
Abstract
Prostate apoptosis response-4 (Par-4) was identified as a tumor suppressor protein that is silenced by promoter methylation in various cancers and has been shown to induce apoptosis selectively in cancer cells but not in normal cells. Par-4 interacts with a variety of partners in cells to mediate various cellular responses and appears to have a pro-apoptotic role in non-hematological tumors. Here, we summarize the literature on the role of Par-4 in hematological cells that is in contrast to its classic pro-apoptotic role. Par-4 is expressed basally in various hematopoietic cells and malignancies at the mRNA and protein level, but is predominant in the early stages of B-cell maturation and specifically in chronic lymphocytic leukemia (CLL). CLL B cells express higher levels of Par-4 than normal B-cell subsets and constitutively active B-cell receptor signaling (BCR) maintains high Par-4 levels in these cells, suggesting a novel regulation of Par-4 through BCR signaling. CLL cell growth is dependent on BCR signaling-mediated Par-4 expression, which is in part due to downregulation of p21 by Par-4. Bcl2 and NF-κB pathways cause differential regulation of apoptotic genes in contrast to non-hematological cancers, and Par-4 may also play a significant role in tumor microenvironment. Thus, Par-4 appears to have unique roles in hematological malignancies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Par-4
- B cells
- B-cell receptor
- Lymphoma
- Chronic lymphocytic leukemia
- Microenvironment
- Tcl1
- Splenectomy
- Stromal cells
- p21
1 Introduction
One of the original hallmarks of cancer is to evade apoptosis and many cancers master this skill by down-regulating tumor suppressors and pro-apoptotic factors [1]. Prostate apoptosis response-4 (Par-4) is a tumor suppressor that is found to be downregulated in many cancers including renal cell carcinoma [2], breast cancer [3], neuroblastoma [4], and also in about 40% of all endometrial cancers, where about 32% of those cases were due to Par-4 promoter hypermethylation and occasionally due to silencing mutations [5, 6]. Early studies in lymphoid cells established that Par-4 expression is deregulated with decreased frequency of expression in immature or less differentiated populations and that an inverse expressional pattern exists between Par-4 and Bcl-2 in leukemic cell lines and acute lymphocytic leukemia (ALL) [6, 7]. Par-4 was originally identified by Sells and colleagues by its upregulation during ionomycin-induced apoptosis of androgen-independent and -dependent rat prostate cancer cells in 1994 [8]. Shortly after, using a yeast two-hybrid assay and HEK-293 mammalian cells, Johnstone et al. discovered that Par-4 interacts with the Wilm’s Tumor-1 protein, a transcriptional suppressor [9]. Additional early studies found that Par-4 also interacts physically with atypical protein kinase c (aPKC) and overexpression of Par-4 in NIH 3T3 fibroblasts led to an apoptotic morphological change [10]. These initial studies defined Par-4 as a pro-apoptotic factor and tumor suppressor primarily in non-hematopoietic cells and suggested a similar role in hematopoietic cells. In fact, the first study to identify Par-4 as a pro-apoptotic protein in lymphatic cells showed that overexpression of Par-4 per se in the Jurkat leukemia cells is not sufficient to induce apoptosis but markedly increased their sensitivity to apoptosis with different chemotherapeutic agents [11].
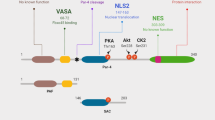
The human Par-4 gene is located on chromosome 12q21 and contains 7 exons, encoding a 340 amino acid, 43-47 kDa protein [12, 13]. Par-4 is ubiquitously expressed in tissues of different species and Johnstone et al. found that mouse Par-4 shows 83% and 91% identity to human and rat Par-4, respectively [12]. Interestingly, the leucine zipper domain, carboxy terminal region, and nuclear localization sequences (NLS) exhibit 100% conservation across species [14]. The leucine zipper domain allows Par-4 to interact with other proteins as either a homo- or a heterodimer. The nuclear localization sequences suggest that Par-4’s function is dependent on nuclear translocation; however, in normal tissues, Par-4 is localized mostly to the cytoplasm [15]. The NLS2 sequence is very interesting in Par-4 as it is sufficient to allow nuclear translocation alone, but it is also part of a domain that is necessary for the apoptosis-inducing properties of Par-4, termed selective for apoptosis of cancer cell (SAC) domain [16]. SAC is a core domain of 59 amino acids in length and includes a threonine residue that is the site of phosphorylation via Protein Kinase A [17]. Activation of Par-4 through phosphorylation indicates that its function is tightly regulated by post-translational modification. PKA, a broad spectrum serine/threonine kinase regulated by cAMP signaling, is associated with cell proliferation, and is frequently overexpressed in cancer cells [17]. Par-4 is able to utilize PKA upregulation in cancer cells to specifically induce apoptosis of cancer but not normal cells [17]. This selective ability of Par-4 makes it an attractive therapeutic target. Additionally, Par-4 is negatively regulated by AKT activity through phosphorylation at serine 249, which is located between the SAC domain and leucine zipper region [13]. Phosphorylation of Par-4 via Akt is required for cancer cell survival, as phosphorylation of Par-4 by Akt leads to binding of the chaperone 14-3-3, retaining[18] Par-4 in the cytoplasm [19]. Many studies have linked the pro-apoptotic activity of Par-4 to its ability to inhibit NF-κB transcriptional activity. Activated Par-4 prevents PKC-ζ from phosphorylating IκB, which is necessary for RelA translocation to the nucleus [10, 18, 20]. Another mechanism of NF-κB inhibition is due to a direct repressive effect of Par-4 in the nucleus but the exact mechanism still needs to be elucidated.
Seminal studies investigating Par-4 function led to the discovery that Par-4 is secreted from most cell types and can induce apoptosis of neighboring cells [21]. PC-3 (prostate cancer) cells transfected with GFP-labeled Par-4 or SAC domain-GFP undergo apoptosis, but GFP-negative cells were also dying as measured through caspase-3 activation [21]. Par-4 secretion is independent of apoptosis. Par-4 secretion occurs through the classical ER-Golgi pathway as inhibition of the network with brefeldin A (BFA) blocked secretion [13, 21]. Par-4 secretion is associated with the ER stress response and was also found to associate with GRP78, a member of the heat shock protein family 70 (HSP70) that works to facilitate proper protein folding, prevent intermediate aggregates, target misfolded proteins for degradation, bind calcium, and serve as an ER stress signal regulator [22]. Burikhanov et al. showed that Par-4 and the SAC domain bind to GRP78 at the plasma membrane in response to TRAIL (tumor necrosis factor-related apoptosis-inducing ligand). TRAIL is a known ER stress-inducing factor and treatment of PC-3 cells with TRAIL led to increased GRP78/Par-4 at the cell surface and induced apoptosis [23]. Initiation of extracellular Par-4-mediated apoptosis results in a feedback loop that promotes more translocation of Par-4 and GRP78 to the surface of the cell. More recently, Hebbar et al. have reported that an N-terminal 15 kDa fragment of Par-4 generated by caspase-3 cleavage is pro-apoptotic to cancer cells and released by cancer cells treated with chemotherapy agents [24]. With respect to hematopoietic cells at least two studies reported in 2004, before the discovery of the role of secreted Par-4, have demonstrated that expression of Par-4 in Jurkat cells promotes a complex interplay between the intrinsic and extrinsic pathway of apoptosis through molecules such as Apaf-1 and survivin [25]. Par-4 was also shown to sensitize neoplastic lymphocytes to ligation of a death receptor CD95 in the extrinsic pathway, thereby activating initiator caspases 8 and 10 which are able to directly activate executioner caspases 6, 7, and 3 [26]. We describe the unique role of Par-4 in normal and cancerous hematological cells in the following sections.
2 Par-4 Expression and Function in Immune Cells
There are now a number of gene expression data sets available from various sequencing studies utilizing normal and malignant hematopoietic cells for data curation of individual genes. We investigated the expression of PAWR (Par-4) gene in normal hematopoietic cells (Bloodspot.eu: Hemaexplorer) [27] and found that it was highly expressed in immature and progenitor stem cells compared to mature counterparts with the exception of B cells (Fig. 1). PAWR expression was significantly higher in B cells compared to other mature lymphoid cells including CD4 and CD8 T cells as well as HSCs (**p < 0.01; ***p < 0.0.001). Consistently, PAWR was found to be most highly expressed in B cells within the hematopoietic hierarchical differentiation tree (Fig. 2). The elevated expression of Par-4 in B cells suggests it may play a unique role in B-cell neoplasms that we will discuss further in detail in Sects. 2, 3 and 4.

