
Abstract
Staphylococci are Gram-positive cocci that appear as grape-like clusters. The genus is comprised of more than 40 species, most of which are harmless and exist on the skin and mucous membranes of humans or other animals. Staphylococci are divided into coagulase-negative (CoNS) and coagulase-positive members, based on their ability to produce the free enzyme coagulase, which causes blood clot formation. While the majority of staphylococcal species are CoNS, few CoNS have been implicated in human disease. This, however, has been changing, with an increasing number of CoNS infections identified, boosting their clinical significance [1, 2]. Staphylococcus aureus (SA), the most notable member of the genus, is coagulase positive and has been the primary focus of clinical identification as it is commonly associated with human infection. Methicillin-resistant Staphylococcus aureus (MRSA), in particular, has garnered much of that attention as it is resistant to all penicillins and most β-lactam drugs and associated with higher morbidity and mortality rates among hospitalized patients and higher patient care costs [3–5].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Staphylococci
- Coagulase-negative staphylococci (CoNS)
- Staphylococcus aureus
- Methicillin-resistant S. aureus (MRSA)
- Phage typing
- Multilocus enzyme electrophoresis (MLEE)
- Random amplification of polymorphic DNA (RAPD)
- Arbitrarily primed PCR (AP-PCR)
- Repetitive element PCR (rep-PCR)
- Amplified fragment length polymorphism (AFLP)
- Accessory gene regulator (agr) typing
- Pulsed-field gel electrophoresis (PFGE) typing
- Staphylococcal protein A (spa) typing
- Multilocus sequence typing (MLST)
- Microarray
- Whole genome sequencing
1 Introduction
Staphylococci are Gram-positive cocci that appear as grape-like clusters. The genus is comprised of more than 40 species, most of which are harmless and exist on the skin and mucous membranes of humans or other animals. Staphylococci are divided into coagulase-negative (CoNS) and coagulase-positive members, based on their ability to produce the free enzyme coagulase, which causes blood clot formation. While the majority of staphylococcal species are CoNS, few CoNS have been implicated in human disease. This, however, has been changing, with an increasing number of CoNS infections identified, boosting their clinical significance [1, 2]. Staphylococcus aureus (SA), the most notable member of the genus, is coagulase positive and has been the primary focus of clinical identification as it is commonly associated with human infection. Methicillin-resistant Staphylococcus aureus (MRSA), in particular, has garnered much of that attention as it is resistant to all penicillins and most β-lactam drugs and associated with higher morbidity and mortality rates among hospitalized patients and higher patient care costs [3,4,5].
MRSA has been shown to asymptomatically colonize 20–30% of the human population [6, 7] but is also responsible for a wide variety of infections, ranging from mild skin and soft tissue infections to life-threatening illnesses such as endocarditis, septicemia, and hemorrhagic pneumonia [8]. MRSA infections were initially associated with hospitals and healthcare settings; however, MRSA has since emerged as a major cause of community-associated infection as well. Adding complexity is the fact that, despite the overwhelming attention given to MRSA, methicillin-sensitive S. aureus (MSSA) infections are increasingly being recognized as presenting a significant threat to public health [9, 10]. With the ever-changing prevalence and epidemiology of S. aureus infections, reliable methods for characterizing strains are essential for outbreak investigations, for tracking clonal spreading, and for the implementation of effective treatment or control measures. At the local level, typing is useful for identifying clones, which aids in disease management and in predicting prognosis. It also helps identify outbreaks and strain spreading within the geographic locale, guiding infection control strategies. At the international level, strain typing aids in investigation related to the evolution and spread of clonal types, both over large areas and over time. Discussed in this chapter are the various phenotypic and molecular methods used to discriminate S. aureus lineages.
2 Identification of Staphylococcal Species
Differentiation of S. aureus from CoNS is accomplished using standard microbiological methods in clinical diagnostic laboratories. Staphylococci are catalase-positive, facultative anaerobes, capable of growing in the presence of bile salts or 6.5% NaCl solution. Columbia or tryptic soy blood agar, with 5% defibrinated sheep or horse blood, is the primary culture plate used for staphylococcal isolation. On blood, S. aureus presents as large, round, golden-yellow colonies that are most often β-hemolytic. CoNS colonies, on the other hand, are typically smaller in size, non-pigmented, smooth, glistening, and opaque, although some species can be gray-yellow to yellow-orange in pigmentation and can also be β-hemolytic. Coagulase tube test or rapid latex and hemagglutination assays allow presumptive identification of S. aureus, while commercial systems can differentiate the staphylococcal species using biochemical procedures. Systems such as Vitek 2 (bioMérieux), the BBL Crystal Identification System’s Rapid Gram-Positive ID Kit (BD Diagnostic Systems, Sparks, MD), the Pos ID Panel family (Siemens Healthcare Diagnostics, Deerfield, IL), the Phoenix Automated Microbiology System (BD Diagnostic Systems), the Biolog systems (Biolog, Hayward, CA), the RapiDEC Staph (bioMérieux), and the API Staph and ID32 Staph strips (bioMérieux, La Balme-les-Grottes, France) are routinely used in clinical laboratories. Antibiotic susceptibility patterns for the staphylococcal species can be obtained on systems such as Vitek 2 (bioMérieux).
While biochemical identification of S. aureus is relatively straightforward, CoNS have proven to be more problematic. Common species such as S. epidermidis, S. saprophyticus, and S. haemolyticus are generally successfully identified by biochemical means, while identification of less common species such as S. warneri and S. hominis shows more variable rates [11,12,13]. Nucleic acid amplification and sequencing of universally occurring genomic regions offer an effective alternative for speciating staphylococci and can be accomplished quickly with minimal cost. Sequencing of a portion of the rpoE gene has been shown to accurately differentiate staphylococcal species [14]; however, sequencing of the 16S rRNA gene is generally considered the gold standard for identification and taxonomic classification of bacterial species. 16S rRNA is the small component of the prokaryotic ribosome that binds to the Shine-Dalgarno sequence, with its gene undergoing slow rates of evolution, making it useful for phylogenetic analysis. The 16S rRNA gene contains highly conserved primer binding regions, as well as nine hypervariable regions (V1–V9), each ranging from 30 to 100 bp in length [15]. Sequencing of the full 16S rRNA gene can be performed; however, more commonly shorter sequences involving the variable regions are targeted. Regions V1–V3, in particular, have been shown to be the most useful in distinguishing among staphylococcal species [16]. Various 16S ribosomal databases exist for analyzing sequencing data, including public databases such as NCBI and secondary ones such as EzBioCloud, Ribosomal Database Project, SILVA, and Greengenes [17,18,19,20]. While the public databases are easily accessible and free, the quality of sequences and taxonomic assignments found on the database are often not validated, making secondary databases that collect and validate 16S rRNA sequences superior choices.
As CoNS are not routinely typed beyond species identification and antibiotic susceptibility, the remainder of this chapter will focus on molecular characterization of S. aureus. Discrimination of isolates based on phenotypic and genotypic characteristics is important for determining clonal relationships between strains and furthering our understanding of the epidemiology of infectious diseases. Presently, classification schemes for Staphylococcus aureus are based less on phenotypic methods and more so on molecular ones. While many of these methods were initially used for research purposes, they are now commonly used in clinical labs as well.
