
Abstract
Small-seeded millets have gained significant research attention in recent years due to their climate-resilient nature. Among millets, foxtail millet [Setaria italica (L.) P. Beauv.] is considered as a model crop due to its rapid life cycle, small genome size, and self-pollinated nature. Its remarkable tolerance to abiotic stresses has invited researches on delineating the molecular machinery underlying tolerance and use the knowledge in developing elite cultivars that could withstand harsh weather and climatic conditions. However, crop improvement in foxtail millet has mostly been made through breeding strategies, but with the release of its draft genome sequence, several genes, QTLs, alleles, and markers were identified that regulate the tolerance traits. The effectual use of this information in crop improvement is yet to be realized as the progress made in foxtail millet research lags behind the major cereals. The genome editing approaches have recently gained importance as they enable precise editing of genes to achieve the desired phenotype. In foxtail millet, efforts are being invested in constructing vectors and optimizing experimental procedures for gene editing. In this context, the present chapter summarizes the progress made in identifying the genomic regions regulating abiotic stress tolerance and elaborates on how genomic designing could enable the development of climate-resilient varieties that could ensure food and nutritional security to the ever-growing population.
Sumi Rana, Lydia Pramitha contributed equally
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Foxtail millet
- Setaria italica
- Genome editing
- Genomic designing
- Climate-resilience
- Abiotic stress tolerance
- Food security
7.1 Introduction
The last few decades have seen various global agriculture challenges due to the exploding population and severe climatic changes. This demands the development of better varieties that could withstand climatic aberrations and provides higher outputs. Climatic changes take a toll on the yield of several staple crops like maize, wheat, and rice. Although these crops are cultivated in more areas, they do not fulfill the major nutrient requirements of the ever-growing population and are easily affected by biotic and abiotic stresses (Lata et al. 2013). Millets, also known as nutricereals, are C4 grasses that belong to the family Poaceae and are mainly known to yield smaller seeds. Finger millet (Eleusine coracana), foxtail millet (Setaria italica), kodo millet (Paspalum scrobiculatum), pearl millet (Pennisetum glaucum), guinea millet (Brachiaria deflexa), tef (Eragrostis tef), and little millet (Panicum sumatrense), etc. are members of this family and used for consumption as food and feed (Dwivedi et al. 2012). They can withstand dry and semi-dry weather conditions, thrives in less fertile soil with minimal nutrients, needs less irrigation, and produces high nutrient containing seeds and hence, mentioned as climate-resilient crops (Muthamilarasan and Prasad 2015).
Among these, foxtail millet [Setaria italica (L.) P. Beauv.] has attracted the attention of researchers worldwide with its distinct traits, viz. morphologically small stature, self-fertilization, small and true diploid genome (~423 Mb) with less repetitive DNA, high photosynthetic efficiency, rapid life cycle, prolific seed yield per plant, and tolerance to abiotic stresses (Lata et al. 2013; Table 7.1). These traits have made it a model crop to study genetic and molecular aspects of biofuel grasses and other C4 crops (Lata et al. 2013). Li and Wu (1996) reported that it was domesticated in Northern China around 8000 years ago, considered one among the oldest grown crops, and is consumed as food and fodder in parts of Asia and Africa (Lu et al. 2009). It is one among the “Five grains of China” due to its higher protein and mineral content than rice, wheat, and maize (Austin 2006). Foxtail millet seeds contain 14–16% of protein, around 8% dietary fiber, 6% of crude fat, antioxidants, and minerals. The glucose release rate after foxtail millet consumption is slow due to the low glycemic index, making it suitable for diabetic patient consumption (Muthamilarasan et al. 2016). Furthermore, morphological features like thickened cell walls, small area of the leaf, alignment of epidermal cells, and compact and deep root architecture have aided the plant to achieve enhanced water use efficiency, nitrogen use efficiency, and even makes it tolerant to various abiotic stresses like heat, salt, and drought (Li 1997; Zhang et al. 2007; Lata et al. 2013; Diao et al. 2014). Foxtail millet holds the second position in world millet production wherein India contributes 32% of it (FAOSTAT 2005; http://faostat.fao/org/) (Table 7.2).
