
Abstract
Wood is a porous, hygroscopic material that can take up water both within cell walls (cell-wall water) and in the macrovoid structure (capillary water). Therefore, moisture transport in wood occurs through multiple pathways and phases of water, that is, both cell-wall water, liquid water, and water vapor can be transported through the material structure. The amount of water in wood is quantified by the moisture content (mass of water in relation to the dry mass), which changes with the surrounding environmental conditions (relative humidity and temperature). This relation is commonly depicted in sorption isotherms that plot the equilibrium moisture content as function of relative humidity for specific, constant temperatures. How water is taken up by wood changes over the relative humidity range. In the hygroscopic range (0% to 97–98% relative humidity), water is predominantly taken up in cell walls, whereas capillary condensation of liquid water in the macrovoid structure becomes dominant in the over-hygroscopic range (>98% relative humidity). The equilibrium illustrated in sorption isotherms for specific environmental conditions is not singular, but depends on the sorption history; this phenomenon is known as sorption hysteresis. Sorption isotherms are generally modeled using mathematical expressions that are fitted to data in the hygroscopic range. Some of these models include quantities describing the physical reality of wood-water interactions; however, these quantities rarely match up to the experimentally determined reality for wood. The same can be said of the mathematical models describing the kinetics of water uptake in wood cell walls.
One of the most well-known concepts concerning water in wood is the fiber saturation point (FSP). It can be found as the threshold moisture content above which physical wood properties do not change significantly with moisture content. The FSP is, however, lower than the maximum moisture content of wood cell walls which occurs in the fully water saturated state. A change in moisture content below the FSP is accompanied by dimensional changes, that is, shrinkage and swelling, which reflect changes in the amount of water within cell walls. While this phenomenon originates at the nanoscale, it is observed on all length scales of wood. Such dimensional changes as well as other physical wood properties and fundamental wood-water interactions can be altered by chemical modification of wood. How these relations are affected depends on the chemical nature of the modification. At the end of this chapter, a broad overview is given of experimental methods for characterizing moisture in wood, including determination of moisture content, sorption isotherms, moisture state and location, and moisture transport in wood.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Moisture
- Sorption
- Cell-wall water
- Capillary water
- Sorption isotherms
- Fiber saturation
- Sorption hysteresis
- Chemical modification
- Swelling
- Diffusion
1 Wood-Water Interactions
In this chapter, the term absorption is used for water uptake. According to the International Union of Pure and Applied Chemistry (IUPAC) [1], absorption is the “process of one material (absorbate) being retained by another (absorbent); this may be the physical solution of a gas, liquid, or solid in a liquid, attachment of molecules of a gas, vapour, liquid, or dissolved substance to a solid surface by physical forces, etc.” For water uptake in wood, the term absorption therefore covers uptake by any mechanism. Adsorption is defined by IUPAC as an “increase in the concentration of a dissolved substance at the interface of a condensed and a liquid phase due to the operation of surface forces.” In the case of water uptake in materials, adsorption describes the adhesion of water molecules to a surface. For wood, this is not a dominating mechanism for water uptake. Moreover, this chapter deals mainly with wood-water relations in wood and modified wood. For detailed information about wood-water relations in wood-based materials, please see [2, 3, 6, 8, 9].
1.1 Cell-Wall Water
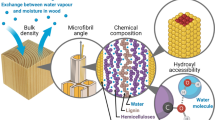
Water molecules within wood cell walls are commonly referred to as “bound water,” and they are closely interacting with the constituent cell-wall polymers. The polar water molecules mainly interact by hydrogen bonding with cell-wall hydroxyl (OH) groups that are found on all cell-wall polymers [4, 5]. The aggregated cellulose microfibrils are, however, impenetrable for water, and it is therefore only hydroxyls on exposed surfaces which can interact directly with water [6, 7]. The hydroxyls capable of interacting with water are often referred to as “accessible hydroxyls,” and the concentration of these in a given material is termed the “hydroxyl accessibility” and can be determined by various methods, see Sect. 7.11.4. Since cellulose is the main cell-wall constituent and the microfibrils have a significant fraction of their hydroxyls within the microfibrillar structure, the relative hydroxyl accessibility (in %) in wood is typically found to be less than half of the total hydroxyl concentration, while the absolute hydroxyl accessibility (in mmol g−1) is in the range 6–10 mmol g−1, see Table 7.1.
The properties of water within cell walls differ from those of water in the solid (ice) and liquid states, which suggests that it is a state distinctly different from other states of water. The confinement of water molecules between the solid cell-wall constituents and the close association by hydrogen bonds between water and the solid substance would suggest a state of cell-wall water comparable to ice. On the other hand, the population of cell-wall water molecules exhibit a larger variation in hydrogen bond lengths and bond angles in the interactions with the cell-wall polymers than in ice as well as retain much higher mobility than in ice. This suggests a state of cell-wall water close to that of liquid water. In fact, the behavior of cell-wall water falls somewhere between that of ice and liquid water, but cell-wall water behaves increasingly more like liquid water as the concentration of water within the cell walls increases.
It has since long been debated whether the overall population of water molecules within cell walls can be described as comprised of two or more distinct subpopulations or types of water. Several theoretical models for describing equilibria between cell-wall water and the ambient environment contain at least two types of water molecules, see Sect. 7.3.2. These interact either directly with the cell-wall constituents or indirectly via bonding to other water molecules within the cell walls. The separation of cell-wall water into several types was in the 1980s corroborated by experimental data from differential scanning calorimetry (DSC) on cellulosic materials that distinguished two types of water within cell walls: freezing and nonfreezing [8,9,24], see Sect. 7.11.1. Based on an observed phase change in the temperature range −20 °C to −10 °C during cooling and a small peak shoulder on the melting peak of liquid water, the freezing cell-wall water was suggested to be less tightly associated with the polymeric material and able to freeze. On the other hand, no phase change was observed for the nonfreezing part of the cell-wall water down to −70 °C [23]. Although a peak shoulder on the water melting peak has been observed with DSC for wood [25, 26], the existence of freezing cell-wall water in wood and other cellulosic materials is controversial. Thus, several studies using DSC have not identified any freezing cell-wall water even for high concentrations of water [23, 27, 28] and other experimental techniques have similarly failed in identifying any phase change of cell-wall water [29, 30]. Recently, Zelinka et al. [28] showed that the occurrence of a freezing peak around −20 °C to −10 °C is an artifact from the specimen preparation and only occurred for isolated ball-milled cellulose and not for solid or milled wood.
Although cell-wall water in wood cannot be partitioned based on phase changes, several experimental techniques have shown the existence of two distinct subpopulations of water within cell walls. The two populations of water encounter different local environments in the cell walls in terms of pore size and chemical interactions as shown by 2D low-field nuclear magnetic resonance (LFNMR) correlation spectroscopy [31, 32], see Sect. 7.11.2. It is, however, not clear what or where these populations are. The presence of different subpopulations of cell-wall water appears to be linked to the presence of the stiff, solid aggregated cellulose microfibrils in cell walls. Close proximity of water molecules to cellulose surfaces reduces the water mobility [5], and the various crystallographic planes of cellulose microfibrils affect the water dynamics differently [4, 19,20,21,36]. Moreover, the water molecules may not be distributed evenly in the regions of the cell walls accessible to water, since some studies find increased concentrations at interfaces between cellulose and the amorphous cell-wall polymers [37, 38]. This might explain why the presence of cellulose microfibrils in the wood cell walls may be able to affect the overall population of water in cell walls so significantly, even though cellulose microfibrils only constitute half of the cell-wall chemistry and only have about 1/3 of their hydroxyls exposed to water [12].
1.2 Capillary Water
At high levels of relative humidity, water is not only present within cell walls, but also in macrovoids in the wood structure such as cell lumina and pit chambers. Here, water uptake is dominated by capillary condensation, that is, water vapor is condensed in pores even below the saturation vapor pressure. At which relative humidity capillary condensation occurs is described by the Kelvin equation:
where ϕ (Pa Pa−1) is the relative humidity, γ (N m−1) is the surface tension of water, Mw (0.018 kg mol−1) is the molar mass of water, θ (rad) is the contact angle, R (8.314 J mol−1 K−1) is the gas constant, ρw (kg m−3) is the density of water, T (K) is temperature, and r (m) is defined by
where r1 and r2 (m) are the two radii of condensation. For surfaces that are wetted by water, the contact angle is zero. For a cylindrical pore, the two radii of condensation equal the pore radius r, that is, r1 = r2 = r, and Eq. (7.1) is then written:
For a slit-shaped pore, r1 = r and r2 = ∞, and Eq. (7.1) is then written:
From Eqs. (7.3) and (7.4), it is seen that capillary condensation occurs at a lower relative humidity in small pores than in large pores. Based on the water properties at a given temperature, the relative humidity at which capillary condensation occurs in different pore sizes can be determined assuming either cylindrical or slit-shaped pores, see Table 7.2. In wood, the macrovoids where capillary condensation occurs are neither cylindrical (apart from vessels) nor slit-shaped. However, to get a sense of the relation between relative humidity and capillary condensation in cell lumina, these can be assumed to be cylindrical. Since cell lumina in wood are typically in the range of 10–40 μm, they are not filled by capillary condensation until above approximately 99.99% relative humidity during absorption, see Table 7.2. This is the reason for the steep absorption isotherm for wood above 99% relative humidity, see Fig. 7.1. However, in smaller voids, such as ends of the tapered tracheids and pit chambers, capillary condensation occurs at slightly lower, but still high, relative humidity levels [39].

(a) Sorption isotherm of Norway spruce (Picea abies (L.) Karst.) as a function of relative humidity based on data from [49]. (b) Over-hygroscopic moisture range plotted as a function of water potential which gives a higher resolution in this range. (c) Schematic softwood tracheid with water located in different parts of the wood structure. The relative humidity levels where water uptake in these parts is significant are indicated by the letters A–D
2 Moisture Content
Within wood science, the moisture content is most commonly determined by the ratio
where ω (g g−1) is the moisture content, mw (g) is the mass of water, and mdry (g) is the dry mass. Since water in wood can be present both in cell walls and in the macrovoid structure, the maximum moisture content is the sum of the amounts of water present in cell walls and macrovoids. The macrovoid porosity, Pmv, is determined by:
where ρb (kg m−3) is the bulk density of wood, and ρs (kg m−3) is the solid (skeletal) density, that is, cell-wall density which is in the range 1430–1530 kg m−3 [40,41,42,43,44,45,46,47,48]. The maximum mass of capillary water in macrovoids in a volume V (m3) of wood is thus:
where ρw (kg m−3) is the density of water. The moisture content of water-filled macrovoids can then be determined as:
where ωcap (g g−1) is the moisture content of water-filled macrovoids. Finally, the maximum moisture content is obtained by adding the contributions from capillary water in voids and the maximum cell-wall moisture content
where ωmax (g g−1) is the maximum moisture content and ωcell wall (g g−1) is the maximum cell-wall moisture content, see Sect. 7.4.
3 Sorption Isotherms
The relation between the moisture content of a material and the state of the water in equilibrium with the ambient climate at constant temperature is described by sorption isotherms. In the hygroscopic moisture range, sorption isotherms are often given as a function of relative humidity. In the over-hygroscopic range, sorption isotherms are generally presented as a function of pore water pressure (Pa) or water potential (J kg−1) on a logarithmic scale, see Fig. 7.1. The latter is related to relative humidity by Eq. (7.57) in Sect. 7.10.2 and gives a higher resolution at high levels of relative humidity.
In the over-hygroscopic range, the water uptake is dominated by capillary condensation in the macrovoid structure (cell lumina and pit chambers). The amount of water taken up in this range is therefore highly related to the bulk density of the wood; the lower density, the more macrovoid volume is available for water. In the hygroscopic range, the moisture concentration (kg m−3) is density dependent since water here is found within cell walls; the more cell-wall material (higher density) the higher moisture concentration (kg m−3). However, since the moisture content of wood most often is related to the dry mass (Eq. (7.5)), the sorption isotherm in the hygroscopic range is not strongly influenced by density [50]. Full sorption isotherms for some wood species and hygroscopic sorption isotherms for several wood species are shown in Figs. 7.2 and 7.3, respectively.



Hygroscopic sorption isotherms for several wood species. (a) Pine (Pinus sylvestris L.), (b) Western white pine (Pinus monticola D. Don), (c) Douglas fir (Pseudotsuga menziesii (Mirb.) Franco), (d) White spruce (Picea glauca (Moench) Voss), (e) Redwood (Sequoia sempervirens (D. Don) Endl.), (f) Birch (Betula sp.), (g) Oak (Quercus robur L.), (h) Teak (Tectona grandis L.f.), (i) Mahogany (Swietenia sp.). (Data from [51] (o) and [53] (+))
The equilibrium moisture content of a material does not only depend on the ambient climate, but also on the moisture history of the material. The sorption isotherm determined in absorption (uptake) from dry state is lower than the sorption isotherm determined in desorption (drying) from the water-saturated state. This phenomenon is termed sorption hysteresis and is observed in many porous materials, see Sect. 7.3.1. Sorption isotherms initiated from any other state than the dry or water-saturated state are generally referred to as scanning isotherms. These connect the desorption and absorption isotherms and describe the equilibrium moisture content of a specimen exposed first to desorption and then to absorption, or vice versa. For example, if a dry specimen is placed in a high relative humidity ϕ1 (e.g., 95%) until equilibrium is reached and then is dried to a lower relative humidity ϕ2 (e.g., 80%), the equilibrium moisture content will be lower than if drying to ϕ2 was initiated from the water-saturated state, see Fig. 7.4. Both moisture contents obtained are, however, equilibrium moisture contents. Many desorption isotherms in literature are in fact scanning isotherms as they are initiated from 90% to 95% reached by absorption [56], whereas less data is available for absorption scanning isotherms and desorption scanning isotherms from lower initial relative humidity levels. At present, no data for scanning isotherms in the over-hygroscopic range is available.

A schematic illustration of a desorption isotherm (upper curve), absorption isotherm (lower curve), and a scanning isotherm. Drying from a high humidity ϕ2 reached by absorption (1) to a lower humidity ϕ1 (2) will give a lower equilibrium moisture content than if drying to ϕ2 is initiated from water saturated state (3)
In older literature, it is sometimes stated that the initial desorption isotherm for wood after harvesting is higher than any desorption isotherm obtained after re-saturating dry specimens [53]. However, Hoffmeyer et al. [57] showed that the initial desorption isotherm can be reproduced after vacuum water-saturation. It is important to keep in mind that initiation of desorption from another state that the water-saturated state yields scanning isotherms and not desorption isotherms. Due to the large sorption hysteresis in wood above 98–99% relative humidity, conditioning to a high moisture state by vapor absorption does not give a high enough moisture content for the specimen to follow the desorption isotherm when drying (Fig. 7.1).
The sorption isotherm also depends on temperature; a higher temperature lowers the sorption isotherm in the hygroscopic [58] and the over-hygroscopic moisture ranges [59], see Fig. 7.5 In the over-hygroscopic range, one possible explanation for this could be the temperature dependence of the surface tension of water. However, this cannot fully explain the temperature dependence of the over-hygroscopic sorption isotherm [59]. The reasons for temperature dependence of the hygroscopic sorption isotherms are further discussed in Sect. 7.3.1.

One of the most prominent datasets concerning wood moisture sorption is the USDA Wood Handbook data presented in numerous publications from the Forest Products Laboratory. The historical origin of the sorption data was thoroughly tracked by Glass et al. [50]. They found that the sorption data is mainly based on experimental measurements performed between 1910 and 1930 with more data occasionally added in the following years. While the original purpose of the data was for practical uses, for example, in wood drying or in conditioning wood before test of mechanical or other physical properties, several studies have used the data for scientific investigations of fundamental wood-water relations. However, Glass et al. [50] clearly document that this data is not reliable for detailed scientific investigations of fundamental wood-water interactions. For instance, the documentation for the experimental methodologies used to obtain the underlying experimental data is often lacking or of poor quality. In addition, the USDA Wood Handbook sorption data represents an undocumented mixture of experimental sorption data for various wood species with extrapolated and interpolated data, and several methods for establishing equilibrium (absorption, desorption, or a mixture of both) have been used. Therefore, this sorption isotherm data should only be used as originally intended by the Forest Products Laboratory, that is, for predicting approximate moisture contents in various environments, and not for detailed scientific studies [50].
3.1 Sorption Hysteresis
Sorption hysteresis is observed as a difference in moisture content between moisture states reached by absorption, desorption, or scanning; for instance, the moisture content reached by desorption is higher than that reached by absorption at similar relative humidity and temperature. The mechanisms behind sorption hysteresis are different in the hygroscopic and over-hygroscopic moisture ranges. In the hygroscopic range, sorption hysteresis for wood decreases with increasing temperature and reportedly vanishes above 75 °C [58]. A similar phenomenon is observed for polymer films of hemicelluloses and cellulose [62, 63]. Sorption hysteresis has been suggested to be related to softening of the amorphous matrix polymers (hemicelluloses and lignin) as they pass their glass transition point and enter the rubbery state [62,63,64]. Hemicelluloses soften above 0.15–0.16 g g−1 moisture content at room temperature which corresponds to about 75% relative humidity, while lignin softens above 60–70 °C at high moisture content [64]. Sorption hysteresis has also been linked with a change in hydroxyl accessibility upon drying; as water is removed and the cell-wall structure shrinks, parts of the constituents have been suggested to form hydrogen-bonded configurations that cannot be re-opened by water [65, 66]. This phenomenon of lower hydroxyl accessibility after drying is known as hornification and has been observed in never-dried, isolated cellulose microfibrils from various origins [67, 68]. In the composite wood cell wall, however, cellulose microfibrils are embedded in hemicelluloses and lignin which prevent hornification [69, 70]. Thus, no change in hydroxyl accessibility is observed in native wood upon full re-saturation with water [12].
Sorption hysteresis is often quantified as either absolute sorption hysteresis or relative sorption hysteresis where the former describes the absolute difference in moisture content between two isotherms (e.g., absorption and desorption), and the latter normalizes this difference with the moisture content reached through absorption. Many studies characterize sorption hysteresis based on absorption and scanning isotherms initiated from 95% relative humidity reached by absorption. Hysteresis curves calculated from these two isotherms exhibit markedly different behavior than sorption hysteresis based on absorption and desorption isotherms, see Fig. 7.6 [52]. In the first case, nonlinear curves are observed with a peak in absolute hysteresis around 75% relative humidity when scanning desorption is initiated from 95% relative humidity. When desorption is initiated from water-saturated conditions, the absolute hysteresis curves are nearly linear (Fig. 7.6).

