
Abstract
Emerging evidence indicates that immune-mediated inflammation plays a key role in the degenerative process of osteoarthritis (OA). There are several types of immune cells that infiltrate the synovial joint promoting inflammation, cartilage degeneration, and joint remodeling. In this chapter, the focus is on a specific type of immune cell; T cells, and most notably, the T helper 17 and Regulatory T cell subsets. T helper 17 cells are found in the synovium and synovial fluid where they contribute to progression of OA through cytokine secretions and cell-to-cell interactions. Conversely, Regulatory T cells are considered immunosuppressive and downregulate the effects of T helper 17 cells. There is plasticity in phenotype and function of these T cell subsets, and transdifferentiation is influenced by joint inflammation in OA. The role of T cells in regulating the inflammatory homeostasis of the joint suggests that there are opportunities for biological therapeutic intervention in targeting T cell effector functions to mitigate OA, particularly if targeted early in disease progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
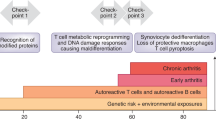
Historically, osteoarthritis (OA) was considered a wear and tear disease initiated and propagated by biomechanical processes resulting in degeneration of articular cartilage. However, there is also strong evidence demonstrating the involvement of inflammation and the immune system in the pathogenesis of OA throughout the disease process. Histological studies reveal that over 50% of patients with OA have a mononuclear cellular infiltrate in the synovial fluid and synovium that consists of lymphocytes, monocytes, and dendritic cells derived from peripheral blood [1, 2]. Acutely following joint injury, pro-inflammatory chemokines and cytokines are released from the cartilage and the synovium and attract a variety of immune cells to the joint, the majority of which are macrophages and T lymphocytes [3]. Through the release of cytokines and chemokines and cell-to-cell interactions, T cells modify the inflammatory joint environment and influence the progression of disease.
This chapter will focus on the role of T cells in early OA, because only in the early phases of OA can true disease intervention and disease prevention occur. Herein, we will lay the foundation for how cytokines and chemokines released from chondrocytes and synoviocytes home T cells to the inflamed joint acutely following injury, and how the specific T cell subtypes can influence disease progression. We will discuss T cell behavior within the synovium, including activation and proliferation in antigen-dependent or independent manners, and why these events in early OA are critical for sustained T cell responses within the joint. We will explore the biology of different T cell subsets within the joint that can act to mitigate or propagate disease progression dependent upon their phenotype, and how the cytokine environment of the joint can reciprocally polarize T cell phenotype, potentially exacerbating the T cell inflammatory response. Finally, we will discuss how further exploration of the interplay between T cells and joint dysfunction will inform the development and utilization of targeted immunotherapies early in disease to mitigate OA. Throughout this chapter, we will convey the need to further explore how T cell functions within the joint during early OA influence disease progression and can potentially be manipulated to mitigate OA to prevent joint destruction.
2 Orchestration of the T Cell Response in OA
T cell infiltration into an inflamed joint is emerging as a hallmark of OA. This infiltration is considered abnormal because there are very few tissue-resident T cells within the synovium or synovial fluid of a healthy joint [4]. While a small population of T cells may reside in a healthy joint and play a role in maintaining joint homeostasis, an inflammatory event is needed to initiate infiltration of pathogenic T cells into the joint. T cell activation can occur in both an antigen-dependent and -independent manner. The presence of mono- and oligo-clonal populations of T cells within the synovium of OA patients points to an antigen-specific proliferation of T cells within the joint itself [2]. Additionally, T cells from the peripheral blood of some patients with OA activate and proliferate in response to chondrocyte and synoviocyte membrane antigens, suggesting that self-specific T cells exist in the circulation of OA patients as well [5]. Moreover, T cells from patients with OA have been found to recognize specific amino acid sequences from aggrecan, which is a major constituent of normal articular cartilage but can also function as an auto-antigen within the joint [6]. Taken together, these data suggest that OA is characterized by aberrant systemic and local joint T cells that are driven by joint-derived antigens.
T cells are part of the adaptive immune system. They are derived from hematopoietic stem cells in the bone marrow that differentiate into lymphoid progenitor cells that migrate to the thymus and commit to the T lymphocyte lineage. During development, diverse T cell receptors are generated through germline DNA rearrangement. These T cell receptors can recognize virtually any antigen. The process of negative selection largely deletes T cells that strongly recognize self-antigen, [7] but this process is not perfect, and some self-reactive T cells can develop [8]. While still in the thymus, T cells either mature into CD4+ helper T (Th) cells, which are the predominant cell type in an OA joint, or CD8+ cytotoxic T cells. These mature T cells that are still naïve to antigen then leave the thymus and travel to secondary lymphoid tissues where they can be activated by an antigen-presenting cell, typically a dendritic cell [9].
Three signals are required for the activation and proliferation of naïve T cells. First is the signal received when a T cell receptor recognizes its cognate antigen presented by an antigen-presenting cell in the context of major histocompatibility complex (MHC). Second is co-stimulatory signaling in which a co-stimulatory molecule on the T cell, like CD28, binds a member of the B7 receptor family on the antigen-presenting cell. Finally, the T cell must also encounter IL-2 for proliferation, and other cytokines that support activation and polarization. This three-step process creates a significant barrier for inappropriate T cell activation to occur, thus preventing the proliferation of T cells that might otherwise recognize auto-antigens. Moreover, if a T cell binds a specific antigen alone without receiving a co-stimulatory signal, it will become anergic and unable to respond to antigen in the future. Interestingly, T cells co-cultured with fibroblast-like synoviocytes that are able to present antigen-loaded MHC II adopt an anergic phenotype. This suggests that, although fibroblast-like synoviocytes are capable of presenting antigen to T cells, they are unable to activate naïve T cells because they lack co-stimulatory molecules [10].