The significant constraints to agricultural production are drought and irrigation; besides, even heat and salt stress significantly impact crop development and production (Ceccarelli and Grando 1996). Concerning the duration of exposure, severity, and time, abiotic stress may have acute and uncertain consequences on crop yield loss. Foxtail millet, being a climate-resilient crop, does not show a reduction in yield due to many abiotic stresses. In comparison to other cereals, the production of seed and yield efficiency of foxtail millet is less. Although rich in nutrients and tolerant to abiotic stress, the major constraints in the production of this crop are lack of awareness among the population. Hence, cultivation has been restricted to only a few parts of the globe. In many places, other crop cultivation has taken over it (Muthamilarasan and Prasad 2015).
On the other hand, the global population is increasing exponentially and might reach 10 billion by 2050. Furthermore, climate change as well as deteriorating soil fertility, expose the plants to several abiotic stresses viz. drought, heat, and salinity, imposing critical pressure on global agricultural productivity that further risks food and nutritional security (Kole et al. 2015). To compensate for climate changes, the input cost for cultivating major cereals has increased, which hikes the crops’ price, making them unavailable to the poor. These major cereals do not meet the nutritional requirements and are susceptible to abiotic and biotic stress (Lata et al. 2013). From 2015 onwards, hunger and malnutrition are increasing in countries like the west of Asia, Africa, and Latin America, wherein the number accounts for 820 million people (1 out of 9 people). Due to the turbulence in climate and economy, and food insecurity, approximately 113 million people face acute hunger. Worldwide around 149 million children under the age of 5 are stunted, and Asia alone houses more than half of that number, i.e., 81.7 million (54.8%) (Global Nutrition Report 2020). Dependency on these major crops (rice, wheat, and maize) is one reason that malnutrition is still prevailing in a substantial level of the global population, mainly in Asia and sub-Saharan Africa (Deaton and Dreze 2009; Hirschi 2009). On the contrary, millets are resilient to heat, drought, and minimal nutrient conditions and can also meet the nutrient requirement of the ever-growing population. Despite having high nutritional values, foxtail millet has been neglected for all these years, but with the change in the climatic conditions, it is again being recognized and utilized.
7.2 Abiotic Stresses Affecting Foxtail Millet Cultivation
Foxtail millet is a crop that inhabits the natural ability to survive under various abiotic stresses. It has a higher water use efficiency than the C4 maize, and its earlier duration makes it more feasible for the farmers to cultivate it in the rainfed zones (Zhang et al. 2007). Although foxtail millet is not affected by abiotic stresses to a greater extent, it is observed to face mild to severe damage depending on the cultivars. However, it faces a yield loss under water stress during its seedling and peak inflorescence stages. This has been brought under control by tracking the drought-tolerant landraces using effective lab screening like polyethylene glycol (PEG) (Wen et al. 2005). Several drought-responsive genes have been molecularly characterized in foxtail millet, and this has been utilized to improvise its drought tolerance at sensitive stages (Muthamilarasan et al. 2014c). Transcriptomic profiling of saline tolerant lines provoked the role of glutamine synthetase and pyrroline-5-carboxylase in saline tolerance (Huang et al. 2013). The role of proline in buffering the existence of foxtail millet under saline conditions proceeded with acceleration in the breeding for salinity-tolerant lines (Veeranagamallaiah and Sudhakar 2017). Other abiotic stresses like cold tolerance, lodging, and waterlogging are being studied in breeding initiatives, and a cold-tolerant foxtail millet variety Liggu No. 26 was developed in China. In foxtail millet, a study of root characters for improving nitrogen use efficiency by Nadeem et al. (2018) and culm characters for enhancing non-lodging efficiency by Dwivedi et al. (2012) was conducted to minimize the influence of abiotic stresses, thereby resulting in expansion of area under cultivation of foxtail millet.
7.3 Molecular Mapping of Resistance/Tolerance Genes and QTLs for Abiotic Stresses
Efforts for mapping traits in foxtail millet began well before the knowledge of markers and genomic sequences. The exploration of collections of several foxtail millet accessions became a base for documenting its variable traits. Thus, traits such as plant height, inflorescence pattern, anthocyanin pigmentation, seed color, length of bristles, and anther and stigma color were used as morphological markers to identify true F1. They also reveal a knowledge of the gene actions; for instance, non-glutinous nature was found to be dominant. Following these processes, the plant’s cytology was studied, which served as a consequent marker for interspecific crosses. The chromosomal abnormalities, chromosomal morphology served as a tool for mapping the traits. Intrusive studies on cell metabolic actions in later stages developed the isozyme markers with cell-specific changes in their expression patterns to identify the genotypes and their cell-based mechanisms. These historical efforts significantly contributed to the initiation of mapping several traits in foxtail millet (Willweber-kishiomoto 1962; Zhang 1980; Radha et al. 2014).