(a) Desorption and absorption isotherms (-), scanning isotherms from 95% (--), and 80% relative humidity (..). (b and c) Absolute and relative sorption hysteresis calculated from the sorption isotherms in a. (Data from [52])
In the over-hygroscopic range, capillary condensation of water first occurs in small voids, for example pit chambers, and in larger voids such as cell lumina at higher relative humidity levels, see Sect. 7.1.2. The sorption hysteresis in this range is large as seen in Fig. 7.1. One well-established theory for explaining sorption hysteresis in porous materials is the ink-bottle or pore blocking effect [71]. That is, when a pore is filled, capillary condensation is controlled by the radius of curvature of the pore (Eq. (7.1)), but if the pore has a narrow neck, the emptying of the pore is controlled by the radius of the neck. Such a pore is thus emptied at a lower relative humidity than the relative humidity at which it is filled during absorption. In wood, cell lumina of adjacent cells are connected by small pit openings and the ink-bottle effect is therefore considered to be the main mechanism behind sorption hysteresis in the over-hygroscopic moisture range [72]. During absorption, capillary condensation occurs in lumina at a relative humidity corresponding to the lumen size (10–40 μm), but the removal of this water during desorption will only occur below a relative humidity corresponding to the size of the pit openings (<4 μm). For example, assuming cylindrical cell lumina and pit openings with radii 20 μm and 2 μm, respectively, the cell lumina would fill at 99.9946% relative humidity during absorption and be emptied at 99.9461% relative humidity during desorption (Table 7.2).
3.2 Sorption Isotherm Models
Theoretical models for describing water sorption isotherms are frequently used to fit experimental hygroscopic sorption data for wood. A large number of sorption isotherm models exist for the hygroscopic range; 77 are listed by Van den Berg and Bruin [73] but many of these are mathematically equivalent [74]. Many empirical sorption isotherm models have been published which only attempt to provide smooth fitting curves between discrete data points; however, a number of sorption isotherm models have been derived from various theoretical frameworks and therefore claim to be able to derive physical quantities related to water uptake in solid materials. Some of the most well-known and commonly used for wood sorption data are the Brunauer-Emmett-Teller (BET), Guggenheim-Anderson-de Boer (GAB), and Hailwood-Horrobin (HH) sorption isotherm models seen in Eqs. (7.10), (7.11), and (7.12). BET was developed based on a statistical mechanical framework which was expanded in the development of the GAB model. The latter model is equivalent to that of Dent [75] that occasionally has been applied to wood sorption data. The HH model was derived from solution thermodynamics. All three models share the common characteristic that they partition water within the cell wall into at least two types. These go under various names, but are commonly described as primary water and secondary water, and they can be thought of as: (1) water molecules interacting directly with functional groups (“sorption sites”) within the cell walls and (2) water molecules interacting with the other water molecules, including those associated with sorption sites. Although experimental evidence points to the possibility of two populations of absorbed water molecules, see Sect. 7.1.1, these are not necessarily equivalent with the types of water suggested in the sorption isotherm models.
where ω is the moisture content (g g−1), ωmono is termed the monolayer moisture content (g g−1), aw is the water activity (similar to relative humidity) (Pa Pa−1), and K, K1, K2, K3, K4, and Mp are model-specific parameters. The monolayer moisture content, ωmono, is a central physical quantity that is commonly derived in literature with sorption isotherm models. It describes the amount of water molecules directly associated with sorption sites, that is, the maximum amount of primary water. For the HH model, the monolayer moisture content, ωmono, is not a parameter in the original equation but can be found as
For comparison with the BET and GAB models, it can be worthwhile to divide Eq. (7.12) with Eq. (7.13) in order to obtain HH in the form
The BET equation is known to only fit well to wood sorption data in the lower (<70% relative humidity) hygroscopic regime, whereas the GAB and HH models fit the experimental sorption data for wood equally well and better than the BET model, typically with R2 > 0.96. The reason for how the three models perform can be found in their mathematical form. Re-arrangement of Eqs. (7.10), (7.11), and (7.12) yields
From Eqs. (7.15), (7.16) to (7.17), it is seen that the models plotted against aw/ω describe a parabola [82] of the form
where C can be freely selected via tuning ωmono. The A/C and B/C in Eq. (7.18) are, however, not equally free to vary in the three sorption isotherm models which is seen from their ratios
These ratios show that the three parameters A, B, and C are described by three independent model parameters in the GAB and HH models, but only two independent model parameters in the BET model. Thus, the GAB and HH models are mathematical equivalent, and it is not surprising that they give better fits to experimental sorption data than the BET model which is constrained by Eq. (7.19). From the fit of the BET, GAB, and HH sorption isotherm models, the monolayer moisture content can be derived from the fitting parameters of the ABC parabola model (Eq. (7.18)) as
The derived monolayer moisture content depends on the sorption isotherm model used for the derivation as evident in Eqs. (7.23), (7.26), and (7.29), where K2 and K4 are seen to be equal. Since the GAB and HH models are mathematically equivalent, they produce the exact same fit to data of the ABC parabola (Eq. (7.18)), and also predict similar monolayer moisture contents. The constraints of the BET model yield a different fit of the ABC parabola to the data.
Another analysis method for characterizing sorption isotherm data which has gained popularity in the last few decades is the “Excess Surface Work” theory (ESW) [83]. In its core, ESW is the depiction of the number of absorbed water molecules per dry mass versus the excess surface work.
where nads (mol g−1) is the number of absorbed water molecules, Φ (J g−1) is the excess surface work, ω (g g−1) is moisture content, Mw (18 g mol−1) is the molar mass of water, R (8.3145 J mol−1 K−1) is the gas constant, and T (K) is temperature. The excess surface work is seen to be defined by the product of the chemical potential of water vapor and the number of absorbed water molecules. A plot of nads versus Φ produces curves starting at the origin and going to negative values with increasing moisture content before reaching a minimum and increasing again. While this procedure is without assumptions regarding the nature of water sorption in wood, typical ESW analysis proceeds to assign the nads value of the minimum Φ, that is, the valley of the plot, to the formation of a complete monolayer of water on internal surfaces [83]. Beyond this moisture content, all absorbed water molecules are assumed to be secondary water. Thus, the monolayer of primary water is completed before secondary water is taken up.
The formation of a complete monolayer of primary water differs between ESW analysis and the BET, GAB, and HH sorption isotherm models. In the latter three models, water molecules absorbed by a material is distributed unevenly among sorption sites, and the proportion of primary and secondary water varies with the moisture content. Only at full saturation is the water monolayer completely filled in the BET model, whereas it remains incompletely filled in the GAB and HH models. None of these two physical images of how water molecules are distributed between primary and secondary water as function of moisture content fits the experimental data of the possible two populations of water molecules. Thus, the experimental evidence suggests that both types of water are present in wood even at low moisture contents [31, 32] contradicting the ESW analysis. Moreover, the relative proportion of these two experimentally found types of water is not reproduced by any of the sorption isotherm models.
A crucial test of the validity of the physical quantities derived from theoretically based sorption isotherm models is how they compare with experimental measurements of the same quantities. For instance, derived monolayer moisture contents can be compared with experimentally determined hydroxyl accessibility, which is the amount of hydroxyls in the wood capable of binding to water molecules, see Sect. 7.11.4. Figure 7.7a shows sorption isotherm data for Klinki pine at four temperatures with fitted BET, GAB, and HH models in Fig. 7.7b, while Fig. 7.7c depicts the derived monolayer moisture content from these three models and ESW analysis. The predicted monolayer moisture contents range from 0.03 g g−1 to 0.06 g g−1, which are commonly reported values in literature for a range of wood species. These values correspond to water contents of 1.5–3.5 mmol g−1, but as described in Sect. 7.1.1, the hydroxyl accessibility for a range of wood species falls within 6–9 mmol g−1. Most of the experimental data for hydroxyl accessibility is based on deuterium exchange which in fact provides a lower limit of the hydroxyl accessibility. This is because one-third of the surface hydroxyls on cellulose microfibrils cannot be probed by deuterium exchange, even though they interact with water molecules [85, 86]. Thus, the four sorption isotherm models described here all predict monolayer moisture contents which are 2–6 times lower than the experimentally determined amount of accessible hydroxyl sorption sites in the wood which is moreover slightly lower than the actual hydroxyl accessibility. This is a significant failure of the sorption isotherm models in providing realistic physical quantities for wood. Other studies have analyzed the predicted thermodynamic quantities of wood based on various theoretically derived sorption isotherm models [87]. Similarly to the failure in predicting correct monolayer moisture contents, these studies have concluded that the physical reality described by the sorption isotherm models does not match up to the experimentally determined reality for wood [87, 88] .

(a) Absorption isotherms for klinki pine (Araucaria klinkii Lauterb.) at four different temperatures [84], (b) example of fit by the GAB, HH, and BET sorption isotherm models to the sorption data at 10 °C, (c) monolayer moisture content derived by the GAB, HH, and BET sorption isotherm models and ESW analysis as function of temperature
4 Fiber-Saturation Point and Maximum Cell-Wall Moisture Content
One of the most well-known concepts concerning water in wood is the fiber saturation point (FSP). It was originally described by Tiemann [89] who investigated the change in mechanical properties for wood with moisture contents from 0.02 g g−1 to above 0.9 g g−1. Like many other properties of wood, the mechanical performance is heavily influenced by moisture as described in Chaps. 8 and 9. Tiemann obtained different moisture contents by drying in steam atmosphere at 27–60 °C for various durations. Although the drying was carefully undertaken to prevent large moisture gradients, the wood was not conditioned to equilibrium in the sense discussed in Sect. 7.10. The results of the mechanical tests showed that the stiffness and strength increased nonlinearly with decreasing moisture content below a certain threshold value, while remaining constant above this value [89]. The nonlinear change in mechanical properties was later shown to be exponential [90]. Therefore, the logarithm of the mechanical properties exhibits a nearly bi-linear behavior with two distinct regimes: one in which properties change with moisture content and one in which they are constant. Tiemann also found that the bulk wood dimensions change nearly linearly with moisture content in a certain intermediate moisture range while remaining constant at high moisture content [89], see Sect. 7.7. The transition point between the regime where properties change with moisture content to the regime with properties independent of moisture content is defined as the FSP. Thus, the FSP can be found as the inflection point between the regimes after linearizations of either dimensions or the logarithm of mechanical properties. In the original definition, it was envisioned that upon drying all liquid water evaporates before cell-wall drying starts, which then causes a change in properties. Similarily, it was envisioned that during absorption, the strength decreases until cell walls were thought to be saturated. Tiemann discussed that the FSP is less distinct if moisture gradients are present in the specimen [89]. Since moisture content influences more than just dimensions and mechanical properties, see Chap. 6, the change in several other properties with moisture content has also been used to determine the FSP. For instance, Stamm measured electrical conductivity as function moisture content upon drying from water saturation [91]. This property decreases by several orders of magnitude upon drying, and an infliction point can be determined from linearization of the nearly constant conductivity at high moisture contents and the sharply changing conductivity in the hygroscopic range [91]. However, Zelinka et al. [92] have shown that the Stamm-data fits a theoretical model for electrical conductivity derived from percolation theory, at least up to a moisture content of 0.63 g g−1. This questions whether a shift in where the water is found within the wood can actually be deduced from electrical conductivity measurements.
The definitions of the FSP based on changes in properties with moisture content have two important problems. Firstly, most of the experimental data is based on specimens conditioned by interrupted drying of the wood at elevated temperature or low humidity. This will inevitably result in moisture gradients within the material, even in the case where the wood is sealed and left to re-distribute the moisture. The outer part will then, unlike the inner parts, reach equilibrium by absorption and this leads, due to sorption hysteresis, to moisture gradients. Measurements of properties determined for a material with moisture gradients will not be representative of the material when conditioned to uniform moisture content. This is because the moisture content in the former case will be either underestimated or overestimated depending on if the moisture gradients were obtained during water uptake or drying. In the hygroscopic moisture range, conditioning can be done with a variety of methods to control the relative humidity in the surrounding climate. However, in the over-hygroscopic moisture range, methods such as the pressure plate or pressure membrane techniques are needed to condition the wood material to equilibrium without generating internal moisture gradients, see Sect. 7.10. By conditioning wood using these techniques, experimental data on changes in mechanical properties, dimensions, and electrical conductivity with moisture content has been obtained for some wood species [55, 93].
The second, and perhaps main problem with the FSP concept, is the picture of how water is removed from the wood structure. In the common definition of the FSP, the transition point between the moisture regime with moisture-dependent properties and the moisture regime with moisture independent properties is often argued to correspond to a transition in how water is taken up or removed from the wood structure. Thus, upon drying from a water-saturated state, water outside cell walls is thought to fully evaporate before moisture in cell walls is removed, while conversely in absorption cell walls are thought to be saturated before significant amounts of water are present outside cell walls. This coupling between a transition point for some physical property and the picture of the sequencing of water uptake or removal in the wood structure is often stated in literature. However, Fredriksson and Thybring [94] found that the cell-wall moisture content of Douglas fir (Pseudotsuga menziesii (Mirb.) Franco) increased during absorption and decreased in desorption, even in the over-hygroscopic moisture range when capillary water was present. Additionally, water within cell walls exhibited sorption hysteresis in the entire moisture range. The study by Fredriksson and Thybring [94] thus showed that water uptake in cell walls occurs simultaneously as capillary condensation occurs in macrovoids in the over-hygroscopic moisture range, and cell walls are not saturated until the whole wood specimen is saturated with water.
As an alternative to deriving the FSP from a change in material properties with moisture content, the maximum cell-wall moisture content, that is, the amount of moisture within cell walls of water-saturated wood, can be measured by, for example, differential scanning calorimetry (DSC), low-field nuclear magnetic resonance (LFNMR), or solute exclusion, see Sect. 7.11. A large amount of experimental data exists for maximum cell-wall moisture content of different wood species and, in general, the moisture content obtained is 0.05–0.15 g g−1 higher than the FSP found by measurements of changes in physical properties, see Table 7.3. In light of the results by Fredriksson and Thybring [94], this difference is logical, since cell walls are not fully saturated until the whole wood specimen is saturated with water.
Another method that has been used in literature for determining the maximum cell-wall moisture content is based on extrapolation of sorption isotherm models such as BET, HH, GAB, see Sect. 7.3.2. After fitting a given model to hygroscopic moisture sorption data, the derived model parameters are used to predict the cell-wall moisture content at saturation by extrapolating the sorption isotherm to 100% relative humidity [100]. While extrapolation is often regarded as highly uncertain, it has been justified by arguing that only minor changes in the shape of the sorption isotherm are likely to occur above 97% relative humidity [100], that is, in the over-hygroscopic moisture range. This is of course incorrect since the actual wood sorption isotherm is steep above 97% relative humidity as seen in Fig. 7.1. Nonetheless, the extrapolation is based on hygroscopic sorption data without significant contributions from water outside cell walls. Therefore, the assumption of only minor changes to the sorption isotherm could be valid for the water within cell walls. Then, predictions using sorption isotherm extrapolation should agree with measured maximum cell-wall moisture contents. This is, however, not the case as the values based on sorption isotherm extrapolations fall within the range of the FSP values determined from changes in physical properties with moisture content [100]. Extrapolation of hygroscopic sorption isotherms is thus not an adequate method for determination of the maximum cell-wall moisture content.
In summary, values of the FSP derived from physical properties do represent an important characteristic which is useful, for example, for engineering calculations of the expected change in properties with moisture. This FSP is, however, not the same moisture content as the maximum moisture content of the cell walls as illustrated in Fig. 7.8. The FSP concept should, tentatively, be used as it originally was defined, that is, the threshold moisture content at which physical properties no longer change significantly with moisture content.

Schematic illustration of the cell-wall moisture content, ωcell wall at three different total moisture contents, ω: below the fiber-saturation point (FSP), at the FSP, and at full saturation, ωmax
5 Effect of Chemical Modification
Modification of the cell-wall chemistry by physical and chemical processes affects the wood-water relations, most notably how much water is absorbed under given environmental conditions.
5.1 Modes of Action of Cell-Wall Modifications
Three modes of action in terms of how cell-wall modifications affect water sorption can be recognized: bulking, cross-linking, and thermal degradation, see Fig. 7.9. Bulking occurs when modification agents are added into cell walls and take up space otherwise available for water, hereby reducing the sorption of water. One example of bulking is in acetylated wood where acetic anhydride is reacted with cell-wall hydroxyls, see Chap. 16. Although this decreases the number of hydroxyls available for interacting with water [10, 19], it has been shown that bulking is the primary effect of acetylation on water sorption [105]. Another example of a modification affecting water sorption by bulking is furfurylation where furfuryl alcohol is polymerized inside cell walls, see Chap. 16. The formed polymer may chemically react with lignin [106, 107], but the reduced water sorption is a consequence of bulking since furfurylation does not affect hydroxyls [107].

Schematic illustration of three general types of chemical modification: bulking, cross-linking, and thermal degradation
For the second mode of action, cross-linking, the modification agents added are capable of reacting with at least two functional groups, for example, hydroxyls. Examples of such modifications include treatments with 1,6-diisocyanatohexane (HDI), 1,3-dimethylol-4,5-dihydroxyethylenurea (DMDHEU), formaldehyde, glutaraldehyde, or glyoxal [108,109,110,111], see Chap. 16. The cross-linking of cell-wall polymers limits the available space for water by increasing the cell-wall stiffness, hereby restraining expansion of the cell wall, see Sect. 7.7. The last mode of action of modification on water sorption is thermal degradation by thermal modification, see Chap. 16. During this type of modification, the cell-wall material is degraded to a certain extent, in particular hemicelluloses, and hereby the number of hydroxyls accessible to water is reduced [15, 21]. Depending on the process conditions, the reaction products formed during thermal modification may re-polymerize inside the cell walls, hereby causing cross-linking of the cell-wall polymers [112, 113]. The final wood material has less affinity for water under hygroscopic conditions (<98% relative humidity) than untreated material to an extent that depends on the process conditions [112].
5.2 Terminology to Describe Effect of Chemical Modification
The effect of chemical modification on wood-water relations depends on both the chemistry itself and the extent of the modification. A common descriptor for the degree of modification is the relative change in mass as a result of the modification process. Modifications that increase the mass of wood are often described by the “weight percent gain” (WPG) in literature, while those modifications that decrease the mass, most notably thermal modification, are described by the “mass loss” (ML). It has, however, been suggested to characterize the degree of modification of all types of modifications by the “modification ratio,” Rmod (−) defined as [114]:
where mmod,d (g) is the dry mass of the wood material after modification and muntr,d (g) is the dry mass of the untreated material, that is, prior to modification.
The effect of modification on water sorption is referred to as the “moisture exclusion efficiency” (MEE) in literature. This is calculated as the relative difference in equilibrium moisture content between unmodified and modified materials under the same environmental conditions
where ξω (−) is the moisture exclusion efficiency, ωmod (g g−1) is the moisture content of the modified wood material, and ωuntr (g g−1) is the moisture content of the untreated material; both moisture contents are determined after conditioning to equilibrium with the same environmental conditions and sorption history (absorption or desorption).
In order to correctly compare the effect on water sorption across different types of modifications or different degrees of modification, it is necessary to correct ωmod for any mass increase related to the modification itself. Otherwise, an added mass due to modification, that is, Rmod > 0, will automatically decrease the moisture content as calculated by Eq. (7.5), even if the mass of water absorbed in the wood material is unaffected by modification, see Fig. 7.10. Therefore, for modifications with Rmod > 0, the moisture content of modified wood is often reported by the “reduced moisture content” [115, 116] found as
where ωmod (g g−1) is the moisture content of modified wood calculated by Eq. (7.5), ωmod,R (g g−1) is the reduced moisture content, mwood,d (g) is the dry mass of the modified material related only to the wood itself, mw (g) is the mass of absorbed water, and Rmod (−) is the modification ratio. No correction for added mass is needed for Rmod < 0.