PAWR expression in normal hematopoietic cells according to BloodSpot Database, Normal Human Hematopoiesis (Hemaexplorer). Log2 expression of the PAWR gene in subsets of immature and mature immune cells. Significance was determined by student t-test (**p < 0.01; ***p < 0.0001) (HSC_BM:Hematopoietic stem cells from bone marrow; early HPC_BM: Early hematopoietic progenitor cells from bone marrow; CMP: Common myeloid progenitor cell; GMP: Granulocyte monocyte progenitors; MEP: Megakaryocyte-erythroid progenitor cell; PM_BM: Promyelocyte from bone marrow; MY_BM: Myelocyte from bone marrow; PMN_BM: Polymorphonuclear cells from bone marrow; PMN_PB: Polymorphonuclear cells from peripheral blood; mDC: CD11c+ myeloid dendritic cells; pDC: CD123+ plasmacytoid dendritic cells)[27]

Similar to its role in non-hematological malignancies, Par-4 in hematological cells is involved in several protein–protein interactions including but not limited to protein kinase A (PKA) [17], atypical protein kinase C [28], Wilm’s tumor 1 (WT-1) [9], death-associated protein (DAXX), DAP-like kinase/ZIP kinase (DLK/ZIPK) [29, 30] and THAP-domain protein 1 (THAP1) [31]. Par-4 interactions with PKA, PKC, and WT-1 are similar to those in non-hematological cells and have been described in above cited references. Analysis of 62 untreated CLL patient peripheral blood and bone marrow samples showed a positive correlation between Par-4 and both DAXX and ZIPK proteins [29]. Phosphorylation of Par-4 and DAXX by ZIPK is involved in the nuclear pathway of apoptosis in promyelocytic leukemia (PML) oncogenic domains (PODs, nuclear domains that exist in all nucleated mammalian cells) through caspase activation. Simultaneous overexpression of DAXX, Par-4, and ZIPK proteins leads to more than a six-fold increase in apoptosis [32]. It has also been shown that a nuclear pro-apoptotic factor THAP-1 co-localizes with Par-4 in PML nuclear bodies (NBs) and that Par-4 is a component of PML NBs in blood vessels, which is a major site of PML expression in vivo [33]. PML nuclear bodies (PML NBs) are discrete membraneless subnuclear domains organized by the promyelocytic leukemia protein, PML, a tumor suppressor, with other client proteins. PML NBs function in promoting apoptosis by recruiting various pro-apoptotic proteins such as DAXX [34, 35] and p53 [36]. PML NBs were discovered through their disorganization in acute promyelocytic leukemia (APL) and arsenic therapy-induced reorganization has been directly implicated in its eradication [37]. PML NBs like nucleoli and Cajal bodies are a type of liquid-like droplets of biomolecules, which self-assemble within another liquid—the cytoplasm or nucleoplasm—and arise from a physicochemical process known as liquid-liquid phase separation, sometimes also called coacervation [38]. At the molecular level, weak, transient interactions between different proteins/RNA molecules with multivalent domains or intrinsically disordered regions are a driving force for this phase seperation [38]. Par4 is well known to interact weakly and transiently with a variety of proteins including various pro-apoptotic proteins including DAXX and p53 [39] which home into PML NBs. The structure of Par-4 has shown that it is basically an intrinsically disordered protein [40,41,42]. All these divergent properties of Par-4 should suggest an emerging theme of molecular interactions in organelles that do not have an enclosing membrane such as PML NBs to remain coherent structures that can compartmentalize and concentrate specific sets of molecules to orchestrate their function.
Par-4 and THAP1 have also been shown recently to form a protein complex by the interaction of their carboxyl termini and this complex competitively with Notch modulated alternative pre-mRNA splicing of cell cycle and apoptosis regulator 1 (CCAR1) inducing cellular apoptosis in Jurkat cells, a human T-ALL cell line [31]. Genome-scale sequencing has revealed that more than 70% of the genome is transcribed into RNAs that do not produce protein. These RNAs are called noncoding RNAs (ncRNAs). Within the last decade, by integrating transcriptome profiles with chromatin state maps, many previously unreported T-ALL-specific lncRNA genes were identified. Notch-regulated LUNAR1 [43] and ARIEL (ARID5B-inducing enhancer-associated) long noncoding RNAs [44] are a few that have garnered interest in T-ALL recently. A novel lncRNA, T-ALL-R-LncR1, discovered with whole-transcriptome deep sequencing from the Jurkat leukemic T-cell line was shown to be markedly expressed in neoplastic T lymphocytes of children with T-ALL. Further studies revealed that knockdown of this T-ALL-R-LncR1 facilitated the formation of a Par-4/THAP1 protein complex, resulting in apoptosis [45]. This suggests a novel role of Par-4 in lncRNA-mediated escape of apoptosis in T-ALL.
To investigate the physiological role of Par-4, Garcia-Cao et al. generated a whole body Par-4 knockout mouse. The average lifespan of Par-4 null mice is 18mo compared to 25mo for Par-4 WT animals with a 87% propensity to develop tumors [18]. These mice also exhibited normal B- and T-cell development but do have a slight increase in total number of lymphocytes leading to an increase in spleen size [46]. The proportions of B and T cells were not changed in young mice lacking Par-4, nor were the memory subsets in each lymphocyte population suggesting that Par-4 does not play a role in B- or T-cell differentiation. Interestingly, the proliferative responses to BCR and TCR cross-linking were increased in Par4−/− compared to WT animals with increased B-cell proliferation associated with an increase in PKC-ζ activity. The lack of Par-4 in these mice led to hyperactivation of atypical protein kinases, blocking JNK signaling in CD4 + T cells that resulted in increased IL-4 production and skewed the null mice towards a Th2 response [46]. Of note, Par-4 deficiency in both CD4+ and CD8+ T cells resulted in increased IL-2 secretion post-TCR stimulation without changes in CD25 expression suggesting Par-4−/− T cells have enhanced functional activity. These observations by Lafuente and colleagues suggest that Par-4 plays a role in regulating B and T lymphocyte function. Par-4 is abundantly expressed in various leukemic/lymphoma cell lines and THP1, a human monocytic leukemia cell line (Fig. 3, left part). However, expression of Par-4 is dramatically decreased with differentiation into macrophages by phorbol-12-myristate-13-acetate (PMA) treatment (Fig. 3, right). When Par-4 is overexpressed in Mycobacterium tuberculosis (Mtb strain: H37Ra) infected macrophages, intracellular survival of Mtb H37Ra was significantly reduced, in part due to increased apoptosis [47].

Expression of Par-4 protein in various hematological cells; Mec1(human CLL); Ly3 and Ly10 (Diffuse Large B-Cell Lymphoma); SUDHL6 (Diffuse Histiocytic Lymphoma); Raji and Ramos (Burkitt’s lymphoma); CLL (Eμ-Tcl1 mouse); Thp1 (human monocytic leukemia); Raw264.7 (murine macrophage cell line). Thp1 monocytes were differentiated into macrophages by treating with PMA which was accompanied by a decrease in Par-4 (Western blot on the right side)
3 Par-4 in B-cell Malignancies
B-cell malignancies encompass both lymphomas and leukemias. According to the 2016 SEER database, leukemia is the ninth most common cancer in the USA contributing to 3.8% of all reported cancer deaths [48]. As a cancer of the blood, abnormal leukemic cells accumulate and do not die, suppress the function of normal immune cells, and eventually out-populate other hematopoietic cell types resulting in anemia. Leukemia may be classified as chronic (slow progression of mature cells) or acute (rapid growth of primarily immature cells) and can affect both the myeloid and lymphoid white blood cells. Patients that are diagnosed with acute leukemia will normally start treatment as soon as possible while patients with chronic leukemia may be placed under a “wait and watch” status until symptoms progress. Subtypes of leukemia include: acute lymphoblastic leukemia, chronic lymphocytic leukemia, acute myelogenous leukemia, and chronic myelogenous leukemia (CML). Specifically, CLL is the most common adult leukemia in the Western world and like all leukemias, patients are grouped into fast or slow progressing disease based on prognostic indicators. Many patients with CLL may live a relatively normal life without symptoms, while others may only survive months to years after diagnosis or treatment initiation. Patients with CLL do have an 82% 5-year survival rate but experts in the field classify CLL as incurable [49].
CLL is a highly heterogeneous disease in terms of clinical course as some patients may live decades past initial diagnosis and likely die from other complications such as infections, while others may progress more rapidly. This heterogeneity can be attributed in part to mutations found within the variable gene segments of the BCR [50]. CLL can be classified into mutated (M-CLL) and unmutated (U-CLL) forms, the latter resulting in increased BCR signaling, more aggressive disease, and worse prognosis. This BCR signaling pathway is a desirable target as it is required for the survival of malignant B cells and is constitutively activated in many CLL cases and B-lymphomas [51, 52]. Additionally, the microenvironment has been found to play a key role in promoting the growth of B-cell malignancies , including CLL, by providing proliferative signals and promoting drug resistance [53, 54]. BCR signaling and microenvironment make CLL a very complex disease to study and treat but also allows for new targets to be explored for therapeutic potential.
Primarily, Par-4 has been characterized in the context of a diseased state rather than healthy tissues but a few studies have investigated its expression in lymphoid cells. Boehrer et al. reported the expression pattern of Par-4 mRNA and protein levels in healthy donor peripheral mononuclear cells compared to patients with ALL and CLL. Par-4 protein expression was detected in 100% of the healthy mononuclear cells and CLL samples [7]. Conversely, Par-4 protein levels were detected in 50% and 70% of pro-lymphocytic leukemia (PLL) and ALL samples, respectively, suggesting that Par-4 protein is downregulated in less differentiated cells comprising PLL and ALL compared to more mature cell populations of peripheral mononuclear cells and CLL [55]. In the same study, the authors also reported that sorted B and T cells expressed the Par-4 protein consistent with the Par-4 mRNA expression patterns from gene expression databases summarized above. Analysis provided by Bloodspot database using the leukemia MILE study shows PAWR to be highly expressed in CLL samples compared to other types of leukemias as well as normal bone marrow (Fig. 4).

