
Abstract
Fungal epidemics, especially in the form of parasitic chytrids, are omnipresent in aquatic environments, infecting diverse organisms. Major target hosts are algae, primarily diatoms, chlorophytes, and colonial or filamentous cyanobacteria. Chytrids are also called ‘zoosporic’ organisms because their life cycle includes dispersal forms, i.e. uniflagellate zoospores, and host-associated infective sporangia as well. They are considered relevant not only for the evolution of their hosts but also for the population dynamics and successions of phytoplankton communities, thus representing an important ecologically driving force in the food web dynamics. However, ecological knowledge of microscopic fungal parasites in aquatic environments is weak, compared to terrestrial ecosystems. We propose a routine protocol based on size fractionation of pelagic samples and the use of the fluorochrome calcofluor white (which binds to β-1,3 and β-1,4 polysaccharides) for diagnosing, identifying, and counting chitinous fungal parasites (i.e. the sporangia of chytrids). The protocol offers a valid method for the quantitative ecology of chytrid epidemics in aquatic ecosystems and food web dynamics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Introduction
Fungal infections are recurrent in aquatic ecosystems [1, 2]. The most described aquatic fungi in freshwater ecosystems belong to Chytridiomycota (or chytrids). Chytrids infect a wide variety of hosts, including fish, eggs, zooplankton, and other aquatic fungi but especially phytoplankton. Typical phytoplankton hosts include prokaryotes and eukaryotes, primarily large-size diatoms and filamentous species [3]. Associated chytrids are external eucarpic parasites which produce a specialized rhizoidal system within host cells, i.e. the diet conveying system that leads to the formation of the chitinous fruit bodies: the sporangium. This parasitic stage produces numerous uniflagellate spores: the zoospores, which constitute the dissemination phase of the life cycle [4].
Various approaches have been used to study fungal parasites, but routine techniques for reliably identifying and counting these organisms are missing in the context of aquatic microbial ecology [5, 6]. So far, observations of parasitic fungi were obtained by using phase contrast light microscopy with live or Lugol’s iodine preserved samples [7,8,9]. Such conventional microscopy allows observation of fungal sporangia or similar forms (especially in laboratory cultures) but is a poor approach for characterizing chytrid parasites in natural samples, at the complex community level. For example, a simple light microscopy observation of fungal rhizoidal systems, i.e. a pertinent criterium for identifying chytrids [4, 10, 11], is very difficult. This situation may help explain the confusion of chytrids with protistan flagellates such as choanoflagellates or other bacterivorous flagellates in the group of Bicosoeca, which are attached to phytoplankton but do not harm their host [5,6,7].
Earlier studies on chytrids were restricted to morphological descriptions and focused on few species [12,13,14,15]. Electron microscopy was used to describe different life stages and the ultrastructural cytology of fungal zoospores and spore differentiation [16,17,18], providing the basis for chytrid taxonomy [19, 20]. Studies on pelagic chytrids started in the British lakes [21], and different authors have provided descriptions of morphological characters [22,23,24,25,26]. However, few attempts have been made to include the related parasitism pathway in the aquatic food web dynamics and to understand environmental factors that trigger epidemics as well [27]. Some authors have also investigated the effects of parasitism on the growth of algal host species and on the genetic structure of infected populations [28]. Parasites are thus considered relevant not only for the evolution of their hosts but also for the population dynamics such as successions of phytoplankton communities and for structuring microbial communities in general [9, 29]. Moreover, chytrids can represent interesting key intermediates in the food chain [30, 31]. The nutrients from infected large-size algae which could not be directly fed by zooplankton can be transferred from sporangium to grazers via fungal zoospore production. Fungal zoospores have suitable dimensions and represent a valuable food source for zooplankton [32]. The activity of zoosporic fungi and the related biogeochemical processes can thus be crucial in matter and energy transfer in aquatic systems [29]. Methodological limitations for the study of the ecological dynamics of chytrid populations can be overcome with epifluorescence microscopy coupled to a specific fluorochrome targeting molecular tracers (i.e. some types of polysaccharides) of the fungal chitinous structures, including sporangium and the rhizoidal system.