3 MRSA Identification and SCCmec Typing
Distinguishing MRSA from MSSA is an important first step in S. aureus classification. MRSA have acquired and integrated into their chromosome a mobile genetic element known as staphylococcal cassette chromosome mec (SCCmec) , which carries the methicillin resistance genes mecA or mecC. mecA was the first methicillin resistance gene identified and encodes an alternative penicillin-binding protein (PBP2a or PBP2’), which has low affinity for semisynthetic penicillins and confers resistance to all β-lactam drugs except ceftaroline and ceftobiprole [21]. mecA remained the only methicillin resistance gene identified in S. aureus until 2011, when the mecC gene was described, sharing 70% identity with mecA, and coding a PBP2a/2′ sharing 63% homology at the amino acid level [22]. A third homologue, mecB, was first identified in 2009 in closely related bacteria, Macrococcus caseolyticus [23]; however, in 2018, it was detected for the first time in S. aureus on a plasmid [24]. The mecB gene shares 60% homology with mecA and confers resistance to methicillin. A fourth homologue, mecD, has been reported on a genomic island (McRImecD-1 and McRImecD-2) in M. caseolyticus but to date has not been detected in S. aureus. The Clinical and Laboratory Standards Institute (CLSI) recommends testing for MRSA using broth microdilution or with cefoxitin disk diffusion or Mueller-Hinton agar plates supplemented with 4% NaCl and 6 μg/ml of oxacillin as alternatives [25]. Chromogenic agars, such as CHROMagar™ MRSA, Oxoid Brilliance™ MRSA, MRSASelect, BBL™ CHROMagar™ MRSA, and ChromID MRSA, are also available for MRSA detection, offering highly sensitive and specific detection [26]. The PBP2a latex agglutination test (Oxoid, Hampshire, UK) is also available as an alternate phenotypic test for detecting PBP2a in S. aureus colonies; however, it suffers from a large variability in performance [27, 28]. No optimal phenotypic method exists for MRSA detection, as they generally require specialized conditions and results are affected by factors such as inoculum size, incubation temperature and time, or pH and salt concentration.
Nucleic acid amplification tests represent a more precise and reliable form of MRSA identification and have become the gold standard for MRSA detection. These assays have traditionally relied on detection of the mecA gene; however, detection of the mecC gene also needs to be considered now. Additionally, while the mecB gene has only been described in one instance, its detection may become important if the gene spreads. Murakami et al. [29] were the first to develop a PCR assay for MRSA detection, targeting the mecA gene, while the first multiplex PCR assay targeting both the mecA and 16S rRNA genes was developed by Geha et al. [30]. Since then, a substantial number of assays have been developed targeting the mecA/mecC genes alone or in conjunction with other targets, such as PVL, fem, nuc, or 16S rRNA, and using both standard and real-time PCR platforms. In 2008, Zhang et al. developed a multiplex PCR assay that could discriminate staphylococci from non-staphylococcal species while simultaneously distinguishing S. aureus from CoNS, identifying MRSA, identifying the Panton-Valentine leukocidin virulence genes, and presumptively identifying USA300 and USA400 epidemic strains [31]. While this assay has been extensively used, it suffers in that it does not detect the mecB or mecC genes. In 2012, Stegger et al. developed a multiplex PCR assay capable of simultaneously detecting both the mecA and mecC genes, along with the PVL genes and the staphylococcal protein A gene (spa) [32]. The assay allows rapid and inexpensive detection of MRSA, with the ability to perform downstream spa typing of isolates, but does not take into account the mecB gene.
As mentioned, the mecA and mecC genes, which confer resistance to β-lactam antibiotics, are carried on a mobile genetic element termed staphylococcal cassette chromosome mec. To date, 13 different SCCmec elements have been described in S. aureus based on the nature of their mec and ccr gene complex and are further divided into subtypes based on differences in their joining regions. These differences provide an important means of classifying MRSA isolates, as even closely related strains can differ in the type of SCCmec element they carry. Initial SCCmec typing schemes involved molecular cloning and sequencing or long-range PCR amplification with multiple sets of primers [33,34,35]. Typing schemes have since improved to include conventional PCR detection of several type-specific loci [36], RFLP analysis [37, 38], multiplex PCR [39], multiplex real-time PCR [40, 41], and targeted DNA microarrays [42]. Multiplex PCR typing is currently the most widely used method of SCCmec typing, with several variations developed. A novel multiplex PCR assay for the characterization and concomitant subtyping of SCCmec I–V was developed by Zhang et al. in 2005 and later updated in 2012 to make it more accurate and reliable [43, 44]. Similarly, in 2007, Milheirico et al. updated a previous multiplex PCR assay to detect SCCmec I–V. These multiplex assays are by far the most commonly used ones for SCCmec typing; however, both are limited to detection of types I–V, requiring other methods for the detection of types VI–XIII. Both are also restricted by their inability to classify newly evolving SCCmec types and subtypes. Unfortunately, to date, no single PCR assay is available to identify all SCCmec types and subtypes. Targeted DNA microarray offers an alternate option for SCCmec typing, simultaneously detecting multiple genes associated with SCCmec, including mecA and its regulatory genes, and sequences in the J regions [42]. As with PCR, only known SCCmec types can be identified with this technique, and it suffers from the added disadvantage that specialized equipment and highly trained personnel are required. As such, multiplex PCR remains the best option for SCCmec typing at present.
4 Historical Typing Methods
In an attempt to understand and track S. aureus (particularly MRSA) infections, numerous typing methods were developed to classify lineages. While these historical methods are rarely used routinely anymore, they still can be of value when typing S. aureus.
Phage Typing
relies on bacterial susceptibility to a defined set of phages, with a set of 23 internationally accepted phages used for typing human strains of S. aureus [45, 46]. While the method was the primary one used for several years, it suffered in that it often lacked reproducibility and was time-consuming and technically challenging and a large percentage of strains remained untypable with the technique [47,48,49,50].
Multilocus Enzyme Electrophoresis (MLEE)
involves the extraction of constitutively expressed proteins from the bacteria and their separation on gels using electrophoresis, with the rate of migration being dependent on amino acid composition in the proteins. Generally, 12–20 proteins are assessed, each being assigned allelic types based on variation in their charge, with the similarity between isolates determined by the proportion of loci which show differences. While MLEE generally has good reproducibility and typability for S. aureus, it is a labor-intensive procedure, and the results are difficult to compare between laboratories [51, 52].
Random Amplification of Polymorphic DNA (RAPD) and Arbitrarily Primed PCR (AP-PCR)
rely on parallel non-stringent amplification of random DNA fragments, resulting in unique gel patterns specific to each bacterial strain [53, 54]. In RAPD, short arbitrary primer sequences and low-temperature, non-stringent annealing conditions allow amplification of multiple PCR products of varying sizes. Amplicons are analyzed either by gel electrophoresis or DNA sequencing, with the number and size of fragments used to define an isolate type [54]. AP-PCR is a variant of RAPD, whereby amplification is done in three parts, each of which has a set stringency and reagent concentrations [53]. While these techniques have been used successfully in outbreaks and are relatively inexpensive and easy, they have lower discriminatory power and lower inter- and intra-laboratory reproducibility [55,56,57].
Repetitive Element PCR (rep-PCR)
employs primers that bind to noncoding repetitive sequences in the bacterial genome, producing fingerprint patterns unique to each isolate [58]. The repetitive palindromic extragenic elements (Rep) are sequences 35–38 bp long that occur in variable positions and numbers. Amplification of the elements creates amplicons of varying lengths, which are separated by electrophoresis, creating fingerprints unique to the strains. For S. aureus, RepMP3 and inter-IS256 and Tn916 are commonly used targets, with RepMP3 showing greater reproducibility and stability [59]. Rep-PCR has high discriminatory power, with good correlation to PFGE; however, reproducibility can suffer from variations in reagents and electrophoresis systems [60].
Amplified Fragment Length POLYMORPHISM (AFLP)
relies on differences in the amplification of digested genomic DNA fragments [61]. Genomic DNA is digested with restriction enzymes, and double-stranded adaptors are ligated to the sticky ends, followed by amplification of the fragments using primers complementary to the adaptors. The primers are generally fluorescently labelled; therefore, after separation of the amplicons based on size, they can be detected with an automated DNA sequencer and compared by computer. Analysis of the high-resolution banding patterns is used to determine the relationship between strains [62]. While this technique is portable and highly reproducible and has high discriminatory power, it is time-consuming and expensive [63, 64].
Accessory Gene Regulator (agr) Typing
is a PCR-based typing method that relies on amplification of hypervariable regions present in the agr locus to classify strains. The accessory gene regulator (agr) is a bacterial regulatory component containing two divergently transcribed units, which has highly conserved and hypervariable regions [65]. Four genes, agrA, agrC, agrD, and agrB, are present in the locus. The C-terminal of agrB and agrD and the N-terminal of agrC are highly divergent and constitute the hypervariable region of the locus, which is used to divide S. aureus into four agr groups (I–IV) [65]. PCR primers for agr group determination were developed by Peacock et al. [66], and a multiplex real-time quantitative PCR assay was developed by Francois et al., targeting the variable region of agrC and offering good specificity [67]. While agr typing is extremely limited in its discriminatory power and would not be useful for defining S. aureus lineages, it does provide additional information about strains that can supplement other typing methods.