Initial T cell priming in OA is likely to occur in a lymph node local to the joint. In this scenario, dendritic cells in the joint carry antigen to the lymph node, or alternatively, dendritic cells in tissues proximal to the damaged joint pick up antigens that have drained out of the damaged joint and then migrate to the lymph node [11]. However, during ongoing disease, there may be other modes of antigen presentation and persistent T cell activation. Lymphoid nodular aggregates and lymphoid follicles containing macrophages, T cells, and B cells can be found in the synovium of patients in all stages of OA [12]. There is evidence in RA that auto-antigens are presented to T cells by antigen-presenting cells within the synovium [13]. A rabbit medial meniscectomy model suggests that this may also be true for OA. At weeks 2 and 4 post-meniscectomy, large numbers of mature dendritic cells were present in lymphoid aggregates within the synovium [14]. A recent study in a mouse model of load-induced arthritis found that the total number of T cells in the inguinal lymph node was significantly increased within 1–2 weeks of loading [15]. This suggests that while initial T cell activation by dendritic cells likely occurs in local lymph nodes, it may then be perpetuated in the synovium [11].
Cytokines in the micro-environment during priming determine the fate of CD4+ T cells and polarize them to one of a number of functional subsets or fates [16]. These CD4+ T cell fates include Th1, Th2, Th17, and Regulatory (Treg) (Fig. 11.1). Th1 cells develop in response to IFN-γ and IL-12, which cause downstream activation of the T-bet transcription factor and induce Th1 cells to secrete IFN-γ and TNF-α. They activate phagocytic cells and are involved in the elimination of intracellular pathogens. IL-4 activates the transcription factor GATA3 and directs naïve cells to a Th2 fate. Th2 cells coordinate the immune response toward extracellular pathogens including helminths and predominantly secrete IL-4, IL-5, and IL-13. Th17 cells are responsible for immunity against extracellular bacteria and fungi through the secretion of IL-17A, IL-21, and IL-22. IL-1β, IL-6, and IL-23 activate the RORγt transcription factor, driving the emergence of the Th17 phenotype. Treg cells develop in one of two ways. Natural Tregs develop in the thymus and Induced Tregs develop in the periphery under the influence of TGF-β and IL-2. Treg development and function is directed by the transcription factor Foxp3 that supports downstream secretion of IL-10 and TGF-β. Tregs are critical for tolerance to self- and foreign-antigen and resolution of inflammation [17]. Activation by antigen recognition initiates T cell proliferation and differentiation prior to homing to sites of inflammation, such as an OA joint, where they carry out their effector functions.

CD4+ T cells fates are determined by cytokine environment during differentiation. Naïve T cells must receive three signals in order to activate and proliferate. This includes binding of the T cell receptor to the appropriate MHC class and cognate antigen for activation, co-receptor signaling to increase survival signal to the T cell, and finally T cells must bind IL-2 in order to proliferate. Subset-specifying cytokines released by mature antigen-presenting cells activate subset-specific master transcription factors, determining T cell fate and effector functions
As T cells are activated and adopt specific fates (see Fig. 11.1) [17], these subsets also express specific chemokine receptors that mediate the ability of T cells to respond to chemokines that direct immune cell migration to and within tissues. In the context of OA, chemokines produced from inflamed cartilage and synovium promote T cell homing to the joint. In addition, the vasculature of the inflamed synovium becomes highly positive for E-selectin, which promotes the extravasation of immune cells from the peripheral blood into the joint [2].
Recent studies have revealed that specific chemokines are key mediators responsible for immune cell homing in early OA, including CCL5, CCL17, CCL20, and CXCL12 [18, 19]. CCL5 is a potent T cell chemoattractant that binds to CCR1, CCR3, and CCR5, all of which can be expressed by T cells [20,21,22]. CCL5 knockout mice are partially protected from cartilage injury as a result of destabilization of the medical meniscus-induced OA compared to wild-type mice [23]. CCL17 induces chemotaxis in T cells through interactions with CCR4, which is expressed only on specific CD4+ T cell subtypes, including Th17 and Tregs [24]. CCL17 blockade in mice with collagenase-induced arthritis resulted in reduced pain and OA [25]. Synoviocytes from OA patients secrete CCL20, which is strongly chemotactic for lymphocytes and binds to CCR6 [26]. CXCL12 is another potent chemokine for lymphocytes that are closely associated with the radiographic severity of OA [27]. Additionally, CXCL12 can enhance the effects of certain pro-inflammatory cytokines, including IL-17A, on fibroblast-like synoviocytes [28]. Because different T cell subsets exhibit specific receptors, the cytokines released by cartilage and synovium during early OA will affect which subtypes are homed to the joint, subsequently playing a role in disease pathogenesis.
The aforementioned chemokines and cytokines orchestrate priming and homing T cells to the joint where they elicit their effector functions through several mechanisms including secretion of cytokines and cell-to-cell interactions (Fig. 11.2). T cells can mediate the progression of OA by affecting both stromal and immune cells within the joint. These effector functions of T cells within the inflamed, early OA joint could be targeted therapeutically on a patient-to-patient basis to interrupt the course of disease before irreversible joint damage has occurred.

T cells alter joint homeostasis in a subset-specific manner. T cells are homed to the joint by inflammatory cytokines released by joint tissues, where they then carry out their effector functions and contribute to loss of joint homeostasis. Cytotoxic T lymphocytes are not abundant in the joint but contribute to increased vascularization and matrix degradation. Th1 cells are the most abundant T cell subtype within the joint but appear to carry out their effector functions mainly thorough macrophage polarization and activation. Th2 cells are found sporadically and sparsely within the OA joint and do not appear to offer protection against cartilage breakdown. Th17 cells contribute significantly to matrix degradation as well as synovial inflammation while further contributing to immune cell homing to the joint. Tregs provide early immunosuppression but are unable to restore joint homeostasis and ultimately cannot sustain their effector functions in order to mitigate OA progression
3 CD8+ Cytotoxic T Cells (CTL)
Cytotoxic T lymphocytes (CTL) express the CD8 co-receptor and perform cell-mediated immunity. CTLs kill harmful cells, including cancer cells and cells carrying intracellular pathogens. CTLs recognizing self- and non-self-antigens presented by MHC class I, which is found on all nucleated cells. CTLs carry out their effector functions through two main actions. First is release of anti-viral and anti-tumor cytokines, primarily IFN-γ and TNF-α. Second is by directly killing cells, either through release of cytotoxic granules or by Fas/FasL interactions [29]. In the synovium of OA joints, CD8+ T cells are present, but at significantly lower numbers than CD4+ T cells [30].
