
Abstract
Mastocytosis is a rare hematologic disorder characterized by abnormal proliferation and accumulation of neoplastic mast cells in various body sites. Isolated skin involvement is termed cutaneous mastocytosis (CM) and the term systemic mastocytosis (SM) refers to multi-organ involvement, most commonly of the bone marrow, skin, liver, and spleen. A subset of patients with SM have an associated clonal hematologic neoplasm which is most commonly myelodysplastic syndrome, chronic myelomonocytic leukemia, or acute myelogenous leukemia and this entity is termed SM with associated hematological neoplasm (AHN). Bone marrow involvement is present in all patients regardless of the subtype of SM. The genetic hallmark of SM is a somatic gain-of-function point mutation within the KIT gene. Other molecular aberrations that have been reported include somatic mutations in TET2, SRSF2, ASXL1, CBL, RUNX1, and RAS and these are common in SM-AHN. The clinical presentation of SM can range from indolent to advanced depending on extent of mast cell burden and genetic profile. In the case of indolent SM, the goal of treatment is to control mediator release-related effects as well as to reduce mast cell burden. In the case of SM-AHN, therapy is primarily that of the AHN and allogeneic hematopoietic stem cell transplantation is the preferred therapy in suitable candidates.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


10.1 Introduction
Mastocytosis is a rare hematologic neoplasm characterized by abnormal proliferation and accumulation of clonal mast cells. Mastocytosis limited to skin is termed cutaneous mastocytosis and will not be discussed further here. Systemic mastocytosis (SM) is characterized by aggregates of atypical mast cells in bone marrow (major criterion for diagnosis defined by World Health Organization) along with infiltration of other organs among which skin, liver, and spleen are most common. Clinical symptoms arise either from release of mediators from mast cells or in advanced cases from organ dysfunction related to mast cell infiltration. In a large subset of cases, SM coexists with an associated hematological neoplasm (AHN) and in such cases features of the hematologic malignancy dominate the clinical picture. Cases of indolent SM (ISM) comprise the other major subset and are characterized by skin involvement and mediator release symptoms. Rare subsets of SM include aggressive SM (ASM) and mast cell leukemia (MCL) [1].
10.2 Biology and Genetics of SM
KIT is a receptor tyrosine kinase expressed on mast cells and interaction with its ligand, namely, stem cell factor (SCF) supports mast cell differentiation, survival, and proliferation. A gain-of-function point mutation (most commonly D816V, though others have been reported) in the tyrosine kinase domain of the KIT gene results in ligand-independent activation of the KIT tyrosine kinase [2]. The presence of an activating point mutation at codon 816 of KIT is seen in more than 90% of patients and fulfills one of the minor criteria for diagnosis of SM [1]. There is a high correlation between KIT mutation detection and the proportion of lesional cells in the samples as well as the sensitivity of the screening method. The question as to whether KIT mutation alone is sufficient to cause neoplastic mast cell transformation remains unsettled.
Recent studies have shown that additional mutations identified in SM include TET2, SRSF2, ASXL1, EZH2, CBL, RUNX1, JAK2, and N-RAS and one or more of these are commonly present in advanced SM which includes SM-AHN, ASM, or MCL and these additional mutations are rare in ISM. The precise prognostic implications of individual mutations are unclear but presence of additional mutations besides KIT is associated with worse overall survival [3,4,5]. In the case of SM-AHN, these additional mutations are acquired at the stem cell level prior to the KIT D816V and are present in the mast cell clone as well as the coexisting myeloid malignancy indicating a stepwise pathogenesis [6, 7]. Single cell sequencing studies have shown the KIT mutation occurs in early hematopoietic stem cells and is present to a variable extent in different hematopoietic lineages in subtypes of SM [8, 9].
Cytogenetic abnormalities are a feature of SM-AHN. Abnormalities identified are often single chromosomal abnormalities including +8, del(5q), del(7), -7, +13, and del(20q). However, these cytogenetic abnormalities are not unique to SM and are the characteristic of the associated myeloid malignancy [10]. In cases of SM-AHN, the mast cells carry the same cytogenetic abnormality as the AHN again showing their common clonal origin [11]. A specific association exists between SM and t(8;21) AML and coexistence of SM confers an adverse prognosis to this otherwise favorable AML subset [12].