PAWR log2 expression in various hematological diseases with indicated mutations. Figure is adapted from Bloodspot.eu analysis of leukemia MILE study [27]. (ALL: Acute Lymphocytic leukemia (ALL); AML: Acute Myeloid Leukemia; CLL: Chronic Lymphocytic Leukemia; CML: Chronic Myeloid Leukemia; MDS: Myelodysplastic syndrome; T-ALL: T cell ALL; B-ALL: B cell ALL; t: translocation; inv: inversion; c:common; pre:precursor)
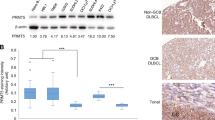
Several studies have examined Par-4 levels in CLL leading to prognostic predictions . Initially, Boehrer et al. examined the levels of Par-4 in normal and neoplastic lymphocytes and found that all patients with CLL (n = 30) expressed Par-4 protein, but only 63% were positive for Par-4 mRNA expression suggesting that there may be a difference in Par-4 regulation in different types of leukemias [7]. Bcl-2 is a well-characterized protooncogene initially identified at the chromosomal breakpoint of t(14;18) bearing B-cell lymphomas. Overexpression of Bcl-2 is to be considered a crucial event in leukemogenesis/lymphomagenesis and is aberrantly overexpressed in CLL, follicular lymphoma (FL), mantle cell lymphoma (MCL), Waldenstrom macroglobulinemia (WM), and one-third of diffuse large B-cell lymphoma (DLBCL) [56]. Bcl-2 is unique among proto-oncogenes in that it is localized to mitochondria as a key regulator of the intrinsic, mitochondrial apoptotic pathway for specifically blocking apoptosis rather than promoting proliferation [57]. Bcl2 family of proteins are also important in inducing drug resistance by many of the chemotherapeutic agents including the most recently approved Bcl2 inhibitor drug Venetoclax [56]. Bcl-2 is directly antagonistic to the actions of Par-4. It is evident that in non-hematopoietic cancer cells Par-4 is consistently downregulated and as such the effects of Par-4 on Bcl2 might be straightforward with respect to the intrinsic mitochondrial pathway of apoptosis. However, in hematopoietic cancer cells with a robust Bcl2 activity, the levels of Par-4 required to counteract it need to be different and this shifts the rheostat of pro- and anti-apoptotic mechanisms intracellularly. Hence, these cancer cells expressing high Bcl2 might express more Par-4, but this amount of Par-4 is not amenable to apoptosis without additional stimuli. It is not surprising that synergistic and antagonistic drug combinations within a single lymphoma model led to uncorrelated levels of Bcl2 and Par-4 [58]. Previous studies had indicated that Par-4 and Bcl-2 are inversely correlated [59], but there was no relationship found between the expression of Par-4 and Bcl-2 protein expression in CLL patients. Next, Chow and colleagues found that CLL patients that lacked the Imatinib targets BCR-ABL, C-Kit and PDGFR were still sensitive to Imatinib treatment and that, the response correlated with Par-4 expression [30]. Additionally, this study confirmed Boehrer et al. findings that there was no relationship between Par-4 expression and Bcl-2 in patients that did or did not respond to Imatinib treatment. Par-4 was also downregulated in the course of the treatment with Imatinib when cells underwent apoptosis after caspase-8 and -3 activation [30]. Lastly, Bojarska-Junak and colleagues assessed the expression of Par-4 in CLL B cells and found a positive correlation of Par-4 with Bcl-2, which is opposite of what is observed in non-hematopoietic cells (Fig. 5).

Cartoon depicting the differences in the role of Par-4 in non-hematological vs hematological malignancies (Created with BioRender.com)
Par-4 was also positively correlated with DAXX (death-associated protein), and ZIPK (zipper-interacting protein kinase) expression in CLL patients [29]. Additionally, Par-4 was found to positively correlate with LDH (lactate dehydrogenase) serum concentrations and was more highly expressed in CD38+ CLL patients who have a more aggressive form of CLL disease [50, 60]. These initial studies suggested that Par-4 in CLL may be regulated differently. Importantly, rather than being downregulated as shown in other cancers, Par-4 was found to be expressed in 30/30 human CLL patient samples [7]. These results were also confirmed in another study showing increased Par-4 expression in peripheral blood mononuclear cells (PBMCs) of human CLL patients, compared to healthy donor PBMCs [61]. These surprising discoveries suggested that aberrant Par-4 expression in CLL is unique and is further discussed below.
4 Intrinsic Role of Par-4 in CLL
4.1 Par-4 Expression in CLL
Even with the incredible discoveries made in the field of chronic lymphocytic leukemia over the last few decades, the cellular origin of the disease is still debated today [62, 63]. CLL cells distinctively express CD19, CD5, CD23, as well as surface Ig molecules [60]. CD19 is a surface antigen that is expressed on both normal and neoplastic B cells and is critical for intrinsic B-cell signaling through BCR interactions as well as BCR-independent signaling [64]. CD5 is also a cell surface molecule that is expressed on thymocytes, mature T cells, and B1 cells but not on conventional B2 population [65]. Previous studies have also indicated that CD5 is found on some activated human B cells that are autoreactive [66]. CD5 is thought to be a negative regulator to mitigate signaling in order to prevent over activation of signaling downstream of the TCR or BCR [67].
The co-expression of low IgM and IgD levels on the surface of CLL cells originally suggested that these cells arise from naïve antigen-inexperienced B cells [68]. Further studies classified CLL into two subgroups, M-CLL and U-CLL defined by mutations in the variable gene segments of the BCR indicating that 50–60% of CLL cells had undergone somatic hypermutation (M-CLL), leading to hypotheses that suggest CLL cells are derived from two cellular origins [69, 70]. Seifert and colleagues suggest that U-CLL cells are derived from unmutated mature CD5+ B cells as their IgV sequence is less than 2% different from germline, whereas M-CLL cells are derived from a distinct CD5 + CD27+ post-germinal center B cell [62]. An additional study investigating phenotypic markers found that CLL cells express more of an activated state (CD69 + CD25 + CD71+) independent of their Ig mutational status when compared to normal CD5+ B cells in humans, suggesting that CLL cells are mature antigen-experienced cells [68]. Antigen-experienced cells can be derived from cells that undergo somatic hypermutation within a germinal center or from extrafollicular responses, but may also develop in a T cell-independent manner which may account for CLL cells that have unmutated Ig variable regions [70, 71]. Further support for antigen-experienced B cells to be CLL precursors comes from studies that have examined the BCR repertoire in multiple CLL samples. It is well accepted that CLL cells have constitutive BCR signaling , but CLL cells also respond to antigen [72, 73]. Recent studies have found that 30% of BCR immunoglobulins within the CLL patient population are quasi-identical resulting in a stereotypy of BCRs [72, 74, 75]. This indicates that the malignant B cells from unrelated patients recognize similar antigens suggesting that there are a few common epitopes which activate CLL cells. Not many antigens have been identified to stimulate CLL cells, but one potential candidate is non-muscle myosin heavy chain IIA which is an intracellular protein that interacts with actin to provide cellular movement and therefore is considered a self-antigen [76]. This is interesting as B1 cells are thought to be self-reactive and respond to autoantigens supporting the idea that CLL cells are derived from B1 cells [70]. Cell autonomous signaling was identified in some CLL patients that express specific immunoglobulin variable regions which also associate with greater severity of the CLL disease. This is thought to be due to homotypic interactions of B-cell receptors with specific V region mutations [77, 78].
B1 cells are primarily found within the peritoneal cavity of mice but are also present in the spleen, albeit at a lower level [79]. As mentioned before, B1 cells express self-reactive BCRs but respond poorly to BCR cross-linking to prevent against self-activation that is suggested to be mediated through CD5 [67, 75, 79, 80]. B1 cells also express restricted BCRs with a predominance of VH12 promoting B1 phenotype [81] and are known to produce antibody quickly in response to infection, primarily IgM, independent of T-cell help (similar to U-CLL) [82]. Additionally, B1 cells are divided into B1a (CD5+) and B1b (CD5-) subsets where B1a cells are the primary source of natural IgM production and B1b cells respond to antigens in mice [63, 79, 83]. It has been suggested that B1a cells serve as the normal counterpart for CLL cells [61]. Elegant studies by Rajewsky and colleagues, where conditional ablation of BCR signaling was combined with conditional activation of candidate downstream signaling pathways of the same cell in vivo , led to the novel revelation that mature B-cell subsets may differ in their dependence on specific signaling pathways. Specifically genetic ablation of canonical NF-κB signaling in mature B cells in mice severely impaired development of marginal zone B cells, but had only mild effects on follicular B cells [84]. Thus, Nf-κB is a critical transcription factor for both B1 cells and CLL cells.