Schematic illustration of the mass of wood, water, and modification agent for different types of modification
In the same manner, the moisture exclusion efficiency needs to be corrected for added mass from the modification itself in order to be comparable across types of modifications and degrees of modification. This is represented in the “reduced moisture exclusion efficiency”
where ξω,R (−) is the reduced moisture exclusion efficiency, ωmod (g g−1) is the moisture content of the modified wood material, ωmod,R (g g−1) is the reduced moisture content of the modified wood material, ωuntr (g g−1) is the moisture content of the untreated material, and Rmod (−) is the modification ratio. Moisture contents must of course be determined after conditioning to equilibrium with the same environmental conditions and sorption history (absorption or desorption). No correction for added mass is needed for Rmod < 0 .
5.3 Effect of Modification on Cell-Wall Water and Capillary Water
The effect of modification on water sorption in the hygroscopic moisture range is illustrated in Fig. 7.11 for three types of wood modifications representing the three modes of action in Fig. 7.9: acetylation (bulking), HDI modification (cross-linking), and thermal modification. The reduced water sorption in modified wood is typically accompanied by a decrease in absolute sorption hysteresis in the hygroscopic range for wood modified by bulking and cross-linking modifications [19, 53, 110]. For thermally modified wood, sorption hysteresis depends on the process conditions, but many studies find only small effects of thermal modifications on absolute sorption hysteresis, although some report increased hysteresis [134] or decreased hysteresis [135] with thermal modification. With very few exceptions, however, sorption hysteresis of the various types of modified wood is evaluated in literature based on the absorption and scanning isotherms rather than absorption and desorption isotherms.

Reduced moisture-exclusion efficiency (ξω,R) in the hygroscopic range above 50% relative humidity for acetylated (closed diamonds), 1,6-diisocyanatohexane (HDI) modified (crosses), and thermally-modified wood (open diamonds) of various softwood and hardwood species as function of modification ratio, Rmod. (Based on experimental data [53, 119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,133])
Few studies are available which investigate the influence of chemical modification on capillary water. Furfurylation has been shown to decrease the lumen volume due to deposits of the polymer and changes in lumen cross-section, especially in rays [136]. Still, the over-hygroscopic sorption isotherm for furfurylated wood is higher than for untreated wood above about 96% relative humidity [29]. The reasons for this are not known, but it is possibly due to capillary condensation in cracks in the cell wall caused by the furfurylation or by capillary condensation between the cell wall and the furfuryl alcohol-based polymer [29]. The over-hygroscopic sorption isotherm of acetylated wood is however lower than for untreated wood [29] even though acetylation does not appear to change the lumen volume [136]. The change in the over-hygroscopic sorption isotherm could be due to weakened interactions between water and lumen surfaces [97, 137]. No or minor effects on the over-hygroscopic sorption isotherm have been observed for thermally modified wood [138] .
6 Cell-Wall Sorption Kinetics
At equilibrium between wood and the ambient environmental conditions (relative humidity and temperature), the constant exchange of water molecules between the absorbed phase and the gaseous phase is not noticeable. However, upon a change in the environmental conditions, a net transport of water molecules is observed as a gradual change in the moisture content as the new equilibrium state is approached. For larger wood specimens, this transport involves the diffusion of water vapor and absorbed water from the macroscopic surface of the bulk specimen, both of which are described in Sect. 7.8. An important part of the response of a wood specimen to a change in environmental conditions is, however, the exchange of water molecules between absorbed water and vapor in adjacent macrovoids in the wood structure in all locations, that is, the sorption process. Since this can easily be masked by macroscopic moisture transport, very thin wood specimens are needed for investigating sorption and eliminating or at least distinguishing it from macroscopic diffusion processes [139,140,141,142,143].
The water sorption process in wood and other polymeric materials after a step change in ambient air humidity is characterized by a gradually decreasing rate of mass change over time. Since sorption involves the transport of water molecules into or from the core of the solid material from/to the interface with the water vapor phase, it is natural to consider diffusion of absorbed water as the process governing sorption. For thin wood specimens or free polymer films of known thickness, it is possible to derive the diffusivity of the material from sorption experiments based on the initial slope of mass gain versus square root of time [144], see Sect. 7.12.2. Diffusivities derived from such an approach are not, however, consistent with results from other investigations. For instance, the sorption process is slower and takes longer time to equilibrium when the initial moisture content is increased with similar steps in relative humidity [139]; this is also seen for thin films of hemicelluloses [145]. This contradicts the finding that the diffusivity of the wood cell wall increases with amount of absorbed water, at least from dry to moderate moisture contents, see Sect. 7.8.1. Moreover, the approach to equilibrium seems to be independent of the wood cell-wall thickness [141], although the sorption process should correlate inversely with the square of the thickness if the process was governed by diffusion. Thus, it seems that diffusion of water molecules into or out of the cell walls is not the rate-limiting process governing sorption. A better indication of this can be seen for free polymer films of known thickness for which the diffusivity can be directly examined in steady-state diffusion experiments, see Sect. 7.12.1. For these, sorption of molecules which cause dimensional changes in the film, that is, shrinkage/swelling, is not related to the diffusivity of the molecules observed in steady-state diffusion experiments. Instead, the sorption process has been suggested to be limited by the relaxation of the swelling stresses related to the dimensional changes induced by sorption [144]; however, the actual mechanism governing sorption has not been convincingly documented yet.
The sorption process in wood is often reported to exhibit two-stage behavior [141, 146] similar to sorption in swelling polymer films [144, 145]. The approach to equilibrium during sorption thus appears to be the sum of at least two processes with different rates. Therefore, it is common in literature to model water sorption in cellulosic materials as a sum of exponentials processes, each responsible for a certain change in mass of absorbed water
where t (s) is time, m(t) (g) is mass of absorbed water at time t, m0 (g) is initial mass of absorbed water content (at time t = 0), Δmi (g) is the change in the ith water component, and τi (s) is the characteristic time constant associated with the ith water component. The most common version of Eq. (7.36) has n = 2 and is termed the Parallel Exponential Kinetics (PEK) model and it usually fits experimental sorption data well with R2 > 0.99. The name was coined by Kohler et al. [147] in a study of cellulosic fibers, but has increasingly been used to describe water sorption in wood as well. The mechanisms associated with the two processes are not agreed upon in literature, and the two components have been claimed to represent, for example, different sorption sites in the material, water molecules bound in different ways, or arising from different mechanical responses of the material. One crucial aspect of acquiring sorption data which influences the fitting of Eq. (7.34) is the duration of the sorption step, also termed hold time, that is, whether data collection is continued until equilibrium is attained or ended before this occurs. A meticulous study by Glass et al. [148] clearly documents that the most commonly employed criterion for ending data collection of 0.002% mass change per minute mischaracterizes the sorption process. In fact, the moisture content when sorption is interrupted may be more than 0.01 g g−1 moisture content away from the true equilibrium value [148]. Of greater concern to the fitting of the PEK model, data from a sorption measurement interrupted prior to equilibrium does not capture all of the kinetic behavior [148]. Thus, Thybring et al. [149] have shown that the PEK model parameters depend strongly on the hold time, and for sorption continued until equilibrium, the PEK model is incapable of mathematically capturing the form of the sorption curve. Therefore, the model cannot be used to obtain insights into moisture relations in wood or other cellulosic materials, and its use as a tool for exploring properties of materials interacting with water is not recommended .
7 Shrinkage and Swelling
The dimensions of wood depend on the moisture content. During desorption from the water-saturated state, the volume decreases, reaching a minimum in the completely dry state. This is called shrinkage and is quantified by the volume ratio
where Ssh (−) is the shrinkage, Vω (m3) is the wood volume at a given moisture content, and Vsat (m3) is the wood volume in water-saturated or green condition. Shrinkage is often used to describe changes in volume upon first drying of wood after harvesting. During absorption of water, the dimensions increase which is known as swelling and is quantified based on the dry wood volume as
where Ssw (−) is the swelling, Vω (m3) is the wood volume at a given moisture content, and Vd (m3) is the wood volume in dry condition.
7.1 Shrinkage and Swelling on Different Length Scales
Shrinkage or swelling upon a change in moisture content reflects a change in the amount of cell-wall water within cell walls. The phenomenon originates at the nanoscale and is observed on all length scales of wood, see Fig. 7.12.

Schematic illustration of dimensional changes in wood on several length scales
As voids are virtually absent in the dry wood cell wall [150, 151], absorption of water results in the creation of nanopores within cell walls, increasing the total wood volume. During desorption, the removal of cell-wall water will cause a collapse of these nanopores. As water molecules are not absorbed in the interior of the cellulose microfibrils, increasing the amount of water results in increased distances between microfibrils [152] and conversely results in a decrease in intermicrofibrillar distances during desorption. However, the microfibrils themselves also undergo deformation during absorption or desorption as a result of the forces associated with the change in cell-wall water content [153, 154].
On the cell wall and tissue levels, a change in moisture content in normal, mature wood cells causes dimensional changes that are much smaller in the longitudinal direction than in the transverse directions. This is due to the orientation of the majority of microfibrils with only a small angle to the longitudinal direction, that is, the microfibril angle (MFA), hereby stiffening the cell wall mechanically in this direction. For wood cells and tissue with a larger MFA, such as compression wood in softwoods, the dimensional changes increases in the longitudinal direction, see Fig. 7.13. At the same time, the transverse shrinkage and swelling decreases with increasing MFA as expected theoretically [155,156,157]. The swelling and shrinkage are equal in the longitudinal and transverse directions at an MFA of 45°. The dimensional changes cause only a very slight shift in the MFA with moisture content [152].

Shrinkage in the longitudinal (open symbols) and tangential direction (closed symbols) from green to oven-dry condition of earlywood (diamonds) and latewood tissue (triangles) as function of microfibril angle. Data for radiata pine (Pinus radiata D. Don) [155]. The gray boxes and arrows around the schematic wood cells indicate the magnitude of shrinkage in different directions
In the transverse directions, wood shows a marked shrinkage and swelling anisotropy, that is, the dimensional changes differ between radial and tangential directions with the latter being about twice as large as the previous, that is, the shrinkage and swelling anisotropy is around two. This may cause distortions in sawn wood, depending on the orientation of growth rings in these, see Fig. 7.14.

Distortions of rectangular, square, and round pieces of wood causes by shrinkage and swelling anisotropy [158]
The shrinkage and swelling anisotropy can be observed on several length scales, but the exact origin is not properly understood. Isolated cell-wall material exhibits larger dimensional changes in directions normal to the interface between lumen and cell walls [159] which may be due to the organization of cellulose microfibrils in concentric lamellas [160]. However, this does not explain why the thickness swelling of tangential cell walls is about twice as large as the swelling in radial cell walls in both earlywood and latewood [161,162,163]. Additionally, isolated tissue of latewood exhibits nearly similar shrinkage and swelling in the transverse directions, whereas the shrinkage and swelling anisotropy in earlywood is higher [163,164,165,166,167]. The higher anisotropy in earlywood may be caused by restraints on the radial swelling from the radially aligned rays that are relatively stiffer compared with thin-walled earlywood cells than thick-walled latewood cells [164, 166]. In bulk wood, the early- and latewood are adjacent to each other and their interaction affects the overall shrinkage and swelling behavior [162, 164, 165]. The shrinkage and swelling anisotropy on the bulk-wood scale is around two. The dimensional changes in the longitudinal direction are for normal wood significantly lower [166] as observed on smaller length scales, but increase with increasing MFA as described previously [155].
The dimensional changes in wood are linearly correlated with the equilibrium moisture content in the range 0.05–0.20 g g−1 [168], although this linearity can extend above this range for wood conditioned without moisture gradients [55, 169], see Fig. 7.15. Table 7.4 gives the maximum swelling as well as the linear swelling coefficient for a range of wood species. The linearity is also observed at the cell-wall level [167]. However, the dimensions of bulk wood do not only depend on the moisture content but also on the moisture history, that is, the swollen volume is slightly larger in desorption than in absorption for the exact same amount of water within the wood [168, 172, 173], see Fig. 7.16.

Volumetric shrinkage (Ssh) of beech (Fagus grandifolia Ehrhart) (black, open diamonds) and yellow birch (Betula alleghaniensis Britton) (gray, closed diamonds) as function of equilibrium moisture content in desorption from water saturation in the hygroscopic and over-hygroscopic moisture range. (Based on experimental data [55, 169])

Swelling (Ssw) in the longitudinal, radial, and tangential directions as function of equilibrium moisture content at 25 °C during absorption (open diamonds) and desorption (closed squares) from nonsaturated condition. Experimental results for spruce [172]
7.2 Effect of Modification on Shrinkage and Swelling
Modification of the cell-wall chemistry affects the shrinkage and swelling of wood. Since the dimensional changes of wood are large compared with those of many other materials, a lot of the early research on wood modifications specifically aimed at reducing the moisture-related dimensional changes. In Sect. 7.5.1, three modes of action of wood modifications on the wood-water relations are described, which also differ in their effect on shrinkage and swelling. Bulking modifications such as acetylation cause an increase in the wood volume, that is, a preswelling or bulking of the material, but do not otherwise prevent swelling. This is apparent when comparing the water-swollen volume of untreated and acetylated wood specimens with similar initial volume. While the total water-swollen volume is similar, the amount of cell-wall water and the volume increase caused by this water is lower for the acetylated wood [117, 122, 174]. Cross-linking modifications reduce shrinkage and swelling beyond what can be expected from pure bulking of the cell wall. This is due to the restricted expansion of the cell wall by cross-linking of adjacent wood polymers. Thus, reductions in water-induced volume changes are gained with less preswelling of the material compared with bulking modifications [115]. Thermal degradation as induced by thermal modification of the wood is highly variable in its effect on shrinkage and swelling. Although the hygroscopic moisture uptake is reduced by thermal modification, the effect of swelling depends on the process parameters. Re-polymerization and cross-linking of cell-wall polymers during modification reduce swelling but in their absence may actually cause the modified material to swell more than untreated wood in the water-saturated state [112]. The initial change in wood volume due to modification is described by the bulking coefficient calculated as
where RV,mod (−) is the bulking coefficient, Vmod,d (m3) is the dry wood volume after modification, and Vuntr,d (m3) is the dry wood volume of the untreated material, that is, prior to modification.
The effect of modification on shrinkage and swelling is referred to as the “antiswelling efficiency” (ASE) in literature. This is calculated as the relative difference in swelling between unmodified and modified materials under similar environmental conditions
where ξsw (−) is the antiswelling efficiency, Ssw,mod (−) is the swelling of the modified wood material, and Ssw,untr (−) is the swelling of the untreated material determined by Eq. (7.38). Swelling is determined after conditioning to equilibrium with the same environmental conditions and sorption history (absorption or desorption). It is often determined in the water-saturated state.
In order to compare the effect of different types of modification or modification intensities on shrinkage and swelling, it is desirable to base the evaluation on a parameter that only includes the effect of modification on wood-water interactions, as described for the moisture exclusion efficiency, see Sect. 7.5.2. Otherwise, two types of modifications that result in similar volume increases upon sorption will be evaluated differently if the initial increase in volume due to modification differs. This is illustrated in Fig. 7.17 for modifications with similar volume increase but different bulking coefficient.

Schematic illustration of swelling of various types of modified wood
The quantification of reduction in swelling should preferably exclude the direct increase in volume that typically accompanies modification. This is done by relating the dimensional change in modified wood to the original wood volume by calculation of the “reduced swelling”
where Ssw,mod (−) is the swelling of the modified wood, Ssw,mod,R (−) is the reduced swelling, Vmod,ω (m3) is the volume of the modified material at given moisture content, Vmod,d (m3) is the dry volume of the modified material, Vuntr,d (m3) is the dry volume of the modified material related only to the wood itself, and RV,mod (−) is the bulking coefficient. The antiswelling efficiency can then be corrected for the increased volume due to modification as done in the “reduced antiswelling efficiency”
where ξsw,R (−) is the reduced antiswelling efficiency, Ssw,mod (−) is the swelling of the modified wood material, Ssw,mod,R (−) is the reduced swelling of the modified wood material, Ssw,untr (−) is the swelling of the untreated material, and RV,mod (−) is the bulking coefficient.
While the mass change due to modification is a commonly reported descriptor for wood modifications in literature, the volume change is rarely reported. This makes it difficult to evaluate the effect of modification on the wood-water relations as described by dimensional changes across different types of modification or modification intensities. However, the antiswelling efficiency calculated by Eq. (7.40) directly reflects the product performance in terms of dimensional stability and is therefore a valuable parameter for application of the modified product. The antiswelling efficiency in the water-saturated state without correction for initial volume increases is illustrated in Fig. 7.18 for three types of wood modifications representing the three modes of action in Fig. 7.9: acetylation (bulking), HDI modification (cross-linking), and thermal modification .