7.3.1 Evolution of Marker Types: RFLPs to SNPs
The molecular markers are an essential tool that led to several identifications in foxtail millet genome-wide studies. Initially, in the foxtail millet, the RFLP probes detected the polymorphic changes in ribosomal DNA. Their amplification length differentiated these probes for ribosomal DNA in varied geographic regions. These variations were used to classify the genotypes into three different sectors (Schontz and Rether 1998). Since the genomic sequence information for foxtail millet was not available during that period, the genetic diversity for conserved regions in the ribosome and mitochondria were employed to classify the genotypes molecularly. Such a study with atp6 genes revealed three kinds of amplified products among the regions. They were designated as Type I, II, and III. The Type I and Type II were distinct due to the recombination between genes of atp6. The Type-I and II were found in Southeast Asian and Afghanistan races. Type I was distinctly present in India, while type-III was in China (Fukunago and Kato 2002). After RFLP, amplified fragment length polymorphism (AFLP) by Key Genes and random amplified polymorphic DNA (RAPD) dominant markers came into the scientific approach in foxtail millet during the 1990s. The historical mapping and analyzing diversity for varied regions with AFLP, RAPD, and nuclear RFLPs were successful in identifying the variations, and it grouped the Eastern Asian accessions from the Indian and Chinese vicinity (Fukunaga et al. 1997; Schonthz and Rether 1998; Fukunaga et al. 2002). These streams of markers also highlighted that the Indian accessions were highly variable. Following these molecular tools, simple sequence repeat (SSR) or microsatellite markers were utilized to cover the nine chromosomes in foxtail millet. One hundred ninety-three primer pairs from two libraries enriched for (GA)n and (CA)n in an F2 population revealed 100 polymorphic primers across all the foxtail millet loci helped develop an SSR linkage map. This map was useful in integrating 81 newly developed SSR markers throughout the genome. The diversity with these markers by UPGMA proposed detecting 228 alleles classified in four clusters (Jia et al. 2009). These SSR markers were then predominantly used in several studies to detect the linkage disequilibrium (LD) values.
Along with these implications, the inter-simple sequence repeat (ISSR) markers in the comparative genomics of the genera in millets displayed the mutual relationship between coix and Setaria (Dvoráková et al. 2015). For identifying the exotic accessions in the Asian regions, the core of the worldwide collections was analyzed with 27 SSR and 4 Expressed sequence tag (EST) markers. These were used to analyze the parts of expressed genes that were complementary to the mRNA (Chander et al. 2017). After the completion of the sequence of the foxtail millet genome, there were several advancements with the identification of novel single nucleotide polymorphisms (SNPs) from mutants. The mutational approaches’ beneficial mutants were compared with the wild types to detect the allele-specific traits, for example, Wp1 for chlorophyll pigmentation (Sun et al. 2019). Genotype-based sequencing approaches by GBddRAD helped identify markers linked to flag leaf width, 1000-grain weight, and yield (Jaiswal et al. 2019). Compared to the SSR markers, the SNP genotyping revealed a higher LD, which presents a higher diversity for SNP alleles across the genome of different accessions. These novel SNP alleles are now ravaging a lot of information in other species. These are now being used to extrapolate the genetic information so that genomic selection techniques could improve foxtail millet.
7.3.2 Mapping Populations and Their Use
A molecular base for any breeding program requires a mapping population. Considering foxtail millet, several linkage maps were constructed, and many quantitative trait loci (QTLs) were mapped. The mapping studies for foxtail millet predominantly included F2 populations. Starting from Sato et al. (2013), who initially employed SSR markers for identifying the gene responsible for spikelet-tipped bristles, the rest of the breeders also adopted this as a base population for QTL mapping. Maintaining an F2 population with higher segregation serves as a base for mapping several key traits, and Fang et al. (2016) used around 10,598 SSR markers to map 29 QTLs. Further, QTL-sequencing by bulk segregant analysis for heading dates with three bulks viz, early heading, late heading, and extremely late heading were used to tap the QTL loci for heading (inflorescence initiation) in this crop (Yoshitsu et al. 2017). A high-density genetic map and QTL analysis with F2 population using RAD-sequencing detected 11 major QTLs for eight agronomic traits (Wang et al. 2017). This projects that the F2 population has been commonly used for mapping traits in foxtail millet and a genome-wide bin analysis with SNP-based markers also developed 11 significant QTLs for eight traits, which could further be utilized as markers (Wang et al. 2019).
