peak age of onset and prognosis vary by fusion partners: EP300-ZNF384 (median age 11, excellent outcome) and TCF3-ZNF384 (median age 5, frequent late relapse) [5, 148, 155]. In contrast, ZNF384-rearranged ALL shows uniformly distinct immunophenotype (weak CD10 and aberrant CD13 and/or CD33 expression) and gene expression profiles [147, 148]. The secondary genomic alterations and gene expression profiles of ZNF384-rearranged BCP-ALL and MPAL cases are similar, and both have lineage plasticity at diagnosis and relapse (lymphoid disease to myeloid disease and vice versa) [147]. Transplantation of purified lymphoid or myeloid subpopulations of cells from ZNF384-rearranged leukemia showed that each subpopulation could reconstitute the immunophenotypic diversity of the primary leukemia, indicating that this plasticity is inherent to all leukemic cells [69]. These data support the notion that ZNF384-rearranged cases should be treated uniformly rather than tailoring therapy according to predominant lineage. In this regard, FLT3 overexpression without mutation is characteristic of ZNF384-rearranged leukemia and in anecdotal reports can be targeted with the multi-kinase inhibitor sunitinib [156]. Due to the propensity of ZNF384-rearranged ALL to change lineage, CD19-directed CAR-T cell therapy may fail due to CD19-negative escape [147, 157, 158].
PAX5-Driven BCP-ALL : PAX5alt and PAX5 P80R
The paired box DNA-binding transcription factor PAX5 is required for B-cell lineage commitment and differentiation. PAX5 alterations are important in the pathogenesis of BCP-ALL as initiating or cooperating lesions. These include (1) disease initiating alterations (PAX5 rearrangements in chimeric fusion oncoproteins and the P80R mutation [5, 16, 159,160,161], rearrangements/focal intragenic amplifications in PAX5-altered ALL [PAX5alt]) [5, 162], (2) secondary lesions (e.g., PAX5 focal deletions in 30% of ETV6-RUNX1 ALL [16, 77] and PAX5 mutations in multiple subtypes), and (3) germline alterations that predispose to ALL [39]. In mouse models, Pax5 heterozygosity cooperates with constitutive activation of the JAK-STAT pathway in the development of BCP-ALL, supporting its role as a haploinsufficient tumor suppressor [163].
PAX5alt is a subtype of BCP-ALL with similar leukemic cell gene expression profiles but diverse nature of underlying PAX5 alterations. These include (1) cases with diverse (>20) PAX5 rearrangements that typically preserve the N-terminal DNA-binding domain of PAX5, but with loss of the C-terminal transactivation domain, (2) cases with focal intragenic amplification of the PAX5 DNA-binding paired domain (PAX5amp), and (3) cases with sequence mutations. Within this group, specific lesions are associated with variation in gene expression profile, for example, cases with PAX5-ETV6 rearrangement, or compound heterozygosity for p.Arg38 and p.Arg140 mutations in the DNA-binding paired domain, have distinct gene expression profiles. PAX5alt is most common in children and the AYA population and is associated with intermediate outcome [5].
The PAX5 P80R subtype is characterized by the presence of the PAX5 P80R mutation with inactivation of the wild-type PAX5 allele by deletion, loss-of-function mutation, or copy-neutral loss of heterozygosity [5, 159, 160]. Notably, heterozygous Pax5P80R/+ knock-in mice develop transplantable BCP-ALL, with genetic inactivation of the wild-type Pax5 allele [5]. Thus, biallelic PAX5 alterations are a hallmark of this subtype, and sequence mutations of lymphoid transcription factors such as PAX5 P80R and IKZF1 N159Y (see below) may be initiating events in leukemogenesis. The prevalence of PAX5 P80R increases with age and is associated with intermediate to favorable prognosis [5, 159, 160]. Additional important cooperating lesions include structural rearrangements of chromosomal arms 9p and 20q, which associate with the presence of dic(9:20). Moreover, mutations in the Ras and JAK-STAT pathway members are particularly enriched, highlighting the potential for targeted therapies.
Other Subtypes of BCP-ALL
BCP-ALL with NUTM1 rearrangements is a rare subtype observed exclusively in children [5, 6]. NUTM1 is a chromatin modifier, recruiting EP300 to increase local histone acetylation [164]. While the common partner, BRD9-NUTM1, is reported in BCP-ALL, BRD4-NUTM1 is a hallmark of NUT midline carcinoma (NMC) and acts to repress differentiation in NMC by widespread repression of histone acetylation, indicating therapeutic approach with bromodomain and HDAC inhibitors. NUTM1 is rearranged to multiple genes in BCP-ALL (and less commonly, T-ALL) [165] in addition to BRD9 [92, 166], including ACIN1 [24, 26, 92, 167, 168], AFF1 [6, 151], BPTF [165], CUX1 [24, 167], IKZF1 [6, 24, 27, 167], SCL12A6 [6, 24, 167], and ZNF618 [6, 24, 29, 151], with emerging evidence that these fusions are enriched in non-KMT2A-rearranged BCR-ALL in infants [92, 168]. The potential for bromodomain inhibition as a therapeutic strategy has not yet been tested in NUTM1-rearranged BCP-ALL.