10.3 Diagnosis and Classification of SM
The WHO diagnostic criteria for systemic mastocytosis are listed in Table 10.1. The major criterion for diagnosis is the presence in the bone marrow of multifocal dense aggregates of mast cells (≥15 cells in aggregates) with spindled morphology, and cytoplasmic hypogranularity, which is in contrast to normal mast cells which are dispersed, round to oval in shape, with prominent cytoplasmic granularity. These aggregates are often seen in a paratrabecular or perivascular location and are accompanied by many small lymphocytes, histiocytes, fibroblasts, and eosinophils [1]. The paratrabecular lesions are sometimes associated with fibrosis and widening of the bony trabeculae and the perivascular forms are often accompanied by hypertrophy of the associated blood vessels. The associated fibrosis renders the mast cells difficult to aspirate and hence a morphologic evaluation of the bone marrow aspirate will not reflect the extent of underlying mast cell proliferation which requires immunohistochemical evaluation of core biopsy specimens.
The stains that are useful in identifying mast cells in bone marrow include tryptase, which is both sensitive and specific for mast cells. Other immunostains that are useful in identifying mast cells include CD117, CD68, CD43, and CD33. Immunophenotypic aberrancy as manifested by mast cell coexpression of CD25 and, less commonly, CD2 is a minor criterion for diagnosis of SM [1]. CD30 (Ki antigen) has been reported to be preferentially expressed in neoplastic mast cells and earlier studies reported its positivity especially in aggressive SM and MCL. However, further studies have reported CD30 expression in ISM as well. Hence, CD30 while useful as a marker for neoplastic mast cells may not be used as a marker for grading SM [13, 14].
The presence of an activating point mutation at codon 816 of KIT fulfills one of the minor criteria for diagnosis of SM. Although this mutation occurs at a high frequency in SM, occurring in more than 90% of SM patients, its detection is dependent on sensitivity of assay used and false negative results may occur [15]. Various factors related to testing can lead to negative results on KIT mutation assays including low sensitivity of next-generation sequencing assays. Less frequently, other mutations outside of tyrosine kinase domain of KIT are present which may be missed in assays directed specifically to KIT D816V. In cases where SM is diagnosed, but KIT D816 mutation is negative, additional sequencing of the KIT gene is recommended since this has therapeutic implications. Since KIT mutations can be found in one-third of cutaneous mastocytosis (CM) cases within lesional skin, the WHO criteria for SM specifies that the KIT mutation must be identified at an extracutaneous site, most commonly within neoplastic mast cells in the bone marrow.
Should the patient meet the WHO criteria for SM, they can be further subclassified based on a combination of histopathologic and clinical features (Table 10.2). Broadly, the multiple variants fall into two general umbrella categories, with different clinical, prognostic, and therapeutic implications: “advanced systemic mastocytosis” versus “indolent” variants. Encompassed in the “advanced systemic mastocytosis” category are SM-AHN, MCL, and aggressive systemic mastocytosis. The “indolent” variants of SM include smoldering SM and indolent SM.
Majority of patients with advanced SM have SM-AHN. Aside from meeting criteria for SM, SM-AHN demonstrates an associated non-mast cell lineage hematologic neoplasm, which can be diagnosed before, with, or after the diagnosis of SM. The associated hematologic neoplasm can be myeloid or lymphoid/plasmacytic in lineage and can range in aggressiveness from an indolent process to acute leukemia. In 90% of cases, the associated hematologic neoplasm is of myeloid lineage including myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPN), chronic myelomonocytic leukemia, or acute myeloid leukemia (AML) [1]. Lymphoid neoplasms associated with SM are not clonally related to the coexisting SM [16]. A subset of AML with the (8;21) RUNX1-RUNX1T1 translocation may show concurrent SM, and the associated mast cell infiltrate may be masked by the acute leukemia infiltrate at initial diagnosis and may not become apparent until following treatment [17]. It is critical to examine bone marrow core biopsy (not aspirate) specimens by immunohistochemistry for mast cell infiltrate if D816 or another activating KIT mutation is detected in myeloid malignancies associated with SM since coexisting SM has adverse prognostic impact especially in AML. Skin involvement as well as symptoms related to mast cell mediator release do not seem to occur in SM-AHN. In most cases, the disease course and prognosis are primarily determined by the associated hematologic neoplasm. Therefore, it is important that the associated hematologic neoplasm is separately designated and classified according to defined WHO criteria.