Adoptive transfer studies of young/early B1a populations into immunocompromised recipient mice led to the development of CLL-like disease [85]. CLL development in this study was independent of oncogene expression but a follow-up study was able to confirm that early B1a cells expressing the oncogene, T-cell leukemia 1 (Tcl1), also led to the development of CLL in recipient mice [86]. These authors did note that not all B1a cells result in CLL development, but were restricted to specific BCRs that were later identified to promote CLL growth [85, 86]. Additional studies favoring the B1a population serving as CLL normal counterpart provide evidence that both B1a cells and CLL cells secrete significant amounts of the cytokine Interleukin-10 that works to suppress the immune response [80].
Controversy regarding the normal counterpart of CLL has been focused on the inability to identify a human B1 population that is similar to mouse B1 cells [87]. Recently, reverse engineering has allowed researchers to identify a human B1 cell population within the umbilical cord blood and adult peripheral blood [88]. Rothstein et al. summarized evidence showing that mouse and human B1 cells share similar phenotypes and also express autoreactive antibodies that protect against infections. Seifert and colleagues compared normal CD5+ B cells from healthy human donors with both populations of CLL cells, M-CLL and U-CLL, and confirmed that CD5+ B cells are the normal B-cell subset that are most similar to CLL [62]. Additionally, a recent review also summarizes evidence that identifies B1 cells as the origin of CLL [89].
Interestingly, a case report involving a 65y male with stage IV CLL identified a “Side Population” of CLL cells identified through flow cytometry that were proposed to be precursors to leukemic development [90]. Ablation of these cells through vaccination after CD40L stimulation diminished the bulk of the disease 12 months after treatment. The “side population” of cells were CD5 and CD19 positive and thought to be similar to the cancer stem cell population characterized in other types of tumor models [91]. True identification of this “side population” would be of great benefit to determine if the likely B1 cells give rise to the malignant counterpart.
Intriguingly, one study investigated if CLL cells could be generated from self-renewing adults HSCs [92]. HSCs from CLL patients developed monoclonal or oligoclonal B cells that frequently expressed CD5. According to the Bloodspot database in Figs. 1 and 2, HSCs, B cells and CLL cells express high levels of Par-4 and it would be of interest to further investigate the role of Par-4 in B-cell development.
Because of these studies, McKenna and colleagues compared Par-4 expression in CLL cells to normal B1 and B2 subsets. Utilizing the Eμ-Tcl1 mouse which is considered to be the most representative model of human CLL [93,94,95,96,97], they measured Par-4 protein and mRNA levels in mouse CLL cells compared to wildtype (WT) mouse B cells. WT B1a cells expressed more Par-4 compared to the other B-cell populations in WT mice but only ~33% of the levels observed in CLL. Par-4 mRNA expression was also elevated in CLL cells compared to B-cell subsets, mirroring the levels of Par-4 protein expression. We further analyzed the B-cell subsets in the Eμ-Tcl1 mouse to confirm that Par-4 expression was not dependent on the overexpression of the Tcl1 oncogene. We isolated different B cell subsets from 2mo old Eμ-Tcl1 mice that had no detectable levels of CLL in the peripheral blood and measured Par-4 levels compared to WT B cell subsets (Suppl. Figure 1C in ref. 61). B1a Eμ-Tcl1 cells expressed higher Par-4 protein levels compared to B1b and conventional B2 Eμ-Tcl1 cells . This finding was similar to what was observed in WT B cell subsets which continues to suggest B1a cells exhibit characteristics similar to CLL cells. Importantly, elevated levels of Par-4 were also detected in human CLL samples compared to normal B cells which are consistent with studies presented by Boehrer and colleagues in that most CLL samples have detectable Par-4 protein levels [7].
Furthermore, CLL is known to be more common in elderly with an average age of CLL patients being 71. Interestingly, we found that B cells from aged mice express more Par-4 than those from young mice (unpublished). This was unique to B cells since there was no such age-related increase in other tissues such as liver and heart but also consistent with other reports that B cells express more Par-4 than other cell types (Fig. 1).
4.2 BCR-Mediated Par-4 Regulation in CLL
High expression levels of Par-4 in CLL led to the investigation of its regulation . In spite of original observation about the increase in Par-4 upon ionomycin treatment, there are very few studies that examine signaling pathways that induce Par-4 expression. Since CLL cells have been shown to have elevated tonic BCR signaling [98, 99], McKenna and colleagues tested the hypothesis that Par-4 expression may be regulated by BCR signaling. The BCR pathway is required for the survival of both normal and malignant B cells despite their oncogenic activation, making it a therapeutic target in B-cell malignancies [51, 98, 100]. Kinase inhibitors targeting Src family kinases (SFK) [101], Syk [102], BTK [103], and PI3K [104] have all been proven effective in the treatment of CLL as each inhibit the required downstream survival signals. Anti-CD20 monoclonal antibodies such as rituximab have also been proven efficient with combination of other chemotherapies [105]. We therefore utilized FDA-approved therapies to target BCR signaling and examined their effects on Par-4 expression. Treatment with dasatinib (SFK inhibitor), fostamatinib (SYK inhibitor), and ibrutinib (BTK inhibitor) all led to a decrease in Eμ-Tcl1 CLL cell survival accompanied by a reduction in Par-4 expression [61]. Par-4 mRNA levels decreased after SFK and BTK inhibition, suggesting regulation at the transcript level. Additionally, Par-4 protein downregulation was replicated in primary human CLL samples after treatment with dasatinib and fostamatinib indicating that this is not unique to mouse CLL cells. shRNA knockdown of Lyn, the most prevalent SFK in B cells, also led to a decrease in Par-4 expression. The most compelling evidence that Par-4 is regulated by BCR signaling is by targeting Igα or CD79a which confirmed that Par-4 is downstream of BCR activation and regulated through this signaling pathway. Additional studies investigating the levels of Par-4 after ERK inhibition in CLL cells showed that Par-4 is further downstream of the BCR signaling cascade. These results provide evidence that a well-defined survival signaling pathway is regulating the expression of Par-4 specifically in B cells since downregulation of Par-4 was not observed after ERK inhibition in PC-3 cells as shown by McKenna and colleagues [61].
4.3 Role of Par-4 in the Regulation of CLL Growth Kinetics
The aberrant expression of Par-4 in CLL and regulation through BCR signaling leads to the question of the true role of Par-4 in CLL. Par-4 knockdown studies in two CLL cell lines (Mec-1 and OSUCLL) resulted in a reduced growth rate in vitro and in xenograft in vivo studies [61]. Par-4 knockdown in these cells lead to increased Akt phosphorylation and reduced Bcl2 levels concordant with previous literature [7, 106] and to promote prosurvival signaling and anti-apoptotic pathways. Studies investigating the reduced growth rate in Par-4 knockdown cells revealed fewer cells entering S phase but more cells in G1 phase suggesting a halt in the G1 to S transition and a unique increase in p21 expression. p21 is involved in different phases of the cell cycle, but primarily works to control the transition from G1 to S [107].
The reduced CLL growth with the loss of Par-4 was also confirmed by crossing the Eμ-Tcl1 mouse with a Par-4−/− mouse. CLL development was significantly delayed in Par-4−/−EμTcl1 mice compared to Par-4+/+EμTcl1 mice leading to an overall improved survival [61]. Indeed, Par-4−/−EμTcl1 spleen cells expressed higher p21 protein levels compared to Par-4+/+EμTcl1 spleen cells providing in vivo confirmation of p21 upregulation observed in vitro using Par-4 knockdown cell lines. In order for p21 to execute its function to block the cell cycle from G1 to S phase, p21 must be found in the nucleus of the cell [108]. Nuclear and cytoplasmic fractions of Par-4+/+EμTcl1 and Par-4−/−EμTcl1 spleen cells were examined and was found that Par-4−/−EμTcl1 cells had greater nuclear p21 levels compared to Par-4+/+EμTcl1 cells, further confirming that Par-4 knockout led to increased levels of functional p21 [61]. This novel finding in CLL is clinically relevant as a study investigating the expression of p21 in CLL cases and patients with Richter’s syndrome found that 80% of CLL cases did not express detectable levels of p21 [109]. Forty-three percent of patients with Richter’s syndrome did express detectable levels of p21. Cobo et al. analyzed the sequence of p21 in three CLL patients and 6 Richter’s syndrome patients to find a germline configuration in all of them indicating that it was not mutated. Sequencing of the p21 gene in the Par-4 knockdown and knockout cells to confirm the mutation was not done, but an increase in nuclear p21 levels in Par4−/−Eμ-Tcl1 CLL cells was observed suggesting that p21 is still able to translocate to the nucleus and function in the regulation of the cell cycle that occurs in the nucleus [61].
Greene and colleagues investigated the effect of overexpression of Par-4 in CLL leukemogenesis in the Eμ-Tcl1 mouse [110]. They generated a B cell-specific human Par-4 overexpressing mouse and crossed it to the Eμ-Tcl1 mouse resulting in reduced accumulation of CD5 + CD19+ CLL cells. They went on to determine that Par-4 overexpression impedes Tcl1-driven NF-κB signaling with reduced nuclear translocation of p65. This finding aligns well with the role of Par-4 and its known interactions with NF-κB [111] and emphasizes the distinct roles of physiological versus increased intracellular levels of Par-4. It also provides further evidence of the pleiotropic roles that Par-4 may play in the development of B cell-specific CLL as well as in its surrounding microenvironment.