IKZF1 alterations, like PAX5, are also common across the spectrum of B-ALL (particularly in BCR-ABL1-positive, Ph-like, and DUX4-rearranged cases), but a specific mutation, IKZF1 p.Asn159Tyr, defines a subtype with gene expression profile [5, 6]. In this subtype, the non-mutated wild-type allele of IKZF1 is retained, and most cases have concurrent gain of chromosome 21. Notably, this mutation is located at a residue that is critical for DNA binding of IKZF1 [169] and is also mutated in germline syndromes with immunodeficiency and autoimmunity [42, 170], although most commonly to serine but not tyrosine, suggesting genotype-phenotype variation of different IKZF1 mutations. The IKZF1 p.Asn159Tyr mutation induces misregulation of IKZF1 transcriptional activation, in part through distinctive nuclear mislocalization and enhanced intercellular adhesion [108].
Relapsed ALL
Genomic analyses of paired primary and relapsed ALL samples have revealed that these secondary mutations are acquired during disease progression with Darwinian patterns of selection, and highly branched clonal architectures, especially in early relapse (9–36 months) [8, 9, 78, 171,172,173,174,175]. Furthermore, chemotherapy of ALL has been postulated to induce bona fide drug resistance mutations including NT5C2, PRPS1, NR3C1, and TP53 [9]. However, recent studies integrating genome sequencing of matched diagnosis and relapse samples, and xenografts propagated from these samples, coupled with drug sensitivity testing of the relapse fated clones have shown that relapse-fated subclones present at diagnosis commonly exhibit drug resistance prior to the administration of any therapy [174] (Fig. 1.6).

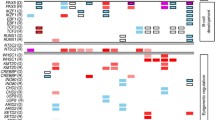
(a) Oncoprint of the most common targets of mutation at relapse in childhood B- and T-ALL. (b) Patterns of clonal evolution in relapsed ALL. (Data taken from Waanders et al. [7])
One of the representative relapse-specific somatic alterations is CREBBP alterations which occur in up to 20% of relapsed B-ALL and impair glucocorticoid sensitivity [60]. Early relapse is commonly associated with 6-MP resistance, as a result of NT5C2 gain-of-function mutations [175,176,177,178], PRPS1 mutations [179], and loss of MSH6 [180]. NT5C2 mutations confer resistance to purine analogs at the cost of impaired tumor cell growth and reduced leukemia-initiating cell activity [175]. While the development of NT5C2 inhibitors may be promising, several problems are anticipated such as the development of mutant specific inhibitors [176]. Importantly, NT5C2 and PRPS1 mutations are not detectable in primary samples even in a minor clone [7, 9, 175]. Other recurrent somatic alterations in relapsed ALL include mutations in [78] SETD2, KDM6, and KMT2D (MLL2) [9, 173, 181]. Tracking of these mutations as MRD may offer the opportunity to identify the relapse-fated clone early in disease evolution and modulate therapy accordingly to circumvent relapse. Detailed, genome-wide analyses of large ALL cohorts have enabled several additional important observations: hypermutation becomes increasingly frequent during disease progression, is enriched in leukemic cells with mutations in mismatch repair genes and hypodiploidy, and results in a predicted increase in expressed neoantigen formation. Thus, strategies to promote autologous T cell reactivity may be efficacious in this setting. Secondly, careful analysis of the nature and structure of coding and non-coding sequence and structural variants has shown that most cases presumed to be second leukemias are indeed clonally related to the primary tumor, including cases with lineage shift/switch, indicating relapse from an ancestral, pre-diagnosis clone [7] (Fig. 1.6b). These observations confirm hypotheses from SNP array analyses of relapsed ALL [78] and are of therapeutic importance for disease monitoring and selection of therapy.