5 Current Molecular Typing Methods
Current typing schemes for S. aureus classification rely predominantly on molecular methods based on DNA sequence variations. A proposal was made that MRSA clones should be defined based on a combination of the genomic type of the strain and the SCCmec type, a nomenclature system that was accepted in 2002 by the subcommittee of the International Union of Microbiology Societies in Tokyo [68]. This system, which can be amended to describe both MRSA and MSSA (e.g., ST8-MRSA-IVa or ST8-MSSA), relies solely on multilocus sequence typing and SCCmec typing (discussed below) to define the strains. While these two methods are important parts of S. aureus classification, the addition of other typing schemes provides more complete information about S. aureus lineages, which are discussed below.
5.1 Pulsed-Field Gel Electrophoresis (PFGE) Typing
PFGE was first described in 1984 and is based on the digestion of bacterial genomes into large fragments with a restriction enzyme and their subsequent separation by gel electrophoresis [69, 70]. Because larger fragments of DNA will co-migrate and appear as a large diffuse band with conventional gel electrophoresis, in PFGE, the voltage direction is periodically switched (pulsed), allowing effective separation of larger DNA pieces. Migration of the DNA fragments produces a DNA fingerprint, which can be used to compare the relatedness of strains.
For PFGE, genomic DNA needs to be intact and free from mechanical shearing; therefore, bacterial cells are incorporated into agarose plugs prior to lysis to protect the DNA from damage [71]. DNA, which is immobilized in the agarose plug, is digested with a rare-cutting restriction endonuclease, at which time the plugs are loaded onto an agarose gel and subjected to PFGE. PFGE protocols for Staphylococcus aureus have been optimized and, with minor variations, include standard features common to typing this species [72,73,74] (https://www.cdc.gov/mrsa/pdf/ar_mras_PFGE_s_aureus.pdf). A number of restriction endonucleases have been used in PFGE typing of bacterial species; however, smaI was found to be the most useful for S. aureus, allowing nearly all isolates to be typed, with reproducible results following repeated subcultures [75,76,77]. S. aureus belonging to the ST398 lineage are the exception, not typable using smaI due to a DNA methyltransferase that modifies the consensus sequence [78]. The restriction enzyme Cfr9I, a neoschizomer of smaI, is able to cleave these strains within the same recognition sequence as smaI and is used for PFGE typing of the ST389 lineage. S. aureus gels are generally run with the contour-clamped homogeneous electric field (CHEF) electrophoresis system, where the current is applied in three directions, offset by 120°, using hexagonally arranged electrodes [52, 79].
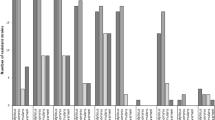
PFGE is a popular technique used by laboratories around the world and is effective for providing local epidemiological information, as well as for identifying epidemics. In experienced hands, the method can provide information related to the presence or absence of some mobile genetic elements such as the SCCmec cassette or phages. The technique has high discriminatory power, and results can be reproducible at both the intra- and inter-laboratory levels when the method is highly standardized [48, 80]. To aid with standardization, the Centers for Disease Control and Prevention in the USA developed PulseNet (https://www.cdc.gov/pulsenet/index.html). It is a national laboratory network that uses bacterial DNA fingerprints (such as PFGE patterns) to detect foodborne illnesses and outbreaks. Standard protocols are available, and data can be shared nationally or internationally. Also helping with standardization is the fact that S. aureus gels are run with the S. braenderup H2812 control standard and the data normalized and analyzed using BioNumerics software (Applied Maths, Sint-Martens-Latem, Belgium). Data analysis criteria set out by Tenover et al. are useful for comparing strains and determining their relatedness [81], and S. aureus PFGE profiles have been assembled into a national database to assist interpretation [72, 82]. In Fig. 9.1, sample PFGE patterns for Canadian and US epidemic reference strains are shown, along with some other common typing information for each strain. PFGE does suffer from limitations, the main ones being the long turnaround time, the high cost for specialized equipment and software, and the skill level required. Without high standardization, data interpretation can be problematic, as differences in electrophoresis equipment and conditions can affect DNA migration, complicating isolate comparisons within and between laboratories [83, 84]. As well, the technique separates DNA based on size, not sequence, and small changes are enough to affect the fingerprint. For example, the acquisition or loss of mobile genetic elements will alter the banding pattern, as will a point mutation in the smaI recognition sequence.

Sample typing results for representative Canadian (CMRSA1-10) and US (USA100-800) epidemic reference strains. Different lineages may share the same type when classified using a single typing method but will become distinguishable from each other when multiple typing schemes are used together. Pulsed-field gel electrophoresis (PFGE) profiles, staphylococcal cassette chromosome mec (SCCmec) type, accessory gene regulator (agr) type, staphylococcal protein A (spa) type (including Ridom repeat pattern and Kreiswirth ID), and multilocus sequence type (MLST) (including MLST profile) are shown
Despite the limitations, PFGE remains a powerful technique for S. aureus typing and classification and is still considered the “gold standard.”
5.2 Staphylococcal Protein A (spa) Typing
The spa gene, coding for protein A, is conserved among S. aureus and has proven to be an effective target for single-locus sequence typing of this species. The gene is approximately 2 kb in length and contains conserved Fc binding regions, a variable X region, and a conserved C-terminal region. The X region (or repeat region) is comprised of polymorphic variable number of tandem repeats (VNTR), generally consisting of 2–18 repetitive sequences of 21–30 bp (most often 24 bp) in length [85]. Each repeat is given an identifier (numerical or letter code), with the number, order, and sequence of these repeats varying between strains, forming the basis for spa typing [86, 87].
Two nomenclature systems, Ridom and Kreiswirth, are used for describing spa types and repeats, with Ridom represented by numerical repeat codes and Kreiswirth represented with alpha numeric repeat codes [86, 88]. Conversion between the two is possible with online tools. The Ridom StaphType software (available for download from www.ridom.de/staphtype/) was developed to ensure uniform assignments of spa repeats and types and is useful for MRSA surveillance. The software synchronizes with the Ridom SpaServer (www.SpaServer.ridom.de), which is a freely accessible server developed to collate and harmonize data from around the world, permitting 100% reproducibility between laboratories and providing public access to typing data. Figure 9.1 shows the spa type, including the Ridom and Kreiswirth profiles, for Canadian and US epidemic reference strains.
spa typing is a reliable way of assigning lineage and has proven to be effective for both short-term and long-term epidemiological studies [80, 86,87,88,89]. The speed and simplicity of targeting a single locus make it favorable for short-term studies, while the stable association of types with lineages over time makes it suitable for long-term studies. Development of the BURP (Based Upon Repeat Pattern) algorithm has provided an automated method to infer clonal relatedness of isolates based on spa repeat patterns and was shown to have high concordance with other typing methods [89, 90]. With a high discriminatory power, spa typing is a cost-effective, easy-to-use method with excellent reproducibility and portability. The major drawback of spa typing is the fact that the method relies on typing a single locus, running the risk that strains can be misclassified due to recombination and/or homoplasy [91]. Strains from different lineages can carry the same spa type (Fig. 9.1), and epidemiologically related strains from a lineage may carry different spa types, varying in as little as a single repeat. spa typing is, consequently, most effective when used in combination with other typing methods.
5.3 Multilocus Sequence Typing (MLST)
MLST is similar in principle to MLEE, but variations are examined directly by DNA sequencing. The method relies on sequencing a 402–516 bp fragment from each of seven essential housekeeping genes, present in all S. aureus isolates. These genes are crucial to cellular function and, therefore, stable and evolve slowly. Based on point mutations, the genes for each locus are assigned numerical allele designations, with the series of seven numbers (one representing each locus) defining the sequence type (ST type) of a strain. For S. aureus, the carbamate kinase (arcC) , shikimate dehydrogenase (aroE) , glycerol kinase (glpF) , guanylate kinase (gmk) , phosphate acetyltransferase (pta) , triosephosphate isomerase (tpi) , and acetyl coenzyme A acetyltransferase (yqiL) genes were selected, as they provided the highest number of alleles with the best resolving power for identifying lineages [92]. The genes are arranged in the abovementioned order (i.e., arcC-aroE-glpF-gmk-pta-tpi-yqiL) to define the ST type (e.g., ST8 has an MLST profile of 3-3-1-1-4-4-3).