5 Par-4 in the Tumor Microenvironment
The original hallmarks of cancer proposed by Hannahan and Weinberg have been expanded to include the tumor microenvironment that promotes growth of cancer cells by avoiding apoptosis and evading immune suppression [112]. However, the dependence of cancer cells on a protective niche is a very old concept dating as far back as 1889 with Stephen Paget’s “seed and soil hypothesis” [113]. Both solid and hematologic tumors are very heterogeneous and comprise of multiple different cell types such as stromal cells, endothelial cells, tumor infiltrating macrophages, and lymphocytes accounting for more than half of the total tumor cell mass. These accessory cells produce vascular growth factors and various cytokines and chemokines that support cancer cell growth [114]. Compelling evidence exists that recognizes the importance of the BCR signaling pathway, Chemokine (C–X–C motif) Receptor 4 (CXCR4) and Chemokine (C–X–C motif) Ligand 12 (CXCL12) axis, which are key pathways of CLL microenvironment cross talk [115]. The role of tumor microenvironment in the form of bone marrow or secondary lymphoid organs that can provide a unique niche for CLL proliferation is based on the following:
-
(a)
Primary CLL cells do not proliferate or survive in long-term in vitro cultures, but undergo spontaneous apoptosis even when conditions that support the growth of other B-cell lines are provided [116].
-
(b)
CLL cells proliferate primarily in secondary lymphatic tissues, where they form characteristic “proliferation centers,” sometimes also referred to as “pseudofollicles” [117].
-
(c)
Deuterium (2H) labeling in patients with CLL demonstrated that lymph nodes are the principle site of proliferation compared to bone marrow or blood [118].
-
(d)
The unique gene expression profile along with Ki67 staining of CLL cells isolated from lymph nodes compared to blood and bone marrow-derived CLL cells [119].
-
(e)
BCR signaling targeted therapies as a drug class effect, induce “redistribution lymphocytosis” causing a rapid shrinkage of primarily lymph nodes with a transient increase in blood leukemic cell counts [120, 121].
The actual site of proliferation and the CLL microenvironment is still debated in the field. This could be because CLL cells are found within the peripheral blood, bone marrow, and other secondary lymphoid organs in which the malignant cell comes into contact with a variety of accessory cells depending on their location. Although studies in human samples find that the lymph node is the site of CLL proliferation; questions are still raised based on the dramatic splenomegaly observed in mouse models [118, 122]. Splenomegaly is indeed observed in human CLL patients at later stages of the disease but role of spleen as a secondary lymphoid organ during earlier stages of human CLL is not known, as it is not amenable to surgical interventions.
The CLL tumor microenvironment provides a physical location supporting the cross talk between malignant cells and accessory cells that inhibit apoptosis and also provide resistance to drug treatment [123]. CLL is a slow progressing disease and was originally thought to simply be an accumulation of cells with defective apoptosis, but recent studies using deuterium labeling have determined that CLL cells proliferate at a rate of 0.1–1% per day suggesting that CLL is a dynamic disease involving cell proliferation [124]. Pseudofollicular proliferation centers that are found throughout infiltrated tissues are the source of newly generated CLL cells [125]. Within this area, CLL cells depend on stimulation through a functioning BCR as discussed above. It is well appreciated that some CLL cells may be activated through antigen-dependent manner and the microenvironment may be the source of antigen/stimulus [126]. Theses antigens are not specifically defined, but may include microbial antigens, natural antibodies, and autoantigens expressed by dying cells. As noted above cell autonomous signaling due to homotypic interactions of BCR may be involved in a subset of CLL patients.
The CLL microenvironment promotes cell-to-cell interactions with a variety of different cell types. Direct interaction between B-CLL cells and T cells via CD40 on B cells and CD40L on T cells provides a proliferative stimulus [127]. CD40 signaling in B cells induces expression of anti-apoptotic molecules and proliferative signaling through AKT, ERK, TRAF, and NF-κB. T cells also secrete cytokines such as IL-4, TNFα, and IL-2 that support CLL proliferation. Alternatively, the CLL microenvironment also supports immune evasion allowing CLL cells to dampen the immune function of cytotoxic T cells by secreting immunosuppressive cytokines like TGFβ and IL-10 [127, 128].
Stromal cells derived from bone marrow or other secondary lymphoid tissues support the survival and proliferation of CLL cells [129]. This interaction provides a bi-directional cross talk that promotes the growth of both CLL and stromal cells. In cell culture, CLL cells actually migrate beneath bone marrow mesenchymal cells, a process known as pseudoemperipolesis, suggesting that this interaction is dependent on cell contact in order for CLL cells to survive. Cells known as nurse-like cells (NLC) can be found in the peripheral blood of patients that are derived from monocytes and become adherent in culture systems [130]. These cells express stromal cell-derived factor-1 (SDF-1) that binds to CXCR4 on CLL cells to prevent spontaneous apoptosis and promotes resistance of CLL cells to chemotherapies. CXCL12 is also secreted by NLCs as well as mesenchymal-derived stromal cells that attract CLL cells via CXCR4 towards proliferation centers within the secondary lymphoid compartments [131]. The phenomenon of “redistribution lymphocytosis” with BCR signaling inhibitors in CLL where mobilized CLL cells, devoid of their nourishing microenvironment in lymph nodes, die gradually has led researchers to propose a novel mechanism of action called “death by neglect” [121]. Similar to CLL cells dying in vitro, CLL cells detached from their supportive tissue microenvironment leads to anoikis, a form of programmed cell death [132]. It is interesting to note that BCR signaling inhibitor, specifically the Bruton tyrosine kinase (Btk) inhibitor, ibrutinib inhibits not only the BCR signaling in the CLL cells but also signaling of other cell surface receptors including chemokine receptors and adhesion molecules [133, 134]. This has been proposed to be the mechanism of redistribution lymphocytosis which is also seen in the other classes of BCR signaling inhibitors such as phosphoinositol 3 kinase (PI3K ) inhibitor, idelalisib, and inhibitors of SYK and PI3Kδ which are involved in signal transduction pathways of chemokine receptors and adhesion molecules [135, 136]. Redistribution lymphocytosis does not cause any adverse symptoms and resolves over time. Ibrutinib is also known to cause redistribution lymphocytosis in mantle cell lymphoma (MCL) [137], Waldenstrom macroglobulinemia (WM) [138] and DLBCL. However, activated B-cell-like (ABC) subtype of DLBCL cells are exquisitely sensitive to ibrutinib but not GCB-DLBCL. This is interesting because ABC-DLBCL are known to have chronic active BCR signaling [139] and use an amplified prosurvival NF-κB signaling [140].
There are very few studies looking at the role of Par-4 in tumor microenvironment of hematological malignancies. In solid tumors, the role of secreted Par-4 is gaining increased attention since it was discovered to be secreted extracellularly and to act exclusively on cancer cells in a paracrine manner and preventing metastasis [141,142,143,144]. Cancer-associated fibroblasts (CAFs) in contrast to normal fibroblasts were modified through an miRNA-dependent (mir-7) pathway to dramatically reduce the secretion of Par-4. Inhibition of mir-7 expression in CAFs induced them to convert back to normal fibroblasts [145].
Chronic active BCR signaling results in constitutive activation of NF-κB and PI3K pathways, both of which are regulated by Par-4 as discussed initially [61, 110]. Antigen-independent tonic BCR signaling supports survival of malignant B cells primarily through the PI3K–AKT–mTOR pathway which is also closely linked to Par-4. These pathways are again involved in regulating homing of malignant cells and retention of proliferating cells in a supportive niche as evidenced by novel BCR inhibitors causing “redistribution lymphocytosis.” Hence, it is not over-arching to hypothesize that aberrant levels of Par-4 in hematological malignancies like CLL alter the balance required to inhibit tumorogenic signals.
As summarized above, CLL cells overexpress Par-4 compared to its levels in normal B-cell subsets [61]. Additionally, CLL cells secrete Par-4 that can induce apoptosis of other cancer cell lines. This led us to question if Par-4 from CLL cells is able to manipulate the microenvironment’s ability to promote or delay CLL growth. Studies in our laboratory have confirmed that the spleen is the primary site of CLL tumor growth in the primary Eμ-Tcl1 mouse model of CLL as well as in adoptive transfer recipients as splenectomy dramatically delayed the development of CLL (manuscript in preparation). We have previously described the difference in CLL development between the Par-4−/−EμTcl1 and Par-4+/+EμTcl1 mice [61]. Par-4−/−EμTcl1 mice exhibited an improved lifespan compared to Par-4+/+EμTcl1 suggesting that the lack of Par-4 intrinsically and/or extracellularly reduced the aggressiveness of the disease. And since elimination of the primary site of CLL growth with splenectomy results in delayed CLL development, we splenectomized Par-4−/− mice to see if the lack of spleen and Par-4 may contribute to changes in CLL growth. Interestingly, we find that absence of the spleen in the Par-4 null background allows for faster growth of CLL cells elsewhere (unpublished), suggesting that Par-4 plays a significant inhibitory role extracellularly in the tumor microenvironment.