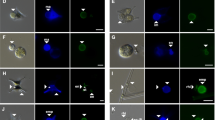
The protein stain fluorescein isothiocyanate (FITC) and, in particular, the chitin stain calcofluor white (CFW) were suggested as good markers that offer useful tools for the investigation of fungal dynamics in aquatic samples [33]. CFW binds to β1–3 and β1–4 polysaccharides such as those found in cellulose or in chitin which commonly occur in the fungal cell wall [1, 2]. It fluoresces when exposed to UV light and is currently used in clinical mycology for direct microscopic examination of skin scrapings, hairs, nails, and other clinical specimens for fungal elements [34, 35]. In contrast to FITC, CFW penetrates into infected host cells and is more efficient for the observation of the complete rhizoidal system of parasites (Fig. 4.1), i.e. a pertinent criterion for chytrid identification [4, 10, 11].

Calcofluor white (CFW) staining of a chytrid infected filamentous cyanobacteria observed under an epifluorescence microscope using the proposed protocol. CFW penetrates well into infected host cells and is more efficient for the observation of both the fungal chitinaceous sporangium and the complete rhizoidal system within the host
The main objective of this chapter is to provide, in a simplified step-by-step format, a routine protocol based on size fractionation of pelagic samples and the use of the fluorochrome calcofluor white for diagnosing, identifying, and counting chitinous fungal parasites (i.e. sporangia of chytrids) within phytoplanktonic communities [3], together with practical advice on how to apply the method.
4.2 Materials
-
1.
25 μm nylon filter
-
2.
0.2 μm filters
-
3.
High-performance concentration/diafiltration system. As an example, we use the system Amicon model DC 10LA (Epernon, France) equipped with a reusable hollow fibre cartridge (0.2 μm cutoff, surface area of 0.45 m2).
-
4.
36% Formaldehyde.
-
5.
Calcofluor white (C40H44N12O10S2 fluorescent brightener 28; Sigma catalog no. F3543).
-
6.
10 N NaOH
-
7.
Balance.
-
8.
Distilled water.
-
9.
15 ml and 0.2 ml tubes
-
10.
Glass slides and coverslips.
-
11.
Epifluorescence microscope equipped with appropriate UV filter sets and Neofluar objective lens (optional).
4.3 Methods
4.3.1 Concentrations of Cells (See Note 1)
-
1.
Pass the sample (ca. 20 L) through the 25 μm pore size nylon filter (see Note 2).
-
2.
Collect large phytoplankton cells in the >25 μm size fraction by washing the filter with 40 mL of 0.2 μm filtered natural sample.
-
3.
Fix the concentrate sample with formaldehyde (2% final conc.), before staining and analysis.
-
4.
Concentrate nanoplanktonic cells in the <25 μm size fraction (i.e. the 20 L filtrate) ca 20x by ultrafiltration to a volume of approximately 1 L, entry pressure 0.9 bar.
-
5.
Fix about 180 mL of the ultrafiltrate retentate with formaldehyde (2% final conc.), before staining and analysis.
4.3.2 Preparation of Calcofluor Stock Solution
-
1.
Weigh 35 mg of calcofluor white into a 15 mL tube.
-
2.
Add 7 mL of sterile distilled water and 2–3 drops of 10 N NaOH (to increase pH to 10–11). Calcofluor does not dissolve well in neutral solutions.
-
3.
Dissolve the calcofluor.
-
4.
Adjust the volume to 10 mL by adding sterile distilled water.
-
5.
Distribute the stock solution in 0.2 ml tubes and store in a lightproof tube at −20 °C.
4.3.3 Staining and Visualization
-
1.
In the dark, stain aliquots (about 200 μl) of concentrated and fixed materials by adding 1–2.5% (vol/vol) of CFW stock solution directly in solution for 10 min.
-
2.
