
Abstract
The heterogeneous group of protracted diarrheas starting in the first months of life traditionally grouped under “intractable diarrhea of infancy” includes many different diseases. Some children are affected by diseases that impair the normal development of intestinal epithelium, causing a severe watery diarrhea that usually requires total parenteral nutrition. The first to be described was microvillus inclusion disease that usually starts in the first days of life with a secretory diarrhea that is worsened by feeding (early-onset microvillus inclusion disease). In a small percentage of cases, diarrhea starts later in life, between 1 and 3 months (late-onset microvillus atrophy). The early form is very severe, and intestinal transplantation should be strongly considered in these cases.
A second congenital epithelial disease is “tufting enteropathy” (intestinal epithelial dysplasia). Tuft enteropathy is associated with severe secretory diarrhea, which worsens with nutrition. That is why affected children have to be treated with total parenteral nutrition. Cases totally dependent on total parenteral nutrition are candidates for intestinal transplantation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Intractable diarrhea of infancy
- Congenital microvillus atrophy
- Intestinal epithelial dysplasia
- Congenital diarrhea
- Secretory diarrhea
- Congenital diseases of intestinal epithelium
Introduction
The Larger Group of “Intractable Diarrheas of Infancy”
Before focusing on microvillus inclusion disease and tufting enteropathy, we will briefly review similar cases in the literature. In 1968, Avery, Villavicencio, and Lilly were the first to describe severe chronic diarrhea in 20 infants and named it “infantile intractable diarrhea”; according to their description, this was prolonged and intractable despite extensive hospital therapy [1].
This syndrome was defined on the basis of some clinical characteristics, namely: (1) Diarrhea of more than 2 weeks duration; (2) Age, less than 3 months; (3) Three or more stool cultures negative for bacterial pathogens; (4) Necessity of intravenous rehydration; and (5) Prolonged and intractable diarrhea despite hospital therapy.
The death rate was very high: 9 out of the 20 babies (45%) in Avery et al.’s report had died, and at 70% it was even higher in Hyman et al [2].
Heterogeneity and lack of specificity are evident in Avery’s original report: different pathologies were grouped in it, some of which with a diagnosis which was well defined even at that time. Only autopsy data were available for the first cases, and only after the introduction of total parenteral nutrition at the beginning of the 1970s [3] was it possible to study the matter in greater depth, thanks to proximal small intestinal biopsy [4] and later on to the development of endoscopic techniques which were safe and adequate for the infant as well. It became consequently possible to discriminate different causes for the so-called intractable diarrhea of infancy [5], but its definition superimposes on the definition of “protracted diarrhea of infancy”: the latter lasts for a similar length of time but a failure to gain weight is enough to define the clinical picture [6].
In 1995 the Pediatric Gastroenterologists of the Federico II School of Medicine of Naples (Italy) observed that in most cases of severe and protracted diarrhea (SPD) an etiological diagnosis was possible and that consequently the term “intractable childhood diarrhea” was now frequently inappropriate. They proposed to limit it to the group that needed total parenteral feeding, defining the clinical picture as “severe diarrhea requiring parenteral nutrition” [7]. In view of the changes in the spectrum of known causes of SPD over the past few decades, the Italian Society of Pediatric Gastroenterology, Hepatology and Nutrition (SIGENP) proposed in 1999 [8] to include in this definition autoimmune enteropathy (severe or partial villus atrophy with crypt hyperplasia and presence of anti-enterocyte antibodies and/or associated autoimmune disorders), congenital microvillus atrophy, tufting enteropathy, epithelial dysplasia, and intestinal microvillus dystrophy (the latter later unified with microvillus inclusion disease).
However, the definition of “protracted diarrhea of infancy” has remained prevalent in clinical practice and in the literature, even compared to the broader definition of “pediatric intestinal failure” [9], an entity resulting from various causes including trichohepatoenteric syndrome, tufting enteropathy, microvillus inclusion disease, and autoimmune enteropathy [10].
Many cases of “protracted diarrhea of infancy” are diet-associated, as a consequence of cow’s milk or lactose intolerance or malnutrition. Malnutrition causes intestinal atrophy and consequently a malabsorption syndrome with diarrhea, apparently improving with fasting. These features have almost disappeared in developed countries.
The main causes of “intractable diarrhea of infancy,” including more severe and longer-lasting forms, can be summed up as follows (Table 1.1):
Autoimmune Enteropathy
The term “autoimmune enteropathy” (AIE) was introduced to describe persistent diarrhea associated with autoimmune diseases with the production of antibodies directed against epithelial cells of the small and large intestine.
This rare disorder (a recent review of the literature found a total of 98 reports published in the form of case reports and case series) [11] is frequently associated with primary immunodeficiencies and mostly occurs in young infants and children (6–18 months old). It is characterized by severe diarrhea and small intestinal mucosal atrophy resulting from immune-mediated injury. A retrospective study on clinical and histological findings from 40 AIE patients showed a prevalent celiac disease pattern (50%), mainly in patients with primary immunodeficiencies, followed by the mixed pattern (35%), chronic active duodenitis (10%), and GVHD-like pattern (5%) [12]. It remains a challenging diagnosis because of its clinical-pathological variability. This entity is dealt with in Chap. 2.
Small Intestinal Enteropathy of Unknown Origin
This entity could be a variation of autoimmune enteropathy, as the increase in inflammatory cells in the lamina propria shows. It appears in infants less than 12 months, with a lower death rate compared to those with autoimmune enteropathy, but it can be very severe. Infants can become TPN-dependent [5].
Intractable Ulcerating Enterocolitis of Infancy
A rare disease initially described in 1991 in five children presenting in the first year of life with intractable diarrhea, ulcerating stomatitis, and large ulcers with overhanging edges throughout the colon within the first year of life [13]. The affected infants can show a colitis whose severity may require a subtotal colectomy, even if the long-term prognosis is good. It has been suggested that affected children have a genetically determined primary immune dysregulation [14].
Congenital Enterocyte Heparan Sulfate Deficiency
Described in 1995 in three infants who within the first weeks of life presented with secretory diarrhea and massive enteric protein loss [15]. The small intestinal mucosa is normal on light microscopy, but histochemical exams show a complete absence of enterocyte heparan sulfate. The sulfated glycosaminoglycans of the basocellular membrane are mostly deficient, particularly heparan sulfate, while the distribution of vascular and lamina propria glycosaminoglycans is normal [15]. Diarrhea is so severe as to make total parenteral nutrition (TPN) necessary, together with repeated albumin infusions because of severe protein-losing enteropathy. Studies in men and mice show that heparan sulfate is essential in maintaining intestinal epithelial barrier function [16], and that the specific loss of heparan sulfate proteoglycans from the basolateral surface of intestinal epithelial cells is common to many forms of protein-losing enteropathy [17].
Congenital Intestinal Integrin Deficiency
In 1999, Lachaux et al. described a case of intractable diarrhea starting 9 days after birth, associated with pyloric atresia and total epithelial detachment of gastric and intestinal mucosa. Immunofluorescence analysis showed α6β4 integrin deficiency at the intestinal epithelium–lamina propria junction [18].
Mutations in α6 or β4 integrins cause junctional epidermolysis bullosa with pyloric atresia. In 2008, two Kuwaiti brothers with pyloric atresia were described, respectively affected by intractable diarrhea and episodes of protein-losing enteropathy, with a novel mutation in β4 integrin that induced a desquamative enteropathy in infancy without significant skin disease [19].
Congenital Secretory Diarrheas
Includes congenital chloridorrhea and congenital sodium diarrhea , dealt with in Chap. 36.
Diseases of the Intestinal Epithelium
Microvillus inclusion disease and tufting enteropathy are the best-known diseases of the intestinal epithelium causing intractable diarrhea of infancy.