6 Summary and Future Outlook
In this chapter, we summarized the expression pattern of Par-4 in normal and malignant immune cells. Unlike its well-established tumor suppressor role in solid cancers, the role of Par-4 in hematological malignancies is complex. In CLL, we have shown that constitutive BCR signaling leads to high levels of Par-4 and a cell intrinsic prosurvival role for Par-4 in CLL cells. This is consistent with a variety of BCR signaling inhibitors that have been shown to be effective in control of CLL disease in patients. Our studies showing an inverse relation between Par-4 and p21 expression suggested that drugs affecting cell cycle could affect CLL despite their low proliferation index. Indeed, Dinaciclib, a CDK inhibitor, has been found to have beneficiary effects in refractory and relapsed CLL patients [146]. This approach may be important for CLL patients with Chr17 deletion (del (17p)) who have a more aggressive form of CLL disease, with a poor prognosis. This deletion leads to the absence of p53, which is known to upregulate p21. Currently, there are no therapies that specifically target this pathway [147]. We have highlighted here the absence of the inverse relation between Par-4 and Bcl2 as well as differential regulation of NF-κB in leukemias, which is in contrast to that seen in non-hematological malignancies (Fig. 5).
Like most cell types, CLL cells secrete Par-4 but are resistant to cytotoxic effects of secreted Par-4. On the other hand, we have discovered that Par-4 has a profound effect on CLL microenvironment. Our studies have shown a unique role for splenic microenvironment for CLL growth. Absence of Par-4 in the microenvironment of splenectomized mice enables better CLL growth. Presently, it is known that chemokines like CxCL12 play a critical role in the interaction between CLL cells and the microenvironment. However, effects of Par-4 expression on these critical chemokines required for CLL homing and survival in secondary lymphoid organs is at present unknown. Interestingly, Par-4 has been linked to the Wnt signaling pathway [148] in breast cancer cells and its overexpression led to downregulation of Frizzled, a Wnt ligand linked to cell proliferation. Expression of Wnt family members is elevated in CLL [149] and frizzled-6 has been shown to be required for CLL growth [150]. Future studies regarding Par-4-mediated gene expression in the CLL microenvironment may enable better Par-4-based treatment strategies for CLL.
References
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Cook J, Krishnan S, Ananth S, Sells SF, Shi Y, Walther MM et al (1999) Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene 18:1205–1208
Zapata-Benavides P, Mendez-Vazquez JL, Gonzalez-Rocha TR, Zamora-Avila DE, Franco-Molina MA, Garza-Garza R et al (2009) Expression of prostate apoptosis response (Par-4) is associated with progesterone receptor in breast cancer. Arch Med Res 40:595–599
Kogel D, Reimertz C, Mech P, Poppe M, Fruhwald MC, Engemann H et al (2001) Dlk/ZIP kinase-induced apoptosis in human medulloblastoma cells: requirement of the mitochondrial apoptosis pathway. Br J Cancer 85:1801–1808
Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I et al (2007) Inactivation of the candidate tumor suppressor par-4 in endometrial cancer. Cancer Res 67:1927–1934
Pruitt K, Ulku AS, Frantz K, Rojas RJ, Muniz-Medina VM, Rangnekar VM et al (2005) Ras-mediated loss of the pro-apoptotic response protein Par-4 is mediated by DNA hypermethylation through Raf-independent and Raf-dependent signaling cascades in epithelial cells. J Biol Chem 280:23363–23370
Boehrer S, Chow KU, Puccetti E, Ruthardt M, Godzisard S, Krapohl A et al (2001) Deregulated expression of prostate apoptosis response gene-4 in less differentiated lymphocytes and inverse expressional patterns of par-4 and bcl-2 in acute lymphocytic leukemia. Hematol J 2:103–107
Sells SF, Wood DP Jr, Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA et al (1994) Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ 5:457–466
Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C et al (1996) A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms' tumor suppressor WT1. Mol Cell Biol 16:6945–6956
Diaz-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L et al (1996) The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell 86:777–786
Boehrer S, Chow KU, Beske F, Kukoc-Zivojnov N, Puccetti E, Ruthardt M et al (2002) In lymphatic cells par-4 sensitizes to apoptosis by down-regulating bcl-2 and promoting disruption of mitochondrial membrane potential and caspase activation. Cancer Res 62:1768–1775
Johnstone RW, Tommerup N, Hansen C, Vissing H, Shi Y (1998) Mapping of the human PAWR (par-4) gene to chromosome 12q21. Genomics 53:241–243
Hebbar N, Wang C, Rangnekar VM (2012) Mechanisms of apoptosis by the tumor suppressor Par-4. J Cell Physiol 227:3715–3721
El-Guendy N, Rangnekar VM (2003) Apoptosis by Par-4 in cancer and neurodegenerative diseases. Exp Cell Res 283:51–66
Boghaert ER, Sells SF, Walid AJ, Malone P, Williams NM, Weinstein MH et al (1997) Immunohistochemical analysis of the proapoptotic protein Par-4 in normal rat tissues. Cell Growth Differ 8:881–890
El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM (2003) Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Mol Cell Biol 23:5516–5525
Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM (2005) Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Mol Cell Biol 25:1146–1161
Garcia-Cao I, Duran A, Collado M, Carrascosa MJ, Martin-Caballero J, Flores JM et al (2005) Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep 6:577–583
Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y et al (2005) Binding and phosphorylation of par-4 by akt is essential for cancer cell survival. Mol Cell 20:33–44
Diaz-Meco MT, Lallena MJ, Monjas A, Frutos S, Moscat J (1999) Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J Biol Chem 274:19606–19612
Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM (2009) The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 138:377–388
Lee AS (2007) GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res 67:3496–3499
Lee AS (2009) The Par-4-GRP78 TRAIL, more twists and turns. Cancer Biol Ther 8:2103–2105
Hebbar N, Burikhanov R, Shukla N, Qiu S, Zhao Y, Elenitoba-Johnson KSJ et al (2017) A naturally generated decoy of the prostate apoptosis response-4 protein overcomes therapy resistance in tumors. Cancer Res 77(15):4039–4050
Boehrer S, Kukoc-Zivojnov N, Nowak D, Bergmann M, Baum C, Puccetti E et al (2004) Upon drug-induced apoptosis expression of prostate-apoptosis-response-gene-4 promotes cleavage of caspase-8, bid and mitochondrial release of cytochrome c. Hematology 9:425–431
Bergmann M, Kukoc-Zivojnov N, Chow KU, Trepohl B, Hoelzer D, Weidmann E et al (2004) Prostate apoptosis response gene-4 sensitizes neoplastic lymphocytes to CD95-induced apoptosis. Ann Hematol 83:646–653
Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sonderby CK et al (2016) BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res 44:D917–D924
Moscat J, Diaz-Meco MT, Wooten MW (2009) Of the atypical PKCs, Par-4 and p62: recent understandings of the biology and pathology of a PB1-dominated complex. Cell Death Differ 16:1426–1437
Bojarska-Junak A, Sieklucka M, Hus I, Wasik-Szczepanek E, Kusz ML, Surdacka A et al (2011) Assessment of the pathway of apoptosis involving PAR-4, DAXX and ZIPK proteins in CLL patients and its relationship with the principal prognostic factors. Folia Histochem Cytobiol 49:98–103
Chow KU, Nowak D, Hofmann W, Schneider B, Hofmann WK (2005) Imatinib induces apoptosis in CLL lymphocytes with high expression of Par-4. Leukemia 19:1103–1105. author reply 1105-1106; discussion 1106-1107
Lu C, Li JY, Ge Z, Zhang L, Zhou GP (2013) Par-4/THAP1 complex and Notch3 competitively regulated pre-mRNA splicing of CCAR1 and affected inversely the survival of T-cell acute lymphoblastic leukemia cells. Oncogene 32:5602–5613
Kawai T, Akira S, Reed JC (2003) ZIP kinase triggers apoptosis from nuclear PML oncogenic domains. Mol Cell Biol 23:6174–6186
Roussigne M, Cayrol C, Clouaire T, Amalric F, Girard JP (2003) THAP1 is a nuclear proapoptotic factor that links prostate-apoptosis-response-4 (Par-4) to PML nuclear bodies. Oncogene 22:2432–2442
Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T et al (1999) PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234
Lindsay CR, Morozov VM, Ishov AM (2008) PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front Biosci 13:7132–7142
Matt S, Hofmann TG (2018) Crosstalk between p53 modifiers at PML bodies. Mol Cell Oncol 5:e1074335
Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de The H (2014) Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med 20:167–174
Shin Y, Brangwynne CP (2017) Liquid phase condensation in cell physiology and disease. Science 357(6357):eaaf4382
Burikhanov R, Shrestha-Bhattarai T, Hebbar N, Qiu S, Zhao Y, Zambetti GP et al (2014) Paracrine apoptotic effect of p53 mediated by tumor suppressor Par-4. Cell Rep 6:271–277
Libich DS, Schwalbe M, Kate S, Venugopal H, Claridge JK, Edwards PJ et al (2009) Intrinsic disorder and coiled-coil formation in prostate apoptosis response factor 4. FEBS J 276:3710–3728