Mount drop (5–10 μl) of the stained samples between glass slides and coverslips for observations and counting.
-
3.
In a dark room, examine the slides under an epifluorescence microscope equipped with an appropriate set of filters and objective lenses. Shift between white and UV light to visualize and determine parasites and phytoplankton cells, and check the viability of the host cell, e.g. presence of chloroplast.
-
4.
Applied a standard procedure for microscopic counting (see Notes 3 and 4).
4.4 Notes
-
1.
Different approaches were tested to concentrate samples: the total community approach and the size-fractionated community approach. For the former approach, 180 ml of experimental samples were fixed with formaldehyde (2% final conc.) and aliquots concentrated in three different ways: (i) by simple gravity following Utermöhl’s [36] method before staining the chyrids, (ii) by vacuum pressure on two different filters before staining directly onto filters, and (iii) by vacuum pressure on the same two types of filters but after staining in solution.
For the Utermöhl method, 100 ml of fixed samples were settled for at least 24 h. For each of the two filter-vacuum pressure methods, 10 ml × 2 of fixed samples were filtered onto polycarbonate white filters (pore size 0.6 μm, catalog no. DTTP02500, Millipore) and nuclepore polycarbonate black filters (pore size 0.8 μm, catalog no. 110659, Whatman), by using gentle vacuum (< 0.2 bar or 20 kPa). For the total community approach using the classical Utermöhl [36] method, visualization of fungal parasites was very difficult and most of the time practically impossible for all the stain concentrations tested. The main reason was that staining directly in the Utermöhl chamber resulted in very poor-quality specimens of parasites observed in any given sample. Other disadvantages of the procedure include the relatively long sedimentation time and the difficulty of increasing the volume analyzed.
The alternative total community approaches based on vacuum pressure concentrations on polycarbonate filters, i.e. white (0.6-μm-pore-size) and black (0.8-μm-pore-size) filters, yielded similar quality images of fungal parasites, either when CFW staining was done before (i.e. in solution) or after (i.e. on filters) concentrating phytoplankton host cells onto filters. However, substantial differences were noted depending both on the type of the filter and on the concentration of the stain. In general, for the two types of filters, high levels of background noises were obtained when using CFW at final concentrations of 3%, 10%, or 20%, precluding any accurate assessment of numerical and phenotypic characteristics of both host cells and their fungal parasites. Staining with 1% CFW final concentration substantially improved the viewing of chytrids on filters, with an increasing contrast from the white DTTP Millipore to the black Whatman filters. However, none of the membrane-retaining approaches yielded satisfactory images of morphological and cellular features of the host cells, e.g. presence of chloroplast and viability of the host cell. Accordingly, the proposed protocol is based on the size-fractionation approach using 1% to 2.5% vol/vol CFW final concentration (from the stock solution), which substantially enhanced the observational results.
-
2.
The approach is efficient since it is based on the concentration of large initial volumes and size partitioning of samples, a step that we judged necessary in order to yield good analytic images of infectious sporangia for an accurate diagnosing and identification of parasites. In addition, this approach yielded satisfactory images of morphological and cellular features of the host cells, for phytoplankton identification based on phenotypic features and viability of the host cell, through the integrity of cell wall and the presence of chloroplasts, which are fundamental parameters to assess the intensity of the disease. We consider this protocol as an optimal for the diagnosis and quantitative assessment of phytoplanktonic chytrid infections in natural samples. Finally, the approach was designed to freeze-conserved particulate DNA samples for quantifying the propagule stages (i.e. zoospores) of chytrids via FISH targeting of specific rRNA oligonucleotide probes (see Chap. 3).
-
3.
To estimate the infectivity parameters of ecological interest in the phytoplankton population, several algorithms are used according to the formula proposed by Bush et al. [37]. These parameters include the prevalence of infection (Pr), i.e. the proportion of individuals in a given the phytoplankton population having one or more sporangia or rhizoids, expressed as Pr (%) = [(Ni/N) × 100], where Ni is the number of infected host cells and N the total number of host cells. The second parameter is the mean intensity of infection (I), calculated as I = Np/Ni where Np is the number of parasites and Ni is the number of infected individuals within a host population.