Summary
Genomic analyses have transformed our understanding of the molecular basis of BCP-ALL, in terms of identification of new subtypes and dysregulated pathways associated with therapeutic targets. Many clinically important alterations are not evident using conventional cytogenetic and molecular approaches, and optimal ALL diagnosis requires next-generation sequencing, with RNA-seq capturing the most relevant information required for risk stratification, disease monitoring, and the development of precision medicine approaches [136]. While clinical implementation of genome and transcriptome sequencing is not trivial, it is now clearly apparent that targeted molecular approaches such as fusion-specific PCR and exome/gene panel capture sequencing are not optimal as they do not capture the diversity of genomic alterations in ALL. Moreover, integrated genome, exome and transcriptome sequencing has been shown to have excellent sensitivity and specificity in detection of the various driver alterations in pediatric cancer [182]. Even if sequencing is not available, several key alterations can be detected by alternative approaches, such as flow cytometry for CRLF2 (which correlates well with CRLF2 overexpression) and FISH assays for gene rearrangements in Ph-like ALL.
These genomic discoveries are partly responsible for a wave of new therapeutic approaches entering the clinic in BCP-ALL including small molecules (TKI, BCL2 inhibitors, MEK inhibitors), antibody-based therapy (blinatumomab, inotuzumab), and cellular immunotherapy. Future challenges and opportunities include (1) determining the tumor intrinsic and extrinsic determinants of response in the era of targeted therapies and immunotherapy, (2) developing efficacious approaches to directly target transcription factor alterations that underlie over 50% of BCP-ALL, and (3) integrating genomic and functional genomic approaches to identify therapeutic vulnerabilities both in the research and clinical setting.
References
Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975–83.
Roberts KG, Mullighan CG. The biology of B-progenitor acute lymphoblastic leukemia. Cold Spring Harb Perspect Med. 2020;10(7):a03483.
Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 2019;16(4):227–40.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52.
Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296–307.
Li JF, Dai YT, Lilljebjorn H, Shen SH, Cui BW, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A. 2018;115(50):E11711–e20.
Waanders E, Gu Z, Dobson SM, Antić Ž, Crawford JC, Ma X, et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020;1(1):96–111.
Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604.
Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood. 2020;135(1):41–55.
Harrison CJ, Foroni L. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Rev Clin Exp Hematol. 2002;6(2):91–113.
Stock W. Adolescents and young adults with acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2010;2010:21–9.
Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. 1997;94(25):13950–4.
Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1(2):133–43.
Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102(8):2951–9.
Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116(23):4874–84.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64.
Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21(6):1258–66.
Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–34.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15.
Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–66.
Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48(5):569–74.
Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48(12):1481–9.
Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331.
Gocho Y, Kiyokawa N, Ichikawa H, Nakabayashi K, Osumi T, Ishibashi T, et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2015;29(12):2445–8.
Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–83.
Lilljebjorn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, von Palffy S, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790.
Lilljebjorn H, Fioretos T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood. 2017;130(12):1395–401.
Zaliova M, Stuchly J, Winkowska L, Musilova A, Fiser K, Slamova M, et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica. 2019;104(7):1396–406.
Roberts KG. Genetics and prognosis of ALL in children vs adults. Hematology Am Soc Hematol Educ Program. 2018;2018(1):137–45.
Reiman A, Srinivasan V, Barone G, Last JI, Wootton LL, Davies EG, et al. Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours. Br J Cancer. 2011;105(4):586–91.
Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet. 2014;51(10):689–97.
Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355(9199):165–9.
Rafei H, DiNardo CD. Hereditary myeloid malignancies. Best Pract Res Clin Haematol. 2019;32(2):163–76.
Plon SE, Lupo PJ. Genetic predisposition to childhood cancer in the genomic era. Annu Rev Genomics Hum Genet. 2019;20:241–63.
Gocho Y, Yang JJ. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood. 2019;134(10):793–7.
Nishii R, Baskin-Doerfler R, Yang W, Oak N, Zhao X, Yang W, et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood. 2021;137(3):364–73.
Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015;16(16):1659–66.
Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226–31.
Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47(5):535–8.
Churchman ML, Qian M, Te Kronnie G, Zhang R, Yang W, Zhang H, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. 2018;33(5):937–48.e8
Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. 2016;374(11):1032–43.
Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4.
Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–10.
Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–5.
Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494–8.
Qian M, Xu H, Perez-Andreu V, Roberts KG, Zhang H, Yang W, et al. Novel susceptibility variants at the ERG locus for childhood acute lymphoblastic leukemia in Hispanics. Blood. 2019;133(7):724–9.
Qian M, Zhao X, Devidas M, Yang W, Gocho Y, Smith C, et al. Genome-wide association study of susceptibility loci for T-cell acute lymphoblastic leukemia in children. J Natl Cancer Inst. 2019;111(12):1350–7.
de Smith AJ, Lavoie G, Walsh KM, Aujla S, Evans E, Hansen HM, et al. Predisposing germline mutations in high hyperdiploid acute lymphoblastic leukemia in children. Genes Chromosomes Cancer. 2019;58(10):723–30.