Interestingly, in OA, there is an increase in the CD4+:CD8+ ratio, and a decrease in the total number of CTLs in the patients’ peripheral blood [30]. In a mouse model of anterior cruciate ligament transection, CD8+ T cells infiltrated synovial fluid of afflicted joints within 30 days and persisted for 90 days. Additionally, CD8+ T cells expressed TIMP1, a regulator of matrix metalloproteinases and disintegrin-metalloproteinases, which helps to maintain extracellular matrix composition, and the number of CD8+ T cells expressing TIMP1 correlated positively with disease severity. Moreover, increased TIMP1, VEGF, and MMP13 in the synovium correlated with CD8+ T cell activation [31]. CD8+ T cells therefore may contribute to imbalance of joint metabolism through dysregulation of both TIMP1 and MMP13, and angiogenesis leading to synovial inflammation.
Aside from these studies, CD8+ T cells in the OA joint have remained somewhat unexplored. In RA, CD8+ T cells are detected in synovium prior to clinical symptoms [32]. Within RA synovial fluid, there is an accumulation of autoreactive CD8+ T cells that are clonally related [33] and are associated with disease severity and breakdown of self-tolerance. Conversely, suppressor CD8+ T cells in the joint may play a role in disease mitigation by inhibiting the functions of autoreactive CD4+ T cells [34]. Additional studies into CD8+ T cells will aid in understanding their contribution to OA initiation and progression, and potentially reveal new therapeutic options for OA patients.
4 T Helper 1 Cells (Th1)
Th1 cells, driven by IL-12 and IFN-γ and controlled by T-bet to produce IFN-γ, [17] are the most abundant T helper cell subset in the synovial fluid and synovium of patients with OA [4]. And although there are fewer Th1 cells in the synovium of patients with OA compared to RA, the Th1 cells present in both types of diseased tissue expressed similar transcript levels of IFN-γ when stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin [35]. Th1 cells in the synovial fluid of patients with OA also secrete higher concentrations of IFN-γ than circulating Th1 cells in the peripheral blood upon PMA and ionomycin stimulation [36]. Within 30 days of anterior cruciate ligament transection in a mouse model of OA, IFN-γ+ cell numbers increased in the synovium, and subsequently decreased by 90 days post-induction. This was associated with an increase in MIP-1γ and number of osteoclasts, while CD4 knockout mice had lower concentrations of MIP-1γ and slower cartilage degeneration [37].
Importantly, while Th1 cells are the most abundant CD4+ T cell subtype in the OA joint, they may not be the most inflammatory. In vitro, Th17 cells induce synthesis of IL-6, IL-8, MMP1, and MMP3 in synovial fibroblasts from patients with early RA, whereas Th1 and naïve CD4+ T cells do not [38]. It has also been reported that in patients with RA, Th1 cells and related cytokines are only significantly increased in peripheral blood in late-stage disease, while Th17 cells and their related cytokines are significantly elevated throughout disease progression [39].
While the role of Th1 cells in the progression of OA remains unclear, their orchestration of the effector functions of macrophages and monocytes is likely partially responsible for continued inflammation within the joint. However, evidence from RA would suggest that the contribution of Th1 cells to rheumatic disease is more prominent during later disease stages and that other CD4+ T cell subtypes, including Th17 cells, are more important drivers of pathogenesis early in the disease process.
5 T Helper 2 Cells (Th2)
Th2 cells that respond to IL-4 and produce IL-4, IL-5, IL-9, and IL-13 under the control of GATA3 are involved in mucosal immunity and the immune responses to extracellular pathogens and tissue repair, [40] but it is currently unclear whether or not Th2 cells are important contributors to the pathogenesis of OA. In the synovium of patients undergoing total knee replacement, neither IL-4 nor IL-5 mRNA transcripts were found in any of the 18 patients [35]. Moreover, synovial fluid cells from OA patients stimulated with PMA and ionomycin did not express levels of IL-4 that were detectable by RT-qPCR after 24 or 72 h of stimulation [41]. Additional studies have failed to find Th2-related transcripts within the joints of OA patients [42]. However, using flow cytometry, low numbers of CD4+IL-4+ cells have been found in the synovial fluid of OA patients [36] and appear in similar frequencies within the synovium when compared to RA patients [43]. IL-4+ cells were also found in all three layers of the synovium using immunohistochemistry, albeit at very low numbers when compared to IFN-γ+ cells or CD4+ cells in total [4].
Therefore, Th2 infiltration into the joints of OA patients appears to be sparse and sporadic, but within these patient subsets, they may aid in disease mitigation through reprogramming of macrophages toward an anti-inflammatory and reparative phenotype, or by secretion of cytokines that can protect tissues of the joints from pro-inflammatory and catabolic cytokines. Overall, further investigation is required to elucidate the role of Th2 cells in early OA.
6 T Helper 17 Cells (Th17)
Th17 cells secrete IL-17 family cytokines, IL-22, and GM-CSF in response to IL-1β, IL-6, and IL-23 and under the control of RORγt, and are implicated in a variety of chronic inflammatory and autoimmune diseases, including rheumatoid arthritis (RA) and psoriatic arthritis (PsA) [44]. Th17 cells were discovered as a distinct T helper subset in 2005, and so have not been well scrutinized under the lens of OA, but in vitro and in vivo models of OA and RA indicate that they play a considerable role in OA initiation and progression [45].
In human patients with OA, IL-17A was significantly increased in synovial fluid compared to undetectable in unmatched, healthy controls [30]. Furthermore, an increase in IL-17A within the joint has been positively correlated with pain and severity of disease in patients with knee OA [46]. In OA patients with inflamed synovium, gene and protein expression of IL-17A and IL-22 were increased in inflamed regions compared to non-inflamed OA synovium, and correlated with the release of IL-6 and IL-23 [47]. This indicates that not only are Th17 cells present and active within the joint but that joint inflammation is associated with their maintenance. Additionally, in early joint trauma (ACL tear), the soluble form of IL-17 receptor A, which transduces IL-17 signaling, was increased 128% in synovial fluid at a mean of 14 days post-injury compared to six days post-injury [48].