Following the F2, recombinant inbred lines were successively used in the foxtail millet to map the agronomic traits (Ni et al. 2016). Around 493 recombinant lines were utilized for developing a high-resolution bin map that facilitated high-density SNP-based markers (Zhang et al. 2017). Similar re-sequencing of 184 RILs was developed to identify sd1 genes beneath the plant height in foxtail millet (Ni et al. 2017). The significant advantage of using these recombinant inbred lines is that it is desirable to screen them across locations. F2 being segregating population results in some difficulties in isolating genetic and environmental variations, and by using 164 RILs across locations, three stable QTLs were observed in chromosomes 3, 6, 7, and 9 by Liu et al. (2020). There were no attempts to map traits with near-isogenic lines as there were not much backcross programs conducted to introgress traits for their improvements. Since foxtail millet had no such demands for trait-specific developments such as disease/pest resistance, future back cross programs could be effected for endemic zones where its cultivation is affected. Similarly, double haploid techniques have not been incorporated due to the lack of proper tissue culture strategy in this crop. Modified populations like pseudo test cross performed in several other crops have some limitations to be encountered as it is a highly self-pollinated crop.
7.3.3 Mapping Approaches and Maps of Different Generations
Different mapping software were employed to identify QTLs and their LOD values. Wang et al. (1998) developed the first linkage map in foxtail millet. Following the same procedure, Doust et al. (2004, 2005) and Jia et al. (2009) adopted Mapmaker v3 in constructing genetic linkage maps in foxtail millet. Later approaches by Ni et al. (2016, 2017) used the MSTmap software package. Following these techniques, MSTMap was also used in performing composite interval mapping (CIM) in the construction of linkage maps (Wang et al. 2017; Zhang et al. 2017). Haldane mapping function was later used in RIL populations to mark the position of the markers to its linkage groups using CarthaGene v1.2.3. (Yoshitsu et al. 2017). Other than CIM, multiple QTL mapping for linkage map was also conducted by R package named one map (Liu et al. 2020). Besides, MSTmap software and JoinMap 4.0 using Haldane and Kosambi mapping function, respectively, were used to obtain precise maps for the mapping of several loci in foxtail millet chromosomes (Sato et al. 2013; Fang et al. 2016; Zhang et al. 2017; Wang et al. 2017; Ni et al. 2017). MSTMap is the most used software for linkage map construction in foxtail millet in comparison with all the packages.
The initial framework for constructing a genetic linkage map in foxtail millet was performed by Wang et al. (1998) with an intervarietal cross for flower color. This map was about 964 cM and was constructed with the help of cytogenetic tools by implying trisomic lines and polymorphic RFLP probes. It depicted chromosome VIII to be the shortest and chromosome IX as the longest chromosome. This map was used as a base by the followers to construct future linkage maps that helped mapping the genes like tb1 in the chromosomes V and VI.
Further, 27 new RFLP probes from maize were mapped to this linkage map by Doust et al. 2004 and as a continuation by Doust et al. (2005), the synteny of rice and other millets on foxtail millet were included to develop a high-density linkage map for the inflorescence patterns. After this, 101 SSR markers were mapped on the nine chromosomes of foxtail millet, wherein chromosome 9 had the densest coverage of markers. This map was confined to the previous map developed by Devos et al. (1998), which was initially constructed with 20 RFLP probes. Later with 86 SSR primers, a maximum coverage in the genome was attained (Jia et al. 2009). Hence, this map was continually refined to a high-resolution map by Sato et al. (2013), who further enriched this map with an additional 26 SSRs and 87 transposon display markers. Altogether finally, this refined map consisted of 13 linkage groups.
With the availability of genome sequence information, a high density and high-resolution map with 1058 SSR markers detecting 1035 loci were constructed, and this was the most saturated map among all the previous foxtail millet studies (Fang et al. 2016). This broader map could be used for functional gene mapping and map-based cloning in the near future. Inclusively, high-density linkage map for 14 agronomic traits under different photoperiods (Zhang et al. 2017), linkage maps for nine phenological traits (Ni et al. 2017), high-density map with 10016 SNP markers (Wang et al. 2017), linkage map by advanced RAD-seq with SNP spanning (Wang et al. 2019) and also binary map construction including 3413 bin markers were developed in foxtail millet. These maps were saturated and more advanced than the RFLP linkage maps probes.
