
Abstract
The genetic material we carry today is different from that we were born with: our DNA is prone to mutations. Some of these mutations can make a cell divide without control, resulting in a growing tumor. Typically, in a cancer sample from a patient, a large number of mutations can be detected, and only a few of those are drivers - mutations that positively contribute to tumor growth. The majority are passenger mutations that either accumulated before the onset of the disease but did not cause it, or are byproducts of the genetic instability of cancer cells. One of the key questions in understanding the process of cancer development is which mutations are drivers, and should be analyzed as potential diagnostic markers or targets for therapeutics, and which are passengers. We propose PathMEx, a novel method based on simultaneous optimization of patient coverage, mutation mutual exclusivity, and pathway overlap among putative cancer driver genes. Compared to state-of-the-art method Dendrix, the proposed algorithm finds sets of putative driver genes of higher quality in three sets of cancer samples: brain, lung, and breast tumors. The genes in the solutions belong to pathways with known associations with cancer. The results show that PathMEx is a tool that should be part of a state-of-the-art toolbox in the driver gene discovery pipeline. It can help detect low-frequency driver genes that can be missed by existing methods.
Supported by NSF grant IIS-1453658.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Human cells are prone to mutations, and some of these may transform the cell into one that divides indefinitely and has the ability to invade other tissues [3], resulting in cancer. For most human cancers to develop, a sequence of between two and eight mutations that target genes involved in specific cell functions is needed [48]. Such mutations, which confer growth advantage to cells and are causally implicated in oncogenesis, are referred to as driver mutations [4]. Known somatic mutations linked to cancer, often with additional information such as known therapies that target the mutation, are being gather in databases [12, 29, 30, 42] that can be used in selecting patient treatment. Newly identified driver genes can also be screened using druggability indices [10], and considered for being targets for drug repositioning [34], leading the way to new therapeutic modalities. Thus, experimental and computational techniques for discovering driver genes are of great interest.
In recent years, the ability to discover driver mutations has advanced greatly due to availability of large datasets generated using second-generation sequencing techniques [41]. The Cancer Genome Atlas (TCGA) [50] and other similar projects perform sequencing of matched tumor and normal samples from hundreds of patients with a given tumor type, allowing for detection of somatic mutations present in tumor tissue. However, even with the increasing availability of data, the problem of identifying driver mutations and genes that harbor them (called driver genes) remains a challenge.
The main issue hampering discovery of driver mutations from sources such as TCGA is that majority of somatic mutations acquired in human cells throughout life are not causally linked to cancer - these are often referred to as passenger mutations. It has been estimated that half or more of all mutations observed in patients’ cancer tissues originate prior to the onset of cancer [44]. In addition to these pre-existing mutations, cancer cells exhibit a mutator phenotype, that is, and increase mutation rate [32]. This further contributes to the dominance of passenger mutations over driver mutations in observed cancer tissue samples. Altogether, while the number of driver mutations in a tumor is typically small – a recent analysis of TCGA data shows it to be between 2 and 6 in most tumors [20] – the total number of somatic mutations present in a single patient can range between 10 to above 100, depending on tumor type and patient age. Most mutations in a cancer tissue sample are thus passenger mutations that do not contribute positively to cancer growth.
On common approach to separate driver from passenger mutations is to calculate the background mutations rate that would be exhibited by passenger mutations, and consider those mutations that are encountered more frequently as drivers. This approach typically considers mutations a result of a Poisson process, which allows for quantifying the statistical significance of any deviations from the background mutation rate. For example, MutSig [9] uses a constant mutation rate across all genes, and can also use methods for functional predictions of mutation significance, such as SIFT [37], CHASM [7], Polyphen-2 [1] and MutationAssessor [40], to account for the fact that mutations differ substantially in their effects on the mutated protein [2]. MutSigCV [24] uses factors such as chromatin state and transcriptional activity to estimate gene-specific background mutation rates. PathScan [51] utilizes a Poissonian mutation model that involves gene lengths, and for a gene set given by the user calculates the probability of observing that many mutations or more under a null hypothesis that the mutations are passengers. If the probability is low across many samples, the genes are considered driver genes. MuSiC [14] extends PathScan by adding knowledge about correlation between mutation rates and factors including clinical variables such as age, molecular variables such as Pfam family to which the genes belong, and sequence correlates such as base composition of the site and proximity among mutation sites. DrGaP tool [17] considers 11 different types of mutation types, with factors including G/C content near the mutation site and methylation status of the site, in estimating the background mutation rate. Detection of driver genes using the gene-centric methods mentioned above is complicated by the fact that rarely a single driver gene is mutated across many patients with a given tumor. Only few genes, such as TP53 or BRCA1, are mutated in large fraction of cases. Most of individual genes are mutated in less than 5% of patients suffering form the same cancer type [39]. Thus, large number of samples is required to detect statistically significant deviations from background mutation rates.