Sequence analysis was initially facilitated by the online server available at MLST.net, a free website which provided the main hub for assigning allele and sequence types, naming new ones, as well as storing other important information related to the clonal types [93]. Now available for analysis is the database at PubMLST (https://pubmlst.org/saureus/), which contains both sequence definition and epidemiological information [94, 95]. To aid with visualizing and analyzing the evolutionary relationship between isolates, the eBURST (Based Upon Related Sequence Type) algorithm was developed [96, 97]. Strains sharing identical allelic profiles are considered as belonging to the same ST type and lineage, while strains differing by one or two loci (single-locus variants or double-locus variants) are considered to be genetically related, belonging to the same clonal complex (CC). The founding genotype for a clonal complex is the one that differs from the highest number of other genotypes by only one locus, assuming strains emerge as dominant clones and then diversify with time. A representative eBURST image showing the relatedness of MLST types from Canadian and US epidemic strains in the global Staphylococcus aureus population is shown in Fig. 9.2.

Demonstration of eBURST analysis showing the relatedness of MLST types identified in the Canadian and US epidemic strains CMRSA1-10 and USA100-800 in the global Staphylococcus aureus population. Clonal complexes are marked in black font for the strains of interest, while ST types are marked in red (Generated on December 1, 2018)
MLST is a useful tool for assigning lineage and has proven to be effective for studying the origin and evolution of S. aureus. The method is unambiguous and portable, making data transfer to, and comparison between, labs around the world simple. The technique is, however, intolerant to sequencing errors, as a single nucleotide change can lead to an incorrect ST assignment. Cost is another drawback to the method, as it requires high-quality sequences for 7 loci, requiring 14 sequencing reads.
This makes it less appealing as a tool for studying outbreaks or for use in smaller facilities with limited sequencing capability. Caution also has to be taken when relating MLST types to epidemiology, as strains with significantly different epidemiological significance can share a common MLST type. For example, the major epidemic strain in North America, USA300, belongs to MLST type ST8, a type also found in the infrequently encountered Canadian lineages, CMRSA9 and CMRSA5 (USA500) (Fig. 9.1). Despite the drawbacks, MLST is highly reproducible with high discriminatory power and, in conjunction with SCCmec type, remains the gold standard for publishing S. aureus epidemiological data.
5.4 Microarray
DNA microarrays use DNA probes attached in a known order to a solid surface to type bacterial isolates [98]. The probes can be oligonucleotides or gene segments (PCR amplicons) and can occur in low (100 s) or high (100,000 s) density. Bacterial DNA is labelled and allowed to hybridize to the microarray, such that complementary sequences present in the strain will bind to the probe. The microarray is scanned, and labelled spots are detected and then compared to known strains.
Microarrays are an effective means of typing and, indirectly, assigning lineage for S. aureus, simultaneously targeting a large number of strain-specific markers such as genes for antimicrobial resistance, exotoxins, surface components, regulators, and hsdS variants [42, 99, 100]. They are also well suited to the detection of complex patterns of virulence genes, mobile genetic elements, and extrachromosomal elements [101, 102] and have been used to understand the molecular mechanisms of pathogenesis, studying regulons such as Agr, Sar, SigB, and Mgr [103,104,105]. As such, microarrays permit strains to simultaneously be assigned to a lineage while having their resistance and virulence capabilities investigated at the same time.
Numerous microarrays have been designed specifically for S. aureus typing, and several companies make it possible to design custom arrays to meet specific needs [106,107,108,109,110]. The Alere StaphType DNA microarray is a commercially available system that covers 334 targets, including 170 genes and their allelic variants [42, 111, 112]. Included are species markers, capsule and agr typing markers, toxin and microbial surface components recognizing adhesive matrix molecule (MSCRAMM) genes, resistance gene markers, and SCCmec markers. On a larger scale, the Sam-62 microarray was developed based on 62 S. aureus whole genome sequencing projects and 153 plasmid sequences. The array targets all open reading frames in the sequences and includes over 29,000 probes, representing 6520 genes and 579 gene variants [113]. Sam-62 has shown potential to identify MRSA, distinguish between extremely similar but non-identical sequences, and be able to identify MRSA transmission events unrecognized using other methods [101].
While DNA microarray is highly accurate, specialized equipment and software are required meaning there is a significant cost associated with their use. Microarrays also suffer in that they cannot directly assign MLST group; strains can only be assigned to a given clonal complex group once the hybridization pattern of a reference strain with known MLST/spa types has been defined.
6 Whole Genome Sequencing (WGS) and the Future of MRSA Typing
WGS is a powerful tool for S. aureus typing, as well as for epidemiological and evolutionary studies, and next-generation sequencing (NGS) has provided a cost-effective means of extracting large amounts of information and identifying genome-wide variations. Today, the most commonly used NGS platform is Illumina (Illumina, Inc., San Diego, CA, USA), which can generate reads up to 300 bp in length. Assembly of a genome can be accomplished via de novo assembly, whereby reads are matched based on overlapping regions, or with reference-guided assembly, where reads are assembled against an existing WGS. De novo assembly in S. aureus is challenging, however, because of the small read sizes and the presence of dispersed or tandemly arrayed repeats in the genome. As such, the resulting genome is not continuous, but rather contains numerous contigs with gaps between assembled regions, due in part to the inability to resolve contig order surrounding these repeat elements. Reference-guided assembly can also be challenging because genomic regions, such as mobile genetic elements (MGEs), that are not present in the reference will be assembled poorly, particularly if they contain repeat elements, such as in SCCmec. Illumina data is still useful for querying genomic traits and variations, as well as for phylogenetic analysis, but for a complete genome assembly, sequencing platforms that generate longer reads are necessary.
Read lengths of >10 kb (and up to 60 kb) are possible with the “third-generation” PacBio sequencing platform (Pacific Biosciences, Menlo Park, CA, USA), while read lengths in the Mbp range have been achieved using nanopore sequencing technology (Oxford Nanopore, Oxford, UK). These systems suffer in that they can be more expensive and have lower read accuracy than Illumina; however, with tailored assembly methods (such as HGAP for PacBio reads), assemblies with higher accuracy are achieved. Hybrid assemblies, combining Illumina short reads and PacBio or Nanopore long reads, currently offer the most accurate and complete genomes.
A major drawback of WGS is the requirement for significant computer resources and bioinformatics support in order to extract meaningful information from the data. Software such as Lasergene exists for assembly and analysis of the genomes; however, in most cases, more complex pipelines are employed and require trained bioinformaticians. For WGS technology to become useful for routine typing of S. aureus, tools for data analysis that are simple enough for use in clinical settings are required, and a number of web-based and downloadable programs are available to help in this regard. The Center for Genomic Epidemiology (Lyngby, Denmark, available at https://cge.cbs.dtu.dk/services/), for example, has web-based analysis tools that are useful for S. aureus WGS analysis and able to extract data from raw reads and assembled or draft genomes generated using Illumina, Ion Torrent, Roche 454, SOLiD, PacBio, or Nanopore platforms. Currently available on the site are MLST, for assigning ST type; spaTyper, for determining spa type; and SCCmecFinder, for classifying SCCmec type. Also available are ResFinder, for identifying acquired antimicrobial resistance genes and/or chromosomal mutations, VirulenceFinder, and Restriction-ModificationFinder. For phylogenetic analysis, CSI Phylogeny will call single-nucleotide polymorphisms (SNPs), filter and validate them, and then infer phylogeny based on the concatenated alignment of the SNPs, generating phylogenetic trees. Also available for phylogenetic analyses are the downloadable software, RAxML (Randomized Axelerated Maximum Likelihood), for sequential and parallel maximum likelihood-based inference of large phylogenetic trees [114], as well as BEAST (Bayesian Evolutionary Analysis Sampling Trees), for inferring rooted, time-measured phylogenies using molecular clock models [115, 116]. Available from the University of Alberta (at http://phaster.ca/) is a web-based tool for rapid identification and annotation of prophage sequences within a bacterial genome, known as PHASTER (PHAge Search Tool – Enhanced Release). The program is able to work on raw DNA sequences as well as annotated GenBank formatted data, providing detailed tables and graphical displays of the phages, with high sensitivity and positive predictive value [117, 118].