In 1994, Girault et al. described eight infants with early-onset severe watery diarrhea associated to facial deformities and unusual tufts of woolly hair with trichorrhexis nodosa. Duodenal biopsies showed moderate to severe villus atrophy, with normal or hypoplastic crypts; colon biopsies were basically normal. As a consequence, severe malabsorption was present. All patients had no antibody response to immunization antigens; the immunological response to vaccinations was poor. Five children died despite TPN [20]. Two children from the series of Girault et al. had hepatic cirrhosis; six additional patients had signs and symptoms compatible with this new “syndromic diarrhea,” associated to hepatic involvement (Trichohepatoenteric syndrome, THES) characterized by fibrotic livers with marked hemosiderosis [21,22,23].
Nine different mutations in TTC37 gene (5q14.3–5q21.2) were found in 12 children from 11 families with classical features of THES. TTC37 codes for a protein that has been named “thespin” (THES ProteIN) [24].
Enlarged platelets with abnormal α-granule secretion can be observed in some patients. The estimated incidence of the syndrome is 1 in 400,000 to 1 in 500,000 live births.
A review of the literature conducted in May 2017 included 80 patients, 40 with mutations of TTC37 and 14 with mutations of SKIV2. This showed that parenteral nutrition was used in the management of 83% of the patients and that it was possible to wean 44% off parenteral nutrition. The mean duration was 14.97 months. Data on the efficacy of immunoglobulins were reported for only six patients, with a diminution of infection or reduced diarrhea. Antibiotics, steroids, and immunosuppressant drugs were used with little efficacy. Hematopoietic stem cell transplantation (HSCT) was performed in four patients, two of whom died [25].
Microvillus Inclusion Disease
In 1978, Davidson et al. described five infants presenting with intractable diarrhea of infancy characterized by secretive diarrhea and malabsorption, starting in the first hours after birth with hypoplastic villus atrophy in the small intestinal biopsy. Four of these infants had a deceased brother who had shown similar symptoms.
In one of these infants, electron microscopy identified the presence of a peculiar abnormality of the microvilli of the enterocytes [26] (Fig. 1.1).

Microvillus inclusion disease. PAS staining highlights abundant PAS-positive material (arrows) in the apical part of the enterocyte cytoplasm. PAS × 260 [20] (Reprinted from Springer and Virchows Archive: Official Journal of the European Society of Pathology, Morroni et al. [99], Fig. 1, with kind permission from Springer Science and Business Media)
Three new cases with the same clinical and histological characteristics as this infant were described in France in 1982, and the four of them were grouped into a new disease called congenital microvillus atrophy [27, 28]. Two new cases were described in Great Britain in 1985 [29], and one in Italy in 1986; a brother of the Italian child, who was born subsequently, was similarly affected [30]. A survey completed in 1987 among centers known for their involvement in pediatric gastroenterology identified more than 30 cases worldwide. Additional cases were later published.
In 1989, Cutz et al. proposed the use of the term “microvillus inclusion disease” to highlight the characteristic ultrastructural lesions of the disease [31].
Clinical Presentation
First child of parents with no blood relation, A.G. was born after 37 weeks of gestation, the pregnancy having been complicated by a risk of miscarriage in the fifth month. His weight at birth was 3500 grams.
The infant was hospitalized when he was 40 days old because of abundant diarrhea (15–20 evacuations a day of liquid stools), which started on the sixth day of life and was resistant to numerous dietary and pharmacological therapies.
On admission to hospital, the patient weighed 2800 grams, and was suffering from dystrophia and dehydration; total parenteral nutrition (TPN) was therefore immediately started. The acid-basic balance showed hyponatremic acidosis (pH 7,2; EB –8,3; Na 128 mEq/1). The secretive nature of diarrhea was confirmed by its entity (about 100 ml/kg/die) with a total absence of oral nutrition and with the persistence of TPN in progress.
Moreover, the typical absence of ionic gap in the stools was present: osmolality 226 mOsm/l, Na 86 mEq/1, K 23.5 mEq/1 (gap 7 mOsm/l).
Loperamide and chlorpromazine increased intestinal absorption but did not change the clinical picture.
Microbiological tests including electron microscopic analysis of the feces for the identification of viruses and the search for enterotoxigenic bacteria and parasites with specific methods were repeatedly negative.
The abdominal ultrasound showed adrenal hyperplasia associated with hyperaldosteronism (1160 ng/ml, v. n. <125 ng/ml).
Jejunal biopsy showed a picture of villus atrophy with no hyperplastic crypts and periodic acid-Schiff (PAS)-positive material stored in the apical cytoplasm of enterocytes. Electron microscopy was diagnostic for microvillus inclusion disease.
Microvillus Inclusion Disease: A Congenital Secretory Diarrhea Starting in Neonatal Age
In most cases, severe diarrhea appears in the first days of life, usually within the first 72 h, and is immediately life threatening. The stools are watery, and the stool output is 100–500 mL/kg/d when the infant is fed, a volume comparable to or higher than that observed in cholera. The diarrhea is of secretory type; therefore, it persists at a stable rate of 50–300 mL/kg/day despite fasting, and the electrolyte content of the stools is increased, without an osmotic gap. However, the mucosal atrophy causes additionally osmotic diarrhea in presence of luminal nutrients. For this reason, feeding increases the fecal output, and oral feeding in nutritionally significant amounts is impossible. Due to the high output, patients can lose up to 30% of their body weight within 24 h, resulting in profound metabolic acidosis and severe dehydration, unless vigorous intravenous rehydration is started.
Microvillus inclusion disease is the leading cause of secretory diarrhea in neonates with onset in most cases within the first few hours after birth [32]. However, in a small percentage of cases (currently considered around 5% of total) [33], diarrhea starts later in life: between 1 and 3 months, and more commonly at 6–8 weeks of age. This less severe form has been denominated late-onset microvillus inclusion disease, while the classical form beginning at neonatal age has been denominated early-onset microvillus inclusion disease [34].
A few cases have been termed atypical microvillus inclusion disease, in which the onset can be early or late, but the histological picture is different, particularly for the absence of detectable microvillus inclusions. The first case was a 5-month-old Navajo with profuse diarrhea beginning on the sixth day of life, who did not have microvillus inclusions in the duodenal tissue; a second biopsy confirmed the absence of classic microvillus inclusions despite the lack of surface microvilli and the presence of cytoplasmic vesicular bodies. However, a few microvilli associated with cytoplasmic inclusions were observed in a third biopsy [35]. In consideration of the typical clinical presentation, a diagnosis of microvillus inclusion disease was made, instead of intestinal microvillus dystrophy proposed for other cases with similar ultrastructural findings but slightly atypical clinical presentation [36]. Other similar cases were observed later.
Therefore, three variants of the disease have been identified: early-onset microvillus inclusion disease, late-onset microvillus inclusion disease, and atypical microvillus inclusion disease.
-
1.
Early-onset MVID presents a complete loss of intestinal proteins, both in the villi and in the crypts.
-
2.
In the late-onset MVID, there are normal microvilli at the base of the villi and in the crypts. The clinical picture is less severe and occurs later, usually from the second month of life.
-
3.
In the atypical MVID, the microvillus proteins are absent or defective only at the crypts, while in the villi there are normal microvilli at the villus surface.
However, because of the sparse distribution of microvillus inclusions, it is not certain that their absence could not be limited to the sample.
The disease is characterized by defective transport of plasma membrane proteins to the apical brush border, due to mutations of the MYO5B gene on chromosome 18q21 [37, 38], encoding myosin Vb motor protein and two small GTP binding proteins, Rab8 and Rab11 [39].
Several mechanisms responsible for the pathological picture of MVID have been suggested, and in particular the presence of defects in vesicle trafficking or delivery (Trafficking model ), in the recycling and delivery of apical recycling endosomes (Recycling model ) or in the colocalization of ezrin and ezrin kinase in apical recycling endosomes (AREs), while ezrin kinases are normally transported to the apical membrane where they activate ezrin (Local induction model ). It is possible that these mechanisms coexist, and so a hybrid model that combines all three models has been proposed [40].
The hallmark of the disease is the electron microscopic finding of disrupted enterocytic microvilli (i.e., digitations of the apical membrane of the intestinal epithelial cell protruding into the lumen) without inflammatory changes and the appearance of characteristic inclusion vacuoles, whose inner surfaces are lined by typical microvilli. Both lesions are seen only with the electronic microscopy.
