
Abstract
Computational prediction of drug-target interaction (DTI) is very important for the new drug discovery. Currently, graph convolutional networks (GCNs) have been gained a lot of momentum, as its performance on non-Euclidean data. Although drugs and targets are two typical non-Euclidean data, many problems are also existed when use GCNs in DTI prediction. Firstly, most state-of-the-art GCN models are prone to the vanishing gradient problem, which is more serious in DTI prediction, as the number of interactions is limit. Secondly, a suitable graph is hardly defined for GCN in DTI prediction, as the relationship between the samples is not explicitly provided. To overcome the above problems, in this paper, a multiple output graph convolutional network (MOGCN) based DTI prediction method is designed. MOGCN enhances its learning ability with many new designed auxiliary classifier layers and a new designed graph calculation method. Many auxiliary classifier layers can increase the gradient signal that gets propagated back, utilize multi-level features to train the model, and use the features produced by the higher, middle or lower layers in a unified framework. The graph calculation method can use both the label information and the manifold. The conducted experiments validate the effectiveness of our MOGCN.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Drug-target interactions
- Graph convolutional network
- Multiple output deep learning
- Auxiliary classifier layer
1 Introduction
The deep learning based DTI prediction method has recently been taken more and more attention, which can be divided into three categories: deep neural networks (DNN) based method, convolutional neural networks (CNN) based method, and graph convolutional networks (GCN) based method.
Many DNN based DTI prediction methods have been proposed. Firstly, different inputs have been designed for DNN [1, 2]. Secondly, different DNN structures have been designed for DTI prediction [3,4,5,6]. Thirdly, some researchers used DNN together with other methods [7, 8]. DNN can effectively improve the effect of DTI prediction. However, a large number of parameters need to be trained. CNN is mainly composed of convolutional layers, which can greatly reduce the parameters.
Many CNN based DTI prediction methods have been proposed, where the main difference is designing different inputs. Firstly, reshaped the extracted feature vector to generate the input is a popular method [9, 10]. Secondly, the sequence of target and the SMILES of drug are used as the input [7, 11, 12]. However, it’s may be difficult for CNN to extract the feature from the drug and the targets, as local features are distributed in different locations in different sequence and SMILES. GCNs have been gained a lot of momentum in the last few years, as the limited performance of CNNs when dealing with the non-Euclidean data. Drugs and targets are two typical non-Euclidean data, and then GCNs could be promising for DTI prediction.
Many GCN based DTI prediction methods have been proposed. Firstly, GCNs have been used to extract features from the drug molecular [13,14,15]. However, the optimization goal of the GCN and the optimization goal of classifier are independent in Ref. [13, 15], and the graph is constructed by atoms but not by molecules in Ref. [14]. Secondly, GCNs have been used for dimensionality reduction [16, 17]. However, GCN optimization goals and classification optimization goals are also independent in these methods. Thirdly, GCNs has been used for classification [18]. However, the GCN structure contains only one GCN layer, and he graph used by GCN is calculated according to DDI and PPI, which is hardly automatically predicted.
Many problems can be concluded from the above methods: Firstly, goals of GCN optimization and classification optimization are optimized independently [13, 15,16,17], and then the classification ability of GCN is not utilized. Secondly, a shallow GCN is used for DTI prediction [18]. Thirdly, the graph could be difficult calculated in real application in Ref. [18]. Furthermore, the graph of many methods is calculated by bonds of drugs [13,14,15,16] but not the relationships among drug-target pairs.
To overcome the first and second problems, a multiple output graph convolutional network (MOGCN) is designed, which contains many auxiliary classifier layers. These auxiliary classifier layers are distributed in the low, middle and high layers. As a result, MOGCN owns following advantages: losses in different layers can increase the gradient signal that gets propagated back; multi classification regularizations can be used to optimize the MOGCN; the model can be trained by multi-level features; features in different levels can be used for classification in a unified framework. To overcome the third and fourth problems, a DNN and k-nearest neighbor (KNN) based method is designed to calculate the graph, where the calculated graph contains both the manifold and the label information. To further overcome the second problem, two auto-encoders are respectively used to learn low-level features for drugs and targets, which can make that the MOGCN can mainly focus on learning high-level features.