Mast cell leukemia is an aggressive variant of SM. It is highly lethal with an overall survival of <1 year. On pathology, it is characterized by the presence of immature mast cells, which are round rather than spindled, and range in levels of immaturity from bilobated/multilobated promastocytes to metachromatically granulated blasts to scantly granular but tryptase-positive blasts. These immature mast cells diffusely infiltrate the bone marrow. By definition, the neoplastic mast cells should comprise ≥20% of cells on aspirate smears [1]. Though it represents the leukemic variant of SM, it is more common for MCL to demonstrate <10% circulating mast cells. Typically, evidence of organ dysfunction secondary to malignant mast cell infiltration is apparent at presentation.
The remaining subtypes of SM are stratified according to clinical aggressiveness and extent of mast cell infiltrate. Provided that MCL and SM-AHN are excluded, if there is evidence of organ dysfunction (defined as the “C findings” in Table 10.3), the diagnosis would be ASM [3]. Aggressive SM most commonly shows organ damage in the form of hematologic or liver dysfunction. Gastrointestinal symptoms (malabsorption and weight loss) count as an organ dysfunction only if biopsies document gastrointestinal mast cell infiltrates as cause of the symptoms. In ASM, the bone marrow aspirate smear (not biopsy) mast cells comprise ≤5% of marrow cellularity. Cases in which marrow aspirate mast cells are >5% but <20% are diagnosed as aggressive SM in transformation (to MCL), whereas cases with marrow aspirate mast cells ≥20% represent frank MCL [1].
Absence of organ dysfunction characterizes smoldering SM, indolent SM, and the very rare entity termed bone marrow mastocytosis. They differ mainly in the extent of mast cell burden in the marrow, spleen, and liver, with a higher burden of disease in smoldering systemic mastocytosis, defined as the presence of two or more “B findings” (Table 10.3). Indolent SM and bone marrow mastocytosis both show low burden of disease, and they differ only in the presence or absence of skin lesions, respectively [1]. Smoldering SM often remains stable for many years, though progression to aggressive SM or MCL can occur.
10.3.1 Case 1. SM-AHN
A 37-year-old male presented with pruritus without a rash and then developed pancytopenia (WBC 4100µL, hemoglobin 10.1 g/dL, platelet count 25,000/µL) for which he underwent a bone marrow biopsy. Bone marrow had 100% cellularity and revealed increase of blasts of 5–10%. In addition, the bone marrow biopsy showed clusters of atypical spindle-shaped mast cells with aberrant expression of CD25 as well as KIT D816V mutation. Tryptase level was 3.1 µg/L. Megakaryocytes were increased in number and showed dysplastic morphology. There was moderate reticulin fibrosis (MF 2 of 3). Next-generation sequencing (NGS) revealed mutations in RUNX1, U2AF1, and CBL in addition to KIT. Cytogenetics revealed trisomy 8. He underwent a haploidentical stem cell transplant from his brother after fludarabine and total body irradiation (1200 cGy) conditioning followed by GVHD prophylaxis with cyclophosphamide, tacrolimus, and mycophenolate mofetil. His bone marrow biopsy on day 34 after transplantation showed persistent mast cell infiltrate (5% of cellularity) but no increased blasts, normal cytogenetics, and absence of previously detected mutations by NGS. Chimerism studies showed 99.14% donor DNA. He continues to remain in remission 2 years after his transplant and is off all immunosuppression with no evidence of graft versus host disease.