WGS is the ultimate tool for the identification of diversity in an organism. In addition to extracting S. aureus typing information, WGS data can be used to track transmission events and outbreaks [119,120,121] and analyze variations between strains within a lineage by SNP analysis [122]. It has shown that related strains have well-conserved core regions but differ in their accessory genetic elements [123] and, likewise, that geographically dispersed isolates of ST239, ST225, and CC30 are stable in their genetic backgrounds, differing by SNPs and MGEs [119, 124, 125]. In the future, we may see the application of extended MLST (eMLST) to S. aureus typing, extending typing beyond the seven housekeeping genes to include a subset or all of the genes in the genome. Ribosomal MLST (rMLST) (adding the ribosomal genes), core genome MLST (cgMLST) (including all core genes present in the majority of isolates, and not subject to selection pressure), whole genome MLST (wgMLST) (also including genes subject to selective pressure), and pan-genome approach (including the full complement of genes within the species) would provide the ultimate high-level genomic epidemiology. Available to facilitate eMLST analysis, the Bacterial Isolate Genome Sequence Database (BIGSdb) software stores and analyzes sequence data for bacterial isolates, allowing a large numbers of loci to be defined and allelic profiles for each strain to be determined. BIGSdb is available within the PubMLST database at https://pubmlst.org/software/database/bigsdb/.
As sequencing costs are reduced and genome analysis tools improve, WGS will almost certainly become the primary tool for S. aureus typing and evolutionary and epidemiological studies.
7 Conclusions
Each typing scheme for S. aureus is met with strengths and limitations, leaving no single method ideal for all situations. PFGE was once considered the gold standard for MRSA typing and remains an effective tool for characterizing outbreaks and understanding S. aureus epidemiology, particularly at the local level. With standardization, it can be expanded to the international level; however, lineage cannot be inferred directly from the PFGE pattern. spa typing is capable of assigning lineage, is useful for analyzing both outbreaks and long-term molecular evolution, and is rapidly becoming the method of choice for clinical laboratories for epidemiological studies of S. aureus. With highly portable and standardized data, it is useful for investigations at both the local and international levels but is not always accurate when assigning lineages. MLST is also an effective tool for assigning lineage and, in combination with SCCmec typing, is considered the gold standard for publishing S. aureus epidemiological data. Similar to spa typing, the data is highly standardized and portable, making it an effective tool for studies at both the local and international levels. However, the cost makes it less appealing for routine use. Microarrays can provide large amounts of strain information within a short timeframe and are well suited for both outbreak investigations and long-term epidemiological studies, particularly at the local level, but suffer in that they cannot directly assign strains to lineages. WGS is the ultimate tool for strain typing and epidemiological studies and will rapidly increase in use as sequencing costs decrease and as easy-to-use data analysis tools are developed.
Ultimately, the technique of choice will depend heavily on the goals and questions that need answering, with a combination of methods offering more detailed information and greater discrimination between isolates. For outbreak situations where speed is important, PCR-based methods may be the better choice, making spa typing an effective tool. However, for routine strain typing and epidemiological monitoring at the local level, PFGE and spa typing complement well, providing better strain and clone discrimination. For international comparisons, spa typing, MLST, and WGS are good for generating highly standardized and portable data, but when detailed strain characterization is desired, a combination of PFGE, agr typing, SCCmec typing, spa typing, and MLST provides a more complete picture. Finally, long-term epidemiological and evolutionary studies benefit from greater detail, making microarrays and WGS attractive options.
8 Summary
Staphylococci are Gram-positive bacteria and commonly divided into coagulase-negative staphylococci (CoNS) and coagulase-positive members, based on their ability to produce the free enzyme coagulase. The majority of staphylococcal species are CoNS, with an increasing number of CoNS infections identified, boosting their clinical significance. Staphylococcus aureus is coagulase positive and has been the primary focus of clinical identification as it is commonly associated with human infection. Methicillin-resistant S. aureus (MRSA ), in particular, has garnered much attention as it is resistant to all penicillins and most β-lactam drugs and is associated with higher morbidity and mortality rates and increasingly being recognized as presenting a significant threat to public health. With the ever-changing prevalence and epidemiology of staphylococcal infections, reliable methods for characterizing strains are essential for outbreak investigations, for tracking clonal spreading, and for the implementation of effective treatment or control measures. In this chapter, we discussed various phenotypic and molecular methods used to discriminate staphylococci and S. aureus lineages. We first described the methods to identify staphylococcal species and to discriminate MRSA from methicillin-susceptible S. aureus (MSSA), including how to characterize different types of staphylococcal cassette chromosome mec (SCCmec) in MRSA. We then discussed various typing methods applied to study the molecular epidemiology and evolutionary nature of S. aureus, starting with the historical methods [phage typing, multilocus enzyme electrophoresis (MLEE), random amplification of polymorphic DNA (RAPD) and arbitrarily primed PCR (AP-PCR), repetitive element PCR (rep-PCR), amplified fragment length polymorphism (AFLP), and accessory gene regulator (agr) typing] and continuing to the current commonly used molecular typing methods [pulsed-field gel electrophoresis (PFGE) typing, staphylococcal protein A (spa) typing, multilocus sequence typing (MLST), and microarray] and to the advanced genome approaches (whole genome sequencing). We also discuss the strengths and limitations for each typing scheme and their suitable applications.
References
Banerjee SN, Emori TG, Culver DH, Gaynes RP, Jarvis WR, Horan T, Edwards JR, Tolson J, Henderson T, Martone WJ (1991) Secular trends in nosocomial primary bloodstream infections in the United States, 1980–1989. National Nosocomial Infections Surveillance System. Am J Med 91:86S–89S
Rupp ME, Archer GL (1994) Coagulase-negative staphylococci: pathogens associated with medical progress. Clin Infect Dis 19:231–243. quiz 244–245
Antonanzas F, Lozano C, Torres C (2015) Economic features of antibiotic resistance: the case of methicillin-resistant Staphylococcus aureus. Pharmacoeconomics 33:285–325
Thampi N, Showler A, Burry L, Bai AD, Steinberg M, Ricciuto DR, Bell CM, Morris AM (2015) Multicenter study of health care cost of patients admitted to hospital with Staphylococcus aureus bacteremia: impact of length of stay and intensity of care. Am J Infect Control 43:739–744
Whitby M, McLaws ML, Berry G (2001) Risk of death from methicillin-resistant Staphylococcus aureus bacteraemia: a meta-analysis. Med J Aust 175:264–267
Kluytmans J, van Belkum A, Verbrugh H (1997) Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10:505–520
Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL (2005) The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762
Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M, Group SP (2001) Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin Infect Dis 32(Suppl 2):S114–S132