-
4.
We propose a third parameter concerning the prevalence of infection of cells in colonial (or filamentous) species (PrCF). PrCF (%) = [(Ni/N) × 100] where Ni is the number of infected host cells in parasitized colonies (or filaments) and N the total number of parasitized host colonies (or filaments).
References
Sigee DC (2005) Freshwater microbiology. Wiley, Chichester
Tsui CKM, Hyde KD (2003) Freshwater mycology. Fungal Diversity Press, Hong Kong
Rasconi S, Jobard M, Jouve L, Sime-Ngando T (2009) Use of calcofluor white for detection, identification, and quantification of phytoplanktonic fungal parasites. Appl Environ Microbiol 75:2545–2553
Sparrow FK (1960) Aquatic Phycomycetes. University of Michigan Press, Ann Arbor
Lefevre E, Bardot C, Noel C, Carrias J-F, Viscogliosi E, Amblard C, Sime-Ngando T (2007) Unveiling fungal zooflagellates as members of freshwater picoeukaryotes: evidence from a molecular diversity study in a deep meromictic lake. Environ Microbiol 9:61–71
Lefèvre E, Roussel B, Amblard C, Sime-Ngando T (2008) The molecular diversity of freshwater picoeukaryotes reveals high occurrence of putative parasitoids in the plankton. PLoS One 3:e2324
Kudoh S, Takahashi M (1990) Fungal control of population-changes of the planktonic diatom Asterionella-formosa in a shallow eutrophic lake. J Phycol 26:239–244
Sen B (1987) Fungal parasitism of planktonic algae in shearwater I. Occurrence of Zygorhizidium affluens Canter on Asterionella formosa Hass in relation to the seasonal periodicity of the alga. Arch Hydrobiol 76:129–144
Van Donk E, Ringelberg J (1983) The effect of fungal parasitism on the succession of diatoms in Lake Maarsseveen I (The Netherlands). Freshw Biol 13:241–251
Canter HM (1950) Fungal parasites of the phytoplankton. I Studies on British chytrids X. Ann Bot 14:263–289
Canter HM, Lund JWG (1951) Studies on plankton parasites. III Examples of the interaction between parasitism and other factors determining the growth of diatoms. Ann Bot 15:359–371
Ingold CT (1940) Endocoenobium Eudorinae gen et sp. nov., a chytridiaceous fungus parasitizing Eudorina elegans ehrenb. New Phytol 39:97–103
Huber-Pestalozzi G (1944) Chytridium Oocystidis (spec. nova?) ein Parasit auf Oocystis lacustris Chodat. Aquat Sci 10:117–120
Canter HM (1953) Annotated list of British aquatic chytrids. Trans Brit Mycol Soc 36:278–303
Canter HM, Lund JWG (1969) The parasitism of planktonic desmids by fungi. Osterr Bot Z 116:351–377
Beakes GW, Canter HM, Jaworski GHM (1992) Comparative ultrastructural ontogeny of zoosporangia of Zygorhizidium affluens and Z. planktonicum, chytrid parasites of the diatom Asterionella formosa. Mycol Res 96:1047–1059
Beakes GW, Canter HM, Jaworski GHM (1992) Ultrastructural study of operculation (discharge apparatus) and zoospore discharge in zoosporangia of Zygorhizidium affluens and Z. planktonicum, chytrid parasites of the diatom Asterionella formosa. Mycol Res 96:1060–1067
Beakes GW, Canter HM, Jaworski GHM (1993) Sporangium differentiation and zoospore fine-structure of the chytrid Rhizophydium planktonicum, a fungal parasite of Asterionella formosa. Mycol Res 97:1059–1074
Powell MJ (1978) Phylogenetic implications of the microbody-lipid globule complex in zoosporic fungi. Biosystems 10:167–180
Barr DJS (1992) Evolution and kingdoms of organisms from the perspective of a mycologist. Mycologia 84:1–11
Cook WRI (1932) An account of some uncommon British species of the Chytridiales found in algae. New Phytol 31:133–144