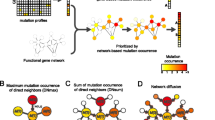
To alleviate the problems associated with relying only on mutation frequencies of individual genes, a new approach of using patterns of mutations spanning multiple genes has emerged in recent years. It has been observed that in many types of tumors, only one mutation per pathway is needed to drive oncogenesis [35, 47, 52]. Thus, the minimal set of mutated genes required for cancer to develop would consist of several sets of genes, each corresponding to a critical pathway such as angiogenesis. Within each gene set, exactly one gene would be mutated in each patient. That is, all patients would be covered by a mutation in a gene from the set, and there would be no excess coverage, that is, no patient will have more mutations than one in the genes from the set. This pattern has been often referred to as mutual exclusivity within a gene set. In actual patient data, additional mutations in driver genes may be present, especially in older patients or in cases of slow growing tumors. Also, some of the mutations may be missed due to observation errors. Thus, instead of detecting the presence or absence of mutual exclusivity in a set of genes covering all patients, driver detection algorithms involve a score that penalizes for deviations from a driver pattern, that is, for zero mutations in a patient, or for more than one mutation. Finding the optimal set of genes with respect to such a score has been shown to be an NP-hard problem [46], and heuristic search procedures are utilized to find a set of genes closest to the high-coverage mutual exclusivity pattern. The approach of finding a gene set through a pattern search procedure has been used by several tools, including Dendrix [46] and Multi-Dendrix [25], and RME [36]. Further methods extend this approach by helping deal with observation errors in the data [43], with cancer subtypes [26], and with computational efficiency of the search for driver genes [6, 53].
Further advances in driver gene detection methods resulted from observations that show that cancer driver mutations are not confined to a specific set of loci but, instead, differ substantially in individual cases. Only when seen from the level of pathways, that is, genes related to a specific cellular process, a clearer picture emerges. This evidence has given rise to network-oriented driver detection methods, such as HotNet [45], which incorporates protein-protein networks and uses a heat diffusion process, in addition to gene mutation frequency, to detect a driver subnetwork. Another network-based technique, MEMo [11], utilizes mutation frequency in individual genes together with gene interactions to form highly mutated cliques, and then filters the cliques using mutual exclusivity principle. We have recently proposed QuaDMutNetEx [5], a method that utilizes human protein-protein interaction networks in conjunction with mutual exclusivity. These methods involve a graph with genes as nodes, and gene-gene or protein-protein interactions as edges, and do not incorporate the existing knowledge of how groups of genes and edges connect into larger functional pathways.
Here, we propose PathMEx, a novel driver gene detection technique that combines the pattern-based and pathway-based detection approaches. It is built around simultaneous optimization of patient coverage, mutual exclusivity and pathway overlap among putative cancer driver genes. We evaluated our method on three cancer mutation datasets obtained from literature and from the Cancer Genome Atlas. Compared to the state-of-the-art tool Dendrix, our method shows higher values of the Dendrix score, a metric used to judge the quality of cancer driver gene sets.