Tiruttani Subhramanyam UK, Kubicek J, Eidhoff UB, Labahn J (2014) Cloning, expression, purification, crystallization and preliminary crystallographic analysis of the C-terminal domain of Par-4 (PAWR). Acta Crystallogr F Struct Biol Commun 70:1224–1227
Clark AM, Ponniah K, Warden MS, Raitt EM, Yawn AC, Pascal SM (2018) pH-Induced folding of the caspase-cleaved Par-4 tumor suppressor: evidence of structure outside of the coiled coil domain. Biomol Ther 8(4):162
Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A et al (2014) Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 158:593–606
Tan SH, Leong WZ, Ngoc PCT, Tan TK, Bertulfo FC, Lim MC et al (2019) The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 134:239–251
Zhang L, Xu HG, Lu C (2014) A novel long non-coding RNA T-ALL-R-LncR1 knockdown and Par-4 cooperate to induce cellular apoptosis in T-cell acute lymphoblastic leukemia cells. Leuk Lymphoma 55:1373–1382
Lafuente MJ, Martin P, Garcia-Cao I, Diaz-Meco MT, Serrano M, Moscat J (2003) Regulation of mature T lymphocyte proliferation and differentiation by Par-4. EMBO J 22:4689–4698
Han JY, Lim YJ, Choi JA, Lee JH, Jo SH, Oh SM et al (2016) The role of prostate apoptosis response-4 (Par-4) in Mycobacterium tuberculosis infected macrophages. Sci Rep 6:32079
Howlader NA, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (2016). SEER Cancer statistics review, 1975–2014. In: Institute, NC (ed). Bethesda, MD
PDQ Adult Treatment Editorial Board (2002) Chronic lymphocytic leukemia treatment (PDQ(R)): Health professional version. PDQ cancer information summaries. National Cancer Institute, Bethesda, MD
Chiorazzi N, Rai KR, Ferrarini M (2005) Chronic lymphocytic leukemia. N Engl J Med 352:804–815
Gururajan M, Jennings CD, Bondada S (2006) Cutting edge: constitutive B cell receptor signaling is critical for basal growth of B lymphoma. J Immunol 176:5715–5719
Burger JA, Chiorazzi N (2013) B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol 34:592–601
Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F (2009) The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood 114:3367–3375
Burger JA, Gribben JG (2014) The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: insight into disease biology and new targeted therapies. Semin Cancer Biol 24:71–81
Boehrer S, Chow KU, Ruthardt M, Hoelzer D, Mitrou PS, Weidmann E (2002) Expression and function of prostate-apoptosis-response-gene-4 in lymphatic cells. Leuk Lymphoma 43:1737–1741
Zhu H, Almasan A (2017) Development of venetoclax for therapy of lymphoid malignancies. Drug Des Devel Ther 11:685–694
Korsmeyer SJ (1992) Bcl-2: an antidote to programmed cell death. Cancer Surv 15:105–118
Chow KU, Nowak D, Boehrer S, Ruthardt M, Knau A, Hoelzer D et al (2003) Synergistic effects of chemotherapeutic drugs in lymphoma cells are associated with down-regulation of inhibitor of apoptosis proteins (IAPs), prostate-apoptosis-response-gene 4 (Par-4), death-associated protein (Daxx) and with enforced caspase activation. Biochem Pharmacol 66:711–724
Qiu G, Ahmed M, Sells SF, Mohiuddin M, Weinstein MH, Rangnekar VM (1999) Mutually exclusive expression patterns of Bcl-2 and Par-4 in human prostate tumors consistent with down-regulation of Bcl-2 by Par-4. Oncogene 18:623–631
Hulkkonen J, Vilpo L, Hurme M, Vilpo J (2002) Surface antigen expression in chronic lymphocytic leukemia: clustering analysis, interrelationships and effects of chromosomal abnormalities. Leukemia 16:178–185
McKenna MK, Noothi SK, Alhakeem SS, Oben KZ, Greene JT, Mani R et al (2018) Novel role of prostate apoptosis response-4 tumor suppressor in B-cell chronic lymphocytic leukemia. Blood 131:2943–2954
Seifert M, Sellmann L, Bloehdorn J, Wein F, Stilgenbauer S, Durig J et al (2012) Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med 209:2183–2198
Haas KM, Poe JC, Steeber DA, Tedder TF (2005) B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 23:7–18
Wang K, Wei G, Liu D (2012) CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol 1:36
Azzam HS, Grinberg A, Lui K, Shen H, Shores EW, Love PE (1998) CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J Exp Med 188:2301–2311
Werner-Favre C, Vischer TL, Wohlwend D, Zubler RH (1989) Cell surface antigen CD5 is a marker for activated human B cells. Eur J Immunol 19:1209–1213
Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S (1996) CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science 274:1906–1909
Damle RN, Ghiotto F, Valetto A, Albesiano E, Fais F, Yan XJ et al (2002) B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood 99:4087–4093
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK (1999) Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94:1848–1854
Gaidano G, Foa R, Dalla-Favera R (2012) Molecular pathogenesis of chronic lymphocytic leukemia. J Clin Invest 122:3432–3438
Carsetti R, Rosado MM, Wardmann H (2004) Peripheral development of B cells in mouse and man. Immunol Rev 197:179–191
Rossi D, Gaidano G (2010) Biological and clinical significance of stereotyped B-cell receptors in chronic lymphocytic leukemia. Haematologica 95:1992–1995
Slupsky JR (2014) Does B cell receptor signaling in chronic lymphocytic leukaemia cells differ from that in other B cell types? Scientifica (Cairo) 2014:208928
Agathangelidis A, Vardi A, Baliakas P, Stamatopoulos K (2014) Stereotyped B-cell receptors in chronic lymphocytic leukemia. Leuk Lymphoma 55:2252–2261
Baliakas P, Hadzidimitriou A, Sutton LA, Minga E, Agathangelidis A, Nichelatti M et al (2014) Clinical effect of stereotyped B-cell receptor immunoglobulins in chronic lymphocytic leukaemia: a retrospective multicentre study. Lancet Haematol 1:e74–e84
Chu CC, Catera R, Hatzi K, Yan XJ, Zhang L, Wang XB et al (2008) Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood 112:5122–5129
Maity PC, Bilal M, Koning MT, Young M, van Bergen CAM, Renna V et al (2020) IGLV3-21*01 is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc Natl Acad Sci U S A 117:4320–4327
Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M et al (2012) Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 489:309–312
Sindhava VJ, Bondada S (2012) Multiple regulatory mechanisms control B-1 B cell activation. Front Immunol 3:372
Alhakeem SS, Sindhava VJ, McKenna MK, Gachuki BW, Byrd JC, Muthusamy N et al (2015) Role of B cell receptor signaling in IL-10 production by normal and malignant B-1 cells. Ann N Y Acad Sci 1362:239–249
Lam KP, Rajewsky K (1999) B cell antigen receptor specificity and surface density together determine B-1 versus B-2 cell development. J Exp Med 190:471–477
Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J (2000) B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med 192:271–280
Alugupalli KR, Gerstein RM (2005) Divide and conquer: division of labor by B-1 B cells. Immunity 23:1–2
Derudder E, Herzog S, Labi V, Yasuda T, Kochert K, Janz M et al (2016) Canonical NF-kappaB signaling is uniquely required for the long-term persistence of functional mature B cells. Proc Natl Acad Sci U S A 113:5065–5070
Hayakawa K, Formica AM, Colombo MJ, Ichikawa D, Shinton SA, Brill-Dashoff J et al (2015) B cells generated by B-1 development can progress to chronic lymphocytic leukemia. Ann N Y Acad Sci 1362:250–255
Hayakawa K, Formica AM, Brill-Dashoff J, Shinton SA, Ichikawa D, Zhou Y et al (2016) Early generated B1 B cells with restricted BCRs become chronic lymphocytic leukemia with continued c-Myc and low Bmf expression. J Exp Med 213:3007–3024
Chiorazzi N, Ferrarini M (2011) Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood 117:1781–1791
Rothstein TL, Griffin DO, Holodick NE, Quach TD, Kaku H (2013) Human B-1 cells take the stage. Ann N Y Acad Sci 1285:97–114
Kikushige Y (2020) Pathophysiology of chronic lymphocytic leukemia and human B1 cell development. Int J Hematol 111:634–641
Foster AE, Okur FV, Biagi E, Lu A, Dotti G, Yvon E et al (2009) Selective depletion of a minor subpopulation of B-chronic lymphocytic leukemia cells is followed by a delayed but progressive loss of bulk tumor cells and disease regression. Mol Cancer 8:106
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414:105–111
Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G et al (2011) Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 20:246–259
Pekarsky Y, Zanesi N, Aqeilan RI, Croce CM (2007) Animal models for chronic lymphocytic leukemia. J Cell Biochem 100:1109–1118
Pekarsky Y, Drusco A, Kumchala P, Croce CM, Zanesi N (2015) The long journey of TCL1 transgenic mice: lessons learned in the last 15 years. Gene Expr 16:129–135
Hamblin TJ (2010) The TCL1 mouse as a model for chronic lymphocytic leukemia. Leuk Res 34:135–136
Simonetti G, Bertilaccio MT, Ghia P, Klein U (2014) Mouse models in the study of chronic lymphocytic leukemia pathogenesis and therapy. Blood 124:1010–1019
Bresin A, D'Abundo L, Narducci MG, Fiorenza MT, Croce CM, Negrini M et al (2016) TCL1 transgenic mouse model as a tool for the study of therapeutic targets and microenvironment in human B-cell chronic lymphocytic leukemia. Cell Death Dis 7:e2071