Pouliot GP, Degar J, Hinze L, Kochupurakkal B, Vo CD, Burns MA, et al. Fanconi-BRCA pathway mutations in childhood T-cell acute lymphoblastic leukemia. PLoS One. 2019;14(11):e0221288.
Winer P, Muskens IS, Walsh KM, Vora A, Moorman AV, Wiemels JL, et al. Germline variants in predisposition genes in children with Down syndrome and acute lymphoblastic leukemia. Blood Adv. 2020;4(4):672–5.
Greaves M. Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol. 2003;7(3):233–45.
Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2003;102(7):2321–33.
Hein D, Borkhardt A, Fischer U. Insights into the prenatal origin of childhood acute lymphoblastic leukemia. Cancer Metastasis Rev. 2020;39(1):161–71.
Ma Y, Dobbins SE, Sherborne AL, Chubb D, Galbiati M, Cazzaniga G, et al. Developmental timing of mutations revealed by whole-genome sequencing of twins with acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2013;110(18):7429–33.
Bueno C, Tejedor JR, Bashford-Rogers R, Gonzalez-Silva L, Valdes-Mas R, Agraz-Doblas A, et al. Natural history and cell of origin of TC F3-ZN F384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood. 2019;134(11):900–5.
Paulsson K, Lilljebjörn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47(6):672–6.
Sutcliffe MJ, Shuster JJ, Sather HN, Camitta BM, Pullen J, Schultz KR, et al. High concordance from independent studies by the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children’s Oncology Group (COG) initiative. Leukemia. 2005;19(5):734–40.
Yang M, Safavi S, Woodward EL, Duployez N, Olsson-Arvidsson L, Ungerback J, et al. 13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking. Blood. 2020;136(8):946–56.
Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471(7337):235–9.
Pui CH, Williams DL, Raimondi SC, Rivera GK, Look AT, Dodge RK, et al. Hypodiploidy is associated with a poor prognosis in childhood acute lymphoblastic leukemia. Blood. 1987;70(1):247–53.
Harrison CJ, Moorman AV, Broadfield ZJ, Cheung KL, Harris RL, Reza Jalali G, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125(5):552–9.
Nachman JB, Heerema NA, Sather H, Camitta B, Forestier E, Harrison CJ, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110(4):1112–5.
Carroll AJ, Shago M, Mikhail FM, Raimondi SC, Hirsch BA, Loh ML, et al. Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: a report from the Children’s Oncology Group. Cancer Genet. 2019;238:62–8.
Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018;36(6):591–9.
Mullighan CG, Jeha S, Pei D, Payne-Turner D, Coustan-Smith E, Roberts KG, et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood. 2015;126(26):2896–9.
Diaz-Flores E, Comeaux EQ, Kim KL, Melnik E, Beckman K, Davis KL, et al. Bcl-2 is a therapeutic target for hypodiploid B-lineage acute lymphoblastic leukemia. Cancer Res. 2019;79(9):2339–51.
Harrison CJ, Haas O, Harbott J, Biondi A, Stanulla M, Trka J, et al. Detection of prognostically relevant genetic abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: recommendations from the Biology and Diagnosis Committee of the International Berlin-Frankfurt-Munster study group. Br J Haematol. 2010;151(2):132–42.
Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98–102.
Harrison CJ, Moorman AV, Schwab C, Carroll AJ, Raetz EA, Devidas M, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28(5):1015–21.
Moorman AV, Richards SM, Robinson HM, Strefford JC, Gibson BE, Kinsey SE, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood. 2007;109(6):2327–30.
Harrison CJ, Schwab C. Constitutional abnormalities of chromosome 21 predispose to iAMP21-acute lymphoblastic leukaemia. Eur J Med Genet. 2016;59(3):162–5.
Heerema NA, Carroll AJ, Devidas M, Loh ML, Borowitz MJ, Gastier-Foster JM, et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk Children’s Oncology Group studies: a report from the Children’s Oncology Group. J Clin Oncol. 2013;31(27):3397–402.
Moorman AV, Robinson H, Schwab C, Richards SM, Hancock J, Mitchell CD, et al. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol. 2013;31(27):3389–96.
Sundaresh A, Williams O. Mechanism of ETV6-RUNX1 leukemia. Adv Exp Med Biol. 2017;962:201–16.
Zaliova M, Kotrova M, Bresolin S, Stuchly J, Stary J, Hrusak O, et al. ETV6/RUNX1-like acute lymphoblastic leukemia: a novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes Chromosomes Cancer. 2017;56(8):608–16.
Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46(2):116–25.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322(5906):1377–80.
Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356–61.
Aspland SE, Bendall HH, Murre C. The role of E2A-PBX1 in leukemogenesis. Oncogene. 2001;20(40):5708–17.
Pui CH, Aur RJ, Bowman WP, Dahl GV, Dodge RK, George SL, et al. Failure of late intensification therapy to improve a poor result in childhood lymphoblastic leukemia. Cancer Res. 1984;44(8):3593–8.
Jeha S, Pei D, Raimondi SC, Onciu M, Campana D, Cheng C, et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia. 2009;23(8):1406–9.
Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS control of childhood acute lymphoblastic leukemia without cranial irradiation: St Jude Total Therapy Study 16. J Clin Oncol. 2019;37(35):3377–91. JCO1901692
Duque-Afonso J, Feng J, Scherer F, Lin CH, Wong SH, Wang Z, et al. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J Clin Invest. 2015;125(9):3667–80.
Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, et al. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 2012;22(5):656–67.
Panagopoulos I, Micci F, Thorsen J, Haugom L, Tierens A, Ulvmoen A, et al. A novel TCF3-HLF fusion transcript in acute lymphoblastic leukemia with a t(17;19)(q22;p13). Cancer Genet. 2012;205(12):669–72.
Hunger SP, Devaraj PE, Foroni L, Secker-Walker LM, Cleary ML. Two types of genomic rearrangements create alternative E2A-HLF fusion proteins in t(17;19)-ALL. Blood. 1994;83(10):2970–7.
Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015;47(9):1020–9.
Huang Y, Mouttet B, Warnatz HJ, Risch T, Rietmann F, Frommelt F, et al. The leukemogenic TCF3-HLF complex rewires enhancers driving cellular identity and self-renewal conferring EP300 vulnerability. Cancer Cell. 2019;36(6):630–44.e9
Winters AC, Bernt KM. MLL-rearranged leukemias-an update on science and clinical approaches. Front Pediatr. 2017;5:4.
Pui CH, Relling MV. Topoisomerase II inhibitor-related acute myeloid leukaemia. Br J Haematol. 2000;109(1):13–23.
Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47(4):330–7.
Valentine MC, Linabery AM, Chasnoff S, Hughes AE, Mallaney C, Sanchez N, et al. Excess congenital non-synonymous variation in leukemia-associated genes in MLL- infant leukemia: a Children’s Oncology Group report. Leukemia. 2014;28(6):1235–41.
Agraz-Doblas A, Bueno C, Bashford-Rogers R, Roy A, Schneider P, Bardini M, et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica. 2019;104(6):1176–88.
Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78.
Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14(5):355–68.
Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med. 2015;21(4):335–43.
Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277–84.
Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A Menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell. 2019;36(6):660–73.e11
Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–30.
Abou Dalle I, Jabbour E, Short NJ, Ravandi F. Treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Curr Treat Options in Oncol. 2019;20(1):4.
Slayton WB, Schultz KR, Kairalla JA, Devidas M, Mi X, Pulsipher MA, et al. Dasatinib plus intensive chemotherapy in children, adolescents, and young adults with Philadelphia chromosome-positive acute lymphoblastic leukemia: results of Children’s Oncology Group trial AALL0622. J Clin Oncol. 2018;36(22):2306–14. Jco2017767228
Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, Slayton WB, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28(7):1467–71.
Shen S, Chen X, Cai J, Yu J, Gao J, Hu S, et al. Effect of dasatinib vs imatinib in the treatment of pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: a randomized clinical trial. JAMA Oncol. 2020;6(3):358–66.
Martinelli G, Iacobucci I, Storlazzi CT, Vignetti M, Paoloni F, Cilloni D, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009;27(31):5202–7.
Chang BH, Willis SG, Stork L, Hunger SP, Carroll WL, Camitta BM, et al. Imatinib resistant BCR-ABL1 mutations at relapse in children with Ph+ ALL: a Children’s Oncology Group (COG) study. Br J Haematol. 2012;157(4):507–10.
Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014;15(3):294–304.
Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell. 2015;28(3):343–56.
Churchman ML, Evans K, Richmond J, Robbins A, Jones L, Shapiro IM, et al. Synergism of FAK and tyrosine kinase inhibition in Ph+ B-ALL. JCI Insight. 2016;1(4):e86082.
Pfeifer H, Cazzaniga G, van der Velden VHJ, Cayuela JM, Schäfer B, Spinelli O, et al. Standardisation and consensus guidelines for minimal residual disease assessment in Philadelphia-positive acute lymphoblastic leukemia (Ph + ALL) by real-time quantitative reverse transcriptase PCR of e1a2 BCR-ABL1. Leukemia. 2019;33(8):1910–22.