2 Methods
The proposed algorithm for detecting driver mutations in cancer operates at the gene level. That is, on input, we are given an n by p mutation matrix G, where n is the number of cancer patients with sequenced cancer tissue DNA and sequenced matched normal tissue, and p is the total number of genes explored. The matrix is binary, that is, \(G_{ij}=1\) if patient i has a non-silent somatic mutation in gene j; otherwise, \(G_{ij}=0\). More generally, \(G_{ij}\) can also be set to one if a gene is part of region with a copy-number alteration, or has a mutation in its regulatory region, although such data is less readily available compared to mutations in gene coding regions. A row vector \(G_i\) represents a row of the matrix corresponding to patient i. The solution we seek is a sparse binary vector x of length p, with \(x_j=1\) indicating that mutations of gene j are cancer driver mutations. In the proposed approach, the solution vector should capture driver genes that are functionally related, for example are all part of a pathway that needs to be mutated in oncogenesis. If we want to uncover all driver genes, we should apply the algorithm multiple times, each time removing the genes found in prior steps from consideration. We will often refer to the nonzero elements of x as the mutations present in x.
In designing the algorithm for choosing the solution vector x, we assumed that any possible vector is penalized with a penalty score based on observed patterns of driver mutations in human cancers. We expect that each patient has at least one mutation in the set of genes selected in the solution; however, in some cases, the mutation may not be detected. Also, while several distinct pathways need to be mutated to result in a growing tumor, typically one mutation in each of those pathways suffices. The chances of accumulating additional mutations in the already mutated pathway before the cancer is detected are low, and decrease with each additional mutation beyond the first one. We capture this decreasing odds through an increasing penalty associated with solution vector x given the observed mutations \(G_i\) in patient i
The term \(G_i x\), that is, the product of row vector \(G_i\) and the solution vector x, captures the number of mutations from solution x present in patient i. We incur no penalty if the number of mutated genes from x in a given patient is one. If the patient is covered by no mutations, the penalty is one. If the patient is covered by more than one mutation, the penalty is equal to the number of mutations in excess of the one required for cancer to develop.
We also expect the genes in the solution to be functionally related, that is, we expect high pathway overlap in the solution. To capture this, we provide a reward (i.e., a negative penalty) for genes in the solution that belong to the same pathway. For a gene j, we denote by \(P_j\) the set of pathways that contain j. Further, for a gene j, we can define the set of co-pathway genes, \(\varPi _j\), that is, the set of genes that share a pathway with j, as
To promote selection of genes from the same pathway, for every gene j we define a pathway overlap term added to the objective function that is being minimized
If gene j is selected to be part of the solution, and it shares pathways with at least one of other genes in the solution, the objective will be decreased by 1.
The final objective function being minimized is a combination of the high-coverage mutual exclusivity terms and the pathway overlap terms
We also introduce a limit on the number of genes in the solution, K, by requiring \(\sum _{j=1}^p x_j \le K\). The solution x is a binary indicator vector, where elements \(x_j=1\) correspond to genes being selected as the set of driver genes. Below, if we need to express solution as a set instead of an indicator vector, we will use \(Z_x = \left\{ j: x_j=1\right\} \).
The problem of minimizing the non-linear objective function L(G, x) over possible solution vectors x can be reformulated into a constrained mixed integer linear program
with p binary variables, \(n + p\) continuous variables, and \(2n+2p+1\) inequality constraints. That is, the size of the problem grows linearly with the number of samples, n, and the number of genes, p.
Mixed-integer linear programs (MILP) are known to be NP-hard in general. However, the optimal solution can be obtained quickly for problems of small size. For cancer driver detection problems involving a large number of genes, where exact solutions are not available in any reasonable time, we designed a meta-heuristic algorithm, PathMEx, that combines network-based exploration of the solution space with optimal search for small subproblems.

If the problem is small enough, PathMEx directly solves the MILP problem and returns a globally optimal solution. In other cases, the main PathMEx algorithm goes through T iterations, as shown in the pseudocode. In each iteration PathMEx considers a candidate set \(\chi \) of s genes, where s is chosen to make the problem tractable for a MILP solver. In our tests, we set \(s=200\). In each iteration, a subproblem involving only genes from \(\chi \) is solved by a MILP solver, and a globally optimal subset \(Z_x\in \chi \) is selected as the current solution. The solution set \(Z_x\) has up to K genes. A new candidate set \(\chi \) is created by keeping all genes in the solution \(Z_x\), and choosing additional genes to make the size of the new candidate set equal to s. These include up to \(s_p\) genes that are either in \(Z_x\) or are randomly selected from all the pathways that contain genes from \(Z_x\). It also includes other genes selected at random until the candidate set size reaches s.