7.3.4 Enumeration of Mapping of Simply-Inherited Stress Tolerance Traits
Different mapping functions were used in developing QTLs and linkage maps that could be used in future molecular breeding. The Kosambi function was initially used for mapping the RFLP probes. The recombination fractions were converted into centimorgans for mapping (Wang et al. 1998). For tapping the genes responsible for plant height and axillary branching in foxtail millet, composite interval mapping was employed later (Doust et al. 2004). Following this, Doust et al. (2005) used three-point linkage analysis to map the markers. Various authors preferred different mapping techniques in constructing genetic maps, and Kosambi function was more used than Haldane (Jia et al. 2009; Fang et al. 2016; Ni et al. 2017). Regarding this, the Haldane mapping function in the construction of genetic maps was implied for agronomic parameters (Yoshitsu et al. 2017; Wang et al. 2017). Composite interval mapping and maximum likelihood calculations were done to segregate markers across nine chromosomes done by Zhang et al. (2017) and Wang et al. (2019). Wang et al. (2019) also used the software Biomercator v3 for integrating selected maps into a reference map by using the applications InforMap and ConsMap in Biomercator. Recently, high-throughput sequencing technologies with SNPs facilitated a desirable multiple QTL mapping with bin markers. These bins are calculated from the recombination breakpoints in which the genotype changes from one type to the other along the chromosomes. This was used as a skeleton to construct a skeleton bin map for the nine chromosomes (Liu et al. 2020). This presents the thrust for developing high-resolution maps with SNPs in the future.
7.3.5 Framework Maps and Markers for Mapping Stress Tolerance QTLs
From the above sections, it is noteworthy that several maps from cytogenetic to high-resolution SNP maps have been generated in foxtail millet. The linkage map constructions proclaim the use of all predominant markers starting from RFLP to cleaved amplified polymorphic sequences (CAPS), transposon display, SSRs, and SNPs obtained by different genotyping techniques. Framework maps were initially constructed by Wang et al. (1998), Doust et al. (2004), and Jia et al. (2009). The first reference genome sequence of foxtail millet was developed in 2012 for the Yugu1 variety, which was employed in several mapping techniques. The segregation of the markers and their polymorphic pattern were the main criteria involved in genetic linkage mapping in foxtail millet.
7.3.6 Depiction of QTL Maps
The LOD in a QTL mapping determines its exact positions in a chromosome, and such maps have been constructed by the authors who conducted a QTL analysis as mentioned above. A LOD score above three is considered to be a major QTL, which can be reliable in molecular breeding, and such QTLs have been enlisted in Table 7.3. The chromosomal positions for the traits, including plant height, heading date, flag leaf length, and width, have attained peaks for chromosomes 5 and 2, respectively. It could be understood that based on the studies carried out by Ni et al. (2017), the loci positions for these traits are present in chromosomes 5 and 2.
7.4 Marker-Assisted Breeding for Resistance Traits
7.4.1 Germplasm Characterization and DUS
Identifying and classifying the germplasm forms the primary dataset for effecting crop improvement in a population. Foxtail millet being a predominant millet across the world, requires an observation of the entities collected around the globe. Reddy et al. (2006) carried out such work in ICRISAT with 1535 accessions collected from 26 countries. They characterized the entire group, and it was found that the Indian accessions had the maximum variability for foxtail millet while the least was for the accessions from Russia. The accessions collected from each country were classified into races and sub-races based on their inflorescence pattern, and the collections from India had all the races except for the sub-race fusiformis. These observations depicted a higher diversity in Indian accessions, like their date of flowering, inflorescence exertion, and plant height. The Chinese accessions were dwarf and decumbent, resembling the S. viridis species, while Indian accessions were taller and erect. The majority of the accessions were taller with medium flowering duration, longer inflorescence, green-leafed, with yellow-colored seeds. By grouping the accessions, it was found that the majority of them belonged to the race indica and maxima. These accessions were further exchanged with several countries like Africa, America, and Europe for their distinct evaluation.