Antiswelling efficiency (ξsw) at water saturation for acetylated (closed diamonds), 1,6-diisocyanatohexane (HDI) modified (crosses), and thermally modified wood (open diamonds) as function of modification ratio, Rmod. (Based on experimental data [111, 117, 121, 122, 125, 126, 128, 129, 133,134,133, 175,176,177,178,179,180,181,182,183,184,185,186,187])
8 Moisture Transport in Wood
Three phases of water can exist in wood above the freezing point of water: cell-wall water and liquid water and water vapor in macrovoids (vessels, lumina, pits), see Sect. 7.1. At lower moisture levels, when no liquid water is present in the wood structure, moisture transport occurs through diffusion including both water vapor diffusion and diffusion of cell-wall water. At higher moisture levels, where liquid water is present, moisture transport also occurs by bulk flow of liquid water.
8.1 Diffusion of Water in Wood
Moisture diffusion in wood involves two of the three phases of water in wood: water vapor and cell-wall water. Diffusion describes a mass flow as a result of a gradient in chemical potential. It is caused by random molecular motion, yet with an overall migration of molecules in the opposite direction of gradients in chemical potential. Even in cases without such gradients, the molecules are continuously performing random walks but no overall net migration can be detected.
Multiple pathways are available for diffusion of moisture in wood as indicated in Fig. 7.19. This is a result of the anatomical structure of wood and the possibility of moisture transport in several phases. Moisture diffusion in the macrovoid structure has much lower diffusion resistance than the diffusion of cell-wall water [188]. Therefore, the diffusion resistance of the various pathways, and thus also the resistance to diffusion in the three anatomical directions, differs widely [189]. The transport of each phase of water should not be viewed as an isolated diffusion process as all diffusion processes are coupled via the continuous exchange of molecules between phases, see Fig. 7.19. This will cause absorption or desorption of cell-wall water if local nonequilibrium conditions occur, for example, when the moisture content in cell walls is different locally from the equilibrium moisture content corresponding to the relative humidity present in adjacent lumina. The kinetics of the absorption or desorption processes following these conditions is governed by the mechanisms discussed in Sect. 7.6.

Transport pathways for water in the longitudinal direction of wood. (Adapted from [190])
8.1.1 Fickian and Non-Fickian Moisture Transport
For isothermal conditions, gradients in chemical potential and concentration are correlated, and the diffusion of moisture can be described by the latter using Fick’s law:
where q (kg m−2 s−1) is the flux, c (kg m−3) is the moisture concentration, Dc (m2 s−1) is the diffusion coefficient with moisture concentration as driving potential, and x (m) is the length. This equation is analogous to, for example, Fourier’s law for heat transport; that is, there is a flux, which is driven by a gradient in concentration, and a transport coefficient. In the case of heat transport, temperature is the only potential that causes flow. However, since the state of water in a material can be described in different ways, there are several possible potentials for describing isothermal moisture diffusion and thus several formulations of Fick’s law:
where p (Pa) is the vapor pressure, v (kg m−3) is the vapor concentration in the gas phase, ω (g g−1) is the moisture content, ϕ (−) is the relative humidity, and D is the diffusion coefficient with units dependent on driving potential for diffusion as indicated by indices p, v, ω, and ϕ. In Eqs. (7.44), (7.45), (7.46), and (7.47), q and dx are the same, but there is one diffusion coefficient for each potential. This is important to keep in mind when comparing diffusion coefficients since comparison requires that the transport coefficients have the same potential. Especially Dv and Dc are easily confused since they have the same unit (m2 s−1). It is however possible to transform a diffusion coefficient based on potential a to a diffusion coefficient based on potential b by setting q = q for two of Eqs. (7.43), (7.44), (7.45), (7.46), and (7.47) above. This gives the relation:
Below are some examples on how to transform between different potentials.
Relation between Dv and Dω:
where dω/dϕ (−) is the slope of the sorption isotherm, and vsat (Pa) is the saturation vapor pressure. Equally, transformation from Dv to Dc can be made by using the sorption isotherm expressed as moisture concentration (kg m−3). The relation between moisture content and moisture concentration is:
where ρ (kg m−3) is the dry density of the wood.
Relation between Dv and Dp:
The flux q in Eq. (7.44) equals q in Eq. (7.45). This gives:
The ideal gas law gives the relation between vapor pressure and vapor content:
where R is the gas constant (8.314 J mol−1 K−1), V (m3) is volume, T (K) is temperature, and n (mol) is the amount of substance:
where m (g) is the mass of water vapor and Mw (g mol−1) is the molar mass of water. Therefore, m/V equals the vapor content and Eq. (7.52) can be written as:
The relation between Dv and Dp can therefore be expressed as:
For isothermal moisture transport, the different potentials give the same direction of the flux. For nonisothermal transport, however, different potentials can give opposite directions of the flux.
Another complexity with moisture transport is that the diffusion coefficient cannot be considered to be constant, as is commonly done for heat transport. The diffusion coefficient depends on the moisture state, and this moisture dependence in turn depends on which potential that is used to express the diffusion coefficient. For example, Dv and Dω are related by the slope of the sorption isotherm (Eq. (7.47)) which is nonlinear (Fig. 7.1) and the shape of the curve describing the diffusion coefficient as a function of moisture content or relative humidity is therefore different for the two potentials, see Table 7.5. For example, a peak in the diffusion coefficient with moisture content as potential has been found at moisture contents around 0.15–0.20 g g−1 [192], that is, in the range where the slope of the hygroscopic sorption isotherm increases. Results from cup measurements, see Sect. 7.12.1, which gives the diffusion coefficient with vapor content as potential, on the other hand yield diffusion coefficients which increase with increasing moisture content. In addition to the moisture dependence, diffusion coefficients are also temperature dependent [193].
Despite the complexity of moisture diffusion in wood with several coupled processes as illustrated in Fig. 7.19, a single diffusion coefficient is often assigned for transport in a given direction using one of Eqs. (7.41), (7.42), (7.43), (7.44), and (7.45). Such overall diffusion coefficients can be determined from steady-state experiments on macroscopic specimens, see Table 7.6. In these, a constant moisture gradient is maintained and the flux of moisture determined, see Sect. 7.12.1. No change in moisture content occurs either globally or locally despite the overall transport of moisture. It is, however, difficult to gain insights about the underlying mechanisms governing the various processes involved in water diffusion from such steady-state experiments. Moreover, the simplification of using a single diffusion coefficient breaks down for transient (nonsteady state) conditions, for example, when a bulk wood specimen in equilibrium with the ambient conditions is exposed to a change in relative humidity. In this case, the moisture content changes both globally and locally by processes governed by sorption kinetics as discussed in Sect. 7.6. It is therefore no surprise that moisture diffusion in cases involving a change in moisture content cannot adequately by described by Fick’s laws [189, 196, 197].
8.1.2 Diffusion of Water Vapor
The wood structure is typically dominated by elongated cells oriented along the longitudinal direction, for example, tracheids in softwoods and vessels in hardwoods. These cells contain long tubular macrovoids available for water vapor diffusion. As the diameter of the tubular elements is much smaller than their length, the unbroken transport pathway in such a macrovoid is shorter in the transverse directions than in the longitudinal direction. Therefore, diffusion coefficients in the longitudinal direction for water vapor and other gases are higher than those in the tangential and radial directions [189, 198,199,200,201]. The water transport pathway of lowest diffusion resistance between adjacent tubular elements is through pits, that is, through apertures and pores in the pit membrane (margo), see Fig. 7.19. The resistance to diffusion through nonaspirated pits is about three times larger than free vapor diffusion in lumina [202]. In aspirated pits, however, the diffusion resistance is significantly higher as the pit border is closed by the membrane and water transport is forced to occur as diffusion of cell-wall water through the solid torus. In the transverse directions, moisture diffusion resistance is dominated by the diffusion resistance of pits and whether these are aspirated or not [199,200,201,202]. The diffusion coefficient of water vapor decreases with increasing vapor pressure due to shorter mean free path of the water molecules [203].
8.1.3 Diffusion of Cell-Wall Water
Water transport inside cell walls occurs as diffusion where the cell-wall water moves by random molecular jumps in the water-accessible parts of the cell-wall structure. The transport is considerably slower than the vapor transport in the porous structure. This complicates the determination of diffusion coefficients for cell-wall water as most experimental setups record contributions from both the slow diffusion of cell-wall water and the much faster water transport in the macrovoid structure. Ideally, transport should be limited to only the cell wall water phase in order to accurately determine the diffusion coefficient for cell-wall water. However, the magnitude of this diffusion coefficient for wood can be estimated based on permeability measurements on thin films of wood polymers and molecular dynamics computer modeling. The water transport in thin films of arabinoxylan or uncoated cellophane has been found to yield diffusion coefficients with moisture content as potential, Dω, in the range 0.1–7·10−12 m2 s−1 [64, 204, 205]. Molecular dynamics simulations of the behavior of absorbed water in amorphous cellulose yield diffusion coefficients with molar concentration as potential increasing from 0.1·10−10 m2 s−1 to 3.2·10−10 m2 s−1 from nearly dry to water-saturation [206]. This corresponds to diffusion coefficients with moisture content as potential, Dω in the range 0.8–26·10−12 m2 s−1. Both experimental results and computer modeling show that the diffusion coefficient for cell-wall water increases with increasing moisture content [204, 206,207,208]. As the moisture content increases, more space within cell walls is taken up by water and the connectivity between patches of absorbed water molecules increases [206]. At a certain moisture content threshold, a long-range network of connected absorbed water molecules is formed which facilitates faster diffusion of cell-wall water [206].
8.2 Liquid Water Transport
At higher moisture contents, liquid water is present in macrovoids within the wood structure, see Sects. 7.1.2 and 7.3. For practical reasons, diffusion coefficients with moisture content or vapor concentration as potentials are sometimes used to describe moisture transport at these high moisture contents. However, the transport of liquid water is rather driven by gradients in capillary pressure [209]. The magnitude of this flow is determined by the permeability, that is, the ease of which a fluid is transported through a porous material under a pressure gradient, and the presence of a flow of water requires that voids are connected. Liquid water transport in wood occurs from cell lumen to cell lumen through pit apertures and pit membrane pores, that is, by only one of the transport pathways in Fig. 7.19.
Since the majority of cells are oriented in the longitudinal direction, liquid water transport is substantially faster in the longitudinal than in the radial and tangential directions, as for diffusion. Due to differences in wood anatomy, the liquid water transport is different for different wood species [210,211,212]. Factors that influence the transport include degree of pit aspiration, presence of tyloses, and deposition of extractives on pit membranes. Wood species with a low permeability are often referred to as refractory species. Within the same wood species, the liquid water transport is generally slower in heartwood than in sapwood [213]. The reason for this is a larger percentage of aspirated pits in heartwood and a higher content of extractives [214].
Softwoods consist predominantly of tracheid cells oriented in the longitudinal direction and ray cells oriented in the radial direction. Tracheid cells in the earlywood have large cell lumina and thin cell walls, while those in latewood have thicker cell walls and smaller cell lumina. In the longitudinal direction, latewood has a more important role in longitudinal liquid water transport than earlywood [210, 213, 215, 216]. Transport in this direction is therefore not uniform since water penetrates faster into latewood than into earlywood [210, 211, 213, 215]. Two reasons are generally given for this: (1) Latewood cells have narrower cell lumina which gives higher negative capillary pressure and (2) due to the thicker cell walls, the bordered pits that connect adjacent cell lumina are not aspirated to the same extent in latewood as in earlywood, thus facilitating transport between cells.
For liquid water transport in the tangential direction, both tracheid cells and ray cells are oriented perpendicular to the transport direction, and the transport thus occurs through pits. Tracheid cells generally have fewer and smaller pits on tangential surfaces and transport between cell lumina through pits is therefore faster in the tangential direction than in the radial direction. As for the longitudinal direction, latewood is the preferable path for liquid water transport in softwoods in the tangential direction [210, 211, 215]. Sedighi-Gilani et al. [210] observed that liquid water transport occurred first in the latewood, and water was then redistributed to earlywood cells by radial water transport. Since transport in the tangential direction is dominated by transport through pits, the faster tangential transport in latewood can be attributed to lower degree of pit aspiration in latewood than in earlywood [217].
In the radial direction, ray cells are aligned with the transport direction while tracheid cells are oriented perpendicular to this direction. Transport therefore occurs predominantly in cell lumina in ray cells and through pits. In some studies, transport in the radial direction was hindered at growth ring boundaries. This was attributed to rays not being continuous over these [210, 211].
Hardwoods have a more complex structure than softwoods with more types of cells and pits that connect adjacent cells. Hardwoods consist of four main cell types: vessels, fibers, longitudinal parenchyma, and ray cells. The different cell types are connected by different types of pits. Flow paths in hardwoods are thus more complex than in softwoods, but the vessels, which are cells with large cell lumina, are the most permeable part unless tyloses are present [214]. Hardwoods are divided into ring-porous and diffuse-porous species. In ring-porous wood, vessels are larger in earlywood than in latewood, while vessels are more evenly sized and distributed in diffuse-porous wood. Unlike softwoods, there is no clear difference in liquid water transport between earlywood and latewood. However, earlywood of ring-porous wood species is generally more permeable than earlywood of diffuse-porous species.
9 Methods to Determine Moisture Content and Moisture-Content Profiles
There are several methods available for determining wood moisture content. Some of these methods give the average moisture content of a whole specimen, while others provide moisture-content profiles, that is, the moisture content at different locations within a specimen. An overview of the most common methods is given below.
9.1 Gravimetric Method
The most basic method for determining the moisture content of a material is by the gravimetric method . First, the mass of the specimen is determined, then the specimen is dried, typically at 103 ± 2 °C for 24 h, and finally the dry mass is determined. From Eq. (7.5), the moisture content can be calculated. Care must be taken for wood species containing substantial amounts of volatile extractives since these may evaporate during drying. This may result in a lower dry mass and seemingly higher moisture content than the actual value. The gravimetric method is mainly used to determine the moisture content of a whole specimen, but is sometimes used to determine moisture content distributions as well. This is done by slicing a specimen into smaller parts and determining the masses and dry masses of the individual pieces [192, 218].
9.2 Electrical Methods
Since the electrical properties of wood are highly dependent on moisture content [219], a common method for determining wood moisture content is by measuring an electric property. This principle of measurement is used both for commercial moisture meters and frequently in scientific studies as well. In the latter, it is most often the electrical resistance that is measured, and the method is used for a range of applications such as moisture content determination in field tests [220, 221], moisture content profiles measurements in specimens in laboratory tests [222], and moisture content determination on cell-wall level [223]. In order to transform electrical resistance to moisture content, the relation between these must be known. Examples of this relationship for Norway spruce (Picea abies (L.) Karst.) are shown in Fig. 7.20. The change in electrical resistance is largest for moisture contents between 0 g g−1 and about 0.25–0.30 g g−1 and the method is therefore more reliable in this moisture range. Changes in moisture content can also be detected above 0.25–0.30 g g−1 moisture content but then the absolute value should be interpreted with care since the measurement error increases significantly with moisture content in this range.