3 Results and Discussion
We evaluated the proposed algorithm using cancer mutation data from the Cancer Genome Atlas (TCGA) [50] and from literature. We used two datasets that were originally used by the authors of Dendrix [46]: somatic mutations in lung cancer (LUNG), and a dataset relating to Glioblastoma Multiforme (GBM) that includes not only somatic mutations but also copy number alternations. We also used a larger dataset of somatic mutations in samples from Breast Invasive Carcinoma (BRCA) downloaded from TCGA, in which, following standard practice [24], we removed known hypermutated genes with no role in cancer, including olfactory receptors, mucins, and a few other genes such as the longest human gene, titin. The characteristics of the datasets are summarized in the Table 1.
In judging the quality of a solution \(Z_x\), that is, a set of putative driver genes, we used two metrics, coverage and excess coverage. Coverage is defined as the number of patients covered by at least one gene from \(Z_x\) divided by the total number of patients. Excess coverage is defined as the number of patients covered by more than one gene from \(Z_x\) divided by the number of patients covered by at least one gene from \(Z_x\). These metrics together capture how well a gene set conforms to the pattern expected of driver genes. Both of the metrics range from 0 to 1. Perfect solution has coverage of 1, and excess coverage of 0, indicating that every single patient has exactly one mutation in genes from solution \(Z_x\). We also used the Dendrix score, the objective function maximized by Dendrix [46], defined as the number of patients covered by at least one gene from \(Z_x\) set minus the coverage overlap, that is, the total count of all mutations in excess of one mutation per patient in genes from \(Z_x\).
We ran PathMEx and Dendrix on the three datasets: GBM, LUNG, and BRCA. For two small datasets, GBM and LUNG, we explored solutions including up to 10 genes; for BRCA, a much larger dataset, we searched for solutions including up to 20 genes. PathMEx automatically picks the best solution with size up to a given range. For Dendrix, which analyzes solutions of a fixed, user-provided size, we performed independent runs for each solution size ranging from 2 to the chosen limit (10 for GBM and LUNG, 20 for BRCA), and picked the solution with the highest value of Dendrix score. Each Dendrix run involved \(10^7\) iterations, as recommended by Dendrix authors. For PathMEx, which is a descent method depending on the randomized initialization, we conducted 10 runs, each consisting of 100 iterations, and picked the solution with the lowest value of the objective function among these 10 runs.
PathMEx relies on prior knowledge of biological pathways, which we obtained from the MSigDB repository of canonical pathways [27]. These include pathways from KEGG, Biocarta, Pathway Interaction Database, and Reactome. We removed 46 pathways related to disease, most notably KEGG_PATHWAYS_IN_CANCER and other cancer-specific pathways, to avoid biasing the method towards re-discovering only known cancer genes. We ended up with 1284 pathways that remained after the filtering step. Each pathway is treated as a set of genes that are members of the pathway.
The results of the tests, presented in Table 2, show that PathMEx consistently returns higher quality solutions than Dendrix. In each of the three datasets, PathMEx reached a higher value of the Dendrix score. Table 3 shows that PathMEx also achieved higher patient coverage, while showing no consistent increase in excess coverage compared to Dendrix across the datasets.
To quantify the statistical significance of the results, we employed the randomization approach used previously [46]. For every gene, the binary column vector describing in which patient the gene is mutated is randomly reshuffled. The results of reshuffling of all genes form a new dataset, that is, a new matrix G. Each randomized dataset preserves the underlying frequencies of mutations of individual genes, but any multi-gene patterns of mutations such as mutual exclusivity may only arise by chance. We created 1024 reshuffled datasets, and ran PathMEx on each of them. As with the original non-randomized dataset, for each reshuffled dataset we performed 10 runs, and picked the solution with the lowest value of the objective function from among them. Finally, as the estimate of the p-value, we quantified the fraction of the 1024 reshuffled datasets in which the value of the objective function minimized by PathMEx (Eq. 5) is lower than or equal to the value obtained on the original non-randomized dataset. As shown in Table 4, the p-values for all three datasets are a magnitude below the 0.05 threshold.