Based on the clinicopathologic and molecular findings, Case 1 can be classified as SM-AHN; in this case, the AHN being MDS with excess blasts-1 which dominated the clinical picture as is typical with most cases of SM-AHN. Although he had symptoms of itching, skin lesions of SM were absent as is typically the case with SM-AHN. The presence of additional mutations besides KIT D816V as well as cytogenetic abnormality is also typical of SM-AHN. The treatment would primarily be that of MDS which would address the associated SM since the two entities are clonally the same. Therefore, the decision was made to proceed to allo-HSCT, given his age as well as excellent performance status and lack or organ dysfunction from the associated SM. Due to the lack of matched sibling or unrelated donor, a decision was made to perform haploidentical HSCT from his brother and patient had an excellent outcome. Although there was a persistent bone marrow infiltrate of neoplastic mast cells at day 34 after HSCT, this is often seen in the case of SM-AHN after HSCT and does not require any intervention as long as there is no evidence of the original AHN.
10.3.2 Case 2. ISM
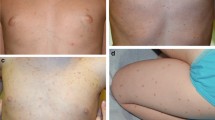
A 61-year-old male developed a pruritic rash of small erythematous to brown papules on his posterior thigh that spread through his trunk which he first noticed in 2013 (Fig. 10.1). He also noticed about 10 lb of weight loss. A skin biopsy from left chest wall was performed in October of 2014 and histopathologic finding was consistent with urticaria pigmentosa. Peripheral blood counts were normal. The bone marrow biopsy in April 2014 showed normocellular bone marrow with involvement by SM (spindle-shaped mast cells with aberrant CD25 expression comprising less than 5% of cellularity) and normal trilineage hematopoiesis. KIT D816V was detected by NGS at a variable allele frequency of 7.4%. There was no hepatosplenomegaly on imaging. He also had elevated serum tryptase levels which was 68.4 µg/L in March 2018. He was started on midostaurin 50 mg twice daily starting October 2018. His pruritus and skin rash improved. Serum tryptase levels are still elevated and was 303 µg/L in May 2020. He is tolerating midostaurin well except for nausea managed with antiemetics.

Urticaria pigmentosa rash in a patient with ISM

Bone marrow features of SM. a Bone marrow biopsy from a patient with ISM showing dense paratrabecular aggregates of spindle-shaped mast cells. Osteosclerosis is evident. Such aggregates fulfill the major WHO criteria for SM. b Immunohistochemistry for mast cell tryptase highlights the mast cell aggregates. c SM-AHN. Bone marrow aspirate showing increased mast cells and myeloblasts in a case of SM associated with MDS with excess blasts-2. d Bone marrow biopsy from SM associated with MDS showing dysplastic megakaryocytes and myeloid precursors
Indolent SM is the second most common form of SM in adults. The term ISM is used for patients with SM without an associated hematologic neoplasm and with <20% mast cells in the bone marrow, low mast cell/disease burden (defined as ≤1 “B findings” as per Table 10.3), and no organ dysfunction (defined as no “C findings” as per Table 10.3). Mast cell infiltrates may be detected in various organs (including liver, spleen, and gastrointestinal tract) in ISM, but by definition there is no organ dysfunction. Otherwise, the diagnosis would be ASM. If ≥2 “B findings” are present, the diagnosis changes to smoldering SM. The clinical course is generally indolent, though severe anaphylaxis can be seen more often compared to more aggressive forms of SM. The KIT D816V mutation is detected in >90% of ISM cases, with other KIT mutations detected in a portion of the remaining patients upon sequencing of the KIT gene.
Case 2 has the typical presentation with urticaria pigmentosa dominating the clinical picture. The patient did not have features related to other organ dysfunction from mast cell infiltration (C findings). Therefore, he would be expected to have a prolonged indolent clinical course and focus of therapy would be to manage symptoms as well as prevent mast cell proliferation. Therapy directed at KIT D816V mutation is appropriate in this case given the dominant role of this mutation in pathogenesis of ISM. Although he responded well to midostaurin clinically with significant improvement in skin rash, this was not reflected in tryptase level which remained high. Therefore, it appears that tryptase level alone cannot be used to follow response of SM to KIT-directed therapy and response should be assessed by a combination of clinical as well as pathologic features. Gastrointestinal side effects from midostaurin can be significant and may require supportive management as in this case.