Hovorkova L, Zaliova M, Venn NC, Bleckmann K, Trkova M, Potuckova E, et al. Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood. 2017;129(20):2771–81.
Herold T, Schneider S, Metzeler K, Neumann M, Hartmann L, Roberts KG, et al. Philadelphia chromosome-like acute lymphoblastic leukemia in adults have frequent IGH-CRLF2 and JAK2 mutations, persistence of minimal residual disease and poor prognosis. Haematologica. 2017;102(1):130–8.
Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK, Chen K, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood. 2017;129(5):572–81.
Loh ML, Zhang J, Harvey RC, Roberts K, Payne-Turner D, Kang H, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children’s Oncology Group TARGET Project. Blood. 2013;121(3):485–8.
Reshmi SC, Harvey RC, Roberts KG, Stonerock E, Smith A, Jenkins H, et al. Targetable kinase gene fusions in high-risk B-ALL: a study from the Children’s Oncology Group. Blood. 2017;129(25):3352–61.
Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, et al. High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol. 2017;35(4):394–401.
Roberts KG, Pei D, Campana D, Payne-Turner D, Li Y, Cheng C, et al. Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol. 2014;32(27):3012–20.
Tasian SK, Hurtz C, Wertheim GB, Bailey NG, Lim MS, Harvey RC, et al. High incidence of Philadelphia chromosome-like acute lymphoblastic leukemia in older adults with B-ALL. Leukemia. 2017;31(4):981–4.
Roberts KG, Reshmi SC, Harvey RC, Chen IM, Patel K, Stonerock E, et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children’s Oncology Group. Blood. 2018;132(8):815–24.
Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood. 2017;130(19):2064–72.
Roberts KG. Why and how to treat Ph-like ALL? Best Pract Res Clin Haematol. 2018;31(4):351–6.
Roberts KG. The biology of Philadelphia chromosome-like ALL. Best Pract Res Clin Haematol. 2017;30(3):212–21.
Russell LJ, Capasso M, Vater I, Moorman AV, Akasaka T, Harder L, et al. IGH translocations involving the pseudoautosomal region 1 (PAR1) of both sex chromosomes deregulate the cytokine receptor-like factor 2 (CRLF2) gene in B cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2008;112:787. (abstract)
Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–6.
Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, et al. Down syndrome acute lymphoblastic leukemia: a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the iBFM Study Group. Blood. 2010;115(5):1006–17.
Russell LJ, Jones L, Enshaei A, Tonin S, Ryan SL, Eswaran J, et al. Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2017;56(5):363–72.
Schwartzman O, Savino AM, Gombert M, Palmi C, Cario G, Schrappe M, et al. Suppressors and activators of JAK-STAT signaling at diagnosis and relapse of acute lymphoblastic leukemia in Down syndrome. Proc Natl Acad Sci U S A. 2017;114(20):E4030–E9.
Perez-Andreu V, Roberts KG, Xu H, Smith C, Zhang H, Yang W, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015;125(4):680–6.
Mullighan CG. How advanced are we in targeting novel subtypes of ALL? Best Pract Res Clin Haematol. 2019;32(4):101095.
Roberts KG, Janke LJ, Zhao Y, Seth A, Ma J, Finkelstein D, et al. ETV6-NTRK3 induces aggressive acute lymphoblastic leukemia highly sensitive to selective TRK inhibition. Blood. 2018;132(8):861–5.
Nardi V, Ku N, Frigault MJ, Dubuc AM, Tsai HK, Amrein PC, et al. Clinical response to larotrectinib in adult Philadelphia chromosome-like ALL with cryptic ETV6-NTRK3 rearrangement. Blood Adv. 2020;4(1):106–11.
Schewe DM, Lenk L, Vogiatzi F, Winterberg D, Rademacher AV, Buchmann S, et al. Larotrectinib in TRK fusion-positive pediatric B-cell acute lymphoblastic leukemia. Blood Adv. 2019;3(22):3499–502.
Tanasi I, Ba I, Sirvent N, Braun T, Cuccuini W, Ballerini P, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood. 2019;134(16):1351–5.
Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120(17):3510–8.
Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129(2):177–87.
Inaba H, Azzato EM, Mullighan CG. Integration of next-generation sequencing to treat acute lymphoblastic leukemia with targetable lesions: the St. Jude Children’s Research Hospital approach. Front Pediatr. 2017;5:258.
Roberts KG, Yang YL, Payne-Turner D, Lin W, Files JK, Dickerson K, et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017;1(20):1657–71.
Moujalled DM, Hanna DT, Hediyeh-Zadeh S, Pomilio G, Brown L, Litalien V, et al. Cotargeting BCL-2 and MCL-1 in high-risk B-ALL. Blood Adv. 2020;4(12):2762–7.