Pathways covering driver genes for GBM dataset (left) and the LUNG dataset (right). Red nodes represent pathways from MSigDB, blue nodes represent genes in the PathMEx solution. For each gene, in parentheses, we provide the number of patients in the dataset that harbor a mutation in that gene. (Color figure online)

Pathways covering driver genes for BRCA dataset. Red nodes represent pathways from MSigDB, blue nodes represent genes in the PathMEx solution. For each gene, in parentheses, we provide the number of patients in the dataset that harbor a mutation in that gene. (Color figure online)
The genes in the solutions are members of pathways known to be associated with cancer. We visualized the most enriched pathways for each dataset in Figs. 1, 2. For each dataset, among all pathways covering the genes in the solution, we first selected the pathway most enriched in solution genes, that is, the pathway with the highest ratio of pathway genes in the solution to all genes in the pathway. We then removed the genes covered by that pathway from consideration, and repeated the process, until only genes that were not members of any pathway remained not covered.
To validate the ability of PathMEx to discover rare putative cancer driver genes, in each of the three datasets we focused on the genes in the solution with the fewest number of mutations. In the brain tumor dataset, six out of ten genes identified by PathMEx are each mutated in only 1 out of 84 patients. Out of these, four have been previously implicated in cancer: IQGAP1 is believed to play a role in cell proliferation and cancer transformation [19] and has been implicated in breast cancer [38], KPNA2 promotes cell proliferation in ovarian cancer [18], TNK2 has been recently recognized as an oncogenic kinase [33], and WEE1 is already a target for cancer therapy [15]. While no cancer role has been so far identified for carboxylesterase 3 (CES3), it is known to be expressed in the source tissue of our samples, the brain [16]. In the lung cancer dataset, out of 10 genes identified by PathMEx, 5 genes have only 2 mutations each in a group of 163 samples. All five genes have been previously linked to various types of cancer. Role of ABL1 in cancer is well established. FES is a known proto-oncogene [28]. PAK6 has been shown to suppress growth of prostate cancer [31]. JUP has been implicated in prostate and breast cancers [23]. Finally, expression of CYSLTR2 gene is a prognostic marker in colon cancer [49] and is causative of melanoma [8]. In the breast cancer dataset, in which we increased the solution size limit to 20, three genes with only 3 mutations each in a cohort of 771 patients were identified by PathMEx as putative cancer genes. All three have been previously linked to cancer: MX2 to lung cancer [21], and CD38 and SLC13A5 to leukemia [13, 22].
4 Conclusions
We have shown that the proposed novel method PathMEx, which combines maximization of patient coverage and gene mutual exclusivity with maximization of pathway overlap is highly successful in detecting rare cancer driver genes. The method shows higher quality scores than existing state-of-the-art mutual exclusivity-based tool Dendrix on three cancer datasets, and has the ability to find genes that are members of pathways with known role in cancer. These results indicate that PathMEx can help detect low-frequency driver genes that may be missed by existing methods, and that it should be part of a state-of-the-art toolbox in the driver gene discovery pipeline.