David MZ, Boyle-Vavra S, Zychowski DL, Daum RS (2011) Methicillin-susceptible Staphylococcus aureus as a predominantly healthcare-associated pathogen: a possible reversal of roles? PLoS One 6:e18217
Jackson KA, Gokhale RH, Nadle J, Ray SM, Dumyati G, Schaffner W, Ham DC, Magill SS, Lynfield R, See I (2019) Public health importance of invasive methicillin-sensitive Staphylococcus aureus infections: surveillance in 8 US Counties, 2016. Clin Infect Dis doi:5477381 [pii]https://doi.org/10.1093/cid/ciz323
Bannerman TL, Kleeman KT, Kloos WE (1993) Evaluation of the Vitek Systems Gram-Positive Identification card for species identification of coagulase-negative staphylococci. J Clin Microbiol 31:1322–1325
Ieven M, Verhoeven J, Pattyn SR, Goossens H (1995) Rapid and economical method for species identification of clinically significant coagulase-negative staphylococci. J Clin Microbiol 33:1060–1063
Spanu T, Sanguinetti M, Ciccaglione D, D’Inzeo T, Romano L, Leone F, Fadda G (2003) Use of the VITEK 2 system for rapid identification of clinical isolates of Staphylococci from bloodstream infections. J Clin Microbiol 41:4259–4263
Mellmann A, Becker K, von Eiff C, Keckevoet U, Schumann P, Harmsen D (2006) Sequencing and staphylococci identification. Emerg Infect Dis 12:333–336
Gray MW, Sankoff D, Cedergren RJ (1984) On the evolutionary descent of organisms and organelles: a global phylogeny based on a highly conserved structural core in small subunit ribosomal RNA. Nucleic Acids Res 12:5837–5852
Conlan S, Kong HH, Segre JA (2012) Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PLoS One 7:e47075
Larsen N, Olsen GJ, Maidak BL, McCaughey MJ, Overbeek R, Macke TJ, Marsh TL, Woese CR (1993) The ribosomal database project. Nucleic Acids Res 21:3021–3023
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617
Chambers HF, Deleo FR (2009) Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641
Garcia-Alvarez L, Holden MT, Lindsay H, Webb CR, Brown DF, Curran MD, Walpole E, Brooks K, Pickard DJ, Teale C, Parkhill J, Bentley SD, Edwards GF, Girvan EK, Kearns AM, Pichon B, Hill RL, Larsen AR, Skov RL, Peacock SJ, Maskell DJ, Holmes MA (2011) Methicillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect Dis 11:595–603
Baba T, Kuwahara-Arai K, Uchiyama I, Takeuchi F, Ito T, Hiramatsu K (2009) Complete genome sequence of Macrococcus caseolyticus strain JCSCS5402, [corrected] reflecting the ancestral genome of the human-pathogenic staphylococci. J Bacteriol 191:1180–1190
Becker K, van Alen S, Idelevich EA, Schleimer N, Seggewiss J, Mellmann A, Kaspar U, Peters G (2018) Plasmid-Encoded Transferable mecB-Mediated Methicillin Resistance in Staphylococcus aureus. Emerg Infect Dis 24:242–248
CLSI (2018) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 11th edn, CLSI Standard M07-A11. Clinical and Laboratory Standards Institute, Wayne
Xu Z, Hou Y, Peters BM, Chen D, Li B, Li L, Shirtliff ME (2016) Chromogenic media for MRSA diagnostics. Mol Biol Rep 43:1205–1212
Sakoulas G, Gold HS, Venkataraman L, DeGirolami PC, Eliopoulos GM, Qian Q (2001) Methicillin-resistant Staphylococcus aureus: comparison of susceptibility testing methods and analysis of mecA-positive susceptible strains. J Clin Microbiol 39:3946–3951
Chapin KC, Musgnug MC (2004) Evaluation of penicillin binding protein 2a latex agglutination assay for identification of methicillin-resistant Staphylococcus aureus directly from blood cultures. J Clin Microbiol 42:1283–1284
Murakami K, Minamide W, Wada K, Nakamura E, Teraoka H, Watanabe S (1991) Identification of methicillin-resistant strains of staphylococci by polymerase chain reaction. J Clin Microbiol 29:2240–2244
Geha DJ, Uhl JR, Gustaferro CA, Persing DH (1994) Multiplex PCR for identification of methicillin-resistant staphylococci in the clinical laboratory. J Clin Microbiol 32:1768–1772
Zhang K, McClure JA, Elsayed S, Louie T, Conly JM (2008) Novel multiplex PCR assay for simultaneous identification of community-associated methicillin-resistant Staphylococcus aureus strains USA300 and USA400 and detection of mecA and Panton-Valentine leukocidin genes, with discrimination of Staphylococcus aureus from coagulase-negative staphylococci. J Clin Microbiol 46:1118–1122
Stegger M, Andersen PS, Kearns A, Pichon B, Holmes MA, Edwards G, Laurent F, Teale C, Skov R, Larsen AR (2012) Rapid detection, differentiation and typing of methicillin-resistant Staphylococcus aureus harbouring either mecA or the new mecA homologue mecA(LGA251). Clin Microbiol Infect 18:395–400
Hiramatsu K, Asada K, Suzuki E, Okonogi K, Yokota T (1992) Molecular cloning and nucleotide sequence determination of the regulator region of mecA gene in methicillin-resistant Staphylococcus aureus (MRSA). FEBS Lett 298:133–136
Ito T, Katayama Y, Asada K, Mori N, Tsutsumimoto K, Tiensasitorn C, Hiramatsu K (2001) Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 45:1323–1336
Ito T, Katayama Y, Hiramatsu K (1999) Cloning and nucleotide sequence determination of the entire mec DNA of pre-methicillin-resistant Staphylococcus aureus N315. Antimicrob Agents Chemother 43:1449–1458
Okuma K, Iwakawa K, Turnidge JD, Grubb WB, Bell JM, O’Brien FG, Coombs GW, Pearman JW, Tenover FC, Kapi M, Tiensasitorn C, Ito T, Hiramatsu K (2002) Dissemination of new methicillin-resistant Staphylococcus aureus clones in the community. J Clin Microbiol 40:4289–4294
van der Zee A, Heck M, Sterks M, Harpal A, Spalburg E, Kazobagora L, Wannet W (2005) Recognition of SCCmec types according to typing pattern determined by multienzyme multiplex PCR-amplified fragment length polymorphism analysis of methicillin-resistant Staphylococcus aureus. J Clin Microbiol 43:6042–6047
Yang JA, Park DW, Sohn JW, Kim MJ (2006) Novel PCR-restriction fragment length polymorphism analysis for rapid typing of staphylococcal cassette chromosome mec elements. J Clin Microbiol 44:236–238
Oliveira DC, de Lencastre H (2002) Multiplex PCR strategy for rapid identification of structural types and variants of the mec element in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 46:2155–2161
Chen L, Mediavilla JR, Oliveira DC, Willey BM, de Lencastre H, Kreiswirth BN (2009) Multiplex real-time PCR for rapid Staphylococcal cassette chromosome mec typing. J Clin Microbiol 47:3692–3706
Francois P, Renzi G, Pittet D, Bento M, Lew D, Harbarth S, Vaudaux P, Schrenzel J (2004) A novel multiplex real-time PCR assay for rapid typing of major staphylococcal cassette chromosome mec elements. J Clin Microbiol 42:3309–3312
Monecke S, Slickers P, Ehricht R (2008) Assignment of Staphylococcus aureus isolates to clonal complexes based on microarray analysis and pattern recognition. FEMS Immunol Med Microbiol 53:237–251
Zhang K, McClure JA, Conly JM (2012) Enhanced multiplex PCR assay for typing of staphylococcal cassette chromosome mec types I to V in methicillin-resistant Staphylococcus aureus. Mol Cell Probes 26:218–221
Zhang K, McClure JA, Elsayed S, Louie T, Conly JM (2005) Novel multiplex PCR assay for characterization and concomitant subtyping of staphylococcal cassette chromosome mec types I to V in methicillin-resistant Staphylococcus aureus. J Clin Microbiol 43:5026–5033
Blair JE, Williams RE (1961) Phage typing of staphylococci. Bull World Health Organ 24:771–784
Weller TM (2000) Methicillin-resistant Staphylococcus aureus typing methods: which should be the international standard? J Hosp Infect 44:160–172
Blair JE (1966) Untypable staphylococci: their identification and possible origin. Health Lab Sci 3:229–234
Bannerman TL, Hancock GA, Tenover FC, Miller JM (1995) Pulsed-field gel electrophoresis as a replacement for bacteriophage typing of Staphylococcus aureus. J Clin Microbiol 33:551–555
Lundholm M, Bergendahl B (1988) Heat treatment to increase phage typability of Staphylococcus aureus. Eur J Clin Microbiol Infect Dis 7:300–302
Mehndiratta PL, Bhalla P (2012) Typing of Methicillin resistant Staphylococcus aureus: a technical review. Indian J Med Microbiol 30:16–23