2 Proposed Method
2.1 Problem Description and Notation Definition
The goal of DTI prediction is to learn a model that takes a pair of drug and target and output the interaction. Before introducing our method, many important notations adopted in this paper are provided in Table 1.
2.2 Overview of Our Method
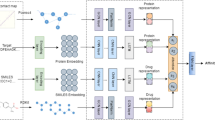
The overview of our method is shown in the Fig. 1, which contains 5 steps, such as feature extraction, low-level features extraction, feature concatenation, graph calculation, and MOGCN. The feature extraction is used to extract the features for drugs and targets. The low-level features extraction is used to learn the low-level features for drugs and targets. The feature concatenation is used to generate the sample of the drug-target pair. The graph calculation is used to calculate the adjacency matrix A for MOGCN. The MOGCN is our newly designed deep learning framework. These steps will be described in detail in the following subsections.

Overview of our method
2.3 Feature Extraction
Drugs features \(D = [d_{1} ,d_{2} , \cdots ,d_{n} ]^{T}\) and targets features \(T = [t_{1} ,t_{2} , \cdots ,t_{m} ]^{T}\) are respectively calculated by PaDEL [19] and the propy tool [20]. The drug features include 797 descriptors (663 1D, 2D descriptors, and 134 3D descriptors) and 10 types of fingerprints. The total number of the drug features is 15329. The target features include five feature groups such as: amino acid composition, autocorrelation, composition, transition and distribution, quasi-sequence order, pseudo-amino acid composition and so on. The total number of the target features is 9543.
2.4 Low-Level Features Extraction and Feature Concatenation
Training GCN directly on high-dimensional drug features and target features will cause too many parameters to be trained in the first layer. Furthermore, with the larger depth of the network, the ability to propagate gradients back through all the layers in an effective manner was a serious concern, which is more serious for the first few layers. This leads to insufficient extraction of low-level features. To overcome the above problem, low-level features are firstly extracted by the stack auto encoder (SAE). Because the goal of the SAE is to extract the low-level features, the shallow encoder and decoder are used in this paper. The encoder and decoder of the SAE contain two linear layers and two activation layers, where the sigmoid function is used for the activation layer. The output of the encoder is the low-level feature.
After training the encoder for drugs by D, the low-level features \(\hat{D}\) of D can be obtained by the encoder. Similarly, the low-level features \(\hat{T}\) of T can be obtained by the encoder trained by T. The u samples \(X = [x_{1} ,x_{2} , \cdots ,x_{u} ]\) can be generated from \(\hat{D}\) and \(\hat{T}\), where \(xk = [\hat{d}_{i} ,\hat{t}_{j} ]^{T}\). The corresponding u labels \(L = [l_{1} ,l_{2} , \cdots ,l_{u} ]\) can be generated from I, where lk is the i-th row and j-th column of I.
2.5 Graph Calculation
An adjacency matrix A should be calculated for MOGCN. Many graph calculation methods have been proposed for DTI prediction. Zhao et.al calculated A by DDI and PPI; however, DDI and PPI are also hardly automatically predicted [18]. And many other A are calculated by bonds of drugs [13,14,15,16,17], however, these graphs are only used for extracting features. Analyzed the function of A, A should represent the relationship between samples, especially the classification relationship between samples. However, the labels of testing samples are unknown. As a result, to calculate the A, a DNN and KNN based method is designed.
Given training samples XL, corresponding label LL, testing samples XT, the DNN and KNN based method owns 4 steps. Firstly, a two layers DNN model is trained. And then XL and XT are projected to \(\hat{Y}_{L}\) and \(\hat{Y}_{T}\) by the first layer of the trained DNN model. Thirdly, \(\hat{Y}\) is calculated by \(\hat{Y} = [\hat{Y}_{L} ;\hat{Y}_{T} ]\). In the last, the A can be calculated by Eq. (1).
A two layers DNN model is trained here, as DNN is only used to learn a low dimensional space that contains both manifold information or label information. Furthermore, low dimensional space is obtained by the first linear sub layer but not by the second linear sub layer, as the result of second linear sub layer is too much affected by the label but the label of the testing sample is unknown. According to above analyze, the proposed graph calculation method can make that \(A_{ij}\) has a greater probability of being set to 1 when xi and xj are near with each other or xi and xj are with the same labels. As a result, A contains both manifold and label information.