The main histological features of the disease include diffuse villus atrophy without inflammatory changes and accumulation of periodic acid-Schiff (PAS)-positive material within the apical cytoplasm of enterocytes. The definitive diagnosis of MID rests on distinctive ultrastructural findings: microvillus inclusions (more frequently in villus enterocytes), increased electron-dense secretory granules (preferentially in crypt epithelial cells), and poorly developed brush border microvilli on the intestinal surface epithelium.
Microvillus inclusion disease is usually characterized by growth retardation and some developmental delay later in infancy. While no other specific findings can be detected, the disease can be associated with other abnormalities, indicated in Table 1.2.
Some cases of microvillus inclusion disease associated with other clinical pictures (for example, cardiac malformations, facial dysmorphia, transient neuronal dysplasia, aganglionic megacolon, Down syndrome, intrahepatic cholestasis, and hypochondroplasia) have been described. In a series of 24 patients with MVID followed up from birth to 23.5 years, liver disease was recorded in 22 patients, kidney disease in 9, and pulmonary disease in 2 cases [41].
An infant who had presented on the second day of life with the first symptoms of necrotizing enterocolitis was diagnosed with congenital microvillus atrophy at the age of 2.5 weeks. The clinical picture of necrotizing enterocolitis was repeated at the age of 4, 7, and 11 weeks of life and was treated with antibiotics on a monthly basis. The authors suggested that the picture of necrotizing enterocolitis was caused by damage to the barrier function of intestinal epithelial cells [42].
A series of eight children aged between 2 days and 14 months at onset, six of whom were homozygotes or compound heterozygotes for MYO5B mutations, were observed with minor microscopy histological abnormalities, sometimes focal or delayed but consistent with MVID. Malformations and severe mental retardation were observed in three cases, and hydrocephaly in one [43].
An infant with microvillus atrophy presented with liver dysfunction , hematuria, and Pneumocystis jiroveci pneumonia during the course of the disease; the child succumbed after massive pulmonary hemorrhage. The authors hypothesized that the coinfections could have been facilitated by an altered MYO5B function [44].
MVID-associated liver dysfunction similar to progressive familial and benign recurrent intrahepatic cholestasis has been described in other cases, to suggest that MVID is not exclusive to the intestine.
Histologic Findings
Findings from duodenal biopsy must not be considered diagnostic. Histologic results of duodenal biopsy samples can range from essentially normal to mildly abnormal, showing the following:
-
Thin mucosa caused by hypoplastic villus atrophy
-
Diffuse villus atrophy (loss of villus height)
-
Crypt hypoplasia
Periodic acid-Schiff (PAS) staining of the intestinal biopsy sample does not show the usual linear staining along the brush border but reveals PAS-positive material in the apical cytoplasm. The PAS staining material corresponds to the increased number of electron-dense secretory granules in the epithelium. The abnormal pattern of staining appears in the upper crypt region and continues over the villus [45] (Fig. 1.2).

Microvillus inclusion disease. Villus enterocytes: the boxed area shows microvilli on the lateral membrane. Inset: Enlargement of the boxed area. ×6200, inset ×22,500 [20] (Reprinted from Springer and Virchows Archive: Official Journal of the European Society of Pathology, Morroni et al. [99], Fig. 5, with kind permission from Springer Science and Business Media)
PAS accumulates in low crypts in atypical microvillus atrophy, in upper crypts in congenital microvillus atrophy, and in low villi in late-onset microvillus atrophy.
Similar results were obtained with anti-CD10 immunohistochemistry: in affected children the normal linear staining in surface enterocytes is absent, while prominent cytoplasmic reactivity is seen. CD10 is a neutral membrane-associated peptidase; thus, abnormal stain findings with PAS or anti-CD10 immunohistochemistry are expressions of the abnormalities in microvillar structure.
Rectal biopsy findings demonstrate microvillus involutions and an increased number of secretory granules. This test has been proposed as a relatively easy method for making an early diagnosis. Anti-CD10 immunohistochemistry can aid in the diagnosis, because abnormal cytoplasmic CD10 staining of absorptive colonocytes has been observed in microvillus inclusion disease [46], although while CD10 immunostaining identifies normal enteric mucosa with 100% specificity, negative staining does not definitively exclude small intestinal mucosa in the setting of active enteritis, a common condition in ileal pouch mucosa.
The diagnosis rests on findings demonstrated by application of immunohistochemical stains for microvilli antigens, such as villin or CD10, and electron microscopy (EM) [47] (see Figs. 1.3 and 1.4).

Microvillus inclusion disease. The apical cytoplasm of villus epithelium shows an increased number of secretory granules associated with microvillus alterations. ×2400 [20] (Reprinted from Springer and Virchows Archive: Official Journal of the European Society of Pathology, Morroni et al. [99], Fig. 4, with kind permission from Springer Science and Business Media)

Microvillus inclusion disease. The villus enterocytes lack brush border microvilli, whereas their apical cytoplasm contains a microvillus inclusion (MI) and numerous lysosomes (L) ×5.500 [20] (Reprinted from Springer and Virchows Archive: Official Journal of the European Society of Pathology, Morroni et al. [99], Fig. 2, with kind permission from Springer Science and Business Media)
Electron microscopy shows well-preserved crypt epithelium with abundant microvilli. Villus enterocytes are severely abnormal, particularly toward the apices of the short villi. The microvilli are depleted in number, short, and irregularly arranged. Some of the enterocytes contain the typical microvillus involutions, which are intracellular vacuoles where microvilli are observed lining the inner surface. Transmission electron microscopy (TEM) can efficiently demonstrate both the absence of surface microvilli, microvillus disorganization, and intracytoplasmic microvillus inclusions in enterocyte cytoplasm containing cryptic microvilli [48].
A striking feature is the finding of several small, membrane-bound vesicles containing electron-dense material (see Figs. 1.3 and 1.4). A few cases have been described in which the classic microvillus inclusions are shadowed by other features, such as large aggregates of electron lucent, vermiform membranous vesicles in enterocyte cytoplasm, corresponding to the PAS-positive material [49].
Epidemiology
Congenital microvillus atrophy is a rare disease. By 2014 only 137 cases had been published or gathered in an online registry [33]; to date around 200 cases of MVID have been reported, with a prevalence <1/1000.000, apparently more numerous in countries where marriages between blood relatives are more frequent [33].
A female preponderance had been observed among the published cases, with a female-to-male ratio of 2:1, but in the total reported 137 cases, there is instead a 1.54 male/female ratio. Consanguinity is present in 41% of the assessable cases with a gender preference for males. A cluster of cases from the Navajo reservation in northern Arizona suggests an incidence as high as 1 case per 12,000 live births [50].
Pathophysiology
Due to their alterations, mature enterocytes inefficiently absorb ions and nutrients, causing a malabsorption syndrome; however, the diarrhea is caused mainly by active secretion of water and electrolytes in the intestinal lumen (secretory diarrhea). The pathogenesis of the secretory diarrhea is unknown; it is believed to result from an unbalance between decreased absorption and unaltered secretion.
Measurement of stool electrolytes and osmolality enables rapid and accurate assessment of the pathogenesis of this chronic diarrhea (osmolar vs secretory) and greatly narrows the differential diagnosis.
Fecal electrolytes demonstrate a typical pattern of secretory diarrhea. Fecal sodium levels are high (approximately 60–120 mEq/L), and no osmotic gap is found. In patients with secretory diarrhea, the following formula applies: 2 × (Na concentration + K concentration) = stool osmolarity± 50. In osmotic diarrhea, stool osmolarity exceeds 2 × (Na concentration + K concentration) by 100 or more.
Secretory diarrhea occurs in the fasting state and is associated with large output losses that cause dehydration and metabolic acidosis.
In osmotic diarrhea, findings on stool microscopy are negative for white blood cells (WBCs), blood (exudative diarrhea), and fat (steatorrhea).
Even if there are data about the anomalies in water and electrolytes transport in the small intestine, it is not known whether and how the colonic mucosa participates in the absorption alterations in the disease.
In one of the Italian cases, we used the technique of rectal perfusion that showed a decrease in sodium absorption, only partially corrected by chlorpromazine administration [51].