Mulligan ME, Arbeit RD (1991) Epidemiologic and clinical utility of typing systems for differentiating among strains of methicillin-resistant Staphylococcus aureus. Infect Control Hosp Epidemiol 12:20–28
Tenover FC, Arbeit R, Archer G, Biddle J, Byrne S, Goering R, Hancock G, Hebert GA, Hill B, Hollis R et al (1994) Comparison of traditional and molecular methods of typing isolates of Staphylococcus aureus. J Clin Microbiol 32:407–415
Welsh J, McClelland M (1990) Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res 18:7213–7218
Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535
Struelens MJ, Bax R, Deplano A, Quint WG, Van Belkum A (1993) Concordant clonal delineation of methicillin-resistant Staphylococcus aureus by macrorestriction analysis and polymerase chain reaction genome fingerprinting. J Clin Microbiol 31:1964–1970
Saulnier P, Bourneix C, Prevost G, Andremont A (1993) Random amplified polymorphic DNA assay is less discriminant than pulsed-field gel electrophoresis for typing strains of methicillin-resistant Staphylococcus aureus. J Clin Microbiol 31:982–985
van Belkum A, Kluytmans J, van Leeuwen W, Bax R, Quint W, Peters E, Fluit A, Vandenbroucke-Grauls C, van den Brule A, Koeleman H et al (1995) Multicenter evaluation of arbitrarily primed PCR for typing of Staphylococcus aureus strains. J Clin Microbiol 33:1537–1547
Versalovic J, Koeuth T, Lupski JR (1991) Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res 19:6823–6831
van der Zee A, Verbakel H, van Zon JC, Frenay I, van Belkum A, Peeters M, Buiting A, Bergmans A (1999) Molecular genotyping of Staphylococcus aureus strains: comparison of repetitive element sequence-based PCR with various typing methods and isolation of a novel epidemicity marker. J Clin Microbiol 37:342–349
Sabat A, Malachowa N, Miedzobrodzki J, Hryniewicz W (2006) Comparison of PCR-based methods for typing Staphylococcus aureus isolates. J Clin Microbiol 44:3804–3807
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M et al (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Mortimer P, Arnold C (2001) FAFLP: last word in microbial genotyping? J Med Microbiol 50:393–395
Duim B, Wassenaar TM, Rigter A, Wagenaar J (1999) High-resolution genotyping of Campylobacter strains isolated from poultry and humans with amplified fragment length polymorphism fingerprinting. Appl Environ Microbiol 65:2369–2375
Zhao S, Mitchell SE, Meng J, Kresovich S, Doyle MP, Dean RE, Casa AM, Weller JW (2000) Genomic typing of Escherichia coli O157:H7 by semi-automated fluorescent AFLP analysis. Microbes Infect 2:107–113
Novick RP (2003) Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 48:1429–1449
Peacock SJ, Moore CE, Justice A, Kantzanou M, Story L, Mackie K, O’Neill G, Day NP (2002) Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect Immun 70:4987–4996
Francois P, Koessler T, Huyghe A, Harbarth S, Bento M, Lew D, Etienne J, Pittet D, Schrenzel J (2006) Rapid Staphylococcus aureus agr type determination by a novel multiplex real-time quantitative PCR assay. J Clin Microbiol 44:1892–1895
Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG (2002) The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci U S A 99:7687–7692
Goering RV (2010) Pulsed field gel electrophoresis: a review of application and interpretation in the molecular epidemiology of infectious disease. Infect Genet Evol 10:866–875
Schwartz DC, Cantor CR (1984) Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell 37:67–75
Matushek MG, Bonten MJ, Hayden MK (1996) Rapid preparation of bacterial DNA for pulsed-field gel electrophoresis. J Clin Microbiol 34:2598–2600
McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC (2003) Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol 41:5113–5120
Murchan S, Kaufmann ME, Deplano A, de Ryck R, Struelens M, Zinn CE, Fussing V, Salmenlinna S, Vuopio-Varkila J, El Solh N, Cuny C, Witte W, Tassios PT, Legakis N, van Leeuwen W, van Belkum A, Vindel A, Laconcha I, Garaizar J, Haeggman S, Olsson-Liljequist B, Ransjo U, Coombes G, Cookson B (2003) Harmonization of pulsed-field gel electrophoresis protocols for epidemiological typing of strains of methicillin-resistant Staphylococcus aureus: a single approach developed by consensus in 10 European laboratories and its application for tracing the spread of related strains. J Clin Microbiol 41:1574–1585
Mulvey MR, Chui L, Ismail J, Louie L, Murphy C, Chang N, Alfa M (2001) Development of a Canadian standardized protocol for subtyping methicillin-resistant Staphylococcus aureus using pulsed-field gel electrophoresis. J Clin Microbiol 39:3481–3485
Carles-Nurit MJ, Christophle B, Broche S, Gouby A, Bouziges N, Ramuz M (1992) DNA polymorphisms in methicillin-susceptible and methicillin-resistant strains of Staphylococcus aureus. J Clin Microbiol 30:2092–2096
Ichiyama S, Ohta M, Shimokata K, Kato N, Takeuchi J (1991) Genomic DNA fingerprinting by pulsed-field gel electrophoresis as an epidemiological marker for study of nosocomial infections caused by methicillin-resistant Staphylococcus aureus. J Clin Microbiol 29:2690–2695
Prevost G, Jaulhac B, Piemont Y (1992) DNA fingerprinting by pulsed-field gel electrophoresis is more effective than ribotyping in distinguishing among methicillin-resistant Staphylococcus aureus isolates. J Clin Microbiol 30:967–973
Argudin MA, Rodicio MR, Guerra B The emerging methicillin-resistant Staphylococcus aureus ST398 clone can easily be typed using the Cfr9I SmaI-neoschizomer. Lett Appl Microbiol 50:127–130
Chu G, Vollrath D, Davis RW (1986) Separation of large DNA molecules by contour-clamped homogeneous electric fields. Science 234:1582–1585
Cookson BD, Robinson DA, Monk AB, Murchan S, Deplano A, de Ryck R, Struelens MJ, Scheel C, Fussing V, Salmenlinna S, Vuopio-Varkila J, Cuny C, Witte W, Tassios PT, Legakis NJ, van Leeuwen W, van Belkum A, Vindel A, Garaizar J, Haeggman S, Olsson-Liljequist B, Ransjo U, Muller-Premru M, Hryniewicz W, Rossney A, O’Connell B, Short BD, Thomas J, O’Hanlon S, Enright MC (2007) Evaluation of molecular typing methods in characterizing a European collection of epidemic methicillin-resistant Staphylococcus aureus strains: the HARMONY collection. J Clin Microbiol 45:1830–1837
Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B (1995) Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33:2233–2239
Swaminathan B, Barrett TJ, Hunter SB, Tauxe RV, Force CDCPT (2001) PulseNet: the molecular subtyping network for foodborne bacterial disease surveillance, United States. Emerg Infect Dis 7:382–389
Cookson BD, Aparicio P, Deplano A, Struelens M, Goering R, Marples R (1996) Inter-centre comparison of pulsed-field gel electrophoresis for the typing of methicillin-resistant Staphylococcus aureus. J Med Microbiol 44:179–184
van Belkum A, van Leeuwen W, Kaufmann ME, Cookson B, Forey F, Etienne J, Goering R, Tenover F, Steward C, O’Brien F, Grubb W, Tassios P, Legakis N, Morvan A, El Solh N, de Ryck R, Struelens M, Salmenlinna S, Vuopio-Varkila J, Kooistra M, Talens A, Witte W, Verbrugh H (1998) Assessment of resolution and intercenter reproducibility of results of genotyping Staphylococcus aureus by pulsed-field gel electrophoresis of SmaI macrorestriction fragments: a multicenter study. J Clin Microbiol 36:1653–1659
Frenay HM, Bunschoten AE, Schouls LM, van Leeuwen WJ, Vandenbroucke-Grauls CM, Verhoef J, Mooi FR (1996) Molecular typing of methicillin-resistant Staphylococcus aureus on the basis of protein A gene polymorphism. Eur J Clin Microbiol Infect Dis 15:60–64
Koreen L, Ramaswamy SV, Graviss EA, Naidich S, Musser JM, Kreiswirth BN (2004) spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro- and macrovariation. J Clin Microbiol 42:792–799
Shopsin B, Gomez M, Montgomery SO, Smith DH, Waddington M, Dodge DE, Bost DA, Riehman M, Naidich S, Kreiswirth BN (1999) Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J Clin Microbiol 37:3556–3563