Qin H, Cho M, Haso W, Zhang L, Tasian SK, Oo HZ, et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood. 2015;126(5):629–39.
Schinnerl D, Mejstrikova E, Schumich A, Zaliova M, Fortschegger K, Nebral K, et al. CD371 cell surface expression: a unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica. 2019;104(8):e352–e5.
Zaliova M, Potuckova E, Hovorkova L, Musilova A, Winkowska L, Fiser K, et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica. 2019;104(7):1407–16.
Zaliova M, Zimmermannova O, Dorge P, Eckert C, Moricke A, Zimmermann M, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014;28(1):182–5.
Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1(plus) defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36(12):1240–9.
Ohki K, Kiyokawa N, Saito Y, Hirabayashi S, Nakabayashi K, Ichikawa H, et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica. 2019;104(1):128–37.
Suzuki K, Okuno Y, Kawashima N, Muramatsu H, Okuno T, Wang X, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol. 2016;34(28):3451–9.
Hirano D, Hayakawa F, Yasuda T, Tange N, Yamamoto H, Kojima Y, et al. Chromosomal translocation-mediated evasion from miRNA induces strong MEF2D fusion protein expression, causing inhibition of PAX5 transcriptional activity. Oncogene. 2019;38(13):2263–74.
Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature. 2018;562(7727):373–9.
Hirabayashi S, Ohki K, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102(1):118–29.
Kim J, Kim HS, Shin S, Lee ST, Choi JR. t(12;17)(p13;q12)/TAF15-ZNF384 rearrangement in acute lymphoblastic leukemia. Ann Lab Med. 2016;36(4):396–8.
Nyquist KB, Thorsen J, Zeller B, Haaland A, Troen G, Heim S, et al. Identification of the TAF15-ZNF384 fusion gene in two new cases of acute lymphoblastic leukemia with a t(12;17)(p13;q12). Cancer Genet. 2011;204(3):147–52.
Qian M, Zhang H, Kham SK, Liu S, Jiang C, Zhao X, et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017;27(2):185–95.
Shago M, Abla O, Hitzler J, Weitzman S, Abdelhaleem M. Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer. 2016;63(11):1915–21.
Yamamoto K, Kawamoto S, Mizutani Y, Yakushijin K, Yamashita T, Nakamachi Y, et al. Mixed phenotype acute leukemia with t(12;17)(p13;q21)/TAF15-ZNF384 and other chromosome abnormalities. Cytogenet Genome Res. 2016;149(3):165–70.
Zhong CH, Prima V, Liang X, Frye C, McGavran L, Meltesen L, et al. E2A-ZNF384 and NOL1-E2A fusion created by a cryptic t(12;19)(p13.3; p13.3) in acute leukemia. Leukemia. 2008;22(4):723–9.
Hirabayashi S, Butler E, Ohki K, Kiyokawa N, Bergmann AK, Boer JM, et al. Acute lymphoblastic leukemia with zinc-finger protein 384 (ZNF384)-related rearrangements: a retrospective analysis from the Ponte Di Legno Childhood ALL Working Group. Blood. 2019;134(Supplement_1):652.
Griffith M, Griffith OL, Krysiak K, Skidmore ZL, Christopher MJ, Klco JM, et al. Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp Hematol. 2016;44(7):603–13.
Oberley MJ, Gaynon PS, Bhojwani D, Pulsipher MA, Gardner RA, Hiemenz MC, et al. Myeloid lineage switch following chimeric antigen receptor T-cell therapy in a patient with TCF3-ZNF384 fusion-positive B-lymphoblastic leukemia. Pediatr Blood Cancer. 2018;65(9):e27265.
Novakova M, Zaliova M, Fiser K, Vakrmanova B, Slamova L, Musilova A, et al. DUX4r, ZNF384r and PAX5-P80R mutated B-cell precursor acute lymphoblastic leukemia frequently undergo monocytic switch. Haematologica. 2021;106(8):2066–75.
Passet M, Boissel N, Sigaux F, Saillard C, Bargetzi M, Ba I, et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood. 2019;133(3):280–4.
Bastian L, Schroeder MP, Eckert C, Schlee C, Tanchez JO, Kampf S, et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2019;33(8):1895–909.
Nebral K, Denk D, Attarbaschi A, Konig M, Mann G, Haas OA, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23(1):134–43.
Schwab C, Nebral K, Chilton L, Leschi C, Waanders E, Boer JM, et al. Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv. 2017;1(19):1473–7.
Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG, et al. Pax5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. 2015;125(23):3609–17.