Pathogenesis
Severe perturbation of the microvillar cytoskeleton may disrupt the transport of brush border components that have to be assembled at the apical membrane. The postulated abnormality in the cytoskeleton causes a block in exocytosis, mainly of periodic acid-Schiff (PAS)-positive material (e.g., polysaccharides, glycoproteins, glycolipids, neutral mucopolysaccharides). Consequently, small secretory granules that contain a PAS-positive material accumulate in the apical cytoplasm of epithelial cells.
Genetic evidence of the link between MVID and apical vesicle trafficking was initially obtained in Rab8 knock-out (KO) mice [52].
In 2008, the presence of mutations in the Myosine Vb (MYO5B) gene was described in seven patients (out of ten tested), predominantly of Turkish origin [37]. Homozygous mutations in the same gene were subsequently found in seven cases of Navajo origin; five parents were heterozygote [38]. A total of 41 unique MYO5B mutations had been identified [33]. Most patients with early-onset MVID display inactivating mutations in the MYO5B gene, one of the three myosin-5 genes (MYO5A, MYO5B, MYO5C) present in mammals, which are implicated in the spatiotemporal segregation and the transportation of organelles.
The MYO5B gene codifies Myosine Vb, an actin-based motor protein which carries the recycling endosomes to the apical plasma membrane along the actin filaments of the microtubules after having bound to a specific small guanosine-5’triphosphatase (GTPase) rab protein, such as Rab 11A, Rab8A, and RAB8A, located on the surface of recycling endosomes [53]. Myosine Vb mediates the tethering of Rab guanosine triphosphates, which determines apical vesicle transport and membrane recycling [54].
Studies in neonatal mouse models have shown that loss of active MYO5B causes early diarrhea, failure to thrive, evident microvillus inclusions, and loss of apical transporters in the duodenum. By contrast, induction of MYO5B loss in adult mice led to the rapid onset of diarrhea, but did not induce the formation of significant numbers of microvillus inclusions, to suggest that the formation of microvillus inclusions in duodenal enterocytes is far more pronounced in neonates, in whom we see a loss of proper trafficking and recycling of transporters to the apical brush border in enterocytes [55].
Thanks to this link the recycling endosomes move along the actine filaments [56] (See Fig. 1.5). The functional deficiency of Myosine Vb causes impaired microvillus formation and defective epithelial polarization [48]. The MYO5B mutant proteins are unable to bind to either RAB8A or RAB11A, with consequent microvillus structural defects, and the recycling endosomes are not carried in a normal way: in the enterocytes of the subjects with microvillus inclusion disease, no regular accumulation of myosine Vb and of the recycling endosome-associated proteins (one of these is Rab 11) can be observed close to the apical membrane, and no specific staining pattern is present [57]. Consequently, liquids and foods are not absorbed sufficiently, resulting in diarrhea, hypo-nutrition, and dehydration.

Endocytic recycling. Myosin Vb is a conformation-dependent binding partner of Rab11-FIP2. Activation of myosin Vb induces translocation of recycling endosomes and their cargo. Final transport from the recycling endosome to the cell surface is mediated by Rab8 [37] (Reprinted by permission from Macmillan Publishers Ltd. and Nature Publishing Group: Nature Reviews Molecular Cell Biology, Grant and Donaldson [56])
Therefore, Rab 11 distribution in the enterocytes can be a helpful diagnostic tool [58]. However, the spectrum of cellular defects in MVID is heterogeneous, and the severity of villus blunting, the ectopic formation of basolateral microvilli, the presence of ultrastructural features of microvillus inclusions (MVIs) may vary significantly. The cellular basis of this variability is still under study. In vitro cell-based models proved useful for greater understanding and showed that the MVID phenotype is correlated with the degree of enterocyte differentiation [59].
Molecular analysis strongly contributes to the unequivocal diagnosis of MVID and even prenatal diagnosis. So far about 60 different mutations have been identified, indicating the strong genetic heterogeneity of the disease.
Other biochemical mechanisms depending on myosin Vb which can produce alterations in the structure of the microvilli are presently being studied [60].
Myosin Vb is expressed in all the epithelial tissues and, as a matter of fact, microvillus inclusions in the stomach and colon, in addition to less well-defined inclusions in gallbladder epithelium and in renal tubular epithelial cells, have been reported in some patients with microvillus inclusion disease (MVID). Nevertheless, no extraintestinal symptoms are generally reported. Two children with renal Fanconi syndrome who carried mutation MYO5B did not show alterations in the apical brush border morphology and the PAS staining pattern in renal tubular epithelial cells, which makes it unlikely for it to be the cause of proximal tubular renal dysfunction [61].
At present it is not possible, from molecular analysis, to predict the outcome of the disease, since the prognosis depends mainly on early treatment, including intestinal transplantation.
Recent evidence shows that the effects of mutations in MYO5B are not limited to the small intestine but extend to other organs such as the liver, colon, pancreas, stomach [62], and bile salt export pump (BSEP) in the canalicular membrane, contributing to cholestasis [63].
In 2014, Dutch investigators found that the mild variant of MVID appears to be caused by loss of function of syntaxin 3 (STX3), an apical receptor involved in membrane fusion of apical vesicles in enterocytes [64]. STX3 mutations were identified by whole exome sequencing in two patients diagnosed with MVID based on clinical symptoms but without MYO5B mutations. In fact, whole exome sequencing of DNA from patients with variant MVID revealed homozygous truncating mutations in STX3, and in addition, patient-derived organoid cultures and overexpression of truncated STX3 in CaCo2 cells recapitulated most characteristics of variant MVID.
Mutations in STXBP2, a gene that encodes the syntaxin-binding protein-2 (also called mammalian uncoupled munc18–2 protein) which as STX3 may have a role in membrane fusion, have also been identified in patients with severe chronic diarrhea starting shortly after birth without signs of infection [65], as well as in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5, OMIM 613101), a hyperinflammatory immune disorder which in 40% of cases is associated with severe chronic diarrhea starting shortly after birth.
It has been suggested that MYO5B, STX3, and STXBP2 are part of a common disease mechanism that unifies a subset of phenotypically linked congenital diarrheal disorders, regulating protein trafficking to the apical brush border [66].
Prenatal Diagnosis
Pregnancy and birth are usually normal in individuals with microvillus atrophy. However, cases with severe prognosis have been reported [67]. Polyhydramnios has been reported very rarely [33], in contrast to the clinical picture of patients with other causes of congenital secretory diarrhea, and antenatal and natal periods are usually uneventful.
Nevertheless, in some cases, polyhydramnios and bowel dilation in the third trimester have been described. In one case, a high fetal alpha-fetoprotein in the second trimester was observed [68]. Authors have speculated that the fetal alpha-fetoprotein elevation might possibly be caused by in utero body fluid leakage into the amniotic fluid through fetal enteropathy.
Identification of the gene responsible for the disease allows its prenatal diagnosis [69].
Treatment
The prognosis of early-onset microvillus inclusion disease is poor. If patients are untreated, the disease is rapidly fatal because of dehydration and malnutrition.
In late-onset microvillus inclusion disease diarrhea tends to be less severe, and some alimentation is possible.
Medical Care
Agents tentatively given to induce a better growth of the intestinal mucosa (e.g., epithelial growth factor, colostrum) are ineffective. Several drugs (e.g., corticosteroids, cromoglycate disodium, cimetidine, somatostatin, octreotide, loperamide, chlorpromazine, urogastrone/epidermal growth factor) have been tried to counteract the massive secretory diarrhea in patients with microvillus atrophy; however, none has proven effective.
In a 4-year-old boy diagnosed with congenital atrophy of the microvilli, after unsuccessful treatment with loperamide (0.2 mg/kg 4 times a day), racecadotril therapy proved to be effective [70]. The drug reduces the degradation of the enkephalins, abundant in the intestinal villi, and has an antisecretory effect through the inhibition of the cyclic adenosine monophosphate (cAMP) [71].
At present, the only available therapy is total parenteral nutrition (TPN). Children with late-onset microvillus inclusion disease usually have less severe diarrhea; as they get older, TPN can be reduced to once or twice per week.