Reynolds N (1940) Seasonal variations in Staurastrum paradoxum eyen. New Phytol 39:86–89
Canter HM, Lund JWG (1948) Studies on plankton parasites. I Fluctuations in the numbers of Asterionella formosa Hass in relation to fungal epidemics. New Phytol 47:238–261
Canter HM, Lund JWG (1953) Studies on plankton parasites. II The parasitism of diatoms with special reference to lakes in the English Lake District. Trans Brit Mycol Soc 36:13–37
Canter HM (1972) A guide to the fungi occurring on planktonic blue-green algae. In: Desikachary TV (ed) Taxonomy and biology of blue-green algae. University of Madras, Chennai, pp 145–159
Pongratz E (1966) De quelques champignons parasites d’organismes planctoniques du Léman. Aquat Sci 28:104–132
Gleason FH, Kagami M, Lefevre E, Sime-Ngando T (2008) The ecology of chytrids in aquatic ecosystems: roles in food web dynamics. Fungal Biol Rev 22:17–25
De Bruin A, Ibelings BW, Kagami M, Mooij WM, van Donk E (2008) Adaptation of the fungal parasite Zygorhizidium planktonicum during 200 generations of growth on homogeneous and heterogeneous populations of its host, the diatom Asterionella formosa. J Eukaryot Microbiol 55:69–74
Rasconi S, Jobard M, Sime-Ngando T (2011) Parasitic fungi of phytoplankton: ecological roles and implications for microbial food webs. Aquat Microb Ecol 62:123–137
Masclaux M, Bec A, Kagami M, Perga ME, Sime-Ngando T, Desvilettes C, Bourdier G (2011) Food quality of anemophilous plant pollen for freshwater zooplankton. Limnol Oceanogr 56:939–946
Gleason FH, Kagami M, Marano AV, Sime-Ngando T (2009) Fungal zoospores are valuable food resources in aquatic ecosystems. Inoculum 60:1–3
Kagami M, von Elert E, Ibelings BW, de Bruin A, van Donk E (2007) The parasitic chytrid, Zygorhizidium facilitates the growth of the cladoceran zooplankter, daphnia in cultures of the inedible alga. Asterionella Proc Biol Sci 274:1561–1566
Muller U, Sengbush P (1983) Visualization of aquatic fungi (Chytridiales) parasitizing on algae by means of induced fluorescence. Archiv Hydrobiol 97:471–485
Hageage GJ, Harrington BJ (1984) Use of calcofluor white in clinical mycology. Lab Med 15:109–112
Harrington BJ, Hageage GJ (2003) Calcofluor white: a review of its uses and application in clinacal micology and parasitology. Lab Med 34:361–367
Utermöhl H (1958) Zur Vervollkommung der quantitative phytoplankton Methodik. Mitt Int Verein Limnol 9:1–38
Bush AO, Lafferty KD, Lotz JM, Shostak AW (1997) Parasitology meets ecology on its own terms: Margolis et al revisited. J Parasitol 83:575–583
Acknowledgements and Additional Information
SR and MG were supported by PhD Fellowships from the French Ministère de la Recherche et de la Technologie (MRT). This study receives grant-aided support from the French ANR Programme Blanc # ANR 07 BLAN 0370 titled DREP: Diversity and Roles of Eumycetes in the Pelagos.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sime-Ngando, T., Rasconi, S., Gerphagnon, M. (2022). Technique for Identifying and Counting Infective Chytrid Sporangia Using the Chitinaceous Fluorochrome Calcofluor White. In: Gupta, V.K., Tuohy, M. (eds) Laboratory Protocols in Fungal Biology. Fungal Biology. Springer, Cham. https://doi.org/10.1007/978-3-030-83749-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-83749-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-83748-8
Online ISBN: 978-3-030-83749-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)