2.6 The Proposed MOGCN Model
Unlike DNN, which are able to take advantage of stacking very deep layers, GCNs suffer from vanishing gradient, over-smoothing and over-fitting issues when going deeper [21]. These challenges are particularly serious in DTI prediction, as the number of interactions is limited in the DTI datasets. Specifically, most interactions focus on only a few targets or a few drugs, which makes that the training samples of most drugs and targets are not enough. As a result, these challenges limit the representation power of GCNs on DTI prediction. For example, Zhao et.al used a GCN structure with one GCN layer [18]. To overcome the above problem, in this paper, a MOGCN for DTI prediction is designed.

Overview of MOGCN
Given the training samples XL, the corresponding labels LL, and the adjacency matrix A, the MOGCN can be shown in the Fig. 2, which contains an input layer, an output layer, an linear layer, many hidden layers and many auxiliary classifier layers. The hidden layer contains a GCN sub layer, an activation sub layer and a dropout sub layer. The auxiliary classifier layer is a new designed layer, which contains a GCN sub layer, an activation sub layer, a linear sub layer, and a loss function.
Given Xg as the input of the g-th layer, the GCN sub layer can be defined as:
Where \(\tilde{A}\) is the graph adjacency matrix with add self-loop for A, \(Z\) is the graph diagonal degree matrix which can be calculated from A, \(A \in R^{u \times u}\) is a graph adjacency matrix that can be calculated by Algorithm 1.
The definition of the activation and dropout sub layers can be found from [22].
For an auxiliary classifier layer, a linear sub layer and a loss function are existed here, which can be respectively defined as Eq. (3) and Eq. (4):
The loss function of the output layer can be defined as:
W and b can be optimized by minimizing Eq. (6) with the Adam optimizer [25].
Where \(J_{g} (W,b,X,L_{L} ) = 0\), if g-th layer is a hidden layer.
It can be seen from Fig. 2 and Eq. (6) that the number of layers of propagate gradients back is smaller when g is smaller. And then the effective of \(J_{g} (W,b,X,L_{L} )\) for calculating Wi and bi is higher. Furthermore, the stronger performance of relatively shallower networks on this task suggests that the features produced by the layers in the middle of the network should be more discriminative. As a result, by adding these auxiliary classifiers layers connected to these intermediate layers, MOGCN would expect to increase discrimination in the lower stages in the classifier, enhance the gradient signal used for propagated back, and provide additional regularization.
2.7 Architectural Parameter
Many architectural parameters are existed in the low-level features extraction, graph calculation and MOGCN.
In the low-level features extraction, output dimensions of two encoder layers should be set, which are set to 800 and 500. Similarly, the output dimensions of two decoder layers are set to 500 and 800. These output dimensions have little effect on the results in a larger range, as encoder layers is only used for low-level features extraction, so that they are simply set to the above values.
In the graph calculation, the output dimensions of two linear layers should be set, which are respectively set to 100, 2, where 2 is the number of the class. k used in Eq. (4) is set to 10. These values are also have little effect on the results in a larger range, as DNN is only used to learn low dimensional space and k is only used to define the manifold, so that they are simply set to the above values.
In the MOGCN, the layer number G, the output dimensions of the GCN sub layer and the linear sub layer should be set. To simplify the parameter search, the output dimensions of all GCN sub layers are set to 300, as the ability of the MOGCN can also be controlled by depth. The output dimensions of all linear sub layers are set to 2, as the number of the class is 2. The layer number G is set to 1, 3, 5, which is chosen by 5-fold cross validation. G is not chosen from a larger range, as a larger MOGCN structure could be hardly trained when the positive examples is limited.
3 Experiments
3.1 Dataset
Nuclear receptors (NR), G protein coupled receptors (GPCR), ion channels (IC), enzymes (E) [23] and drug bank (DB) [24] are used here. Simple statistics of them are represented in the Table 2. The 2-th to 4-th rows respectively represents the number of drugs, targets and interactions, which shows that the number of interactions is small and the number of drug target pairs is far more than that of interactions.
3.2 Compared Methods
MOGCN is a GCN based method, so that it is compared with a GCN based method, which is come from MOGCN by replacing each auxiliary classifier layer of MOGCN with a hidden layer. MOGCN is also compared with two DNN structures [3, 5], as these two DNN structures may be good and both DNN and GCN belong to deep learning. These two DNN structures are defined as DNN1 [3] and DNN2 [5].