If patients are treated with TPN, their prognosis entirely depends on the complications of this approach. These complications include cholestasis with subsequent liver damage leading to cirrhosis, catheter-related sepsis due to infection with bacterial or fungal agents, and progressive lack of vascular access.
In the observed cases, cholestasis appears to be worsened by transplantation.
The study of eight patients who developed cholestatic liver disease suggests that cholestasis is enhanced by the impairment of the MYO5B/RAB11A apical recycling endosome pathway in hepatocytes [72].
Surgical Care
Successful outcomes of small intestinal transplantation have been reported, and evidence suggests that an early transplant might be beneficial. The limited experience accumulated in a few centers worldwide reflects an overall survival rate of approximately 50% at 5 years after small-bowel transplantation; this is a much better outcome than is seen with other indications for intestinal transplantation [73]. Patients who did not receive colonic transplant weaned later from parenteral nutrition.
The analysis of 16 patients who underwent a small-bowel transplantation shows a lower death rate compared to those who did not (23% versus 37%) after an average 3.5-year observation period (but variable between 3 months and 14 years). In all the cases, apart from the first two, the colon had been transplanted too [74].
Although only small series have been reported, evidence suggests that early small-bowel transplantation should be performed, at least in children with early-onset microvillus inclusion disease. Patients with late-onset microvillus atrophy appear to have an improved prognosis.
Transplantation appears to be the only option for patients who do not fare well with long-term TPN (e.g., because of sepsis, liver damage, lack of vascular access). For patients in whom transplantation is successful, a gradual return to a normal diet is considered possible.
In the observed cases, TPN-related cholestasis appears to be made worse by transplant. Therefore, in children with cholestasis, the worsening of this picture after the transplant points to a combined liver-intestinal transplantation.
Tufting Enteropathy (or Intestinal Epithelial Dysplasia)
In 1994, Reifen et al. described two infants less than a month old with protracted diarrhea. The diarrhea was so profuse to make total parenteral nutrition (TPN) necessary but it improved when enteral nutrition was interrupted. The jejunal biopsies showed a peculiar picture characterized by the presence of focal aggregations of packed enterocytes in the shape of a teardrop, as a consequence of an apical rounding of the plasma membrane. These focal areas looked like tufts and that is why the term “tufting enteropathy” was coined [75]. Curiously, a case with the same characteristics was identified among those presented by Davidson et al. in the same paper where the first case of microvillus inclusion disease had been described [26].
According to current criteria, diagnosis is based on the presence of total or partial villus atrophy associated with crypt hyperplasia, in the absence of signs of inflammation, associated with the characteristic focal localized epithelial tufts, whose presence is an element of distinction from two other enteropathies that directly affect enterocytes, the microvillus inclusion disease, and the trichohepatoenteric syndrome. The tufts are formed by enterocytes enclosed in a plasma membrane, located in the duodenum and jejunum.
Clinical Expression
The incidence of the disease has been estimated to be rare, with a prevalence of 1:50.000–1:100,000 live births in Western Europe [76], but it seems higher in people of Arab origin.
The vast majority of the mutations in the EpCAM gene that cause the disease have been identified in patients originating from Europe, North West Africa, and the Mediterranean area and from Saudi Arabia in particular. The incidence rate is higher in areas with high proportion of close relatives and in the Arab region than in other regions [77]. However, reports from Asia are very scarce, and the incidence of tufting enteropathy in this area is unknown; the cases published so far include only two patients from South Korea and one from China [78].
The clinical picture is characterized by a severe secretory diarrhea, generally with loose stools, starting in the first weeks of life. During pregnancy, there is no polyhydramnios, as in the microvillus inclusion disease and differently from congenital sodium diarrhea and congenital chloridorrhea.
The alterations in the enterocytes in any case cause an accentuation of the diarrhea with nutrition, including total enteral nutrition, as had already been observed from the very first cases described. The histological analysis of the intestine shows anomalies of the basement membrane, disorganization of the enterocytes, and a “crowding” at the apex of the villi which are arranged like tufts.
There are two different clinical forms: one is isolated and the other is syndromic, associated with various anomalies, particularly facial dysmorphism with choanal atresia and superficial punctuated keratitis [79, 80], together with reduced body size and immunodeficiencies.
However, the clinical picture can be confusing. Three cases of tufting enteropathy associated with chronic arthritis and one diagnosed with juvenile rheumatoid arthritis, treated with prednisolone, have been described [81, 82].
Pathophysiology
The clinical picture is mainly caused by an abnormal development of intestinal epithelial cells, which are destroyed and grouped into clusters.
In 2008, a biallelic mutation of the gene for the epithelial cell adhesion molecule (EpCAM gene) was identified in five affected children, two of whom belonged to the same family [83].
Subsequently, other mutations were identified, the main ones of which are located in exons 3, 4, and 5, and cause a deletion of the extracellular and transmembrane regions of the EpCAM protein [84]. The EpCAM is a Type I superficial glycoprotein that is expressed on the surface of the basolateral membrane of many epithelial cells, with a fundamental role in the structural integrity and adhesion of epithelial tissues [85]. The mutant EpCAM accumulated in the endoplasmic reticulum is co-localized with GRP78/BiP, a reticulum chaperon. It has therefore been hypothesized that a response through a protein pathway may be induced in the endoplasmic reticulum [86].
In 2010, a mutation in the SPINT2 gene was found in a case affected by a syndromic form of tufted enteropathy. SPINT2 is a transmembrane protein which seems to be involved in epithelial regeneration [87], whose mutations may result in an indirect loss of EpCAM protein, due to activation of matriptase, a type II transmembrane serine protease expressed in most human epithelia, which causes its proteolysis [88]. Mutations in SPINT2 (MIM# 605124) have been implicated in a syndromic form of the disease, which may cause an indirect loss of EpCAM protein due to proteolysis by the activation of matriptase [89].
It is interesting to note how mutations in the SPINT 2 gene are also present in the syndromic congenital sodium diarrhea, where choanal atresia, hypertelorism, and corneal erosions are particularly frequent and anal atresia can be found in certain cases [90].
The analysis of 57 patients revealed mutations in the gene for EpCAM in 73% of the cases, all of them presenting with an isolated intestinal disease, but in 21% of cases, all with a syndromic form of the disease, mutations of the SPINT2 gene were present [90].
According to this study, tufting enteropathy could be separated into at least three genetic classes, each with specific phenotypes.
However, it seems impossible at present to distinguish between tufting enteropathy and syndromic enteropathy, even from a genetic point of view.
Histological Features
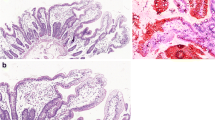
Jejunal biopsy shows a picture of partial villus atrophy together with crypt hyperplasia. The most characteristic feature, the one which gave the name to the disease, is the presence of “tufts,” small focal aggregates of teardrop-shaped enterocytes with apical rounding (see Fig. 1.6a, b) [91], in addition to characteristic focal epithelial tufts composed of enterocytes with plasma membrane rounding found in the duodenum and jejunum.

(a) Numerous tufts of enterocytes (*) on the mucosal surface of the duodenum. (b) A characteristic tear-drop-shaped structure (arrow) in an epithelial tuft (H&E stain; original magnification: a – ×80; b – ×400) [58] (Reprinted from Springer and European Journal of Pediatrics, El-Matary et al. [91], Fig. 1, with kind permission from Springer Science and Business Media)
The “tufts” are not a characteristic exclusive to intestinal epithelial dysplasia, because they have been observed in other mucosal enteropathies and in normal jejunum. In the latter cases anyway, they were present in <10% of the epithelial surface, while in “tufting enteropathy,” they are present in more than 80% of the jejunal surface. But the picture is not always so evident in the earliest period of the disease. Attempts at immunohistochemical analysis (including beta-catenin, E-cadherin, desmoglein, and laminins) have not been easy applicable [92]. On the contrary, staining with EpCAM/MOC31 antibody, an EpCAM antibody clone, showed a sensitivity and specificity of 100% for loss of staining in 15 studied patients [93].