Apart from moisture content, the relation between electrical resistance and wood moisture content is affected by a variety of factors [227]. These factors might therefore need to be taken into account when using electrical resistance measurements for moisture content determination. Examples of such factors are temperature [219], wood species [228], and anatomical direction [219]. In addition, the relation between moisture content and electrical resistance is influenced by wood modification [226, 229].
The electrical resistance is determined between two electrodes in contact with the wood specimen. In general, two principal types of electrodes are used: contact electrodes and pin electrodes. Contact electrodes are metal plates which are pressed against or glued to the wood by an electrically conductive adhesive. Such electrodes can be used to measure the moisture content of whole wood specimens [219]. Alternatively, the electrical resistance is measured by inserting two pin electrodes in the wood; the design and size of these electrodes depend on the purpose of the measurement. The most simple electrode type is uninsulated pin electrodes. The measured resistance then mainly reflects the maximum moisture content between the electrodes, that is, where the electrical resistance is lowest. For measurement of the moisture content at a specific depth, insulated pin electrodes can be used. The measured electrical resistance then mainly reflects the moisture content in the vicinity of the electrode tips [230]. One factor that needs to be considered when using pin electrodes is that the measured electrical resistance is affected if electrodes are used close to a surface or in a specimen of small volume [231]. This yields lower determined moisture contents than the actual values. Another factor, which also produces seemingly lower moisture contents, is if the electrodes loose contact with the wood. This can be a problem during long-term measurements where the wood is exposed to fluctuating moisture contents. Here, the electrodes should be designed to retain contact during shrinkage and swelling, for example, by using glued electrodes [93, 228] or screws [225].
9.3 Low Field Nuclear Magnetic Resonance
Low field nuclear magnetic resonance (LFNMR) can be used for moisture content determination [232, 233]; however, the main use of LFNMR is to separate water in different states in the wood structure. The principles of LFNMR are therefore further described in Sect. 7.11.2. For moisture content determination, a free induction decay (FID) pulse sequence is used. The amplitude of the signal obtained is linearly proportional to the water content of the wood specimen, but with an additional signal from the protons in the wood itself. After calibration on specimens with known moisture contents for the specific instrument, the moisture content can be calculated by the amplitude of the FID and the specimen mass [232].
9.4 Imaging Methods
9.4.1 X-ray Computed Tomography
X-ray computed tomography (XCT) can determine moisture content or moisture-content distributions within a whole log or in wood specimen of larger size such as boards [213, 234]. A specimen is exposed to an X-ray beam which is attenuated to different degree depending on the density of the specimen. The X-ray linear attenuation coefficient is calculated in small volume elements and is then normalized to the X-ray absorption coefficient of water [235]. This normalized value is called the CT-number and is related to the wood density. Since wood density depends on moisture content, the moisture content distribution within a wood specimen can be evaluated from the CT-number if measurements are made for both a moist and a dry specimen [235]. Since changes in moisture content in the hygroscopic range result in dimensional changes, swelling/shrinkage during measurements needs to be accounted for. This is done by geometrically transforming the images by using an image processing algorithm.
9.4.2 Neutron Imaging
Neutron imaging can be used to measure moisture content profiles [210, 236, 237]. As for X-ray imaging, neutron imaging is based on transmission measurements. A specimen is exposed to a neutron beam which is attenuated to different extents depending on specimen composition and structure. However, unlike X-rays, neutrons interact with atomic nuclei rather than the surrounding electrons and have a high interaction with, for example, hydrogen. Since hydrogen is a main component in water, neutron imaging is well suited to study water distributions within a material. The procedure used to evaluate moisture content within a wood specimen is described in [210, 236].
9.4.3 Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) is based on the same principle as nuclear magnetic resonance (NMR), see Sect. 7.11.2. However, in addition to information of moisture content or physical and chemical environment, MRI also provides spatial information. This is accomplished by varying the resonance frequency with position by applying a linear magnetic field gradient. Hereby, MRI can be used to determine moisture content profiles [238, 239] and to obtain spatial information on relaxation time distributions in different parts of the wood structure [240,241,242].
10 Methods to Determine Sorption Isotherms
Determination of sorption isotherms includes two steps: (1) conditioning in a specific constant climate and (2) determination of the equilibrium moisture content. Which conditioning technique that is appropriate depends both on the moisture range (hygroscopic or over-hygroscopic) and the size of the specimens since some techniques require specimens of a certain size or mass. The moisture content is typically determined by the gravimetric method, see Sect. 7.9.1.
Irrespective of the conditioning technique or moisture range, it is important to either dry or water-saturate specimens before conditioning to get well-defined absorption and desorption isotherms, respectively. If the absorption isotherm is to be determined, the specimens should be dried before conditioning to the intended relative humidity level. If the desorption isotherm is to be determined, the specimens should be saturated with water, preferably by vacuum saturation to avoid entrapped air, before conditioning to the intended relative humidity level. It is important to be aware of this since if specimens with an unknown moisture history are conditioned to a certain relative humidity, the moisture content distribution will not be uniform within the specimen after conditioning. This is due to sorption hysteresis effects, see Sect. 7.3.1, as some parts of the specimen will reach equilibrium by absorption and other parts by desorption.
10.1 Hygroscopic Range
10.1.1 Saturated Salt Solutions
A common method to equilibrate specimens to various moisture levels is to use saturated salt solutions which generate different relative humidity levels [243]. Table 7.7 shows examples of salts that can be used for conditioning of materials and the relative humidity they generate at 20 °C. Conditioning of specimens can be done either by keeping several specimens in larger boxes or desiccators [246] or by keeping the specimens in individual containers [247]. For the former, it is beneficial to have a small fan inside the box to reduce the diffusion resistance within the air and hence the time to equilibrium. The sorption isotherm can be determined by conditioning specimens above several saturated salt solutions generating different relative humidities. This can be done either by conditioning different specimens to different relative humidity levels or by moving the same specimen between different relative humidity levels. When the specimens have reached equilibrium, the moisture content is determined, most often by the gravimetric method, see Sect. 7.9.1. An alternative to using saturated salt solutions is to condition specimens in climate chambers.
Instead of saturated salt solutions, pure water is sometimes used with the purpose of generating 100% relative humidity. Conditioning above pure water does not, however, yield equilibrium moisture contents corresponding to 100% relative humidity. At such high humidity levels, even very small temperature fluctuations influence the relative humidity substantially, and there is a high risk for condensation to occur. It is therefore practically impossible to condition specimens to 100% relative humidity above pure water. In reality, an uncertain high relative humidity is obtained. Since the wood moisture content is very sensitive to relative humidity at levels close to 100%, even very small deviations from 100% relative humidity will cause large changes in moisture content, see Fig. 7.1. Other methods are therefore used to condition specimens to relative humidity levels close to 100%, see Sect. 7.10.2.
Irrespective of conditioning method, a criterion defining when equilibrium is reached is needed. Such criteria most often relate to when the change in mass over a certain period of time is small enough. Many studies use the criterion that the change in mass in three consecutive mass readings, each made at least 24 h apart, should be less than 0.1% [248]. There are however also other equilibrium criteria which take into account the moisture capacity and slope of the sorption isotherm of the material [247].
10.1.2 Automated Sorption Balances
Automated sorption balances, also referred to as dynamic vapor sorption (DVS), can be used to determine sorption isotherms in the relative humidity range 0% to 95–97%. A small specimen (typically 5–20 mg) is exposed to a series of preprogrammed relative humidity levels while the specimen mass is monitored continuously with a high resolution (typically 0.1–1 μg). A certain relative humidity is generated by mixing different proportions of dry and water saturated carrier gas, for example, nitrogen. The relative humidity is controlled either by the apparatus continuously monitoring the relative humidity by a sensor and adjusting the mass flow regulators automatically or the mass flow controllers are assumed to generate the correct relative humidity based on an initial calibration. Irrespective of the method used, it is important to regularly validate the relative humidity generation of the instrument using saturated salt solutions, which can be done in different ways [249].
A consideration that needs to be made is the initial moisture content of the specimen, that is, whether measurements should begin with dry or water-saturated specimens. This is the case irrespective of conditioning method, but particularly studies using automated sorption balances tend to start with initially drying the specimen before measuring the absorption isotherm, typically up to 95% relative humidity. The desorption isotherm is subsequently determined by stepwise decreasing the relative humidity down to 0%. However, this measurement protocol does not yield desorption isotherms, since initiating desorption from a relative humidity of 95% reached by absorption will give a scanning isotherm and not the desorption isotherm, see Sect. 7.3. As seen in Fig. 7.1, due to the large hysteresis in the over-hygroscopic range, the moisture content at 95% relative humidity reached by absorption is not high enough for the specimen to follow the desorption isotherm during drying. The only way to construct desorption isotherms is to start with water-saturated specimens [52].
As for all conditioning methods, a criterion for when equilibrium is obtained is needed. In an automated sorption balance, this can principally be done in two ways: either as a fixed period of time at each relative humidity level or by a mass stability criterion. Most studies on wood use a mass stability criterion of 0.002% min−1 (20 μg g−1 min−1) over 15 min, but Glass et al. [148] show that this criterion is not sufficient. A mass stability criterion of 3 μg g−1 min−1 calculated over a period of 2 h was instead suggested [250]. Alternatively, a criterion of 5 μg g−1 min−1 can be used if a special correction of the moisture content is applied subsequently [147].
Different brands of automated sorption balances calculate the mass stability criterion differently, but the mass stability criterion is often related to the dry mass of the specimen. For determination of absorption isotherms, the reference mass is the specimen dry mass. Using a mass stability criterion for determining desorption isotherms can, however, be problematic as the measurements should start with water-saturated specimens. The presence of large amounts of liquid water within and on the outside of small wood specimens makes the mass in the water-saturated condition much more variable between specimens. Rigorous assignment of a reference mass for water-saturated specimens is therefore difficult. Instead, it is advisable to use a fixed period of time of conditioning, that is, hold time. Starting with desorption, it is important to consider that the equilibration time needed at the first relative humidity step (e.g., 95%) needs to be substantially longer than for the other steps [52]. A time criterion can be used at all relative humidity levels, but requires initial experiments to determine which equilibration times that are sufficient for the specific material at the different relative humidity levels. Irrespective of the criterion used to define equilibrium, it is important to make sure that the chosen criterion is good enough for the purpose of the measurement. Special care should be taken to obtain a sufficiently equilibrated dry mass since a too high dry mass affects the whole sorption isotherm. Drying at elevated temperature is therefore preferable, but depending on the type of automated sorption balance, this is not always possible.
10.2 Over-Hygroscopic Range
10.2.1 Pressure Plate and Pressure Membrane
In the over-hygroscopic moisture range, the most common methods for determining sorption isotherms are the pressure plate and pressure membrane techniques. Here, specimens are placed on a porous ceramic plate in a pressure plate container and are exposed to a gas pressure. This pressure corresponds to a certain relative humidity level by:
where ϕ (Pa Pa−1) is the relative humidity, Mw (0.018 kg mol−1) is the molar mass of water, ΔP (Pa) is the pressure relative to the atmospheric pressure, R (8.314 J mol−1 K−1) is the gas constant, T (K) is temperature, and ρw (kg m−3) is the density of water. Sorption isotherms in this range are generally given as a function of pressure or water potential on a logarithmic scale since this gives a better resolution of the over-hygroscopic part, see Fig. 7.1, Sect. 7.3. The water potential ψ (J kg−1) and relative humidity are related by:
For more details on the water potential concept, see [251]. Examples of pressures and corresponding water potentials and relative humidity levels are given in Table 7.8. By applying several pressure levels and determining the equilibrium specimen mass at these, the over-hygroscopic sorption isotherm can be determined. From Table 7.8, it can be seen that a rather large change in pressure only results in a small change in relative humidity. Thus, conditioning by this technique enables relative humidity control with high resolution close to 100% relative humidity. Pressure plate equipment can be used for pressures up to 15 bar and pressure membrane equipment up to 100 bar, corresponding with the relative humidity ranges 98.98–100% and 92.88–100%, respectively. The method was originally developed for soil [252], but has been widely used for wood [29, 49, 59, 253].
A schematic illustration of the pressure plate equipment is shown in Fig. 7.21. The pressure vessel is connected to a pressure source of nitrogen gas or air. To avoid mold growth, nitrogen is preferred for measurements on wood. Inside the pressure vessel, a pressure plate cell consisting of a porous ceramic plate enclosed by a neoprene diaphragm is placed and connected to the outlet. The upper side of the porous ceramic plate is exposed to the pressure applied in the pressure vessel. The underside is exposed to atmospheric pressure since the outlet is connected to the space formed between the ceramic plate and the neoprene diaphragm. The pore size distribution of the porous ceramics is designed to withstand the applied pressure, otherwise there would be a gas leakage during the measurement.

A pressure plate vessel with the pressure plate cell consisting of a porous ceramic plate and an internal screen enclosed by a neoprene diaphragm. The outflow tube is connected to a burette to enable monitoring of the water outflow
When determining desorption isotherms, both the pressure plate cell and the specimens are initially vacuum saturated with water. The specimens are then placed on the ceramic plate and sometimes cotton cloth or kaolin clay is placed between the specimens and the ceramic plate. After applying pressure, the outflow of water from the pressure plate cell is registered in a burette. Equilibrium is considered to be reached when there is no longer a water flow out from the cell, and the pressure is then released and the mass of each specimen is determined. This should be done rapidly, however, to avoid loss of water during handling of specimens, and they can be placed in small boxes on a grid above a moist cloth. The moisture state in such a box is close to the moisture state of the specimens, and drying is therefore slower than if the specimens are kept in laboratory climate during weighing. After weighing, the specimens can either be dried to determine the dry mass and subsequently the moisture content of each specimen or the experiment can continue at another pressure level. In the latter case, the specimens are placed in the pressure plate vessel again and another, higher pressure is applied. Since it is unavoidable that the specimens dry during handling, it is important that the next pressure level is not too close to the previous one. Otherwise, the risk is that specimens have dried to a moisture content lower than the equilibrium moisture content at the next pressure level. They will therefore not reach equilibrium by desorption.
The measurement principle of pressure membrane equipment is the same as for the pressure plate equipment. The design of the pressure membrane container is similar except that it is designed for a higher pressure. Instead of a pressure plate cell with porous ceramic plate, pressure membrane equipment contains a cellulose membrane placed on a plate with a metal grid. The pores of the cellulose membrane are much smaller than those in the porous ceramic plate used in the pressure plate equipment. This is necessary to withstand pressures up to 100 bar.
For most other conditioning techniques, equilibrium is defined as a mass stability criterion, but since the specimens are inside the pressure vessel, this is not possible for the pressure plate and pressure membrane techniques. Instead, equilibrium is considered to be obtained when there is no longer an outflow of water. In the standard ASTM C1699-09 [254], equilibrium is defined as when the outflow of water is less than 0.05 ml in 48 h. Tougher criteria are usually used in literature, for example, no outflow of water for 3 days [61], 7 days [253], 12 days [49], up to 5 weeks [93]. The resolution of the burette in which the outflow is registered is, however, seldom reported.
The equilibration time depends on factors such as specimen thickness, orientation of the anatomical directions of the specimen, pressure level, and moisture transport properties of the material. In the literature, reported equilibration times for wood ranges from days [255] to 1–3 months [49, 253] to several months [93]. Since moisture transport in wood is fastest in the longitudinal direction, it is desirable to have the longitudinal direction of the specimen perpendicular to the plate to shorten the equilibration time. The time of measurement can also be reduced by reducing specimen thickness; the shortest equilibration time in literature is reported for microtomed specimens [255]. It is, however, important to consider the representativeness of specimens when choosing the specimen thickness. If specimens are cut so that the transport direction in the experiment is in the longitudinal direction, it is important to make sure that the specimens are thick enough to maintain the ink-bottle effect, that is, a representative specimen should be thicker than the cell length. If specimens are cut thinner than the cell length, lumina will be emptied at a lower pressure since the pits then do not act as ink-bottle necks.
Both the pressure plate and membrane techniques are originally designed for desorption measurements. The pressure plate method has, however, been modified for use in absorption as well. Two main problems have to be solved in order to perform absorption experiments. First, the ceramic plate and the specimens need to be supplied with water, which requires adjustments of the pressure plate cell. Second, it is important to ensure that all parts of the specimen reach equilibrium from absorption. This is problematic since the ceramic plate always needs to be water saturated at the onset of the experiment to avoid gas leakage. Efforts have been made to solve one [256] or both [49, 94] of these problems in order to enable measurements of absorption isotherms in the over-hygroscopic range.
11 Methods to Measure Where in the Wood Structure Water is Present
Water in wood is present either in cell walls or in macrovoids such as cell lumina and pit chambers, see Sect. 7.1, and there are a range of techniques available to separate water in different locations within the wood structure. These techniques include determination of cell-wall moisture content, hydroxyl accessibility, that is, how many hydroxyl groups in cell walls that are accessible for water molecules, separation of cell-wall water and capillary water, as well as separation of different pools of capillary water. The principles of these techniques and how they are used in wood science are presented below.
11.1 Differential Scanning Calorimetry
Low-temperature differential scanning calorimetry (DSC) can be used to determine the amount of freezable water. Based on the assumption that all water except cell-wall water freezes at or slightly below the freezing point of liquid water, the method can be used to determine the amount of cell-wall water. It was first applied to textile fibers [257], but has since been used on both untreated and modified wood [25, 26, 94, 103]. The protocol consists of cooling moisture conditioned or water-saturated specimens to a temperature below 0 °C; literature studies often go down to between −65 °C and −20 °C. The temperature is then increased, most often at constant rate, to a temperature above 0 °C, typically 10–25 °C. The heat needed to the melt the ice formed during cooling, that is, the melting enthalpy, is evaluated, and the amount of freezing water is determined as [25]:
where mfw (g) is the mass of the freezing water, mtot (g) is the total mass of the specimen, mdry (g) is the dry mass of the specimen, Q (J) is the integrated heat of melting (Fig. 7.22), and ΔHw (333.6 J g−1) is the enthalpy of melting pure water. The cell-wall moisture content can be determined by:
where ωcell wall (g g−1) is the cell-wall moisture content, mdry (g) is the dry mass, and mw (g) is the total mass of water in the specimen. Determination of the cell-wall moisture content by Eq. (7.59) is based on the attribution of freezable and nonfreezable water to capillary and cell-wall water, respectively. Studies have, however, documented freezable water tightly associated with polymeric materials [24, 258], as well as nonfreezable water adsorbed on void surfaces [259]. Nonetheless, the use of Eq. (7.59) based on the melting peak around 0 °C is justified based on the following arguments. Firstly, water within solid cell walls is located in pores of the order of 2 nm [260,261,262] and would therefore melt at a much lower temperature [263, 264]. Secondly, the thin layers of nonfreezable water on macrovoid surfaces in the wood contribute insignificantly to the total amount of nonfreezable water [94]. Therefore, the error associated with attributing nonfreezable water to cell-wall water within solid wood specimens has been found to be small [94]. However, care must be taken if measuring on powdered wood, since freezable water held between particles may obscure the results [28].

Schematic illustration of a heating curve for water-saturated wood. Q is the integrated heat of melting in Eq. (7.58)
11.2 Low Field Nuclear Magnetic Resonance
1H nuclear magnetic resonance (NMR) relaxometry at low magnetic field is generally called low field nuclear magnetic resonance (LFNMR), time-domain NMR, or NMR relaxometry. LFNMR can be used for moisture content determination, see Sect. 7.9.3, but the main area of usage is to differentiate between different water states or populations within a material. For the latter, the method has been used for studies on both untreated wood of several wood species [98, 233, 265,266,267] and modified wood [97, 137, 268, 269].
In short, when a specimen is exposed to a static magnetic field, the spin of the protons orients along the magnetic field direction. This causes a small polarization of the specimen in the direction along the static magnetic field, that is, a magnetization vector. Short-lived radio-frequency pulses are then applied with a direction perpendicular to the static magnetic field which causes the magnetization vector to fall out of alignment with the static field. After these radio-frequency pulses end, the LFNMR equipment records the gradual re-orientation of the magnetization vector back to being aligned with the static magnetic field. The re-orientation is characterized by two characteristic relaxation processes: one is known as T1 relaxation, spin-lattice relaxation or longitudinal relaxation, and the other as T2 relaxation, spin-spin relaxation, or transverse relaxation. Longitudinal relaxation (T1) relates to the increase in the component of the magnetization vector in the direction along the static magnetic field. Transverse relaxation (T2) relates to the decrease of the transverse component of the magnetization vector, that is, the component perpendicular to the static magnetic field. Both types of relaxation processes are first-order rate processes which can be described by exponential decay functions with characteristic relaxation times T1 and T2, respectively, where T2 is always less than or equal to T1. Different radio-frequency pulses can be applied, but most commonly the Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence is used to determine T2 relaxation times [270, 271].
For water confined in a porous material such as wood, the LFNMR relaxation times of the spins of water nuclei depend on the physical and chemical environment of the specific pore. For instance, the T2 relaxation time for capillary water is related to the surface-to-volume ratio of pores by [272]:
where T2bulk (s) is the relaxation time of the bulk liquid, ρ (m s−1) is the surface relaxivity, and S/V (m−1) is the surface area-to-volume ratio of the pore. Therefore, if a material contains water in a range of different pore sizes, this will be reflected in the recorded T1 and T2 relaxation times which will contain contributions from water in each of the different pore sizes present. The recorded relaxation curve is therefore a sum of exponential decay functions with different relaxation times, representing water in each of the different pore environments. Analyzing such composite relaxation curves requires deconvolution in order to separate the contributions of water in different pores from each other. This deconvolution can be done by different methods of which the simplest is to use discrete multiexponential fitting of the form:
where y (−) is the composite signal, t (s) is the time, C (−) and T2 (s) are the amplitude and relaxation time of each water component, respectively, and n (−) is the number of exponential components needed to describe the recorded composite relaxation curve, thus corresponding to the number of different water pools present in the material. This method provides a discrete number of water pools with characteristic relaxation times and amplitudes. Another method is to use multiexponential decay analysis where a large number of exponential decays are fitted to the recorded composite relaxation curve [273]. This can be done with algorithms found in computer software for LFNMR data analysis, for example, the CONTIN algorithm [274] or the nonnegative least squares (NNLS) algorithm by Lawson and Hanson [275]. In this method, continuous distributions of T2 relaxation times and amplitudes are obtained, and no assumption on the number of water pools in the material needs to be made.
The analysis of LFNMR data, either by discrete exponential fitting or multiexponential decay analysis, allows the total moisture content of a wood specimen to be divided into different water populations. The moisture content representing a given pore environment can be quantified by the relative amplitude of a given component in discrete exponential fitting or by the relative sum of amplitudes for a given peak in multiexponential decay analysis. The moisture content represented by a specific component or peak can be determined as
where ωi (g g−1) is the moisture content of the given component or peak, ω (g g−1) is the total moisture content of the specimen. For discrete exponential fitting, Si (−) is the amplitude of component i, and Stot (−) is the sum of amplitudes. For multiexponential decay analysis, Si (−) is the sum of amplitudes for peak i and Stot (−) is the total sum of amplitudes for the continuous T2 distribution.
Figure 7.23 shows an example of a continuous T2 distribution for water-saturated Norway spruce. For water-saturated or green wood specimens, often 3–4 peaks are seen in LFNMR measurements [97, 137, 233, 265,266,267]. The peak with the shortest T2, typically in the range of 1–3 ms, is attributed to water in cell walls. Peaks with longer relaxation times, typically in the range of 10–300 ms, are attributed to water in voids in the wood structure such as cell lumina and pit chambers where smaller voids has shorter T2. For example, LFNMR can be used to separate capillary water in differently sized cell types in hardwoods [266] and a shorter T2 has been observed for water in latewood lumina than in earlywood lumina [267]. Although Eqs. (7.61) and (7.62) could be used to determine the amount of cell-wall water, LFNMR measurements performed above 0 °C underestimate the cell-wall moisture content [97, 98]. Therefore, accurate quantification of the cell-wall moisture content requires a different analysis protocol and measurements performed at subzero temperatures in which case only one peak, attributed to cell-wall water, is observed [97, 98].