The presence of elevated IL-17A concentrations in the joint is thought to contribute directly to joint inflammation, tissue remodeling, and loss of function. In vitro, IL-17A treatment of human cartilage explants or synoviocytes activated NF-κB and led to increased synthesis of MMP1, MMP13, [49] and collagenase-3, all of which contribute to matrix loss [50]. Importantly, in a co-culture system of T cells and synovial fibroblasts from early RA patients, Th1 cells did not elicit the same catabolic responses that Th17 cells did, suggesting that Th17, not Th1, responses may be more responsible for joint destruction in OA [38]. In addition, IL-17A enhanced the expression of IL-6 and IL-8 from synovial fibroblasts, aiding in the maintenance of the Th17 phenotype and perpetuating immune cell homing to the inflamed joint [51]. Moreover, IL-17A further promoted T cell recruitment by upregulating the expression of CCL2 and CCL20 expression in synovial fibroblasts [52]. These data suggest that Th17 cells, particularly through their ability to produce large amounts of IL-17, are important players in the loss of joint homeostasis in OA.
Studies in murine models are consistent with these in vitro and ex vivo findings in humans. In murine models of collagen-induced arthritis, treatment with anti-IL-17A neutralizing antibody reduced, though did not eliminate, synovitis, and cartilage damage [53]. Furthermore, arthritis was considerably diminished in IL-17A-deficient mice compared to wildtype mice. Not only did fewer IL-17A-deficient mice develop arthritis, but those than did had lower arthritis scores [54]. Of note, other immune cell types, such as local synovial macrophages, participate in the orchestration of Th17 responses, where they promote differentiation and maintenance of Th17 cells within the synovium [55]. Mouse models have also shed light on the contribution of other Th17-derived factors, such as IL-22, to arthritic disease. For example, IL-22 mRNA and protein expression were increased during onset of antigen-induced arthritis in mice, while the use of an anti-IL-22 antibody and IL-22 deficiency in mice attenuated pain and reduced synovitis, suggesting that targeting of multiple cytokines released by Th17 can reduce arthritis symptoms [56].
Taken together, data from human OA and RA patients, as well as in vitro and in vivo models, suggest that Th17 cells and cytokines play a role in the establishment and progression of OA. IL-17A has been shown to have a direct inflammatory role on synoviocytes and chondrocytes by initiating and perpetuating catabolism and homing of additional immune cells to the inflamed joint. It will be critical to continue investigation of Th17 cells in early OA in order to find targets for which new therapeutics can be made, or for which existing therapeutics can be implemented to mitigate OA progression.
7 Regulatory T Cells (Tregs)
TGF-β and IL-2 induce activation of the Foxp3 transcription factor in Regulatory T cells, which can occur in the thymus or periphery to give rise, respectively, to natural or induced Tregs. Tregs produce immunosuppressive cytokines such as IL-10 and TGF-β and dampen immune activation through cell-to-cell interactions and by acting as an IL-2 “sink” to prevent IL-2-associated activation of auto-reactive naïve T cells. Tregs are key players in a multitude of autoimmune and inflammatory diseases, with disease emergence and progression often associated with a lack of Tregs at critical sites or a failure of Tregs to control or arrest ongoing T cell activation [57].
In patients with mild to severe OA, there is an increase in the percentage of cells in peripheral blood that exhibit a Treg phenotype. However, when stimulated with PMA and ionomycin, these cells were significantly inhibited in their ability to secrete IL-10 [58]. The inability of T cells to carry out their effector functions can be indicative of overstimulation and subsequent exhaustion. Within the context of OA, this could be a potential consequence of chronic inflammation in the joint. Evidence from RA patients would also suggest that peripheral Tregs have a reduced ability to suppress aberrant activation of effector CD4+ T cells through cell-to-cell interactions. This is due to defects in expression of the immune checkpoint molecule CTLA-4, which competitively binds to B7 family members on antigen-presenting cells, blocking effector T cell activation [59].
Not only are Tregs enriched within the peripheral blood of OA patients, they are enriched within the synovium and synovial fluid. There is evidence that Tregs infiltrate the joint during the acute phase of inflammation and are highly active in this phase. Following acute ACL tear, IL-10 increased in synovial fluid, but waned as early as 3 months post-injury [60]. Tregs within the synovium of patients with chronic OA displayed an activated effector memory phenotype compared to peripheral blood Tregs, which displayed a resting central memory phenotype [61]. Moreover, IL-10 transcripts were detected in the synovium of nearly all OA patients [35]. Taken together, these data indicate that Tregs are present during initiation of inflammation, persist in the joint, and may actively attempt to suppress inflammation but are unable to return to joint homeostasis.
Animal models support the role of Tregs and IL-10 in chondroprotection. IL-10 knockout mice with collagen-induced arthritis developed more severe arthritis scores than wildtype mice, which was associated with an increase in production of Th1 and Th17 cytokines, and polarization of macrophages toward an M1 phenotype [62, 63]. In a rabbit model of OA, intra-articular injection of synoviocytes overexpressing IL-10 through retroviral gene transfer five days post-excision of the medial collateral ligament plus medial meniscectomy improved histological scores compared to controls [64]. While absence of IL-10 leads to more severe arthritis, presence and over-expression of IL-10 do not appear to mitigate disease progression in the long term, suggesting that Treg cytokines alone are not sufficient to resolve inflammation.
The continued progression of OA suggests that Treg secreted factors and Treg cell-to-cell contact-mediated suppressor functions are not sufficient to mitigate disease progression. This is in spite of early Treg migration to and activation within the inflamed joint. Furthermore, evidence suggests that Treg activity is dampened as disease progresses, rendering these cells unable to mount suppressive functions that could help control inflammation in the joint to promote repair, thus contributing to OA pathogenesis and failure of disease mitigation.