Electronic microscopy shows relatively normal microvilli, and it may be useful as a diagnostic tool only to exclude microvillus inclusion disease.
Mild inflammation of the lamina propria is also present. Infiltration of T lymphocytes within the lamina propria had always been observed since the original description, even if inferior to celiac disease, but it sometimes gives rise to a suspicion of autoimmune enteropathy [75].
Treatment
Tuft enteropathy is associated with severe secretory diarrhea, which worsens with nutrition. That is why affected children have to be treated with total parenteral nutrition (TPN).
Some cases seem to have a less severe course and they can be given a partial parenteral nutrition [94].
There is currently no specific treatment for tufting enteropathy. Many cases are treated with long-term parenteral nutrition, which it may be possible to reduce or suspend with increasing age [95]. Twelve patients survived for 8–30 years under long-term parenteral nutrition therapy. In those cases where it is impossible to continue parenteral nutrition, or when there were other serious issues, bowel transplantation was attempted [96].
Cases totally dependent on total parenteral nutrition are candidates for intestinal transplantation.
Tufting enteropathy is often associated with other pathologies related to epithelial cells or malformations. More than 60% of patients have punctate keratitis [97] or other eye diseases [98], associated with other changes, such as nostril atresia, esophageal atresia, or absence of the anus [79].
In some cases where there was a risk of liver failure as a result of TPN, an intestinal or combined liver / intestinal transplant was performed, with satisfactory results in a case where the transplant was performed before aggravation. Repeated biopsies are sometimes required for diagnosis.
References
Avery GB, Villavicencio O, Lilly JR, Randolph JG. Intractable diarrhea in early infancy. Pediatrics. 1968;41:712–22.
Hyman CJ, Reiter J, Rodnan J, Drash AL. Parenteral and oral alimentation in the treatment of the nonspecific protracted diarrheal syndrome of infancy. J Pediatr. 1971;78:17–29.
Shwachman H, Filler RM, Khaw KT. A new method of treating malnourished infants with severe chronic diarrhea. Acta Pediatr Scand. 1970;59:446–7.
Shwachman H, Lloyd-Still JD, Khaw KT, Antonowicz I. Protracted diarrhea of infancy treated by intravenous alimentation. II studies of small intestinal biopsy results. Am J Dis Child. 1973;125:365–8.
Walker-Smith JA. Intractable diarrhea of infancy. Saudi J Gastroenterol. 1995;1:152–6.
Larcher VF, Shepherd R, Francis DE, Harries JT. Protracted diarrhoea in infancy. Analysis of 82 cases with particular reference to diagnosis and management. Arch Dis Child. 1977;52:597–605.
Guarino A, Spagnuolo MI, Russo R, Albano F, Guandalini S, et al. Etiology and risk factors of severe and protracted diarrhea. J Pediatr Gastroenterol Nutr. 1995;20:173–8.
Catassi A, Fabiani E, Spagnuolo MI, et al. Severe and protracted diarrhea: results of the 3-year SIGEP multi-center survey. J Pediatr Gastroenterol Nutr. 1999;29:63–8.
Duggan CP, Jaksic T. Pediatric intestinal failure. N Engl J Med. 2017;377(7):666–75.
Wong T, Gupte G. Intestinal failure in children. Indian J Pediatr. 2016;83:1436–43.
Ahmed Z, Imdad A, James A, Connelly JA, Acra S. Autoimmune Enteropathy: an updated review with special focus on stem cell transplant therapy. Dig Dis Sci. 2019;64(3):643–54.
Villanacci S, Lougaris V, Ravelli A, Buscarini E, Salviato T, Lionetti P, Salemme M, Martelossi S, De Giacomo C, Falchetti D, Pelizzo G, Bassotti G. Clinical manifestations and gastrointestinal pathology in 40 patients with autoimmune enteropathy. Clin Immunol. 2019;207:10–7.
Sanderson IR, Risdon RA, Walker-Smith JA. Intractable ulcerating enterocolitis of infancy. Arch Dis Child. 1991;66:295–9.
Thapar N, Shah N, Ramsay AD, Lindley KJ, Milla PJ. Long-term outcome of intractable ulcerating enterocolitis of infancy. J Pediatr Gastroenterol Nutr. 2005;40:582–8.
Murch SH, Winyard PJ, Koletzko S, Wehner B, Cheema HA, Risdon RA, et al. Congenital enterocyte heparan sulphate deficiency with massive albumin loss, secretory diarrhoea, and malnutrition. Lancet. 1996;347:1299–301.
Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y, et al. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118:229–38.
Bode L, Freeze HH. Applied glycoproteomics – approaches to study genetic-environmental collisions causing protein-losing enteropathy. Biochim Biophys Acta. 1760;2006:547–59.
Lachaux A, Bouvier R, Loras-Duclaux I, Chappuis JP, Meneguzzi G, Ortonne JP. Isolated deficient alpha6beta4 integrin expression in the gut associated with intractable diarrhea. J Pediatr Gastroenterol Nutr. 1999;29:395–401.
Salvestrini C, McGrath JA, Ozoemena L, Husain K, Buhamrah E, Sabery N, et al. Desquamative enteropathy and pyloric atresia without skin disease caused by a novel intracellular beta4 integrin mutation. J Pediatr Gastroenterol Nutr. 2008;47:585–91.
Girault D, Goulet O, Le Deist F, Brousse N, Colomb V, Césarini JP, et al. Intractable infant diarrhea associated with phenotypic abnormalities and immunodeficiency. J Pediatr. 1994;125:36–42.
Stankler L, Lloyd D, Pollitt RJ, Gray ES, Thom H, Russell G. Unexplained diarrhoea and failure to thrive in two siblings with unusual facies and abnormal scalp hair shafts: a new syndrome. Arch Dis Child. 1982;57:212–6.
Verloes A, Lombet J, Lambert Y, Hubert AF, Deprez M, Fridman V, et al. Tricho-hepato-enteric syndrome: further delineation of a distinct syndrome with neonatal hemochromatosis phenotype, intractable diarrhea, and hair anomalies. Am J Med Genet. 1997;68:391–5.
Fabre A, André N, Breton A, Broué P, Badens C, Roquelaure B. Intractable diarrhea with "phenotypic anomalies" and tricho-hepato-enteric syndrome: two names for the same disorder. Am J Med Genet A. 2007;143A:584–8.
Hartley JL, Zachos NC, Dawood B, Donowitz M, Forman J, Pollitt RJ, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology. 2010;138:2388–98, 2398.e1–2.
Fabre A, Bourgeois P, Coste M-E, Roman C, Barlogis V, Badens C. Management of syndromic diarrhea/tricho-hepato-enteric syndrome: a review of the literature. Rare Dis Res. 2017;6:152–7.
Davidson GP, Cutz E, Hamilton JR, Gall DG. Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology. 1978;75:783–90.
Schmitz J, Ginies JL, Arnaud-Battandier F, et al. Congenital microvillous atrophy, a rare cause of neonatal intractable diarrhoea. Pediatr Res. 1982;16:1014.
Goutet JM, Boccon-Gibod L, Chatelet F, Ploussard JP, Navarro J, Polonovski CI. Familial protracted diarrhoea with hypoplastic villous atrophy: report of two cases. Pediatr Res. 1982;16:1045.
Phillips AD, Jenkins P, Raafat F, Walker-Smith JA. Congenital microvillous atrophy: specific diagnostic features. Arch Dis Child. 1985;60:135–40.
Guarino A, Nocerino A, Cinti S, Berni Canani R, Terracciano L, Raimondi F, Guandalini S. Atrofia congenita dei microvilli intestinali. Riv Ital Ped. 1992;18:150–3.
Cutz E, Rhoads JM, Drumm B, Sherman PM, Durie PR, Forstner GG. Microvillus inclusion disease: an inherited defect of brush-border assembly and differentiation. N Engl J Med. 1989;320:646–51.