Example of continuous T2 distributions obtained from LFNMR measurements on Norway spruce (Picea abies (L.) Karst.) earlywood and the attributions of the different peaks [267]
Additional information about the state of water in wood can be obtained by performing 2D LFNMR measurements. Here, both T1 and T2 are determined simultaneously allowing an analysis of their correlation [276]. By performing a T1–T2 2D LFNMR measurement, it is possible to separate effect of physical (surface-to-volume-ratio) and effect of chemical environment (surface chemistry and presence of solutes). Pores of similar chemical environment but different pore size are seen in T1–T2 2D LFNMR to have similar ratios of relaxation times, that is, T1/T2. On the other hand, pores of different chemical environments appear as peaks with different T1/T2 ratios. 2D T1–T2 LFNMR on softwoods has shown two peaks assigned to cell-wall water [31, 32], that is, with similar T2 time around 1–3 ms as observed in 1D LFNMR measurements but with different T1 times.
Before analysis of LFNMR relaxation curves, it is important that the output data are properly recorded. Since LFNMR data consists of exponentially decaying components, it is important that the composite relaxation curve is fully decayed. For wood species with large voids such as hardwoods with water-filled vessels, longer measurement times are required than for wood species with smaller voids. Additionally, to observe water pools with short relaxation times such as cell water, the choice of CPMG pulse separation should be such that a number of data points are acquired before the shortest expected relaxation time. It is, however, important to consider that the choice of settings influences how much the specimen is heated by the CPMG radio-frequency pulses during the measurement. The settings therefore need to be optimized for the specimens to be measured so that all relevant information is captured without substantial heating of the specimen. This can also be achieved by using logarithmic pulse spacing during data acquisition [31].
11.3 Solute Exclusion Technique
The solute exclusion technique can be used to determine the pore size distribution of water-filled pores of small sizes (0.3–60 nm) as well as the amount of cell-wall water in wood. It was originally developed to characterize pore size distributions and pore volumes in pulps [277], but has since then also been used to characterize wood [278]. The measurement protocol involves an aqueous solution of molecular probe solutes with known concentration and known size of the probe molecules. By placing a water-saturated specimen with known moisture content in the solution, solutes will diffuse into the specimen causing a decrease in their concentration in the solution. This change in concentration can be related to the volume of water within the specimen into which these molecules can diffuse. The specimen is kept in the solution until equilibrium is reached, and the change in concentration is measured using, for example, high performance liquid chromatography (HPLC). The inaccessible mass of water, that is, water in pores that are too small for the probe molecules to enter, can be determined by:
where mw,inaccess (g) is the mass of water inaccessible to the probe molecules, mw (g) is the total mass of water in the water-saturated specimen, msol (g) is the mass of the solution added, cinit (g L−1) is the initial concentration of probe molecules in the solution, and c∞ (g L−1) is the concentration of probe molecules after equilibrium has been attained.
By using probe solutions with probe molecules of different sizes and calculating the volume accessible to the probe molecule of each size, the pore size distribution can be determined. When molecules above a certain size are used, the mass of inaccessible water becomes independent of the size. In this case, it can be inferred that the probe molecules are not diffusing into the cell walls, but only access the macrovoid structure, which has significantly larger pore sizes than the size of any probe molecule. The mass of inaccessible water found when this occurs corresponds with the maximum cell-wall moisture content. There is, however, one problem with determining pore size distributions with the solute exclusion technique: A basic assumption is that the solute concentration in the accessible pores is equal to that in the solution surrounding the specimen. However, the solute concentration within pores decreases as the size of probe molecule approaches the size of the pores [279]. If the probe molecule is about half the size of the pore, the solute concentration is about 30% of the concentration in the solution surrounding the specimen [279]. This causes an error in the calculated pore size distribution which needs to be corrected for. If the method is used solely to determine maximum cell-wall moisture content, this issue is, however, not a problem due to the large difference in size between macrovoid pores and probe molecules.
The solute exclusion technique has been used both for solid, small wood specimens [119, 261, 262, 280] and shavings, pulp or milled wood [95, 101, 278]. The time period for which specimens are kept in the solutions varies in literature between a few minutes for pulp specimens [278] to 1–14 days for solid wood specimens [119, 261, 280]. The probe molecules used are typically small sugars, polyethylene glycol (PEG) or dextrans, and their size is evaluated from the Stokes-Einstein equation. An empirical correlation between probe diameter and molecular weight for PEGs and dextrans has been determined based on a compilation of literature data [114], see Fig. 7.24. The correlation can be described by
where d (nm) is molecular diameter of probe molecule, M (Da) is the molecular weight, and a and b are fitting parameters. The regression line for PEGs in Fig. 7.24 yields values of a and b of 0.539 and 5.394 ·10−2, respectively, while the line for dextrans results in a and b of 0.454 and 7.460 ·10−2, respectively. A summary of considerations to make when choosing probe molecules is given in [278].

Common probe molecules and their sizes evaluated from the Stokes-Einstein equation. Insert: Schematic illustration of the principle behind solute exclusion where a water-saturated wood specimen is exposed to a solution of probe molecules of two sizes. Probe molecules of a given size can enter water-filled pores larger than themselves but are excluded from smaller pores. (Based on [114])
11.4 Hydroxyl Accessibility
Hydroxyl accessibility is a measure of how many hydroxyl groups on cell wall polymers that interact with water molecules. It can be determined by two principally different methods: by using an automated sorption balance or by spectroscopic methods. Both are based on deuterium exchange where specimens are exposed to deuterium oxide in vapor or liquid form. This causes the hydrogens of hydroxyl groups interacting with absorbed water to be exchanged from protium (1H) to deuterium (2H).
11.4.1 Automated Sorption Balances with Deuterium Oxide
Automated sorption balances are commonly used to determine sorption isotherms, see Sect. 7.10.1, but can also be used for measuring hydroxyl accessibility. The hydroxyl accessibility is measured by conditioning specimens for sufficiently long time using heavy water (deuterium oxide, 2H2O or D2O) instead of normal water. This causes deuteration of accessible hydroxyls, which can be measured as a change in dry mass. This is then used to calculate the hydroxyl accessibility. A measurement is performed by first drying a specimen and determining the dry mass. The specimen is then exposed to deuterium oxide vapor for a certain period of time before being dried a second time, see Fig. 7.25. The hydroxyl accessibility can be calculated based on the change in dry mass before and after conditioning with deuterium oxide vapor as:
where OHaccess (mol g−1) is the hydroxyl accessibility, Δmdry (g) is the difference in dry mass before and after deuteration, m0,dry (g) is initial dry mass, and ΔMhydrogen (g mol−1) is molar mass difference (1.006 g mol−1) between deuterium (2H or D) and protium (1H).

Schematic illustration of hydroxyl accessibility determination in an automated sorption balance. The specimen is dried, exposed to deuterium oxide vapor, and then dried a second time. The difference in dry mass, Δm, is then used to evaluate the hydroxyl accessibility by Eq. (7.65)
Since the measurement is done in an automated sorption balance, an equilibration criterion is needed to determine when the specimen is dry, just as when measuring sorption isotherms, see Sect. 7.10.1. For hydroxyl accessibility measurements, it is very important to have equilibrated dry masses since those are the basis for the calculations. The use of a mass stability criterion is therefore not recommended, and a fixed period of time should be used instead, see also discussion about equilibrium criteria in Sect. 7.10.1.
As when measuring sorption isotherms using an automated sorption balance, the specimen mass is in the range of 5–20 mg. In order to increase the surface area exposed to deuterium oxide vapor, it is advisable to cut the wood specimens into thin slices with a razor blade or a microtome, rather than having one solid specimen. It is also advisable to dry the carrier gas used in the automated sorption balance since even high purity gas may contain enough water to contaminate the deuterium oxide vapor and cause re-protonation during specimen drying. Drying of the carrier gas can for example be achieved by installing a desiccant filter containing molecular sieves.
11.4.2 Spectroscopic Methods
Hydroxyl accessibility can be determined using attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) or Fourier transform near infrared (FT-NIR) [14, 66]. This is done by exposing wood specimens to deuterium oxide which causes an exchange of the hydrogens of hydroxyl groups from protium (1H) to deuterium (2H). The deuterium exchange can be done by either keeping specimens in liquid deuterium oxide or by conditioning them in deuterium oxide vapor. Hydroxyl accessibility is evaluated by comparing the stretching vibrations of the hydroxyls in the FTIR or FT-NIR spectra, that is, the OH and OD stretching vibrations, see Fig. 7.26. The relative hydroxyl accessibility can be determined by relating the area below the OD stretching peak to the sum of the areas below the OH and OD stretching peaks, or relating it to the area below the OH stretching peak before deuteration [14] .

Effect of deuteration on the hydroxyl vibrational peak in ATR-FTIR. As a result of deuteration the size of the O-H peak decreases while an O-D peak appears
12 Methods to Measure Moisture-Transport Coefficients
The transport of moisture in wood occurs by the overall net migration of water in different phases: water within cell walls and liquid water and vapor in macrovoids, see Sects. 7.8.1 and 7.8.2. For moisture transport in the hygroscopic range, the moisture transport properties of wood are typically derived based on either the steady-state cup method or unsteady-state sorption methods. In the previous, transport properties are based on a constant rate of transport through the wood material, whereas the latter methods derive transport properties from a changing rate of transport. Liquid water transport through wood can be characterized by the capillary absorption method. In all of these methods, moisture transport occurs in several phases simultaneously and with exchange between phases (sorption).
12.1 Steady-State Cup Method
A schematic illustration of the set-up used in the cup method is shown in Fig. 7.27. Here, a specimen is placed as a lid on a cup containing a saturated salt solution which generates a certain relative humidity, ϕ0, inside the cup. Alternatively, the cup can contain a desiccant resulting in 0% relative humidity inside. Placing the cup in a different relative humidity, ϕ2, will cause a moisture flux through the material (the lid). For example, if ϕ2 is lower than ϕ0, the moisture flux will be directed out of the cup, and the mass of the cup will decrease over time. By regularly determining the mass of the cup, steady state flow can be observed after a while as a linear change in mass over time. When this occurs, the moisture transport coefficient can be evaluated by:
where Dv (m2 s−1) is the moisture transport coefficient with vapor concentration as driving potential, q (kg m−2 s−1) is the flux which is determined from the mass change and the surface area of the specimen, dspec (m) is the specimen thickness, and Δv (kg m−3) is the difference in vapor content over the specimen:
where Δϕ is the difference between ϕ1 and ϕ2, and vsat is the saturation vapor content at the specific temperature (vsat at 20 °C is 17.28 g m−3). Due to the air gap between the salt solution and the underside of the specimen, the relative humidity to which the specimen surface is exposed, ϕ1, is not equal to the relative humidity of the salt solution. A correction for the air gap needs to be made by:

Schematic illustration of the experimental setup for the cup method
The relative humidity at the underside of the specimen surface, ϕ1, can be solved as:
where dair (m) is the distance between the surface of the salt solution and the underside of the specimen, and Dv,air (m2 s−1) is the diffusion coefficient for air. Additionally, corrections for resistance on the external specimen surface can be necessary, especially if air circulation is not promoted by a fan. Moreover, a correction for the error caused by the two-dimensional flux that occurs in the region of masked edges of the specimen may be necessary [281].
Both Dv and Dc depend on relative humidity and needs to be related to the relative humidity range for which they have been determined. Diffusion coefficients are typically determined for several relative humidity intervals, and the results are then related to the average relative humidity of each interval.
The cup method has been widely used for wood [194, 282] with various designs of the cup as well as choice of sealing material. Special care should be taken to make sure that the there is no leakage through the sealing since that can significantly affect the results [283].
12.2 Unsteady-State Sorption Method
An unsteady state method that is widely used within wood science for measurement of moisture diffusion coefficients is the sorption method . In this, a specimen is first equilibrated to a certain relative humidity, after which the relative humidity is changed and the resulting change in mass over time is recorded [284]. In order to simplify the evaluation of the data, moisture transport is usually limited to one anatomical direction, for example, by sealing four sides of a cuboid specimen. The experiment can be performed in either absorption or desorption and between different moisture levels. The change in mass is plotted as a function of the square root of time as shown in Fig. 7.28. For specimens with only transport in one direction, the diffusion coefficient can be evaluated from [285]:
where Dc (m2 s−1) is the diffusion coefficient with moisture concentration as driving potential, L (m) is half the specimen thickness, t (s) is the time, and E (−) is the fractional mass change defined as:
where Δmt (g) is the mass change at time t, and Δmeq (g) is the final mass change at equilibrium. The slope of the sorption curve (dE/d√t) can be defined in different ways. Often, the time corresponding with E = 0.5 is used, but it is also possible to evaluate, for example, the maximum slope which is beneficial if there is a surface resistance [286]. A comparison of different methods to evaluate unsteady state sorption experiments is given in [286].

A schematic illustration of the fractional mass change E as a function of square root of time
Apart from the sorption method, there are other approaches to evaluate diffusion coefficients from unsteady-state measurements. One of these is to evaluate diffusion coefficients from moisture distributions in wood after different times of one-directional drying or wetting [192]. The moisture distribution can be approximated by cutting wood specimens into slices and determining the moisture content in each slice. In this way, diffusion coefficients as a function of moisture content up to 1.1–1.4 g g−1 in the three directions have been obtained [192], although at these high moisture levels, water transport occurs as a combination of moisture diffusion and liquid water transport, see Sect. 7.8.2. Another example of diffusion coefficients derived from unsteady-state measurements is the determination of moisture profiles by neutron imaging [288] .
12.3 Comparison of Diffusion Coefficients from the Cup and Sorption Methods
The diffusion coefficient obtained by the cup method is with vapor content as potential. Therefore, comparison with diffusion coefficients obtained by the sorption method requires transformation since diffusion coefficients obtained with the sorption method has moisture concentration or moisture content as potential. The diffusion coefficients Dv and Dc can easily be confused since they have the same unit (m2 s−1). For conversion between different potentials, see Sect. 7.8.1.
Provided that the rate of diffusion is governed by Fickian diffusion, the diffusion coefficients obtained by steady-state and unsteady-state methods should be the same. Several studies have, however, shown that this is not the case. Diffusion coefficients obtained with the cup method (steady-state) and the sorption method (unsteady-state) differ [189]. The reason is that sorption curves obtained in unsteady-state sorption experiments are not accurately described by Fick’s law. The non-Fickian sorption behavior of wood is especially pronounced at higher levels of relative humidity than lower levels [189]. In addition, for specimens with fast absorption rates, the sorption curve may have initial disturbances caused by surface resistance and heat effects [289]. In general, there are multiple factors of uncertainty involved in un-steady state sorption methods, some of which can be reduced by careful design of the experiment [289, 290].
12.4 Capillary-Water Absorption Method
Liquid water transport in materials is commonly characterized by capillary absorption experiments [291]. A material is then placed in contact with liquid water and the mass of the specimen is regularly determined. The sides of the specimens are sealed to ensure a one-dimensional flow and as for cup measurements, the choice of sealant is important since it can affect the results [287, 292]. Also the upper surface is generally sealed to prevent evaporation, but a hole is left in the sealing to maintain atmospheric pressure at the top of the specimen [293]. Since the water uptake in wood typically is fast during the first minutes, it can be challenging to collect a sufficient amount of data points in the beginning of the experiment; the time frame is short and a minimum of 5 data points is needed [291]. In addition, in the most commonly used experimental set-up [291, 293, 294], the specimen is removed from the water while weighing. However, Zelinka and co-workers solved this problem by designing an automated experimental set-up where the specimen was suspended from a load cell and data was collected continuously without needing to remove the specimen from the water.
The data is evaluated by plotting the water uptake (mass per surface area) as function of square root of time. For most porous materials, a bilinear behavior is then seen where the first part has a larger slope than the second part. The capillary absorption coefficient, Acap (kg m−2 s−0.5), is then evaluated as the slope of the initial part of the curve [292]. However, for wood, this clear bi-linear behavior is not seen in any of the three anatomical directions [293]. This is visualized in Fig. 7.29 where capillary absorption data for pine wood are shown together with data for calcium silicate; while the data for calcium silicate has a clear bilinear shape, the slope of the wood data keeps changing over time. This nonlinear behavior for wood is attributed to swelling due to moisture uptake in cell walls since a bi-linear behavior is seen if a nonswelling liquid (cyclohexane) is used instead of water [293]. The nonbilinear behavior is, however, a problem when evaluating the data since that is done by performing a straight-line fit to the initial part of the data, see example in Fig. 7.29. In the literature, this problem has been handled differently; Zelinka and co-workers used the data for the first hour of the test, while Zillig [293] excluded the first part of the curve .