8 Th17: Treg Phenotype Plasticity
CD4+ T helper cell lineages were originally thought to be stable; however, plasticity between Th17 and Treg phenotypes has now been described in multiple contexts including uveitis and scleritis, as well as RA [65]. This instability, within the context of normal physiological conditions, aids in overcoming infections, preventing collateral tissue damage, and resolution of inflammation [66]. However, when plasticity becomes unregulated, it can lead to uncontrolled inflammatory T-cell responses. While transcription factors RORγt and Foxp3, respectively, drive Th17 and Treg phenotype and function, the cytokine microenvironment can activate the reciprocal transcription factor, leading to phenotype plasticity (Fig. 11.3) [67, 68]. The result is that T cell function is altered through simultaneous activation of both transcription factors, and cells are subsequently able to acquire the capabilities of both subsets; secreting Th17 cytokines while eliciting Treg suppressor functions [69].

Th17:Treg plasticity may contribute to disease pathogenesis. Although T helper cell phenotypes were thought to be terminal and stable following naïve T cell differentiation, there can be plasticity between Th17 and Tregs. Cytokines within the OA joint can activate the reciprocal transcription factor, leading to an intermediate cell type that secretes Th17 cytokines and, in some contexts, is also capable of carrying out Treg suppressor functions. Instability in Treg phenotype may play a role in loss of joint homeostasis and continued catabolism
Evidence from diseases specifically affecting the joint, including RA and PsA, suggests this plasticity could be involved in the failure of OA resolution as well.
In chronic diseases, persistence of inflammatory cytokines, including IL-1β, IL-6, and IL-23, can lead to destabilization of the Foxp3 transcription factor in Tregs. These cytokines promote the expression of RORγt, yet the resultant Th17-like Tregs also maintain Foxp3 expression, though these cell do not always fully maintain the effector functions of true Tregs [69]. Evidence in mice supports a role for synoviocytes during the induction of Treg phenotype plasticity. For example, in a mouse model of collagen-induced arthritis, Foxp3+ T cells secrete IL-17 following incubation with rheumatoid fibroblast-like synoviocytes, indicating that fibroblast-like synoviocytes from the inflamed joint are sufficient to induce conversion of local Foxp3+CD4+ T cells to Foxp3+CD4+IL-17A+ cells, exacerbating early inflammation [70].
Increased Th17-like Treg cells can be found in the blood of RA patients and is positively correlated with an increase in Th17 cells in the peripheral blood [71]. A parallel enrichment of Th17 cells in the peripheral blood of OA patients suggests that they also exhibit an increase in peripheral blood Th17-like Tregs [72]. Although these Th17-like Tregs begin to secrete IL-17A, they are still capable of suppressing effector T cell proliferation ex vivo. Conversely, Th17-like Tregs within the joint of RA patients do not maintain suppressor functions, and through secretion of IL-17A, likely contribute to disease progression. There is an upregulation of IL-1β and IL-6 in the synovial tissues following injury, and an upregulation of IL-23 in the peripheral blood of OA patients, further suggesting that Treg phenotypic switching is involved in the pathogenesis of OA [73, 74].
While pro-inflammatory cytokines can lead to Treg phenotype switching, during the resolution phase of inflammation, anti-inflammatory cytokines can induce Th17 cells to convert to a regulatory phenotype through upregulation of Foxp3. In a mouse model of colitis, during resolution of inflammation, high concentrations of TGF-β1 decreased RORγt activity in a dose-dependent manner and caused Th17 cells to transdifferentiate into IL-17A+Foxp3+ cells, which simultaneously secrete IL-17A and IL-10 [75]. Furthermore, TGF-β1 and PGE2 secreted by mouse and human tumor cells induced Foxp3 expression and subsequent suppressor functions in Th17 cells [76]. Retinoic acid, a driver of Foxp3 activation in inducible Tregs, has also been implicated in suppression of Th17 phenotype [77]. However, it is not clear how retinoic acid concentrations vary within the joint, and whether increased retinoic acid contributes more strongly to cartilage destruction or immunomodulation in OA [78]. Regardless, increased concentrations of TGF-β1 and PGE2 observed in joints of OA patients suggest that there is potential to drive infiltrating Th17 cells to express Foxp3 and limit the pro-inflammatory and pro-catabolic functions of Th17 cells [18, 79].
Observations made of IL-17A+Foxp3+ cells and Th17-like Tregs in other diseases suggest that phenotype plasticity between Th17 and Tregs may play a role in OA pathogenesis. Investigating plasticity in Th17 and Treg phenotype will potentially increase our understanding of how T cells respond to the local joint environment in a context-dependent manner, which will be an important step toward developing and applying immunotherapies for early OA.
9 T Cell-Targeted Immunotherapies for OA
During progression of OA, homeostasis is lost in favor of a catabolic state, where catabolism is defined as progressive and irreversible joint destruction and pain. It is now accepted that disease modification must occur early before this destruction becomes irreversible [80, 81]. Immunotherapy is the use of treatment that targets the immune response, which can include stimulation or suppression, in order to modify disease progression. In the case of autoimmune disease, including RA, immunotherapies that suppress and block aberrant immune function have been successfully implemented for several decades to protect the patient from chronic pain and joint destruction [82]. While some of the key pathways in OA could similarly be targeted using existing immunotherapies, thus far, immunotherapy has not been a mainstay of OA treatment due to inconsistent patient results. For example, use of the anti-TNF-α monoclonal antibody (mAb) therapy adalimumab failed to reduce pain and symptoms in patients with erosive hand OA but did improve joint stiffness and WOMAC pain in patients with knee OA [83]. Currently, there are therapies available and in use for other diseases that target several key areas of the T cell response, including T cell homing, T cell activation and maintenance, and T cell effector functions (Fig. 11.4). A number of these could be leveraged to treat various aspects of the inflammatory response in OA to limit or perhaps even reverse joint destruction. However, understanding more about T cells in the pathogenesis of OA will be important for the targeted use of immunotherapies in OA.