Furthermore, SAE is used to learn the low level features in this paper. To prove that SAE is benefit for MOGCN, low level features and the original features are respectively inputted to the compared methods. As a result, after defining the original features as SRC and the low level features as SAE, following methods are compared: SRC+DNN1, SRC+DNN2, SRC+GCN, SRC+MOGCN, SAE+DNN1, SAE+DNN2, SAE+GCN and SAE+MOGCN.
3.3 Experimental Setting
In this work, three experimental settings that are shown in the Table 3 are evaluated. CVD and CVT and CVP respectively represent the corresponding DTI values of certain drugs, targets and interactions in the training set are missed but existed in the test sets, where CVD can be used for new drug development, CVT can be used to find effective drugs from known drugs for new targets, CVP can be used to find new interactions from known drugs and known targets.
A standard 5-fold cross validation is performed. More precisely, drugs, targets and interactions are respectively divided into 5 parts in CVD, CVT and CVP, where one part is used for testing and other parts are used for training. Furthermore, for the training samples, not all negative samples but five times more negative samples than positive samples are used, as using too more negative samples will make the data imbalance problem too prominent and using too few negative samples will result in too much negative sample information loss. The used negative samples are randomly selected all negative samples. The batch size is set to 128 for DNN1 and DNN2. The batch size is set to 1 for GCN and MOGCN, as GCN based methods trained by the batched training samples is still difficult. Furthermore, two evaluation metrics named AUC and AUPR are used in the experiments, where AUC is the area under the ROC and AUPR is the area under the precision-recall curve.
3.4 The CVD Experiments
CVD can be used for new drug development. The experiment results are presented in the Fig. 3. Following concludes can be obtained from the Fig. 3:
Firstly, GCN based methods are better than DNN based methods. It can be seen from Fig. 3 that most evaluation metrics of GCN based methods are higher than that of DNN based methods, and all evaluation metrics of the SRC+MOGCN and SAE + MOGCN are higher than that of the DNN based methods. They show that using GCN based methods to improve the DTI prediction is necessary.
Secondly, the low level features are better than the original features for GCN based methods. It can be seen from Fig. 3 that AUCs and AUPRs of SAE+GCN and SAE+MOGCN are higher than that of SRC+GCN and SRC+MOGCN on IC, GPCR, E and DB, and most evaluation metrics of SAE+MOGCN are higher than that of SRC+MOGCN on NR. They prove that using SAE to learn low level features is benefit for GCN and MOGCN.

Results of CVD experiments
Thirdly, MOGCN is better than GCN. It can be seen from Fig. 3 that AUCs and AUPRs of SRC+MOGCN and SAE+MOGCN are higher than that of SRC+GCN and SAE+GCN on all datasets. It shows that using the multiple output graph convolutional network structure can improve the effectiveness of DTI prediction.
Fourthly, SAE+MOGCN is the best. It can be seen from Fig. 3 that most evaluation metrics of SAE+MOGCN are higher than that of the compared methods. Specially, AUCs of SAE+MOGCN are respectively 2.56%, 1.58%, 0.75%, 1.36%, and 0.92% higher than that of the second best methods on datasets. AUPRs of SAE+MOGCN are respectively 3.27%, 2.14%, 0.39%, 1.25%, 1.36%, and 1.55% higher than that of the second best methods on datasets. They show that using SAE together with MOGCN is a good method for DTI prediction.
3.5 The CVT Experiments
CVT can be used to find effective drugs for new targets. The experiment results are presented in the Fig. 4. Following concludes can be obtained from the Fig. 4:
Firstly, GCN based methods are better than DNN based methods. It can be seen from Fig. 4 that nearly all evaluation metrics of GCN based methods are higher than that of DNN based methods on IC, GPCR, E and DB, where only some evaluation metrics of SRC+GCN lower than that of SRC+DNN2. Furthermore, it can be seen from Fig. 4 that AUCs and AUPRs of the SAE+MOGCN are higher than that of the DNN based methods on NR. They prove that using GCN based methods to improve the DTI prediction is necessary.