Data from capillary absorption experiments for pine wood and calcium silicate [292]. Unlike calcium silicate, the wood does not show a clear bilinear behavior
References
Leigh, G.J.: Nomenclature of Inorganic Chemistry: Recommendations, p. 1990. Blackwell Scientific Publications, Oxford/Boston (1990)
Suchland, O.: The Swelling and Shrinkage of Wood. A Practical Technology Primer. Forest Products Society, Madison (2004)
Niemz, P., Sonderegger, W.: Holzphysik – Physik des Holzes und der Holzwerkstoffe. Fachbuchverlag Leipzig im Hanser Verlag, Munich (2017)
Matthews, J.F., Skopec, C.E., Mason, P.E., Zuccato, P., Torget, R.W., Sugiyama, J., Himmel, M.E., Brady, J.W.: Carbohydr. Res. 341(1), 138–152 (2006)
Lindh, E.L., Terenzi, C., Salmén, L., Furo, I.: Phys. Chem. Chem. Phys. 19(6), 4360–4369 (2017)
Hofstetter, K., Hinterstoisser, B., Salmén, L.: Cellulose. 13(2), 131–145 (2006)
Salmén, L., Bergström, E.: Cellulose. 16(6), 975–982 (2009)
Hill, C.A.S., Jones, D.: Holzforschung. 53(3), 267–271 (1999)
Taniguchi, T., Harada, H., Nakato, K.: Mokuzai Gakkaishi. 10, 215–220 (1966)
Beck, G., Strohbusch, S., Larnøy, E., Militz, H., Hill, C.: Holzforschung. 72(1), 17–23 (2017)
Sumi, Y., Hale, R.D., Meyer, J.A., Leopold, B., Rånby, B.G.: Tappi J. 47(10), 621–624 (1964)
Thybring, E.E., Thygesen, L.G., Burgert, I.: Cellulose. 24(6), 2375–2384 (2017)
Tarmian, A., Burgert, I., Thybring, E.E.: Wood Sci. Technol. 51(4), 845–853 (2017)
Fackler, K., Schwanninger, M.: J. Near Infrared Spectrosc. 19(5), 359–368 (2011)
Digaitis, R., Thybring, E.E., Künniger, T., Thygesen, L.G.: Wood Sci. Technol. 51(5), 1067–1080 (2017)
Altaner, C., Apperley, D.C., Jarvis, M.C.: Holzforschung. 60(6), 665–673 (2006)
Fernandes, A.N., Thomas, L.H., Altaner, C.M., Callow, P., Forsyth, V.T., Apperley, D.C., Kennedy, C.J., Jarvis, M.C.: Proc. Natl. Acad. Sci. U. S. A. 108(47), E1195–E1203 (2011)
Chow, S.Z.: Tappi J. 55(4), 539–544 (1972)
Popescu, C.M., Hill, C.A.S., Curling, S., Ormondroyd, G.A., Xie, Y.: J. Mater. Sci. 49(5), 2362–2371 (2014)
Taniguchi, T., Harada, H., Nakato, K.: Nature. 272(5650), 230–231 (1978)
Phuong, L.X., Takayama, M., Shida, S., Matsumoto, Y., Aoyagi, T.: Holzforschung. 61(5), 488–491 (2007)
Berthold, J., Desbrieres, J., Rinaudo, M., Salmén, L.: Polymer. 35(26), 5729–5736 (1994)
Berthold, J., Rinaudo, M., Salmén, L.: Colloids Surf., A. 112(2–3), 117–129 (1996)
Nakamura, K., Hatakeyama, T., Hatakeyama, H.: Text. Res. J. 51(9), 607–613 (1981)
Simpson, L.A., Barton, A.F.M.: Wood Sci. Technol. 25(4), 301–308 (1991)
Repellin, V., Guyonnet, R.: Holzforschung. 59(1), 28–34 (2005)
Kärenlampi, P.P., Tynjälä, P., Ström, P.: J. Wood Sci. 51(2), 118–123 (2005)
Zelinka, S.L., Lambrecht, M.J., Glass, S.V., Wiedenhoeft, A.C., Yelle, D.J.: Thermochim. Acta. 533(0), 39–45 (2012)
Thygesen, L.G., Engelund, E.T., Hoffmeyer, P.: Holzforschung. 64(3), 315–323 (2010)
Czihak, C., Muller, M., Schober, H., Vogl, G.: Physica B. 276, 286–287 (2000)
Cox, J., McDonald, P.J., Gardiner, B.A.: Holzforschung. 64(2), 259–266 (2010)
Bonnet, M., Courtier-Murias, D., Faure, P., Rodts, S., Care, S.: Holzforschung. 71(6), 481 (2017)
Bergenstråhle, M., Wohlert, J., Larsson, P.T., Mazeau, K., Berglund, L.A.: J. Phys. Chem. B. 112(9), 2590–2595 (2008)
Wickholm, K., Hult, E.L., Larsson, P.T., Iversen, T., Lennholm, H.: Cellulose. 8(2), 139–148 (2001)
Hult, E.L., Larsson, P.T., Iversen, T.: Nordic Pulp Pap. Res. J. 16(1), 33–39 (2001)
Malm, E., Bulone, V., Wickholm, K., Larsson, P.T., Iversen, T.: Carbohydr. Res. 345(1), 97–100 (2010)
Kulasinski, K., Guyer, R., Derome, D., Carmeliet, J.: Biomacromolecules. 16(9), 2972–2978 (2015)
Terenzi, C., Prakobna, K., Berglund, L.A., Furo, I.: Biomacromolecules. 16(5), 1506–1515 (2015)
Engelund, E.T., Thygesen, L.G., Hoffmeyer, P.: Holzforschung. 64(3), 325–330 (2010)
Kellogg, R.M., Wangaard, F.F.: Wood Fiber Sci. 1(3), 180–204 (1969)
Christensen, G.N., Hergt, H.F.A.: Holzforschung. 22(6), 165 (1968)
Hill, C.A.S., Ormondroyd, G.A.: Holzforschung. 58(5), 544–547 (2004)
Pfriem, A., Zauer, M., Wagenführ, A.: Holzforschung. 63(1), 94–98 (2009)
Scholz, G., Zauer, M., Van den Bulcke, J., Van Loo, D., Pfriem, A., Van Acker, J., Militz, H.: Holzforschung. 64(5), 587–593 (2010)
Plötze, M., Niemz, P.: Eur. J. Wood Wood Prod. 69(4), 649–657 (2011)
Zauer, M., Pfriem, A., Wagenführ, A.: Wood Sci. Technol. 47(6), 1197–1211 (2013)
Stamm, A.J., Hansen, L.A.: J. Phys. Chem. 41(7), 1007–1016 (1937)
Stamm, A.J.: J. Phys. Chem. 33(3), 398–414 (1929)
Fredriksson, M., Johansson, P.: Dry. Technol. 34(1), 132–141 (2016)
Glass, S.V., Zelinka, S.L., Johnson, J.A.: Investigation of Historic Equilibrium Moisture Content Data from the Forest Products Laboratory. US Department of Agriculture, Forest Service, Forest Products Laboratory, Madison (2014)
Ahlgren, L.: Moisture Fixation in Porous Building Materials, pp. 1–200. Division of Building Technology, The Lund Institute of Technology, Lund (1972)
Fredriksson, M., Thybring, E.E.: Cellulose. 25(8), 4477–4485 (2018)
Spalt, H.A.: For. Prod. J. 8, 288–295 (1958)
Fortin, Y.: Moisture content-matric potential relationship and water flow properties of wood at high moisture contents. In: Department of Forestry, p. 187. University of British Columbia, Vancouver (1979)
Almeida, G., Hernández, R.E.: Wood Sci. Technol. 40(7), 599–613 (2006)
Hill, C.S., Norton, A., Newman, G.: Wood Sci. Technol. 44(3), 497–514 (2010)
Hoffmeyer, P., Engelund, E.T., Thygesen, L.G.: Holzforschung. 65, 875–882 (2011)
Weichert, L.: Holz Roh Werkst. 21, 290–300 (1963)
Defo, M., Fortin, Y., Cloutier, A.: Wood Fiber Sci. 31(1), 62–70 (1999)
Djolani, B.: Ann. Sci. Forestieres (Paris). 29(4), 465–474 (1972)
Tremblay, C., Cloutier, A., Fortin, Y.: Wood Sci. Technol. 30(5), 361–371 (1996)
Salmén, L., Larsson, P.A.: Carbohydr. Polym. 182, 15–20 (2018)
Keating, B.A., Hill, C.A.S., Sun, D., English, R., Davies, P., McCue, C.: J. Appl. Polym. Sci. 129(4), 2352–2359 (2013)
Engelund, E.T., Thygesen, L.G., Svensson, S., Hill, C.A.S.: Wood Sci. Technol. 47(1), 141–161 (2013)
Suchy, M., Kontturi, E., Vuorinen, T.: Biomacromolecules. 11(8), 2161–2168 (2010)
Suchy, M., Virtanen, J., Kontturi, E., Vuorinen, T.: Biomacromolecules. 11(2), 515–520 (2010)
Atalla, R.S., Crowley, M.F., Himmel, M.E., Atalla, R.H.: Carbohydr. Polym. 100(0), 2–8 (2014)
Lee, K.Y., Bismarck, A.: Cellulose. 19(3), 891–900 (2012)
Laivins, G.V., Scallan, A.M.: J. Pulp Pap. Sci. 22(5), J178–J184 (1996)
Oksanen, T., Buchert, J., Viikari, L.: Holzforschung. 51(4), 355–360 (1997)
McBain, J.W.: J. Am. Chem. Soc. 57(4), 699–700 (1935)
Salin, J.-G.: Dry. Technol. 26(5), 560–567 (2008)
van den Berg, C., Bruin, S.: Water activity and its estimation in food systems: theoretical aspects. In: Stewart, G.F. (ed.) Water Activity: Influences on Food Quality, pp. 1–61. Academic Press (1981)
Boquet, R., Chirife, J., Iglesias, H.A.: J. Food Technol. 15(3), 345–349 (1980)
Dent, R.W.: Text. Res. J. 47(2), 145–152 (1977)
Brunauer, S., Emmett, P.H., Teller, E.: J. Am. Chem. Soc. 60, 309–319 (1938)
Anderson, R.B.: J. Am. Chem. Soc. 68(4), 686–691 (1946)
Anderson, R.B., Hall, W.K.: J. Am. Chem. Soc. 70(5), 1727–1734 (1948)
Guggenheim, E.A.: Applications of Statistical Mechanics. Oxford University Press, New York (1966)
De Boer, J.H.: The Dynamical Character of Adsorption, p. 239. Clarendon Press, Oxford (1953)
Hailwood, A.J., Horrobin, S.: Trans. Faraday Soc. 42(0), B084–B092 (1946)
Zelinka, S.L., Glass, S.V., Thybring, E.E.: Wood Sci. Technol. 52(6), 1701–1706 (2018)
Adolphs, J., Setzer, M.J.: J. Colloid Interface Sci. 180(1), 70–76 (1996)
Kelsey, K.E.: Aust. J. Appl. Sci. 8(1), 42–54 (1957)
Lindh, E.L., Bergenstråhle-Wohlert, M., Terenzi, C., Salmén, L., Furó, I.: Carbohydr. Res. 434, 136–142 (2016)
Lindh, E.L., Salmén, L.: Cellulose. 24(1), 21–33 (2017)
Simpson, W.: Wood Fiber Sci. 12(3), 183–195 (1980)
Willems, W.: Holzforschung. 69(1), 67 (2015)
Tiemann, H.D.: In: Pinchot, G. (ed.) Effect of Moisture upon the Strength and Stiffness of Wood. US Department of Agriculture, Forest Service, Washington, DC (1906)
Wilson, T.R.C.: Strength-Moisture Relations for Wood, pp. 1–88. US Department of Agriculture, Madison (1932)
Stamm, A.J.: Ind. Eng. Chem. Process Des. Dev. 1(2), 94–97 (1929)
Zelinka, S.L., Glass, S.V., Stone, D.S.: Wood Fiber Sci. 40(4), 544–552 (2008)
Fredriksson, M., Wadsö, L., Johansson, P.: Eur. J. Wood Wood Prod. 71(4), 515–524 (2013)
Fredriksson, M., Thybring, E.E.: PLoS One. 14(11), e0225111 (2019)
Ahlgren, P.A., Wood, J.R., Goring, D.A.I.: Wood Sci. Technol. 6(2), 81–84 (1972)
Pang, S., Herritsch, A.: Holzforschung. 59(6), 654 (2005)
Beck, G., Thybring, E.E., Thygesen, L.G., Hill, C.: Holzforschung. 72(3), 225 (2018)
Telkki, V.V., Yliniemi, M., Jokisaari, J.: Holzforschung. 67(3), 291–300 (2013)
Gao, X., Zhuang, S., Jin, J., Cao, P.: Bioresources. 10(4), 8208–8224 (2015)
Stamm, A.J.: Wood Sci. 4(2), 114–128 (1971)
Feist, W.C., Tarkow, H.: For. Prod. J. 17(10), 65–68 (1967)
Myer, J.E., Rees, L.W.: Electrical Reistance of Wood with Special Reference to the Fiber Saturation Point, pp. 1–22. Syracuse University, Syracuse (1926)
Zauer, M., Kretzschmar, J., Groβmann, L., Pfriem, A., Wagenführ, A.: Wood Sci. Technol. 48(1), 177–193 (2014)
Barkas, W.W.: Proc. Phys. Soc. 48, 576–588 (1936)
Papadopoulos, A.N., Hill, C.A.S., Gkaraveli, A.: Holz Roh Werkst. 62(2), 107–112 (2004)
Nordstierna, L., Lande, S., Westin, M., Karlsson, O., Furo, I.: Holzforschung. 62(6), 709–713 (2008)
Barsberg, S.T., Thygesen, L.G.: ChemistrySelect. 2(33), 10818–10827 (2017)
Yasuda, R., Minato, K.: Wood Sci. Technol. 28(2), 101–110 (1994)
Yasuda, R., Minato, K.: Wood Sci. Technol. 29(4), 243–251 (1995)
Himmel, S., Mai, C.: Holzforschung. 69(5), 633–643 (2015)
Ellis, W.D., Rowell, R.M.: Wood Fiber Sci. 16(3), 349–356 (1984)
Altgen, M., Hofmann, T., Militz, H.: Wood Sci. Technol. 50(6), 1181–1195 (2016)
Tjeerdsma, B.F., Boonstra, M., Pizzi, A., Tekely, P., Militz, H.: Holz Roh Werkst. 56(3), 149–153 (1998)
Thybring, E.E., Kymäläinen, M., Rautkari, L.: Wood Sci. Technol. 52(2), 297–329 (2018)
Thybring, E.E.: Int. Biodeterior. Biodegradation. 82(0), 87–95 (2013)
Akitsu, H., Norimoto, M., Morooka, T., Rowell, R.M.: Wood Fiber Sci. 25(3), 250–260 (1993)
Chang, H.T., Chang, S.T.: Bioresour. Technol. 85(2), 201–204 (2002)
Chauhan, S.S., Aggarwal, P., Karmarkar, A., Pandey, K.K.: Holz Roh Werkst. 59(4), 250–253 (2001)
Forster, S.: The Decay Resistance of Chemically Modified Softwood, pp. 1–252. University of Wales, Bangor (1998)
Ibach, R.E., Rowell, R.M.: Mol. Cryst. Liq. Cryst. 353, 23–33 (2000)
Minato, K., Takazawa, R., Ogura, K.: J. Wood Sci. 49(6), 519–524 (2003)
Obataya, E., Minato, K.: Wood Sci. Technol. 42(7), 567–577 (2008)
Papadopoulos, A.N., Hill, C.A.S.: Wood Sci. Technol. 37(3–4), 221–231 (2003)
Rowell, R.M.: Physical and mechanical properties of chemically modified wood. In: Hon, D.N.S. (ed.) Chemical Modification of Lignocellulosic Materials, pp. 295–310. Marcel Dekker, New York (1996)
Yasuda, R., Minato, K., Norimoto, M.: Holzforschung. 49(6), 548–554 (1995)
Almeida, G., Brito, J.O., Perre, P.: Holzforschung. 63(1), 80–88 (2009)
Borrega, M., Kärenlampi, P.P.: Eur. J. Wood Wood Prod. 68(2), 233–235 (2010)
Esteves, B.M., Domingos, I.J., Pereira, H.M.: Bioresources. 3(1), 142–154 (2008)
Kamdem, D.P., Pizzi, A., Jermannaud, A.: Holz Roh Werkst. 60(1), 1–6 (2002)
Metsä-Kortelainen, S., Viitanen, H.: Wood Mater. Sci. Eng. 4(3–4), 105–114 (2009)
Welzbacher, C.R., Brischke, C., Rapp, A.O.: Wood Mater. Sci. Eng. 2(2), 66–76 (2007)
Martins, V.A.: Dimensional stabilisation by chemical modification of wood. In: Department of Forestry and Agriculture. University College of North Wales, Bangor (1992)
Chen, G.C.: Wood Fiber Sci. 24(3), 307–314 (1992)
Olek, W., Majka, J., Czajkowski, L.: Holzforschung. 67(2), 183–191 (2013)
Hosseinpourpia, R., Adamopoulos, S., Holstein, N., Mai, C.: Polym. Degrad. Stab. 138, 161–168 (2017)
Moghaddam, M.S., Wålinder, M.E.P., Claesson, P.M., Swerin, A.: Holzforschung. 71(2), 119 (2017)
Thygesen, L.G., Elder, T.: Wood Fiber Sci. 40(3), 309–320 (2008)
Zauer, M., Meissner, F., Plagge, R., Wagenführ, A.: Holzforschung. 70(2), 137 (2016)
Christensen, G.N.: Appita J. 13(3), 112–123 (1959)
Christensen, G.N., Kelsey, K.E.: Holz Roh Werkst. 17(5), 178–188 (1959)
Christensen, G.N.: The rate of sorption of water vapor by thin materials. In: Winn, P.N. (ed.) Volume Four: Principles and Methods of Measuring Moisture in Liquids and Solids, pp. 279–293. Reinhold Publishing Corporation, New York (1965)
Christensen, G.N.: Nature. 213(5078), 782–784 (1967)
Kelly, M.W., Hart, C.A.: Wood Fiber Sci. 1(4), 270–282 (1970)
Newns, A.C.: Trans. Faraday Soc. 52(11), 1533–1545 (1956)
Sadoh, T., Christensen, G.N.: Aust. J. Appl. Sci. 15(4), 297–308 (1964)
Christensen, G.N., Hergt, H.F.A.: J. Polym. Sci., Part A: Polym. Chem. 7(8PA1), 2427–2430 (1969)