Immunotherapies that target different aspects of T cell response offer new intervention options for OA mitigation. There may be missed opportunities to rapidly translate existing immunotherapies for use in OA. Therapies that target antigen presentation to T cells, T cell trafficking, activation, phenotype plasticity, and effector cytokines are already available and approved for use in other T cell-mediated diseases and could be used to limit or mitigate progression of OA if applied at the right time during disease imitation
As stated previously, acutely following joint damage, chondrocytes and synoviocytes release a cascade of cytokines and chemokines that not only affect the local joint environment but also home immune cells, including T cells, to the damaged joint. One potential method to reduce T cell-induced inflammation within the joint is to stop T cell trafficking to the joint by either blocking chemokines or their receptors. In RA, treating patients with an antagonist against CCR1, which is expressed by T cells and binds CCL5, reduced the number of CD4+ and CD8+ T cells within the synovium after only 14 days of treatment and significantly reduced the number of tender and swollen joints [84]. Although all patients in the study had firmly established disease, application of such a therapy to early OA may restore homeostasis and promote complete repair of damaged tissue. In a murine model of collagenase-induced OA, treatment with mAb therapy targeting the T cell chemoattractant CCL17 ameliorated the pain and significantly reduced histological score and osteophyte size [25]. Thus, blockade of T cell trafficking chemokines and their receptors may present viable options for OA mitigation.
Another approach to modifying disease progression in OA is to use therapies that target T cell activation. This can be achieved through several pathways, including blockage of extracellular signaling, or stopping downstream transcription factor activation. One approach to limit T cell activation is to block co-stimulation. Used for the treatment of RA, abatacept is a fusion protein composed of the extracellular domain of the immune checkpoint molecule CTLA-4 and the Fc portion of IgG1. Abatacept shuts down T cell activation by antigen-presenting cells, and within the context of RA, helps to prevent T cell recognition of self-antigen, a driving factor of the disease [85]. It is also possible to target T cell activation more directly. For example, cyclosporin is an immunosuppressant used in the treatment of chronic diseases, such as RA and Crohn’s disease, that targets calcineurin, which is a signaling molecule critical for T cells to elicit effector functions [86]. Calcineurin activates nuclear factor of activated T cell cytoplasmic (NFATc), which upregulates downstream T cell responses. These therapies hold potential to mitigate and alleviate OA symptoms within the context of aberrant T cell activation. However, considering previous failures and mixed results of immunotherapy in OA patients, it will be pertinent to target patients who actively present with T cell-driven OA or else intervention is likely to be ineffective.
A third approach is to target inflammatory cytokines that are secreted by activated T cells. For example, anti-IL-17A therapy has been met with success in the treatment of RA and PsA where, as with OA, IL-17A secreted by Th17 cells is increased in the synovial fluid compared to healthy patients. In PsA patients, treatment with anti-IL-17A therapy reduced radiographic disease progression, effectively inhibiting structural degeneration of the joint [87]. Furthermore, RA patients in some clinical trials who did not respond to anti-TNA-α antibody had reduced disease severity following treatment with anti-IL-17A mAbs [88]. Although mAb therapy targeting the Th1 cytokine TFN-α has previously produced mixed results in OA patients, it is possible that this is because treatment was applied too late in the progression of OA [83]. While treatment of anti-TNF-α therapy did not appear to mitigate disease nor reduce clinical symptoms of several cohorts of patients with end-stage hand OA, it did yield promising results in patients with knee OA with a Kellgren–Lawrence grade of 2–3. Treated patients had significant improvements in WOMAC pain score, stiffness, and function [89].
A final approach is the targeting of cytokines that maintain T cell phenotype and/or promote plasticity toward a pro-inflammatory phenotype. Of note, anti-IL-23 and combination anti-IL-12/23 antibodies that target the p40 region common to both cytokines have undergone phases II and III clinical trials in RA and PsA patients. IL-23 and IL-12 drive and maintain Th17 and Th1 phenotypes, respectively. In PsA patients, anti-IL-12/23 therapy inhibited radiographic progression of joint damage. However, in patients with RA, while there was numerically higher improvement in tender and swollen joints following anti-IL-12/23 treatment, neither the aforementioned treatment nor anti-IL-23 treatment significantly improved RA symptoms [90]. This may be partially explained by the findings that, in a murine model of collagen-induced arthritis, IL-12-driven Th1 activity was not responsible for collagen-induced arthritis, but IL-23 was responsible for T cell-mediated flare-ups, indicating that timing of anti-IL-23 is critical for mitigation of inflammation driven by T cells [91].
Taken together, findings from use of immunotherapies in OA and related diseases warrant further exploration of their application early in the OA disease process. This is before irreversible joint destruction has occurred when there is still the possibility of mitigating catabolism and returning the joint to homeostasis. However, the dynamic nature of OA also calls for a better understanding of T cell involvement during early stages and progression of disease, so that we are able to not only to identify targets for immunotherapies but also timing of when those therapies will be most effective at mitigating disease.
10 Conclusion
The immune response is one of a number of critical factors that contribute to disease pathogenesis of OA. There is mounting evidence that T cell populations are altered not only in the synovium and synovial fluid of those afflicted with OA but also within the peripheral blood. Although a variety of CD8+ and CD4+ T cells infiltrate the joint acutely following injury, work in human patients and in animal models indicates that Th17 and Treg effector functions and phenotype plasticity within the joint environment play critical roles in the balance between catabolism and anabolism subsequent to joint damage.
Understanding how T cells contribute to OA initiation and progression presents the opportunity to use immunotherapies that successfully modulate T cell activities as has been done in other inflammation-mediated diseases, including RA and PsA. To date, there have been mixed outcomes of clinical trials using immunotherapies in OA patients, perhaps because the application of these therapies targeted the wrong T cell population or because of T cell plasticity, or more simply, they were used too late in the disease process, when cartilage damage is complete and irreversible. Therefore, it is important that the role of T cells in early OA continues to be investigated to yield new insights into OA as an immune-mediated disease. This will be critical for identifying novel immunotherapies that can truly modify the course of OA and mitigate disease progression.
References
Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr Cartil. 2013;21:16–21.
Sakkas LI, Platsoucas CD. The role of T cells in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007;56(2):409–24.
Lopes EBP, et al. Immune contributions to osteoarthritis. Curr Osteoporos Rep. 2017;15(6):593–600.
Ishii H, et al. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthr Cartil. 2002;10:277–81.
Alsalameh S, et al. Cellular immune response toward human articular chondrocytes. T cell reactivities against chondrocyte and fibroblast membranes in destructive joint diseases. Arthritis Rheum. 1990;33:1477–86.
De Jong H, et al. Cartilage proteoglycan aggrecan epitopes induce proinflammatory autoreactive T-cell responses in rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 2010;69:255–62.