10.4 Clinical Features of SM
The clinical features of SM result from release of mast cell mediators, infiltration of organs, or the associated neoplasm in case of SM-AHN. Constitutional symptoms like fever, night sweats, and weight loss are common. Mediators induce a wide array of allergic-type symptoms including urticaria, episodic flushing, hypotension, tachycardia, and bronchospasm [18]. The classic clinical finding of indolent forms of SM is skin infiltration with mast cells manifesting as urticaria pigmentosa which is a common presenting feature (Fig. 10.1). Patients may have bone lesions on X-ray which includes osteosclerosis, osteoporosis, or lytic lesions and pathologic fractures may occur. Hepatosplenomegaly is not uncommon, however lymphadenopathy is rare. Bone marrow involvement may manifest as peripheral blood cytopenia which could be from mast cell infiltration or from the associated AHN. Gastrointestinal symptoms include abdominal pain, nausea, diarrhea, and heartburn and does not always denote infiltration of gastrointestinal tract with mast cells. Neurologic symptoms of headache, cognitive impairment, and depression have been observed [18]. In the case of SM-AHN, symptoms of mediator release as well as urticaria pigmentosa and other organ infiltration are unusual and the diagnosis is often made concurrently with or during the course of treatment of the associated hematologic neoplasm, most commonly AML or MDS.
Laboratory studies for serum tryptase level are helpful in the initial diagnostic work-up. In SM, a persistently elevated serum tryptase level (>20 ng/ml) is a minor diagnostic criterion as per the WHO [1]. The levels vary according to the type of SM; a greater proportion of ASM and SM-AHN patients exhibit a markedly elevated serum tryptase level (>200 ng/ml) compared to the ISM. Levels over 200 ng/ml are also seen in smoldering SM which is characterized by a higher mast cell burden compared to ISM [15]. However, serum tryptase levels are not specific to SM and may be elevated in the presence of other myeloid neoplasms such as AML, MDS, and CML. In the absence of another myeloid malignancy, serum tryptase is a relatively specific marker for mast cell burden. In reactive mast cell activation, serum tryptase is elevated during and within several hours of the patient’s symptoms but returns to a normal level when the patient is asymptomatic, indicating an episode of mast cell activation without an increase in total body mast cell number. On the other hand, persistent elevation of baseline serum total tryptase >20 ng/mL drawn while the patient is asymptomatic indicates an increase in total body mast cell number. If baseline tryptase is elevated on two occasions, an evaluation for SM should be pursued.
10.5 Treatment of SM
The goals of treatment in SM are to improve mediator release symptoms as well as to decrease mast cell burden in order to improve urticaria pigmentosa rash and other organ dysfunction if present. In the case of SM-AHN, the primary goal is to treat the AHN.
Attempts have been made to develop prognostic scoring systems for SM based on clinical, laboratory, and molecular features. Details of these scoring systems have been published elsewhere [15]. Some of the adverse prognostic factors include age over 60 years and elevated alkaline phosphatase level (for non-advanced SM), and anemia, thrombocytopenia, high tryptase level, lack of skin involvement as well as presence of high-risk mutations in genes including ASXL1, RUNX1, DNMT3A, SRSF2, and NRAS [15]. These mutations are a feature of advanced forms of SM including SM-AHN where the AHN determines prognosis.