Kohler, R., Dück, R., Ausperger, B., Alex, R.: Compos. Interfaces. 10(2-3), 255–276 (2003)
Glass, S.V., Boardman, C.R., Zelinka, S.L.: Wood Sci. Technol. 51(2), 243–260 (2017)
Thybring, E.E., Boardman, C.R., Glass, S.V., Zelinka, S.L.: Cellulose. 26(2), 723–735 (2019)
Fahlén, J., Salmén, L.: Biomacromolecules. 6(1), 433–438 (2005)
Salmén, L., Fahlén, J.: Cellul. Chem. Technol. 40(3–4), 181–185 (2006)
Leppänen, K., Bjurhager, I., Peura, M., Kallonen, A., Suuronen, J.P., Penttilä, P., Love, J., Fagerstedt, K., Serimaa, R.: Holzforschung. 65(6), 865–873 (2011)
Zabler, S., Paris, O., Burgert, I., Fratzl, P.: J. Struct. Biol. 171(2), 133–141 (2010)
Toba, K., Yamamoto, H., Yoshida, M.: Cellulose. 19(4), 1405–1412 (2012)
Harris, J.M., Meylan, B.A.: Holzforschung. 19(5), 144 (1965)
Barber, N.F., Meylan, B.A.: Holzforschung. 18(5), 146 (1964)
Meylan, B.A.: Wood Sci. Technol. 6(4), 293–301 (1972)
Glass, S.V., Zelinka, S.L.: Moisture relations and physical properties of wood. In: Wood handbook : wood as an engineering material, pp. 4.1–4.19. U.S. Dept. of Agriculture, Forest Service, Forest Products Laboratory, Madison (2010)
Rafsanjani, A., Stiefel, M., Jefimovs, K., Mokso, R., Derome, D., Carmeliet, J.: J. R. Soc. Interface. 11(95), 20140126 (2014)
Fahlén, J., Salmén, L.: Plant Biol. 4(3), 339–345 (2002)
Gu, H., Zink-Sharp, A., Sell, J.: Holz Roh Werkst. 59(6), 436–442 (2001)
Ishimaru, Y., Iida, I.: J. Wood Sci. 47(3), 178–184 (2001)
Sakagami, H., Matsumura, J., Oda, K.: IAWA J. 28(1), 29–37 (2007)
Patera, A., et al.: J. Struct. Biol. 182(3), 226–234 (2013)
Patera, A., Van den Bulcke, J., Boone, M.N., Derome, D., Carmeliet, J.: Wood Sci. Technol. 52(1), 91–114 (2018)
Derome, D., Griffa, M., Koebel, M., Carmeliet, J.: J. Struct. Biol. 173(1), 180–190 (2011)
Murata, K., Masuda, M.: J. Wood Sci. 52(4), 283–289 (2006)
Hartley, I.D., Avramidis, S.: J. Inst. Wood Sci. 14(2), 83–88 (1996)
Almeida, G., Hernandez, R.E.: Wood Fiber Sci. 38(1), 74–83 (2006)
Noack, D., Schwab, E., Bartz, A.: Wood Sci. Technol. 7(3), 218–236 (1973)
Keylwerth, R.: Holz Roh Werkst. 20(7), 252–259 (1962)
Seifert, J.: Holz Roh Werkst. 30(8), 294–303 (1972)
Hernandez, R.E.: Wood Sci. Technol. 27(5), 337–345 (1993)
Obataya, E.: Wood Sci. Technol. 39(6), 472–483 (2005)
Cetin, N.S., Özmen, N.: Wood Sci. Technol. 35(3), 257–267 (2001)
Goethals, P., Stevens M.: In: 25th Annual Meeting of the International Research Group on Wood Protection (1994)
Hill, C.A.S., Jones, D.: Holzforschung. 50(5), 457–462 (1996)
Ohmae, K., Minato, K., Norimoto, M.: Holzforschung. 56(1), 98–102 (2002)
Stamm, A.J., Tarkow, H.: J. Phys. Colloid Chem. 51(2), 493–505 (1947)
Tarkow, H., Stamm, A.J., Erickson, E.C.O.: Acetylated Wood, pp. 1–15. Forest Products Laboratory, Madison (1955)
Temiz, A., et al.: J. Appl. Polym. Sci. 102(5), 4506–4513 (2006)
Yasuda, R., Minato, K., Norimoto, M.: Wood Sci. Technol. 28(3), 209–218 (1994)
Özmen, N.: J. Appl. Sci. 7(5), 710–714 (2007)
Korkut, S., Budakci, M.: Wood Res. 55(1), 67–78 (2010)
Korkut, S., Karayilmazlar, S., Hiziroglu, S., Sanli, T.: For. Prod. J. 60(5), 473–480 (2010)
Stamm, A.J., Baechler, R.H.: For. Prod. J. 10(1), 22–26 (1960)
Tuong, V.M., Li, J.A.: Bioresources. 5(2), 1257–1267 (2010)
Stamm, A.J.: For. Prod. J. 10(12), 644–648 (1960)
Wadsö, L.: Wood Fiber Sci. 26(1), 36–50 (1994)
Krabbenhøft, K., Damkilde, L.: Mater. Struct. 37(273), 615–622 (2004)
Bertelsen, N.H.: Diffusionsmåling med kopmetoden på rødgran [Diffusion measurements with the cup method on Norway spruce], p. 18. Technical University of Denmark, Lyngby (1984)
Koponen, H.: Paperi ja Puu – Papper och trä. 12, 740–745 (1984)
Avramidis, S., Siau, J.F.: Wood Sci. Technol. 21(3), 249–256 (1987)
Sonderegger, W., Vecellio, M., Zwicker, P., Niemz, P.: Holzforschung. 65(6), 819–828 (2011)
Vanek, M., Teischinger, A.: Holzforschung und Holzverwertung. 1, 3–6 (1989)
Wadsö, L.: J. Mater. Sci. 29(9), 2367–2372 (1994)
Wadsö, L.: Wood Sci. Technol. 28(1), 59–65 (1993)
Choong, E.T.: For. Prod. J. 13, 489–498 (1963)
Sorz, J., Hietz, P.: Trees. 20(1), 34–41 (2006)
Tarkow, H., Stamm, A.J.: For. Prod. J. 10(5), 247–250 (1960)
Tarkow, H., Stamm, A.J.: For. Prod. J. 10(6), 323–324 (1960)
Petty, J.A.: Wood Sci. Technol. 7(4), 297–307 (1973)
Schirmer, R.: Z. Ver. Dtsch. Ing. 70(6), 170–177 (1938)
Stamm, A.J.: J. Phys. Chem. 60(1), 83–86 (1956)
Peroval, C., Debeaufort, F., Despre, D., Voilley, A.: J. Agric. Food Chem. 50(14), 3977–3983 (2002)
Kulasinski, K., Keten, S., Churakov, S.V., Guyer, R., Carmeliet, J., Derome, D.: ACS Macro Lett. 3(10), 1037–1040 (2014)
Topgaard, D., Söderman, O.: Langmuir. 17(9), 2694–2702 (2001)
Topgaard, D., Söderman, O.: Cellulose. 9(2), 139–147 (2002)
Spolek, G.A., Plumb, O.A.: Wood Sci. Technol. 15(3), 189–199 (1981)
Sedighi-Gilani, M., Griffa, M., Mannes, D., Lehmann, E., Carmeliet, J., Derome, D.: Int. J. Heat Mass Transf. 55(21–22), 6211–6221 (2012)
Desmarais, G., Sedighi-Gilani, M., Vontobel, P., Carmeliet, J., Derome, D.: Transp. Porous Media. 113(2), 383–404 (2016)
Siau, J.F.: Transport Processes in Wood. Springer-Verlag, Berlin (1984)
Fredriksson, M., Lindgren, O.: Wood Mater. Sci. Eng. 8(4), 245–252 (2013)
Hansmann, C., Wimmer, W.G.R., Teischinger, A.: Drevarsky Vyskum. 47(4), 1–16 (2002)
Sedighi-Gilani, M., Vontobel, P., Lehmann, E., Carmeliet, J., Derome, D.: Mater. Struct. 47(6), 1083–1096 (2014)
Almeida, G., Leclerc, S., Perre, P.: Int. J. Multiphase Flow. 34(3), 312–321 (2008)
Siau, J.F.: Wood structure and chemical composition. In: Transport Processes in Wood, pp. 35–72. Springer, Heidelberg (1984)
Segerholm, I., Claesson, J.: Wood Mater. Sci. Eng. 3(3&4), 109–118 (2008)
Stamm, A.J.: Ind. Eng. Chem. 19(9), 1021–1025 (1927)
Brischke, C., Rapp, A.O., Bayerbach, R., Morsing, N., Fynholm, P., Welzbacher, C.R.: Build. Environ. 43(10), 1575–1582 (2008)
Niklewski, J., Isaksson, T., Frühwald Hansson, E., Thelandersson, S.: Wood Mater. Sci. Eng. 13(3), 129–140 (2017)
Fredriksson, M., Wadsö, L., Johansson, P., Ulvcrona, T.: Wood Mater. Sci. Eng. 11(4), 189–200 (2016)
Zelinka, S.L., Passarini, L., Quintana, J.C., Glass, S.V., Jakes, J.E., Wiedenhoeft, A.C.: Wood Fiber Sci. 48, 54–61 (2016)
Du, Q.P., Geissen, A., Noack, D.: Holz Roh Werkst. 49(7–8), 305–311 (1991)
Hjort, S.: J. Coatings Technol. 68(856), 31–39 (1996)
Otten, K.A., Brischke, C., Meyer, C.: Constr. Build. Mater. 153, 640–646 (2017)
Vermaas, H.F.: Holzforschung. 29(4), 140–144 (1975)
Brischke, C., Rapp, A.O., Bayerbach, R.: Build. Environ. 43(10), 1566–1574 (2008)
Brischke, C., Kathrin, A.S., Christian, R.W.: Modeling the influence of thermal modification on the electrical conductivity of wood. In: Holzforschung, p. 185 (2014)
Zelinka, S.L., Wiedenhoeft, A.C., Glass, S.V., Ruffinatto, F.: Wood Mater. Sci. Eng. 10(2), 189–196 (2015)
Fredriksson, M., Claesson, J., Wadsö, L.: Math. Probl. Eng. 2015, 7 (2015)
Merela, M., Primoz, O., Sersa, I., Mikac, U.: Holzforschung. 63(3), 348–351 (2009)
Menon, R.S., MacKay, A.L., Hailey, J.R.T., Bloom, M., Burgess, A.E., Swanson, J.S.: J. Appl. Polym. Sci. 33(4), 1141–1155 (1987)
Watanabe, K., Lazarescu, C., Shida, S., Avramidis, S.: Dry. Technol. 30(3), 256–262 (2012)
Lindgren, O., Davis, J., Wells, P., Shadbolt, P.: Holz Roh Werkst. 50(7–8), 295–299 (1992)
Sedighi Gilani, M., Abbasion, S., Lehmann, E., Carmeliet, J., Derome, D.: Int. J. Therm. Sci. 81(0), 1–12 (2014)
Rosner, S., Riegler, M., Vontobel, P., Mannes, D., Lehmann, E.H., Karlsson, B., Hansmann, C.: Holzforschung. 66(6), 751–756 (2012)
Rosenkilde, A., Glover, P.: High resolution measurement of the surface layer moisture content during drying of wood using a novel magnetic resonance imaging technique. In: Holzforschung, p. 312 (2002)
Dvinskikh, S.V., Henriksson, M., Mendicino, A.L., Fortino, S., Toratti, T.: Eng. Struct. 33(11), 3079–3086 (2011)
Javed, M.A., Kekkonen, P.M., Ahola, S., Telkki, V.V.: Holzforschung. 69(7), 899–907 (2015)
Gezici-Koç, Ö., Erich, S.J.F., Huinink, H.P., van der Ven, L.G.J., Adan, O.C.G.: Cellulose. 24(2), 535–553 (2016)
Passarini, L., Malveau, C., Hernández, R.E.: Wood Sci. Technol. 49(6), 1251–1268 (2015)
Greenspan, L.: J. Res. Nat. Bur. Stand. 81A(1), 89–96 (1977)
Mozurkewich, M.: Atmos. Environ. Part A. 27(2), 261–270 (1993)
Nyqvist, H.: Int. J. Pharm. Technol. Prod. Manuf. 4(2), 47–48 (1983)
Neimsuwan, T., Wang, S., Taylor, A.M., Rials, T.G.: Wood Sci. Technol. 42(6), 493–506 (2008)
Wadsö, L., Svennberg, K., Dueck, A.: Dry. Technol. 22(10), 2427–2440 (2004)
ISO: ISO 12571:2013 Hygrothermal Performance of Buildign Materials and Products – Determination of Hygroscopic Properties. ISO International Organization for Standardization (2013)
Wadsö, L., Anderberg, A., Åslund, I., Söderman, O.: Eur. J. Pharm. Biopharm. 72(1), 99–104 (2009)
Glass, S.V., Boardman, C.R., Thybring, E.E., Zelinka, S.L.: Wood Sci. Technol. 52(4), 909–927 (2018)
Cloutier, A., Fortin, Y.: Wood Sci. Technol. 25(4), 263–280 (1991)
Richards, L.A.: Soil Sci. 66(2), 105–110 (1948)
Almeida, G., Hernandez, R.E.: Wood Mater. Sci. Eng. 2(1), 33–44 (2007)
ASTM: Standard Test Method Fo Moisture Retention Curves of Porous Building Materials Using Pressure Plates. ASTM International (2015)
Stone, J.E., Scallan, A.M.: Tappi J. 50(10), 496–501 (1967)
Cloutier, A., Fortin, Y.: Dry. Technol. 12(8), 1793–1814 (1994)
Magne, F.C., Portas, H.J., Wakeham, H.: J. Am. Chem. Soc. 69(8), 1896–1902 (1947)
Hatakeyama, T., Hirose, S., Hatakeyama, H.: Macromol. Chem. Phys. 184(6), 1265–1274 (1983)
Zanotti, J.M., Bellissent-Funel, M.C., Kolesnikov, A.I.: Eur. Phys. J. Spec. Top. 141(1), 227–233 (2007)
Hill, C.A.S., Forster, S.C., Farahani, M.R.M., Hale, M.D.C., Ormondroyd, G.A., Williams, G.R.: Int. Biodeterior. Biodegradation. 55(1), 69–76 (2005)
Flournoy, D.S., Kirk, T.K., Highley, T.L.: Holzforschung. 45(5), 383–388 (1991)
Flournoy, D.S., Paul, J.A., Kirk, T.K., Highley, T.L.: Holzforschung. 47(4), 297–301 (1993)
Borrega, M., Kärenlampi, P.P.: Wood Fiber Sci. 43(2), 206–214 (2011)
Maloney, T.C., Paulapuro, H.: J. Pulp Pap. Sci. 25(12), 430–436 (1999)
Araujo, C.D., MacKay, A.L., Hailey, J.R.T., Whittall, K.P., Le, H.: Wood Sci. Technol. 26(2), 101–113 (1992)
Almeida, G., Gagné, S., Hernández, R.E.: Wood Sci. Technol. 41(4), 293–307 (2007)
Fredriksson, M., Thygesen, L.G.: Holzforschung. 71(1), 77–90 (2017)
Thygesen, L.G., Elder, T.: Wood Fiber Sci. 41(2), 194–200 (2009)
Kekkonen, P.M., Ylisassi, A., Telkki, V.-V.: J. Phys. Chem. C. 118(4), 2146–2153 (2014)
Carr, H.Y., Purcell, E.M.: Phys. Rev. 94(3), 630–638 (1954)
Meiboom, S., Gill, D.: Rev. Sci. Instrum. 29(8), 688–691 (1958)
McDonald, P.J., Rodin, V., Valori, A.: Cem. Concr. Res. 40(12), 1656–1663 (2010)
Whittall, K.P., Bronskill, M.J., Henkelman, R.M.: J. Magn. Reson. 95(2), 221–234 (1991)
Provencher, S.W.: Comput. Phys. Commun. 27(3), 229–242 (1982)
Lawson, C.A., Hanson, R.: Solving Least Squares Problems. Prentice-Hall, Englewood Cliffs (1974)
Song, Y.Q., Venkataramanan, L., Hürlimann, M.D., Flaum, M., Frulla, P., Straley, C.: J. Magn. Reson. 154(2), 261–268 (2002)
Aggebrandt, L.G., Samuelson, O.: J. Appl. Polym. Sci. 8(6), 2801–2812 (1964)
Stone, J.E., Scallan, A.M.: Cellul. Chem. Technol. 2, 343–358 (1968)
Alince, B.: Tappi J. 74(11), 200–202 (1991)
Farahani, M.R.M.: Decay Resistance of Modified Wood, in School of Agriculture and Forestry, p. 279. University of Wales, Bangor (2003)
Claesson, J., Hagentoft, C.E., Wadsö, L.: Polym. Eng. Sci. 34(10), 821–826 (1994)
Zelinka, S.L., Glass, S.V., Boardman, C.R., Derome, D.: Wood Mater. Sci. Eng. 11(4), 228–238 (2016)
Zelinka, S., Glass, S., Boardman, C.: J. Test. Eval. 44(6), 2396–2402 (2016)
Wadsö, L.: Wood Sci. Technol. 27(5), 396–400 (1993)
Crank, J.: The Mathematics of Diffusion, 2nd edn. Clarendon Press, Oxford (1975)
Wadsö, L.: Dry. Technol. 12(8), 1863–1876 (1994)
Bomberg, M., Pazera, M., Plagge, R.: J. Therm. Envel. Build. Sci. 28(3), 227–243 (2005)
Mannes, D., Sonderegger, W., Hering, S., Lehmann, E., Niemz, P.: Holzforschung. 63(5), 589 (2009)
Wadsö, L.: Holzforschung. 48, 133–138 (1994)
Wadsö, L.: Holzforschung. 48, 75–81 (1994)
Hall, C., Hoff, W. D.: Water transport in brick, stone and concrete. CRC Press, Abingdon, UK (2012)
Feng, C., Janssen, H.: Build. Environ. 134, 21–34 (2018)
Zillig, W.: Moisture transport in wood using a multiscale approach. In: Building Physics. Katholieke Universiteit Leuven, Leuven (2009)
Niemz, P., Mannes, D., Herbers, Y., Koch, W.: Bauphysik. 32(3), 149–153 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Thybring, E.E., Fredriksson, M. (2023). Wood and Moisture. In: Niemz, P., Teischinger, A., Sandberg, D. (eds) Springer Handbook of Wood Science and Technology. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-030-81315-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-81315-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-81314-7
Online ISBN: 978-3-030-81315-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)