Borowski C, et al. On the brink of becoming a T cell. Curr Opin Immunol. 2002;14:200–6.
Enouz S, et al. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med. 2012;209:1769–79.
Smith-Garvin JE, et al. T cell activation. Annu Rev Immunol. 2009;27:591–619.
Tran CN, et al. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology. 2005;12:183–9.
Wehr P, et al. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin Exp Immunol. 2019;196:12–27.
Revell PA, et al. The synovial membrane in osteoarthritis: a histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis. 1988;47:300–7.
Sarkar S, Fox DA. Dendritic cells in rheumatoid arthritis. Front Biosci. 2005;10:656–65.
Xiaoqiang E, et al. Dendritic cells of synovium in experimental model of osteoarthritis of rabbits. Cell Physiol Biochem. 2012;30:23–32.
Wheeler TA, et al. T cells mediate progression of load-induced osteoarthritis. bioRxiv. 2020.
Zhu J, et al. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–89.
Luckheeram RV, et al. CD4 +T cells: differentiation and functions. Clin Dev Immunol. 2012;2012:925135.
Haseeb A, Haqqi TM. Immunopathogenesis of osteoarthritis. Clin Immunol. 2013;146:185–96.
Mellado M, et al. Targeting cell migration in rheumatoid arthritis. Front Immunol. 2015;6:1–12.
Schaller MA, et al. A key role for CC chemokine receptor 1 in T-cell-mediated respiratory inflammation. Am J Pathol. 2008;172:386–94.
Danilova E, et al. A role for CCL28-CCR3 in T-cell homing to the human upper airway mucosa. Mucosal Immunol. 2015;8:107–14.
Veazey RS, et al. Dynamics of CCR5 expression by CD4+ T cells in lymphoid tissues during simian immunodeficiency virus infection. J Virol. 2000;74:11001–7.
Takebe K, et al. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthr Cartil. 2015;23:454–61.
Chung L, et al. Interleukin-17 and senescence regulate the foreign body response. bioRxiv. 2019.
Lee MC, et al. CCL17 blockade as a therapy for osteoarthritis pain and disease. Arthritis Res Ther. 2018;20:1–10.
Alaaeddine N, et al. CCL20 stimulates proinflammatory mediator synthesis in human fibroblast-like synoviocytes through a MAP kinase-dependent process with transcriptional and posttranscriptional control. J Rheumatol. 2011;38:1858–65.
Xu Q, et al. Association of CXCL12 levels in synovial fluid with the radiographic severity of knee osteoarthritis. J Investig Med. 2012;60:898–901.
Kim KW, et al. Up-regulation of stromal cell-derived factor 1 (CXCL12) production in rheumatoid synovial fibroblasts through interactions with T lymphocytes: role of interleukin-17 and CD40L-CD40 interaction. Arthritis Rheum. 2007;56:1076–86.
Halle S, et al. Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. 2017;38:432–43.
Hussein MR, et al. Alterations of the CD4+, CD8+ T cell subsets, interleukins-1β, IL-10, IL-17, tumor necrosis factor-α and soluble intercellular adhesion molecule-1 in rheumatoid arthritis and osteoarthritis: preliminary observations. Pathol Oncol Res. 2008;14:321–8.
Hsieh JL, et al. CD8+ T cell-induced expression of tissue inhibitor of metalloproteinses-1 exacerbated osteoarthritis. Int J Mol Sci. 2013;14:19951–70.
De Hair MJH, et al. Features of the synovium of individuals at risk of developing rheumatoid arthritis: implications for understanding preclinical rheumatoid arthritis. Arthritis Rheumatol. 2014;66:513–22.
Carvalheiro H, et al. CD8+ T cell profiles in patients with rheumatoid arthritis and their relationship to disease activity. Arthritis Rheumatol. 2015;67:363–71.
Kang YM, et al. CD8T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med. 2002;195:1325–36.
Sakkas LI, et al. T cells and T-cell cytokine transcripts in the synovial membrane in patients with osteoarthritis. Clin Diagn Lab Immunol. 1998;5:430–7.
Dolganiuc A, et al. Shift toward T lymphocytes with Th1 and Tc1 cytokine-secretion profile in the joints of patients with osteoarthritis. Roum Arch Microbiol Immunol. 1999;58:249–58.
Shen PC, et al. T helper cells promote disease progression of osteoarthritis by inducing macrophage inflammatory protein-1γ. Osteoarthr Cartil. 2011;19:728–36.
Van Hamburg JP, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum. 2011;63:73–83.
Chen J, et al. Comprehensive evaluation of different T-helper cell subsets differentiation and function in rheumatoid arthritis. J Biomed Biotechnol. 2012;2012:535361.
Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. 2018;18:121–33.
Haynes MK, et al. Phenotypic characterization of inflammatory cells from osteoarthritic synovium and synovial fluids. Clin Immunol. 2002;105:315–25.
de Lange-Brokaar BJE, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthr Cartil. 2012;20(12):1484–99.
Yudoh K, et al. Reduced expression of the regulatory CD4+ T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis Rheum. 2000;43:617–27.
Tesmer LA, et al. Th17 cells in human disease. Immunol Rev. 2008;223:87–113.
Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32.
Liu Y, et al. Correlation of IL-17 level in synovia and severity of knee osteoarthritis. Med Sci Monit. 2015;21:1732–6.
Deligne C, et al. Differential expression of interleukin-17 and interleukin-22 in inflamed and non-inflamed synovium from osteoarthritis patients. Osteoarthr Cartil. 2015;23:1843–52.
King JD, et al. Joint fluid proteome after anterior cruciate ligament rupture reflects an acute posttraumatic inflammatory and chondrodegenerative state. Cartilage. 2018;11(3):329–37.
Moran EM, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-α, Oncostatin M and response to biologic therapies. Arthritis Res Ther. 2009;11:1–12.
Benderdour M, et al. Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: differential activation of AP-1 members by IL-17 and IL-1β. J Rheumatol. 2002;29(6):1262–72.
Hwang S-Y, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6:R120–8.
Hattori T, et al. Gene expression profiling of IL-17A-treated synovial fibroblasts from the human temporomandibular joint. Mediators Inflamm. 2015;2015:1–12.