Pecache N, Patole S, Hagan R, Hill D, Charles A. J M Papadimitriou neonatal congenital microvillus atrophy. Postgrad Med J. 2004;80:80–3.
van der Velde KJ, Dhekne HS, Swertz MA, Sirigu S, Ropars V, Vinke PC, Rengaw T, van den Akker PC, Rings EH, Houdusse A, van Ijzendoorn SC. An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations. Hum Mutat. 2013;34:1597–605.
Phillips AD, Schmitz J. Familial microvillous atrophy: a clinic pathological survey of 23 cases. J Pediatr Gastroenterol Nutr. 1992;14:380–96.
Mierau GW, Wills EJ, Wyatt-Ashmead J, Hoffenberg EJ, Cutz E. Microvillous inclusion disease: report of a case with atypical features. Ultrastruct Pathol. 2001;25:517–21.
Raafat F, Green NJ, Nathavitharana KA, Booth IW. Intestinal microvillous dystrophy: a variant of microvillous inclusion disease or a new entity? Hum Pathol. 1994;25:1243–8.
Müller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz-Erian P, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40:1163–5.
Erickson RP, Larson-Thomé K, Valenzuela RK, Whitaker SE, Shub MD. Navajo microvillous inclusion disease is due to a mutation in MYO5B. Am J Med Genet A. 2008;146A(24):3117–9.
Vogel GF, Janecke AR, Krainer IM, Gutleben K, Witting B, Mitton SG, Mansour S, Ballauff A, Roland JT, Engevik AC, Cutz E, Müller T, Goldenring JR, Huber LA, Hess MW. Abnormal Rab11-Rab8-vesicles cluster in enterocytes of patients with microvillus inclusion disease. Traffic. 2017;18:453–64.
Schneeberger K, Roth S, Nieuwenhuis EES, Middendorp S. Intestinal epithelial cell polarity defects in disease: lessons from microvillus inclusion disease. Dis Model Mech. 2018;11(2):dmm031088.
Halac U, Lacaile F, Joly F, Hugot JP, Talbotec C, Colomb V, Ruemmele FM, Goulet O. Microvillous inclusion disease: how to improve the prognosis of a severe congenital enterocyte disorder. J Pediatr Gastroenterol Nutr. 2011;52:460–5.
Ruemmele FM, Bindl L, Woelfle J, Buderus S, Phillips AD, Lentze MJ. Recurrent episodes of necrotizing enterocolitis complicating congenital microvillous atrophy. Dig Dis Sci. 2001;46:1264–9.
Perry A, Bensallah H, Martinez-Vinson C, Berrebi D, Arbeille B, Salomon J, Goulet O, Marinier E, Drunat S, Marie-Elisabeth Samson-Bouma ME, Gérard B, Hugot JP. Microvillous atrophy: atypical presentations. J Pediatr Gastroenterol Nutr. 2014;59:779–85.
Siahanidou T, Koutsounaki E, Skiathitou AV, Stefanaki K, Marinos E, Panajiotou I, Chouliaras G. Extraintestinal manifestations in an infant with microvillus inclusion disease: complications or features of the disease? Eur J Pediatr. 2013;172(9):1271–5.
Phillips AD, Szafranski M, Man LY, Wall WJ. Periodic acid-Schiff staining abnormality in microvillous atrophy: photometric and ultrastructural studies. J Pediatr Gastroenterol Nutr. 2000;30(1):34–42.
Groisman GM, Amar M, Livne E. CD10: a valuable tool for the light microscopic diagnosis of microvillous inclusion disease (familial microvillous atrophy). Am J Surg Pathol. 2002;26(7):902–7.
Koepsell SA, Talmon G. Light microscopic diagnosis of microvillus inclusion disease on colorectal specimens using CD10. Am J Surg Pathol. 2010;34:970–2.
Bell SW, Kerner JA, Sibley RK. Microvillous inclusion disease. The importance of electron microscopy for diagnosis. Am J Surg Pathol. 1991;15:1157–64.
Weeks DA, Zuppan CW, Malott RL, Mierau GW. Microvillous inclusion disease with abundant vermiform, electron-lucent vesicles. Ultrastruct Pathol. 2003;27:337–40.
Pohl JF, Shub MD, Trevelline EE, Ingebo K, Silber G, Rayhorn N, Holve S, Hu D. A cluster of microvillous inclusion disease in the Navajo population. J Pediatr. 1999;134(1):103–6.
Guandalini S, Nocerino A, Saitta F, Fasano A, Ascione G, De Curtis M, et al. Valutazione dell’assorbimento di elettroliti ed acqua nel colon di un lattante affetto da atrofia congenita dei microvilli. Riv Ital Ped. 1987;13:76.
Sato T, Mushiake S, Kato Y, Sato K, Sato M, Takeda N, Ozono K, Miki K, Kubo Y, Tsuji A, Harada R, Harada A. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature. 2007;448:366–9.
Schafer JC, Baetz NW, Lapierre LA, McRae RE, Roland JT, Goldenring JR. Rab11-FIP2 interaction with MYO5B regulates movement of Rab11a-containing recycling vesicles. Traffic. 2014;15:292–308.
Knowles BC, Roland JT, Krishnan M, et al. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J Clin Invest. 2014;124:2947–62.
Weis VG, Knowles BC, Choi E, Goldstein AE, Williams JA, Manning EH, Roland JT, Lynne A, Lapierre LA, Goldenring JR. Loss of MYO5B in mice recapitulates microvillus inclusion disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell Mol Gastroenterol Hepatol. 2016;2(2):131–57.
Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Moll Cell Biol. 2009;10:597–608.
Szperl AM, Golachowska MR, Bruinenberg M, Prekeris R, Thunnissen A-MWH, Karrenbeld A, Dijkstra G, Hoekstra D, Mercer D, et al. Functional characterization of mutations in the myosin Vb gene associated with microvillus inclusion disease. J Pediatr Gastroenterol Nutr. 2011;52(3):307–13.
Geoffrey TG, Melissa HM, DiMaio DJ, Muirhead D. Rab11 is a useful tool for the diagnosis of microvillous inclusion disease. Int J Surg Pathol. 2012;20(3):252–16.
Mosa MH, Nicolle O, Maschalidi S, et al. Dynamic formation of microvillus inclusions during Enterocyte differentiation in Munc18–2-deficient intestinal organoids. Cell Mol Gastroenterol Hepatol. 2018;6(4):477–493.e1.
Dhekne HS, Pylypenko O, Overeem AW, et al. MYO5B, STX3, and STXBP2 mutations reveal a common disease mechanism that unifies a subset of congenital diarrheal disorders: a mutation update. Hum Mutat. 2018;39(3):333–44.
Golachowska MR, van Dael CM, Keuning H, Karrenbeld A, Hoekstra D, Gijsbers CF, et al. MYO5B mutations in patients with microvillus inclusion disease presenting with transient renal Fanconi syndrome. J Pediatr Gastroenterol Nutr. 2012;54:491–8.
Schlegel C, Weis VG, Knowles BC, et al. Apical membrane alterations in non-intestinal organs in microvillus inclusion disease. Dig Dis Sci. 2018;63:356–65.
Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, Sauvat F, Irtan S, Davit-Spraul A, Jacquemin E, Ruemmele F, Rainteau D, Goulet O, Colomb V, Chardot C, Henrion-Caude A, Debray D. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology. 2014;60(1):301–10.
Wiegerinck CL, Janecke AR, Schneeberger K, Vogel GF, van Haaften-Visser DY, Escher JC, et al. Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology. 2014;147:65–8.
Vogel GF, van Rijn JM, Krainer IM, Janecke AR, Posovszky C, Cohen M, Searle C, Jantchou P, Escher JC, Patey N, Cutz E, Müller T, Middendorp S, Hess MW, Huber LA. Disrupted apical exocytosis of cargo vesicles causes enteropathy in FHL5 patients with Munc18-2 mutations. JCI Insight. 2017;2(14):e94564.
Stepensky P, Bartram J, Barth TF, Lehmberg K, Walther P, Amann K, Philips AD, Beringer O, Zur Stadt U, Schulz A, Amrolia P, Weintraub M, Debatin KM, Hoenig M, Posovszky C. Persistent defective membrane trafficking in epithelial cells of patients with familial hemophagocytic lymphohistiocytosis type 5 due to STXBP2/MUNC18-2 mutations. Pediatr Blood Cancer. 2013;60(7):1215–22.