French CA. NUT carcinoma: clinicopathologic features, pathogenesis, and treatment. Pathol Int. 2018;68(11):583–95.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–8.
Nordlund J, Backlin CL, Zachariadis V, Cavelier L, Dahlberg J, Ofverholm I, et al. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin Epigenetics. 2015;7(1):11.
Hormann FM, Hoogkamer AQ, Beverloo HB, Boeree A, Dingjan I, Wattel MM, et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica. 2019;104(10):e455–e9.
Pincez T, Landry JR, Roussy M, Jouan L, Bilodeau M, Laramée L, et al. Cryptic recurrent ACIN1-NUTM1 fusions in non-KMT2A-rearranged infant acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2020;59(2):125–30.
Cobb BS, Morales-Alcelay S, Kleiger G, Brown KE, Fisher AG, Smale ST. Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev. 2000;14(17):2146–60.
Boutboul D, Kuehn HS, Van de Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. 2018;128(7):3071–87.
Kimura S, Seki M, Yoshida K, Shiraishi Y, Akiyama M, Koh K, et al. NOTCH1 pathway activating mutations and clonal evolution in pediatric T-cell acute lymphoblastic leukemia. Cancer Sci. 2019;110(2):784–94.
Ferrando AA, Lopez-Otin C. Clonal evolution in leukemia. Nat Med. 2017;23(10):1135–45.
Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2016;113(40):11306–11.
Dobson SM, Garcia-Prat L, Vanner RJ, Wintersinger J, Waanders E, Gu Z, et al. Relapse-fated latent diagnosis subclones in acute B lineage leukemia are drug tolerant and possess distinct metabolic programs. Cancer Discov. 2020;10(4):568–87.
Tzoneva G, Dieck CL, Oshima K, Ambesi-Impiombato A, Sanchez-Martin M, Madubata CJ, et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature. 2018;553(7689):511–4.
Dieck CL, Ferrando A. Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. Blood. 2019;133(21):2263–8.
Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45(3):290–4.
Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19(3):368–71.
Li B, Li H, Bai Y, Kirschner-Schwabe R, Yang JJ, Chen Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21(6):563–71.
Evensen NA, Madhusoodhan PP, Meyer J, Saliba J, Chowdhury A, Araten DJ, et al. MSH6 haploinsufficiency at relapse contributes to the development of thiopurine resistance in pediatric B-lymphoblastic leukemia. Haematologica. 2018;103(5):830–9.
Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469.
Rusch M, Nakitandwe J, Shurtleff S, Newman S, Zhang Z, Edmonson MN, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun. 2018;9(1):3962.
Karvonen H, Perttilä R, Niininen W, Hautanen V, Barker H, Murumägi A, et al. Wnt5a and ROR1 activate non-canonical Wnt signaling via RhoA in TCF3-PBX1 acute lymphoblastic leukemia and highlight new treatment strategies via Bcl-2 co-targeting. Oncogene. 2019;38(17):3288–300.
Churchman ML, Jones L, Evans K, Richmond J, Shapiro IM, Pachter JA, et al. Efficacy of focal adhesion kinase inhibition in combination with dasatinib in BCR-ABL1 acute lymphoblastic leukemia. Blood. 2015;126(23):3766.
Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740–53.
Jung M, Schieck M, Hofmann W, Tauscher M, Lentes J, Bergmann A, et al. Frequency and prognostic impact of PAX5 p.P80R in pediatric acute lymphoblastic leukemia patients treated on an AIEOP-BFM acute lymphoblastic leukemia protocol. Genes Chromosomes Cancer. 2020;59(11):667–71.
Acknowledgments
The authors thank colleagues at St Jude Children’s Research Hospital, the Children’s Oncology Group, the NCI TARGET initiative, and colleagues for collaborative research described in this review. C.G.M. is supported by the St Jude Comprehensive Cancer Center Support Grant NCI P30 CA021765 and an NCI Outstanding Investigator Award R35 CA197695, the Henry Schueler 41&9 Foundation, a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (to C.G.M.), and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital.
Declaration of Competing Interest
Charles Mullighan has received research funding from AbbVie, Pfizer, and Loxo Oncology and consulting fees from Illumina.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Roberts, K.G., Mullighan, C.G. (2022). Molecular Pathways and Targets in B-Cell Progenitor Acute Lymphoblastic Leukemia. In: Litzow, M.R., Raetz, E.A. (eds) Clinical Management of Acute Lymphoblastic Leukemia. Springer, Cham. https://doi.org/10.1007/978-3-030-85147-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-85147-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-85146-0
Online ISBN: 978-3-030-85147-7
eBook Packages: MedicineMedicine (R0)