References
Adzhubei, I.A., et al.: A method and server for predicting damaging missense mutations. Nat. Methods 7(4), 248–249 (2010)
Arodź, T., Płonka, P.M.: Effects of point mutations on protein structure are nonexponentially distributed. Proteins Struct. Funct. Bioinf. 80(7), 1780–1790 (2012)
Bertram, J.S.: The molecular biology of cancer. Mol. Aspects Med. 21(6), 167–223 (2000)
Bignell, G.R., et al.: Signatures of mutation and selection in the cancer genome. Nature 463(7283), 893–898 (2010)
Bokhari, Y., Alhareeri, A., Arodz, T.: QuaDMutNetEx: a method for detecting cancer driver genes with low mutation frequency. BMC Bioinf. 21(1), 1–12 (2020)
Bokhari, Y., Arodz, T.: QuaDMutEx: quadratic driver mutation explorer. BMC Bioinf. 18(1), 1–15 (2017)
Carter, H., et al.: Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Can. Res. 69(16), 6660–6667 (2009)
Ceraudo, E., et al.: Direct evidence that the GPCR CysLTR2 mutant causative of uveal melanoma is constitutively active with highly biased signaling. J. Biol. Chem. 296, 100163 (2021)
Chapman, M.A., et al.: Initial genome sequencing and analysis of multiple myeloma. Nature 471(7339), 467–472 (2011)
Chen, Y., et al.: Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One 9(5), e98293 (2014)
Ciriello, G., Cerami, E., Sander, C., Schultz, N.: Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 22(2), 398–406 (2012)
Damodaran, S., et al.: Cancer Driver Log (CanDL): catalog of potentially actionable cancer mutations. J. Mol. Diagn. 17(5), 554–559 (2015)
Deaglio, S., Mehta, K., Malavasi, F.: Human CD38: a (r)evolutionary story of enzymes and receptors. Leuk. Res. 25(1), 1–12 (2001)
Dees, N.D., et al.: MuSiC: identifying mutational significance in cancer genomes. Genome Res. 22(8), 1589–1598 (2012)
Do, K., Doroshow, J.H., Kummar, S.: Wee1 kinase as a target for cancer therapy. Cell Cycle 12(19), 3348–3353 (2013)
Holmes, R.S., Cox, L.A., VandeBerg, J.L.: Mammalian carboxylesterase 3: comparative genomics and proteomics. Genetica 138(7), 695–708 (2010)
Hua, X., Xu, H., Yang, Y., Zhu, J., Liu, P., Lu, Y.: DrGaP: a powerful tool for identifying driver genes and pathways in cancer sequencing studies. Am. J. Hum. Genet. 93(3), 439–451 (2013)
Huang, L., et al.: KPNA2 promotes cell proliferation and tumorigenicity in epithelial ovarian carcinoma through upregulation of c-Myc and downregulation of FOXO3a. Cell Death Dis. 4(8), e745 (2013)
Johnson, M., Sharma, M., Henderson, B.R.: IQGAP1 regulation and roles in cancer. Cell. Signal. 21(10), 1471–1478 (2009)
Kandoth, C., et al.: Mutational landscape and significance across 12 major cancer types. Nature 502(7471), 333–339 (2013)
Kobayashi, K., et al.: Identification of genes whose expression is upregulated in lung adenocarcinoma cells in comparison with type II alveolar cells and bronchiolar epithelial cells in vivo. Oncogene 23(17), 3089–3096 (2004)
Kuang, S., et al.: Genome-wide identification of aberrantly methylated promoter associated CpG islands in acute lymphocytic leukemia. Leukemia 22(8), 1529–1538 (2008)
Lai, Y.H., et al.: SOX4 interacts with plakoglobin in a Wnt3a-dependent manner in prostate cancer cells. BMC Cell Biol. 12(1), 50 (2011)
Lawrence, M.S., et al.: Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499(7457), 214–218 (2013)
Leiserson, M.D., Blokh, D., Sharan, R., Raphael, B.J.: Simultaneous identification of multiple driver pathways in cancer. PLoS Comput. Biol. 9(5), e1003054 (2013)
Leiserson, M.D., Wu, H.T., Vandin, F., Raphael, B.J.: CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 16(1), 1 (2015)
Liberzon, A., Subramanian, A., Pinchback, R., Thorvaldsdóttir, H., Tamayo, P., Mesirov, J.P.: Molecular signatures database (MSigDB) 3.0. Bioinformatics 27(12), 1739–1740 (2011)
Lionberger, J.M., Smithgall, T.E.: The c-Fes protein-tyrosine kinase suppresses cytokine-independent outgrowth of myeloid leukemia cells induced by Bcr-Abl. Can. Res. 60(4), 1097–1103 (2000)