Ruemmele FM, Schmitz J, Goulet O. Microvillous inclusion disease (microvillous atrophy). Orphanet J Rare Dis. 2006;1:22.
Kennea N, Norbury R, Anderson G, Tekay A. Congenital microvillous inclusion disease presenting as antenatal bowel obstruction. Ultrasound Obstet Gynecol. 2001;17:172–4.
Chen CP, Chiang MC, Wang TH, et al. Microvillus inclusion disease: prenatal ultrasound findings, molecular diagnosis and genetic counseling of congenital diarrhea. Taiwan J Obstet Gynecol. 2010;49(4):487–94.
Gordon M, Akobeng A. Racecadotril for acute diarrhoea in children: systematic review and meta-analyses. Arch Dis Child. 2016;101:234–40.
Tran LC, Lazonby G, Ellis D, Goldthorpe J, Iglesias N, Steele J, Zamvar V, Puntis JWL, Vora R. Racecadotril may reduce diarrhoea in microvillous inclusion disease. J Pediatr Gastroenterol Nutr. 2017;64(1):e25–6.
Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, Sauvat F, Irtan S, Anne D-S, Jacquemin E, Ruemmele F, Rainteau D, Goulet O, Colomb V, Chardot C, Henrion-Caude A, Debray D. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology. 2014;60(1):301–10.
Ruemmele FM, Jan D, Lacaille F, Cézard JP, Canioni D, Phillips AD, et al. New perspectives for children with microvillous inclusion disease: early small bowel transplantation. Transplantation. 2004;77:1024–8.
Halac U, Lacaille F, Joly F, Hugot JP, Talbotec C, Colomb V, et al. Microvillous inclusion disease: how to improve the prognosis of a severe congenital enterocyte disorder. J Pediatr Gastroenterol Nutr. 2011;52:460–5.
Reifen RM, Cutz E, Griffiths AM, Ngan BY, Sherman PM. Tufting enteropathy: a newly recognized clinicopathological entity associated with refractory diarrhea in infants. J Pediatr Gastroenterol Nutr. 1994;18:379–85.
Goulet O, Salomon J, Ruemmele F, de Serres NP, Brousse N. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;2:20.
Goulet O. Intestinal epithelial dysplasia: a new entity. Arch Pediatr. 1996;3(suppl 1):324s–5s.
Tang W, Huang T, Xu Z, Huang Y. Novel mutations in EPCAM cause congenital tufting Enteropathy. J Clin Gastroenterol. 2018;52(1):e1–6.
Bird LM, Sivagnanam M, Taylor S, Newbury RO. A new syndrome of tufting enteropathy and choanal atresia, with ophthalmologic, hematologic and hair abnormalities. Clin Dysmorphol. 2007;16:211–21.
Roche O, Putterman M, Salomon J, Lacaille F, Brousse N, Goulet O, Dufier JL. Superficial punctate keratitis and conjunctival erosions associated with congenital tufting enteropathy. Am J Ophthalmol. 2010;150:116–21.
Al-Mayouf S, Alswaied N, Alkuraya F, et al. Tufting enteropathy and chronic arthritis: a newly recognized association with a novel EpCAM gene mutation. J Pediatr Gastroenterol Nutr. 2009;49:642–4.
Azzopardi C, Pullicino E, Coleiro B, Galea SS. Congenital tufting enteropathy and chronic arthritis: a clinical and radiological perspective. BMJ Case Rep. 2016;2016:bcr2016215252.
Ko JS, Seo JK, Shim JO, Hwang SH, Park HS, Kang GH. Tufting Enteropathy with EpCAM mutations in two siblings. Gut Liver. 2010;4(3):407–10.
Sivagnanam M, Mueller JL, Lee H, Chen Z, Nelson SF, Turner D, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135:429–37.
Yahyazadeh Mashhadi SM, Kazemimanesh M, Arashkia A, Azadmanesh K, Meshkat Z, Golichenari B. Sahebkar a shedding light on the EpCAM: an overview. J Cell Physiol. 2019;234(8):12569–80.
Das B, Okamoto K, Rabalais J, Ronald R, Marchelletta RR, Barrett KE, Das S, Niwa M, Sivagnanam M. Congenital tufting enteropathy-associated mutant of epithelial cell adhesion molecule activates the unfolded protein response in a murine model of the disease. Cell. 2020;9(4):946.
Sivagnanam M, Janecke AR, Müller T, Heinz-Erian P, Taylor S, Bird LM. Case of syndromic tufting enteropathy harbors SPINT2 mutation seen in congenital sodium diarrhea. Clin Dysmorphol. 2010;19(1):48.
Wu C-J, Feng X, Lu M, Morimura S, Udey MC. Cleavage of EpCAM destabilizes claudins and dysregulates intestinal epithelial homeostasis. J Clin Investig. 2017;127(2):623–34.
Salomon J, Goulet O, Canioni D, Brousse N, Lemale J, Tounian P, et al. Genetic characterization of congenital tufting enteropathy: EpCAM associated phenotype and involvement of SPINT2 in the syndromic form. Hum Genet. 2014;133:299–310.
Heinz-Erian P, Müller T, Krabichler B, Schranz M, Becker C, Rüschendorf F, et al. Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am J Hum Genet. 2009;84:188–96.
El-Matary W, Dalzell AM, Kokai G, Davidson JE. Tufting enteropathy and skeletal dysplasia: is there a link? Eur J Pediatr. 2007;166:265–8.
Patey N, Scoazec JY, Cuenod-Jabri B, Canioni D, Kedinger M, Goulet O, Brousse N. Distribution of cell adhesion molecules in infants with intestinal epithelial dysplasia (tufting enteropathy). Gastroenterology. 1997;113:833–43.
Ranganathan S, Schmitt LA, Sindhi R. Tufting enteropathy revisited: the utility of MOC31 (EpCAM) immunohistochemistry in diagnosis. Am J Surg Pathol. 2014;38:265–72.
Lemale J, Coulomb A, Dubern B, Boudjemaa S, Viola S, Josset P, et al. Intractable diarrhea with tufting enteropathy: a favorable outcome is possible. J Pediatr Gastroenterol Nutr. 2011;52:734–9.
Ashworth I, Wilson A, Aquilina S, Parascandalo R, Mercieca V, Gerada J, Macdonald S, Simchowitz V, Hill S. Reversal of intestinal failure in children with tufting enteropathy supported with parenteral nutrition at home. J Pediatr Gastroenterol Nutr. 2018;66(6):967–71.
Paramesh AS, Fishbein T, Tschernia A, Leleiko N, Magid MS, Gondolesi GE, Kaufman SS. Isolated small bowel transplantation for tufting enteropathy. J Pediatr Gastroenterol Nutr. 2003;36(1):138–40.
Roche O, Putterman M, Salomon J, Lacaille F, Brousse N, Goulet O, Dufier JL. Superficial punctate keratitis and conjunctival erosions associated with congenital tufting enteropathy. Ophthalmology. 2010;150(1):116–121.e1.
Hirabayashi KE, Moore AT, Mendelsohn BA, Taft RJ, Chawla A, Perry D, Henry D, Slavotinek A. Congenital sodium diarrhea and chorioretinal coloboma with optic disc coloboma in a patient with biallelic SPINT2 mutations, including p.(Tyr163Cys). Am J Med Genet A. 2018;176(4):997–1000.
Morroni M, Cangiotti AM, Guarino A, Cinti S. Unusual ultrastructural features in microvillous inclusion disease: a report of two cases. Virchows Arch. 2006;448(6):805–10.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nocerino, A., Guandalini, S. (2022). Microvillus Inclusion Disease and Tufting Enteropathy. In: Guandalini, S., Dhawan, A. (eds) Textbook of Pediatric Gastroenterology, Hepatology and Nutrition. Springer, Cham. https://doi.org/10.1007/978-3-030-80068-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-80068-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-80067-3
Online ISBN: 978-3-030-80068-0
eBook Packages: MedicineMedicine (R0)