Given the universal presence of KIT mutation in SM and its role as the primary driver of mast cell proliferation and survival, KIT inhibitors are the logical choice to lower mast cell burden. Midostaurin (PKC 412) a multikinase inhibitor with known targets including FLT3 and KIT was evaluated for SM. The phase II trial of midostaurin in advanced SM showed an overall response rate (ORR) of 60% and a major response was seen in 45% of patients. Complete remissions, however, were not observed [20]. Based on these results, midostaurin received FDA approval for advanced SM. The more selective KIT inhibitor avapritinib (BLU 285) is a potent inhibitor of D816-mutated KIT and has shown higher response rate than midostaurin in an ongoing Phase 1 study although it must be pointed out that the responses were assessed by different criteria. The ORR for avapritinib was 77% which included 13% CR and CR with partial hematologic recovery [21]. Response to therapy in the avapritinib trial was assessed by the stringent International Working Group—Myeloproliferative neoplasms Research and Treatment and the European Competence Network on Mastocytosis criteria where a CR was defined as resolution of all symptoms and end-organ damage with blood counts as follows: ANC >1 x 109/l with normal differential, hemoglobin level 11 g/dl, and platelet count (100 x 109/l), resolution of hepatosplenomegaly and other end-organ damage, a tryptase level less than 20 ng/ml, and resolution of neoplastic cells in the bone marrow or organ of known involvement [22]. Midostaurin and avapritinib are FDA approved for advanced SM. Gastrointestinal side effects, fatigue, and edema are commonly observed side effects with these agents and often requires dose reduction.
In the very rare situation where D816 mutation in KIT is not detected, it is important to sequence other exons of the KIT gene for mutations in its extracellular, transmembrane, or juxtamembrane domains since some of these mutations are sensitive to imatinib which can induce clinical response in such cases [18]. Cladribine has been used in advanced SM and has an ORR of around 50% with a higher response rate in more indolent forms of SM [18, 23]. It may be a choice for patients who fail KIT inhibitor therapy or need more rapid debulking of mast cell burden. Interferon alpha in combination with corticosteroids has also shown activity in SM but this therapy has more side effects and poor tolerability [18, 24].
Relief of symptoms due to mediator release can be addressed by histamine receptor blockers, corticosteroids, cromolyn sodium, and leukotriene antagonists. Treatment of osteoporosis with biphosphonates has also been recommended [18, 19].
Majority of patients with advanced SM have associated AHN and curative therapy for the AHN by allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the preferred approach in suitable candidates. Allo-HSCT for ASM has been attempted as a potentially curative therapy for patients with advanced SM. There are no randomized studies to show benefit and the largest experience is from a retrospective series of 57 patients with following SM subtypes: SM-AHN (38 patients), MCL (12 patients), and ASM (7 patients). The overall survival at 3 years was 57% but depended on type of SM (MCL: 17%, ASM: 43%, SM-AHN: 74%). Diagnosis of MCL and use of reduced intensity conditioning were associated with worse outcomes after HSCT [25]. The role and timing of allo-HSCT, as well as optimal conditioning regimen, remain to be clarified in other forms of advanced SM besides SM-AHN [26]. It is important to recognize that in case of SM-AHN, the residual bone marrow mast cell infiltrate may persist after successful allo-HSCT and does not mean failure of HSCT provided there is no evidence of the AHN.