Lubberts E, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–9.
Nakae S, et al. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–7.
Egan PJ, et al. Promotion of the local differentiation of murine Th17 cells by synovial macrophages during acute inflammatory arthritis. Arthritis Rheum. 2008;58:3720–9.
Pinto LG, et al. Joint production of IL-22 participates in the initial phase of antigen-induced arthritis through IL-1β production. Arthritis Res Ther. 2015;17:1–13.
Sharabi A, et al. Regulatory T cells in the treatment of disease. Nat Rev Drug Discov. 2018;17(11):823–44.
Li S, et al. Downregulation of IL-10 secretion by Treg cells in osteoarthritis is associated with a reduction in Tim-3 expression. Biomed Pharmacother. 2016;79:159–65.
Flores-Borja F, et al. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2008;105:19396–401.
Bigoni M, et al. Acute and late changes in intraarticular cytokine levels following anterior cruciate ligament injury. J Orthop Res. 2013;31:315–21.
Moradi B, et al. CD4+CD25+/highCD127low/- regulatory T cells are enriched in rheumatoid arthritis and osteoarthritis joints-analysis of frequency and phenotype in synovial membrane, synovial fluid and peripheral blood. Arthritis Res Ther. 2014;16(2):R97.
Finnegan A, et al. Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res Ther. 2003;5:R18–24.
Ye L, et al. Interleukin-10 attenuation of collagen-induced arthritis is associated with suppression of interleukin-17 and retinoid-related orphan receptor γt production in macrophages and repression of classically activated macrophages. Arthritis Res Ther. 2014;16:1–14.
Zhang X, et al. Suppression of early experimental osteoarthritis by gene transfer of interleukin-1 receptor antagonist and interleukin-10. J Orthop Res. 2004;22:742–50.
Diller ML, et al. Balancing inflammation: the link between Th17 and regulatory T cells. Mediators Inflamm. 2016;2016:1–8.
Sehrawat S, Rouse BT. Interplay of regulatory T cell and Th17 cells during infectious diseases in humans and animals. Front Immunol. 2017;8:341.
Zhou L, et al. TGF-β-induced Foxp3 inhibits Th17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–40.
Amarnath S, et al. Endogenous TGF-β activation by reactive oxygen species is key to Foxp3 induction in TCR-stimulated and HIV-1-infected human CD4+CD25-T cells. Retrovirology. 2007;4:1–16.
Qiu R, et al. Regulatory T cell plasticity and stability and autoimmune diseases. Clin Rev Allergy Immunol. 2018;58(1):52–70.
Komatsu N, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–8. https://doi.org/10.1038/nm.3432
Wang T, et al. Regulatory T cells in rheumatoid arthritis showed increased plasticity toward Th17 but retained suppressive function in peripheral blood. Ann Rheum Dis. 2015;74:1293–301.
Qi C, et al. Circulating T helper 9 cells and increased serum interleukin-9 levels in patients with knee osteoarthritis. Clin Exp Pharmacol Physiol. 2016;43:528–34.
Wojdasiewicz P, et al. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459.
Askari A, et al. Increased serum levels of IL-17A and IL-23 are associated with decreased vitamin D3 and increased pain in osteoarthritis. PLoS One. 2016;11:1–8.
Gagliana N, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–5.
Downs-Canner S, et al. Suppressive IL-17A+ Foxp3+ and ex-Th17 IL-17Aneg Foxp3+ Treg cells are a source of tumour-associated Treg cells. Nat Commun. 2017;8:14649.
Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (80- ). 2007;317:256–9.
Blasioli DJ, Kaplan DL. The roles of catabolic factors in the development of osteoarthritis. Tissue Eng Part B Rev. 2014;20:355–63.
van der Kraan PM. Factors that influence outcome in experimental osteoarthritis. Osteoarthr Cartil. 2017;25:369–75.
Van Steenbergen HW, et al. Preventing progression from arthralgia to arthritis: targeting the right patients. Nat Rev Rheumatol. 2018;14:32–41.
Mahmoudian A, et al. Towards secondary prevention of early knee osteoarthritis. RMD Open. 2018;4:1–12.
Meier FM, et al. Current immunotherapy in rheumatoid arthritis. Immunotherapy. 2013;5:955–74.
Zheng S, et al. Monoclonal antibodies for the treatment of osteoarthritis. Expert Opin Biol Ther. 2016;16:1529–40.
Haringman JJ, et al. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62:715–21.
Cutolo M, et al. CTLA-4 blockade in the treatment of rheumatoid arthritis: an update. Expert Rev Clin Immunol. 2016;12:417–25.
Perry M. Management of rheumatoid arthritis in primary care. Pract Nurs. 2017;28:1337–8.
Garcia-Montoya L, Marzo-Ortega H. The role of secukinumab in the treatment of psoriatic arthritis and ankylosing spondylitis. Ther Adv Musculoskelet Dis. 2018;10:169–80.
Huang Y, et al. Efficacy and safety of secukinumab in active rheumatoid arthritis with an inadequate response to tumor necrosis factor inhibitors: a meta-analysis of phase III randomized controlled trials. Clin Rheumatol. 2019;38:2765–76.
Maksymowych WP, et al. Targeting tumour necrosis factor alleviates signs and symptoms of inflammatory osteoarthritis of the knee. Arthritis Res Ther. 2012;14:1–7.
Frieder J, et al. Anti-IL-23 and anti-IL-17 biologic agents for the treatment of immune-mediated inflammatory conditions. Clin Pharmacol Ther. 2018;103:88–101.
Furst DE, Emery P. Rheumatoid arthritis pathophysiology: update on emerging cytokine and cytokine-associated cell targets. Rheumatology (Oxford). 2014;53:1560–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 ISAKOS
About this chapter

Cite this chapter
Keller, L.E., Fortier, L.A., Tait Wojno, E.D. (2022). T Cells in Early Osteoarthritis. In: Lattermann, C., Madry, H., Nakamura, N., Kon, E. (eds) Early Osteoarthritis. Springer, Cham. https://doi.org/10.1007/978-3-030-79485-9_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-79485-9_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-79484-2
Online ISBN: 978-3-030-79485-9
eBook Packages: MedicineMedicine (R0)