Liu, E.M., Martinez-Fundichely, A., Bollapragada, R., Spiewack, M., Khurana, E.: CNCDatabase: a database of non-coding cancer drivers. Nucleic Acids Res. 49(D1), D1094–D1101 (2021)
Liu, S.H., et al.: DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res. 48(D1), D863–D870 (2020)
Liu, T., et al.: p21-Activated kinase 6 (PAK6) inhibits prostate cancer growth via phosphorylation of androgen receptor and tumorigenic E3 ligase murine double minute-2 (Mdm2). J. Biol. Chem. 288(5), 3359–3369 (2013)
Loeb, L.A.: Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer 11(6), 450–457 (2011)
Mahajan, K., Mahajan, N.: ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene 34(32), 4162–4167 (2015)
Martinez-Ledesma, E., de Groot, J.F., Verhaak, R.G.: Seek and destroy: relating cancer drivers to therapies. Cancer Cell 27(3), 319–321 (2015)
McCormick, F.: Signalling networks that cause cancer. Trends Biochem. Sci. 24(12), M53–M56 (1999)
Miller, C.A., Settle, S.H., Sulman, E.P., Aldape, K.D., Milosavljevic, A.: Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC Med. Genomics 4(1), 1 (2011)
Ng, P.C., Henikoff, S.: SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31(13), 3812–3814 (2003)
Osman, M.A., Antonisamy, W.J., Yakirevich, E.: IQGAP1 control of centrosome function defines distinct variants of triple negative breast cancer. Oncotarget 11(26), 2493 (2020)
Pon, J.R., Marra, M.A.: Driver and passenger mutations in cancer. Annu. Rev. Pathol. 10, 25–50 (2015)
Reva, B., Antipin, Y., Sander, C.: Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, gkr407 (2011)
Schuster, S.C.: Next-generation sequencing transforms today’s biology. Nature 200(8), 16–18 (2007)
Sondka, Z., Bamford, S., Cole, C.G., Ward, S.A., Dunham, I., Forbes, S.A.: The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18(11), 696–705 (2018)
Szczurek, E., Beerenwinkel, N.: Modeling mutual exclusivity of cancer mutations. PLoS Comput. Biol. 10(3), e1003503 (2014)
Tomasetti, C., Vogelstein, B., Parmigiani, G.: Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad. Sci. 110(6), 1999–2004 (2013)
Vandin, F., Upfal, E., Raphael, B.J.: Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol. 18(3), 507–522 (2011)
Vandin, F., Upfal, E., Raphael, B.J.: De novo discovery of mutated driver pathways in cancer. Genome Res. 22(2), 375–385 (2012)
Vogelstein, B., Kinzler, K.W.: Cancer genes and the pathways they control. Nat. Med. 10(8), 789–799 (2004)
Vogelstein, B., Papadopoulos, N., Velculescu, V.E., Zhou, S., Diaz, L.A., Kinzler, K.W.: Cancer genome landscapes. Science 339(6127), 1546–1558 (2013)
Wang, D., DuBois, R.N.: Eicosanoids and cancer. Nat. Rev. Cancer 10(3), 181–193 (2010)
Weinstein, J.N., et al.: The Cancer Genome Atlas pan-cancer analysis project. Nat. Genet. 45(10), 1113–1120 (2013)
Wendl, M.C., et al.: PathScan: a tool for discerning mutational significance in groups of putative cancer genes. Bioinformatics 27(12), 1595–1602 (2011)
Yeang, C.H., McCormick, F., Levine, A.: Combinatorial patterns of somatic gene mutations in cancer. FASEB J. 22(8), 2605–2622 (2008)
Zhao, J., Zhang, S., Wu, L.Y., Zhang, X.S.: Efficient methods for identifying mutated driver pathways in cancer. Bioinformatics 28(22), 2940–2947 (2012)
Acknowledgements
TA is supported by NSF grant IIS-1453658.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Bokhari, Y., Arodz, T. (2021). PathMEx: Pathway-Based Mutual Exclusivity for Discovering Rare Cancer Driver Mutations. In: Paszynski, M., Kranzlmüller, D., Krzhizhanovskaya, V.V., Dongarra, J.J., Sloot, P.M.A. (eds) Computational Science – ICCS 2021. ICCS 2021. Lecture Notes in Computer Science(), vol 12743. Springer, Cham. https://doi.org/10.1007/978-3-030-77964-1_43
Download citation
DOI: https://doi.org/10.1007/978-3-030-77964-1_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-77963-4
Online ISBN: 978-3-030-77964-1
eBook Packages: Computer ScienceComputer Science (R0)