Davids MS, Brown JR (2012) Targeting the B cell receptor pathway in chronic lymphocytic leukemia. Leuk Lymphoma 53:2362–2370
Yan XJ, Albesiano E, Zanesi N, Yancopoulos S, Sawyer A, Romano E et al (2006) B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 103:11713–11718
Bojarczuk K, Bobrowicz M, Dwojak M, Miazek N, Zapala P, Bunes A et al (2015) B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis 55:255–265
Song Z, Lu P, Furman RR, Leonard JP, Martin P, Tyrell L et al (2010) Activities of SYK and PLCgamma2 predict apoptotic response of CLL cells to SRC tyrosine kinase inhibitor dasatinib. Clin Cancer Res 16:587–599
Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T et al (2010) Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood 115:4497–4506
Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA et al (2014) Bruton's tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 123:1207–1213
Jamroziak K, Pula B, Walewski J (2017) Current treatment of chronic lymphocytic leukemia. Curr Treat Opt Oncol 18:5
Vitale C, Burger JA (2016) Chronic lymphocytic leukemia therapy: new targeted therapies on the way. Expert Opin Pharmacother 17:1077–1089
Joshi J, Fernandez-Marcos PJ, Galvez A, Amanchy R, Linares JF, Duran A et al (2008) Par-4 inhibits Akt and suppresses Ras-induced lung tumorigenesis. EMBO J 27:2181–2193
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82:675–684
Cmielova J, Rezacova M (2011) p21Cip1/Waf1 protein and its function based on a subcellular localization [corrected]. J Cell Biochem 112:3502–3506
Cobo F, Martinez A, Pinyol M, Hernandez L, Gomez M, Bea S et al (2002) Multiple cell cycle regulator alterations in Richter's transformation of chronic lymphocytic leukemia. Leukemia 16:1028–1034
Greene JT, Mani R, Ramaswamy R, Frissora F, Yano M, Zapolnik K et al (2019) Par-4 overexpression impedes leukemogenesis in the Emicro-TCL1 leukemia model through downregulation of NF-kappaB signaling. Blood Adv 3:1255–1266
Chendil D, Das A, Dey S, Mohiuddin M, Ahmed MM (2002) Par-4, a pro-apoptotic gene, inhibits radiation-induced NF kappa B activity and Bcl-2 expression leading to induction of radiosensitivity in human prostate cancer cells PC-3. Cancer Biol Ther 1:152–160
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Langley RR, Fidler IJ (2011) The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer 128:2527–2535
Ansell SM, Vonderheide RH (2013) Cellular composition of the tumor microenvironment. Am Soc Clin Oncol Educ Book. https://doi.org/10.1200/EdBook_AM.2013.33.e91
ten Hacken E, Burger JA (2014) Microenvironment dependency in chronic lymphocytic leukemia: the basis for new targeted therapies. Pharmacol Ther 144:338–348
Collins RJ, Verschuer LA, Harmon BV, Prentice RL, Pope JH, Kerr JF (1989) Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br J Haematol 71:343–350
Stein H, Bonk A, Tolksdorf G, Lennert K, Rodt H, Gerdes J (1980) Immunohistologic analysis of the organization of normal lymphoid tissue and non-Hodgkin's lymphomas. J Histochem Cytochem 28:746–760
Herndon TM, Chen SS, Saba NS, Valdez J, Emson C, Gatmaitan M et al (2017) Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia 31:1340–1347
Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B et al (2011) The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 117:563–574
Herman SE, Niemann CU, Farooqui M, Jones J, Mustafa RZ, Lipsky A et al (2014) Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia 28:2188–2196
Burger JA, Montserrat E (2013) Coming full circle: 70 years of chronic lymphocytic leukemia cell redistribution, from glucocorticoids to inhibitors of B-cell receptor signaling. Blood 121:1501–1509
Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R et al (2002) Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A 99:6955–6960
Munk Pedersen I, Reed J (2004) Microenvironmental interactions and survival of CLL B-cells. Leuk Lymphoma 45:2365–2372
Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D et al (2005) In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest 115:755–764
Herreros B, Rodriguez-Pinilla SM, Pajares R, Martinez-Gonzalez MA, Ramos R, Munoz I et al (2010) Proliferation centers in chronic lymphocytic leukemia: the niche where NF-kappaB activation takes place. Leukemia 24:872–876
Caligaris-Cappio F, Bertilaccio MT, Scielzo C (2014) How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin Cancer Biol 24:43–48
Herishanu Y, Katz BZ, Lipsky A, Wiestner A (2013) Biology of chronic lymphocytic leukemia in different microenvironments: clinical and therapeutic implications. Hematol Oncol Clin North Am 27:173–206
Alhakeem SS, McKenna MK, Oben KZ, Noothi SK, Rivas JR, Hildebrandt GC et al (2018) Chronic lymphocytic leukemia-derived IL-10 suppresses antitumor immunity. J Immunol 200:4180–4189
Burger JA (2011) Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2011:96–103
Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell'Aquila M, Kipps TJ (2000) Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 96:2655–2663
Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG et al (2017) Chronic lymphocytic leukaemia. Nat Rev Dis Primers 3:17008
Frisch SM, Screaton RA (2001) Anoikis mechanisms. Curr Opin Cell Biol 13:555–562
Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG et al (2012) The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 119:1182–1189
de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ et al (2012) The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 119:2590–2594
Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S et al (2003) The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med 198:1539–1550
de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW et al (2007) Bruton's tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 26:93–104
Chang BY, Francesco M, De Rooij MF, Magadala P, Steggerda SM, Huang MM et al (2013) Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 122:2412–2424
de Rooij MF, Kuil A, Kraan W, Kersten MJ, Treon SP, Pals ST et al (2016) Ibrutinib and idelalisib target B cell receptor- but not CXCL12/CXCR4-controlled integrin-mediated adhesion in Waldenstrom macroglobulinemia. Haematologica 101:e111–e115
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB et al (2010) Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463:88–92
Yang Y, Shaffer AL 3rd, Emre NC, Ceribelli M, Zhang M, Wright G et al (2012) Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 21:723–737
Solvason N, Wu WW, Parry D, Mahony D, Lam EW, Glassford J et al (2000) Cyclin D2 is essential for BCR-mediated proliferation and CD5 B cell development. Int Immunol 12:631–638
Burikhanov R, Hebbar N, Noothi SK, Shukla N, Sledziona J, Araujo N et al (2017) Chloroquine-inducible Par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep 18:508–519
Wang P, Burikhanov R, Jayswal R, Weiss HL, Arnold SM, Villano JL et al (2018) Neoadjuvant administration of hydroxychloroquine in a phase 1 clinical trial induced plasma Par-4 levels and apoptosis in diverse tumors. Genes Cancer 9:190–197
Rasool RU, Nayak D, Chakraborty S, Katoch A, Faheem MM, Amin H et al (2016) A journey beyond apoptosis: new enigma of controlling metastasis by pro-apoptotic Par-4. Clin Exp Metastasis 33:757–764
Shen Z, Qin X, Yan M, Li R, Chen G, Zhang J et al (2017) Cancer-associated fibroblasts promote cancer cell growth through a miR-7-RASSF2-PAR-4 axis in the tumor microenvironment. Oncotarget 8:1290–1303
Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S et al (2015) Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 29:1524–1529
Aitken MJL, Lee HJ, Post SM (2019) Emerging treatment options for patients with p53-pathway-deficient CLL. Ther Adv Hematol 10:2040620719891356
de Bessa Garcia SA, Pavanelli AC, Cruz EMN, Nagai MA (2017) Prostate apoptosis response 4 (PAR4) expression modulates WNT signaling pathways in MCF7 breast cancer cells: a possible mechanism underlying PAR4-mediated docetaxel chemosensitivity. Int J Mol Med 39:809–818
Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM et al (2004) Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 101:3118–3123
Wu QL, Zierold C, Ranheim EA (2009) Dysregulation of Frizzled 6 is a critical component of B-cell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood 113:3031–3039
Acknowledgments
This work was supported by the National Institutes of Health, National Cancer Institute (CA 165469) (N.M., V.M.R., and S.B.). M.K.M. was supported by National Institutes of Health, National Cancer Institute T32 grant CA165990. We also acknowledge the support from the National Center for Advancing Translational Sciences (UL1TR001998 and UL1TR000117), the University of Kentucky Markey Cancer Center and Markey Cancer Center’s Flow Cytometry and Immune Monitoring Shared Resource Facility (P30CA177558).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Noothi, S.K. et al. (2021). Role of Par-4 in B-Cell Hematological Malignancies. In: Rangnekar, V.M. (eds) Tumor Suppressor Par-4. Springer, Cham. https://doi.org/10.1007/978-3-030-80558-6_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-80558-6_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-80557-9
Online ISBN: 978-3-030-80558-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)