Secondly, the low level features are better than the original features for GCN based methods. It can be seen from Fig. 4 that AUCs and AUPRs of SAE+GCN and SAE+MOGCN are all higher than that of SRC+GCN and SRC+MOGCN. It shows that using SAE to learn low level features is benefit for GCN and MOGCN.

Results of CVT experiments
Thirdly, MOGCN is better than GCN. It can be seen from Fig. 4 that AUCs and AUPRs of SRC+MOGCN and SAE+MOGCN are higher than that of SRC+GCN and SAE+GCN. It shows that using the multiple output graph convolutional network structure can improve the effectiveness of DTI prediction.
Fourthly, SAE+MOGCN is the best. It can be seen from Fig. 4 that most evaluation metrics of SAE+MOGCN are higher than that of the compared methods. Specially, AUCs of SAE+MOGCN are respectively 0.70%, 1.05%, 2.88%, 1.02% and 0.97% higher than that of the second best methods. AUPRs of SAE+MOGCN are respectively 5.16%, -0.46%, 3.20%, 0.68%, and 2.11% higher than that of the second best methods. They prove that using SAE together with MOGCN is also a good method for DTI prediction.
3.6 The CVP Experiments
CVP can be used to find effective drugs from known drugs for new targets. The experiment results of all compared methods are presented in the Fig. 5. Following concludes can be obtained from the Fig. 5:
Firstly, the GCN based methods are better than the DNN based methods. It can be seen from Fig. 5 that the results of all evaluation metrics of GCN based methods are higher than that of DNN based methods. It proves that GCN based methods are better than the DNN based methods.
Secondly, the low level features are better than the original features for GCN based methods. It can be seen from Fig. 5 that most AUCs and AUPRs of SAE+MOGCN are higher than that of SRC+MOGCN on all databases, and most evaluation metrics of SAE+GCN are higher than that of SRC+GCN on IC and GPCR. They show that using SAE to learn low level features is benefit for GCN and MOGCN.
Thirdly, MOGCN is better than GCN. It can be seen from Fig. 5 that most evaluation metrics of SRC+MOGCN and SAE+MOGCN are higher than that of SRC + GCN and SAE+GCN. It proves that using the multiple output graph convolutional network structure can improve the effectiveness of DTI prediction.
Fourthly, SAE+MOGCN is the best. It can be seen from Fig. 5 that most evaluation metrics of SAE+MOGCN are higher than that of the compared methods. Specially, AUCs of SAE+MOGCN are respectively 0.81%, 0.34%, 0.91%, 0.50% and 0.85% higher than that of the second best methods on datasets. AUPRs of SAE+MOGCN are respectively 1.16%, 1.49%, −0.29%, 0.62% and 1.55% higher than that of the second best methods on datasets. They prove that using SAE together with MOGCN is a good method for DTI prediction.

Results of CVP experiments
4 Conclusion
This paper presents a MOGCN for DTI prediction. In MOGCN, SAE is used to learn low level features, a DNN and KNN based method is used to calculate the graph, and many auxiliary classifier layers are added to connect to the intermediate layers and the parameters are trained by multi loss functions. SAE makes that MOGCN can mainly focus on learning high-level features. The graph calculation method can makes that the graph contains both the label information and the manifold. The auxiliary classifier layers can increase discrimination in the lower stages in the classifier, enhance the gradient signal used for propagated back, and provides additional regularization. The conducted experiments validate that MOGCN is a competitive method compared to the previous ones.
As one of the further work, residual network family can used to increase the width and depth of the GCN. As another further work, a new GCN framework is used to calculate the graph, which can minimizing the distances among samples whose drugs and targets are with interactions and maximizing the distances among samples whose drugs and targets are without interactions.