Harmsen D, Claus H, Witte W, Rothganger J, Claus H, Turnwald D, Vogel U (2003) Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J Clin Microbiol 41:5442–5448
Strommenger B, Kettlitz C, Weniger T, Harmsen D, Friedrich AW, Witte W (2006) Assignment of Staphylococcus isolates to groups by spa typing, SmaI macrorestriction analysis, and multilocus sequence typing. J Clin Microbiol 44:2533–2540
Mellmann A, Weniger T, Berssenbrugge C, Rothganger J, Sammeth M, Stoye J, Harmsen D (2007) Based Upon Repeat Pattern (BURP): an algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol 7:98
Nubel U, Roumagnac P, Feldkamp M, Song JH, Ko KS, Huang YC, Coombs G, Ip M, Westh H, Skov R, Struelens MJ, Goering RV, Strommenger B, Weller A, Witte W, Achtman M (2008) Frequent emergence and limited geographic dispersal of methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci USA 105:14130–14135
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG (2000) Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38:1008–1015
Aanensen DM, Spratt BG (2005) The multilocus sequence typing network: mlst.net. Nucleic Acids Res 33:W728–W733
Jolley KA, Maiden MC (2010) BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595
Jolley KA, Bray JE, Maiden MCJ (2018) Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG (2004) eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518–1530
Spratt BG, Hanage WP, Li B, Aanensen DM, Feil EJ (2004) Displaying the relatedness among isolates of bacterial species -- the eBURST approach. FEMS Microbiol Lett 241:129–134
Ramsay G (1998) DNA chips: state-of-the art. Nat Biotechnol 16:40–44
Sanchini A, Campanile F, Monaco M, Cafiso V, Rasigade JP, Laurent F, Etienne J, Stefani S, Pantosti A (2011) DNA microarray-based characterisation of Panton-Valentine leukocidin-positive community-acquired methicillin-resistant Staphylococcus aureus from Italy. Eur J Clin Microbiol Infect Dis 30:1399–1408
Lindsay JA, Moore CE, Day NP, Peacock SJ, Witney AA, Stabler RA, Husain SE, Butcher PD, Hinds J (2006) Microarrays reveal that each of the ten dominant lineages of Staphylococcus aureus has a unique combination of surface-associated and regulatory genes. J Bacteriol 188:669–676
McCarthy AJ, Breathnach AS, Lindsay JA (2012) Detection of mobile-genetic-element variation between colonizing and infecting hospital-associated methicillin-resistant Staphylococcus aureus isolates. J Clin Microbiol 50:1073–1075
McCarthy AJ, Lindsay JA (2012) The distribution of plasmids that carry virulence and resistance genes in Staphylococcus aureus is lineage associated. BMC Microbiol 12:104
Bischoff M, Dunman P, Kormanec J, Macapagal D, Murphy E, Mounts W, Berger-Bachi B, Projan S (2004) Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J Bacteriol 186:4085–4099
Dunman PM, Murphy E, Haney S, Palacios D, Tucker-Kellogg G, Wu S, Brown EL, Zagursky RJ, Shlaes D, Projan SJ (2001) Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J Bacteriol 183:7341–7353
Luong TT, Dunman PM, Murphy E, Projan SJ, Lee CY (2006) Transcription Profiling of the mgrA Regulon in Staphylococcus aureus. J Bacteriol 188:1899–1910
Dunman PM, Mounts W, McAleese F, Immermann F, Macapagal D, Marsilio E, McDougal L, Tenover FC, Bradford PA, Petersen PJ, Projan SJ, Murphy E (2004) Uses of Staphylococcus aureus GeneChips in genotyping and genetic composition analysis. J Clin Microbiol 42:4275–4283
Monecke S, Ehricht R (2005) Rapid genotyping of methicillin-resistant Staphylococcus aureus (MRSA) isolates using miniaturised oligonucleotide arrays. Clin Microbiol Infect 11:825–833
Otsuka J, Kondoh Y, Amemiya T, Kitamura A, Ito T, Baba S, Cui L, Hiramatsu K, Tashiro T, Tashiro H (2008) Development and validation of microarray-based assay for epidemiological study of MRSA. Mol Cell Probes 22:1–13
Saunders NA, Underwood A, Kearns AM, Hallas G (2004) A virulence-associated gene microarray: a tool for investigation of the evolution and pathogenic potential of Staphylococcus aureus. Microbiology 150:3763–3771
Strommenger B, Schmidt C, Werner G, Roessle-Lorch B, Bachmann TT, Witte W (2007) DNA microarray for the detection of therapeutically relevant antibiotic resistance determinants in clinical isolates of Staphylococcus aureus. Mol Cell Probes 21:161–170
Monecke S, Jatzwauk L, Weber S, Slickers P, Ehricht R (2008) DNA microarray-based genotyping of methicillin-resistant Staphylococcus aureus strains from Eastern Saxony. Clin Microbiol Infect 14:534–545
Monecke S, Coombs G, Shore AC, Coleman DC, Akpaka P, Borg M, Chow H, Ip M, Jatzwauk L, Jonas D, Kadlec K, Kearns A, Laurent F, O’Brien FG, Pearson J, Ruppelt A, Schwarz S, Scicluna E, Slickers P, Tan HL, Weber S, Ehricht R (2011) A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6:e17936
McCarthy AJ, Witney AA, Gould KA, Moodley A, Guardabassi L, Voss A, Denis O, Broens EM, Hinds J, Lindsay JA (2011) The distribution of mobile genetic elements (MGEs) in MRSA CC398 is associated with both host and country. Genome Biol Evol 3:1164–1174
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchene S, Fourment M, Gavryushkina A, Heled J, Jones G, Kuhnert D, De Maio N, Matschiner M, Mendes FK, Muller NF, Ogilvie HA, du Plessis L, Popinga A, Rambaut A, Rasmussen D, Siveroni I, Suchard MA, Wu CH, Xie D, Zhang C, Stadler T, Drummond AJ (2019) BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol 15:e1006650
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A (2018) Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4:vey016
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS (2011) PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352
Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD (2010) Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474
Harrison EM, Paterson GK, Holden MT, Larsen J, Stegger M, Larsen AR, Petersen A, Skov RL, Christensen JM, Bak Zeuthen A, Heltberg O, Harris SR, Zadoks RN, Parkhill J, Peacock SJ, Holmes MA (2013) Whole genome sequencing identifies zoonotic transmission of MRSA isolates with the novel mecA homologue mecC. EMBO Mol Med 5:509–515
Price JR, Golubchik T, Cole K, Wilson DJ, Crook DW, Thwaites GE, Bowden R, Walker AS, Peto TE, Paul J, Llewelyn MJ (2014) Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of Staphylococcus aureus in an intensive care unit. Clin Infect Dis 58:609–618
Koser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, Sanders M, Enright MC, Dougan G, Bentley SD, Parkhill J, Fraser LJ, Betley JR, Schulz-Trieglaff OB, Smith GP, Peacock SJ (2012) Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med 366:2267–2275
Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, Barron A, Bason N, Bentley SD, Chillingworth C, Chillingworth T, Churcher C, Clark L, Corton C, Cronin A, Doggett J, Dowd L, Feltwell T, Hance Z, Harris B, Hauser H, Holroyd S, Jagels K, James KD, Lennard N, Line A, Mayes R, Moule S, Mungall K, Ormond D, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Sharp S, Simmonds M, Stevens K, Whitehead S, Barrell BG, Spratt BG, Parkhill J (2004) Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci USA 101:9786–9791
McAdam PR, Templeton KE, Edwards GF, Holden MT, Feil EJ, Aanensen DM, Bargawi HJ, Spratt BG, Bentley SD, Parkhill J, Enright MC, Holmes A, Girvan EK, Godfrey PA, Feldgarden M, Kearns AM, Rambaut A, Robinson DA, Fitzgerald JR (2012) Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci USA 109:9107–9112
Nubel U, Dordel J, Kurt K, Strommenger B, Westh H, Shukla SK, Zemlickova H, Leblois R, Wirth T, Jombart T, Balloux F, Witte W (2010) A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog 6:e1000855
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
McClure, J.M., Zhang, K. (2022). Staphylococci . In: de Filippis, I. (eds) Molecular Typing in Bacterial Infections, Volume II. Springer, Cham. https://doi.org/10.1007/978-3-030-83217-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-83217-9_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-83216-2
Online ISBN: 978-3-030-83217-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)