References
Swerdlow SH et al (2017) WHO classification of tumours of haematopoietic and lymphoid tissues, revised 4th edn. International Agency for Research on Cancer, Lyon, France
Nagata H, Worobec AS, Oh CK et al (1995) Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A 92:10560
Ustun C, Arock M, Kluin-Nelemans HC et al (2016) Advanced systemic mastocytosis: from molecular and genetic progress to clinical practice. Haematologica 101(10):1133–1143
Schaab J, Schnittger S. Sotlar K et al (2013) Comprehensive mutational profiling in advanced systemic Mastocytosis. Blood 12(14):460–466
Jawhar M, Schwaab J, Schnittger S et al (2016) Additional mutations in SRSF2, ASXL1 and or RUNX1identify a high risk group of patients with KIT D816V(+) advances systemic mastocytosis. Leukemia 30(1):136–143
Jawhar m, Schwaab J, Schnittger S et al (2015) Molecular profiling of myeloid progenitor cells in multimutated advanced systemic mastocytosis identifies KITD8116V as a distinct and late event Leukmaia 29(5):1115–1122
Hanssens K, Brenet F, Agopian J et al (2014) SRSF2-p95 hotspot mutation is hifhly associated with advanced forms of mastocytosis and mutations in epigenetic regulator genes. Haematologica 99(5):830–835
Yavuz AS, Lipsky PE, Yavuz S et al (2002) Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood 100:661
Grootens J, Ungerstedt JS, Ekoff M et al (2019) Single cell analysis reveals the KIT D816V mutation in hematopoietic stem and progenitor cells in systemic mastocytosis. EBioMedicine 43:150–158
Naumann N, Jawhar M, Schwaab J (2018) Incidence and prognostic impact of cytogenetic aberrations in patients with systemic Mastocytosis. Genes Chromosomes Cancer 57(5):252–259
Pullarkat V, Bedell V, Kim Y et al (2007) Neoplastic mast cells in systemic mastocytosis associated with t(8;21) acute myeloid leukemia are derived from the leukemic clone. Leuk Res 31(2):261–265
Pullarkat VA, Bueso-Ramos C, Lai R et al (2003) Systemic mastocytosis associated with clonal hematological non mast cell lineage disease: analysis of clinicopathologic features and activating c-kit mtations. Am J Hematol 73(1):12–17
Morgado JM, Perbellini O, Johnson RC et al (2013) CD30 expression by bone marrow mast cells from different diagnostic variants of systemic mastocytosis. Histopathology 63:780–787
Sotlar K, Cerny-Reiterer S, Petat-Dutter K et al (2011) Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol 24:585–595
Reiter A, George TI, Gotlib J (2020) New developments in diagnosis, prognostication and treatment of advanced systemic mastocytosis. Blood 35(16):1365–1376
Kim Y, Weiss LM, Chen YY, Pullarkat V (2007) Distinct clonal origins of systemic mastocytosis and associated B-cell lymphoma. Leuk Res 31(12):1749–1754
Johnson RC, Savage NM, Chiang T et al (2013) Hidden mastocytosis in acute myeloid leukemia with t(8;21)(q22;q22). Am J Clin Pathol 140(4):525–535
Pardanani A (2019) Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management. Am J Hematol 94:363–377
Pardanani A (2013) How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage). Blood 121:3085–3094
Gotlib J, Kluin-Nelemans HC, George TI et al (2016) Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med 374:2530–2541
Radia D, Deininger M, Gotlib J et al (2019) Avapritinib, a potent and selective inhibitor of KIT D816V induces complete and durable responses in patients with advanced systemic mastocytosis European Hematology Association Congress. Amsterdam, The Netherlands
Gotlib J, Pardanani A, Akin C et al (2013) International working group-myeloproliferative neoplasms research and treatment (IWG-MRT) & European competence network on mastocytosis (ECNM) consensus response criteria in advanced systemic mastocytosis. Blood 121
Kluin –Nelemans C, Oldhoff JM, Van Doormaal JJ (2003) Cladaribine therapy for systemic mastocytosis. Blood 102: 4270–4276
Casassus P, Caillat-Vigneron N, Martin A et al (2002) Treatment of adult systemic mastocytosis with interferon-alpha: results of a multicentre phase II trial on 20 patients. Br J Haematol 119:1090–1097
Ustin C, Reiter A, Scott BL (2014) Hematopoietic stem cell transplantation for advanced systemic mastocytosis. J Clin Oncol 32:3264–3274
Ustun C, Gotlib J, Popat U et al (2016) Consensus opinion on allogeneic hematopoietuc cell transplantation in advanced systemic mastocytosis. Biol Blood Marrow Transplant 22:1348–1356
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pullarkat, S.T., Wu, W., Pullarkat, V. (2021). Systemic Mastocytosis: Advances in Diagnosis and Current Management. In: Pullarkat, V., Marcucci, G. (eds) Biology and Treatment of Leukemia and Bone Marrow Neoplasms . Cancer Treatment and Research, vol 181. Springer, Cham. https://doi.org/10.1007/978-3-030-78311-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-78311-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78310-5
Online ISBN: 978-3-030-78311-2
eBook Packages: MedicineMedicine (R0)