References
Wang, H., Wang, J., Dong, C.: A novel approach for drug-target interactions prediction based on multimodal deep autoencoder. Front. Pharmacol. 10, 1592 (2020)
Sturm, N., et al.: Industry-scale application and evaluation of deep learning for drug target prediction. J. Cheminform. 12(1), 1–13 (2020). https://doi.org/10.1186/s13321-020-00428-5
Xie, L.W., et al.: Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genom. 19(667), 93–102 (2018)
Wang, L., et al.: Computational methods for the prediction of drug-target interactions from drug fingerprints and protein sequences by stacked auto-encoder deep neural network. Bioinform. Res. Appl. 10330, 46–58 (2017)
Wen, M., et al.: Deep learning-based drug-target interaction prediction. J. Proteome Res. 16(4), 1401–1409 (2017)
Wang, Y.B. et al. A deep learning-based method for drugtarget interaction prediction based on long short-term memory neural network, BMC Medical Informatics and Decision Making, 20(49), 1–9 (2020)
Lee, I., Keum, J., Nam, H.: DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences, PLOS Comput. Biol. 15(6) (2019)
Zhang, Y.F., et al.: SPVec: A Word2vec-inspired feature representation method for drug-target interaction prediction. Front. Chem. 7, 1–11 (2020)
Rayhan, F. et al. FRnet-DTI: Deep convolutional neural network for drug-target interaction prediction, Heliyon. 6(3), e03444 (2020)
Hu, S.S. et al. A convolutional neural network system to discriminate drug-target interactions, IEEE/ACM Trans. Comput. Biol. Bioinform. (2019), in press.
Monteiro, N.R.C., Ribeiro, B., Arrais, J.: Drug-target interaction prediction: end-to-end deep learning approach. IEEE/ACM Trans. Comput. Biol. Bioinform. (2020). https://doi.org/10.1109/TCBB.2020.2977335
Rifaioglu, A.S. et al. DEEPScreen: high performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations, Chem. Sci. 9, 1–68 (2020)
David, D. et al. Convolutional networks on graphs for learning molecular fingerprints. In: Proceedings of the 28th International Conference on Neural Information Processing Systems, pp. 2224–2232 (2015)
Feng, Q.Y. et al. PADME: A Deep Learning-based Framework for Drug-Target Interaction Prediction. Comput. Sci. arXiv:1807.09741 (2019)
Gao K.Y. et al. Interpretable drug target prediction using deep neural representation. In: International Joint Conference on Artificial Intelligence, pp. 3371–3377 (2018)
Torng, W., Altman, R.B.: Graph convolutional neural networks for predicting drug-target interactions. J. Chem. Inform. Model. 59(10), 4131–4149 (2019). https://doi.org/10.1021/acs.jcim.9b00628
Sun, C., Xuan, P., Zhang, T., Ye, Y.: Graph convolutional autoencoder and generative adversarial network-based method for predicting drug-target interactions. IEEE/ACM Trans. Comput. Biol. Bioinform. (2020). https://doi.org/10.1109/TCBB.2020.2999084
Zhao, T.Y. et al. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief. Bioinform. bbaa044, 1–10 (2020)
.Yap, C.W.: PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 32(7), 1466–1474 (2011)
Sheng, D., Liang, Q.S., Zeng, Y.: Propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 29(7), 960–962 (2013)
Li, G.H., Xiong, C.X., Thabet, A., Bernard Ghanem. DeeperGCN: All You Need to Train Deeper GCNs. arXiv:2006.07739 (2020)
Bengio, Y.: Learning deep architectures for AI, in Learning Deep Architectures for AI (2009)
Yamanishi, Y., Araki, M., Gutteridge, A., et al.: Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 24(13), 1232–1240 (2008)
DrugBank Release Version 5.1.7, https://go.drugbank.com/releases/latest
Kingma, D.P., Ba, L.J. Adam: A method for stochastic optimization. In: International Conference on Learning Representations (2015)
Acknowledgments
The authors thank the members of Machine Learning and Artificial Intelligence Laboratory, School of Computer Science and Technology, Wuhan University of Science and Technology, for their helpful discussion within seminars. This work was supported by, National Natural Science Foundation of China (No.61972299, 61502356), Zhejiang Provincial Natural Science Foundation (No.LQ18F020006, LQ18F020007), Hubei Province Natural Science Foundation of China (No. 2018CFB526).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Ye, Q., Zhang, X., Lin, X. (2021). Drug-Target Interaction Prediction via Multiple Output Graph Convolutional Networks. In: Huang, DS., Jo, KH., Li, J., Gribova, V., Premaratne, P. (eds) Intelligent Computing Theories and Application. ICIC 2021. Lecture Notes in Computer Science(), vol 12838. Springer, Cham. https://doi.org/10.1007/978-3-030-84532-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-84532-2_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84531-5
Online ISBN: 978-3-030-84532-2
eBook Packages: Computer ScienceComputer Science (R0)