Regular characterizations in different collections across countries is a laborious process that also involves the possibility of experimental errors. To overrule, this a subset of such collections representing the overall population is essential to preserve the complete genetic richness of conserved germplasm. Plant genetic resources portray the development of core, mini-core, and reference set collections in conserving the overall variability. The core collections are observed to have a population size of 10% of the whole base population. They are entitled to represent the entire genetic base of the population. For manipulation across locations, a more desirable mini-core concept was developed, and this is a representation of the entire core subset that is formed. The mini-core comprises 1% of the core collection with maximum representation for all the traits. Favoring trait-specific breeding programs, a reference set/collection is also formulated from the base. This is formed so that the overall variability of the base population for a particular trait is represented. They have a population that follows a normal distribution for a specific trait from the base (Upadhyaya et al. 2009a, b; Pramitha et al. 2020).
These conservation techniques are also adopted in conserving the foxtail millet genetic diversity. ICRISAT is one of the organizations that preserve worldwide collections for foxtail millet, and around 1474 accessions from 23 countries are being characterized and conserved. Initially, a core collection with 155 accessions representing the entire 1474 accessions was developed to ease handling and multi-environmental trials. For the formation of this core initially, a principal component analysis separated the base accessions into 29 clusters, and 10% of the accessions from each cluster constituted this core. X2 and Shannon-Weaver diversity index indicated the null difference between the core and the base set. The accessions represented a maximum diversity, and they were grouped based on 12 qualitative and 11 quantitative traits following a continuous variation with descriptors of S. italica and S. pumila from IBPGR,1985. The core was further stratified based on its races, and similar to the base population, a maximum variability was observed in indica and maxima (Upadhyaya et al. 2008). These collections could be improvised later with reference set screening for nutritional traits, abiotic and biotic stress tolerances in the future.
As a support to international characterizations, nation-wide diversity studies concentrating on yield contributing traits on foxtail millet were performed in India (Upadhyaya et al. 2008). Nirmalakumari and Vetriventhan (2010) conducted initial characterizations for 741 accessions from Tamil Nadu Agricultural University for seven traits. This presented the highest heritability and genetic advance for yield with minimum values for days to 50% flowering. Also the traits, days to 50% flowering, plant height, number of tillers, number of productive tillers, panicle length, and days to maturity were identified as possible indicators for higher yield by its correlation. Parallel to these characterizations, China is a water-scarce country and has predominant importance for foxtail millet cultivation from its domestication era. A base population of 3356 accessions was initially studied, and these vast collections were from the Northwest, Northeast, and Northern parts of China. From this base, 128 accessions were sub-setted and subjected to 79 genome-wide SSR markers for characterization. The population structure indicated the highest diversity among the panel and clustered the accessions into six groups. The second group had unique geographic origins with a higher intra-population diversity from central and southern Shanxi province, which could be further utilized in breeding programs. Thus, it revealed that the clustering pattern was dependent on spring and summer type adaptations in China’s foxtail millet collections (Liu et al. 2011). Studies were made in a foxtail millet for two seasons to enumerate the environmental influence, and a higher yield of the accessions in summer was recorded (Brunda et al. 2015).
Comprehensive analysis with core and reference populations was later conducted in China with the base population of 27,509 accessions. From this, China has identified several drought-tolerant, rust-resistant, and high protein compassing genotypes. This developed core and reference set in china have also revealed the inheritance of many morphological characteristics. For example, seed color was found to be controlled by three loci (BBIIKK). BB causes gray, II enhances color, and KK produces yellow seeds. The order of dominance of seed color is light yellow > red > white > yellow color. The waxy trait in the collections was dominant (chromosome 1 and 9), and the disruption of the GBSS1 gene causes it. Similarly, long bristles, purple bristles (chromosome 4), orange anther color (chromosome 6), purple leaf sheath (chromosome 7), palmate panicle, and plant height (chromosome 3) were dominantly inherited in the foxtail millet germplasm collections, and they were successfully used as morphological markers to identify hybrids (Diao and Jia 2017). Thus, the concept of sub-setting a larger population was well-utilized in China.
The utility of germplasm relies on its variability; thus, screening a more extensive base population earlier will be more effective and helpful in developing core samples for vast collections. Core collections for foxtail millet were formed only in India and China. Both the countries have documented the passport data of the accessions collected across countries. In India, the AICRP project characterized 1312 germplasm accessions under the small millets improvement initiative. This described the importance of documenting the quantitative traits like days to 50% flowering, peduncle length, flag leaf length, and width among the 37 clusters formed by D2 analysis. The array of observations revealed a higher variability with a negative correlation for plant height and flowering with yield. In contrast, the traits—number of basal tillers and peduncle length were positively associated with increasing the yield pattern (Nandini et al. 2018). These accessions could be further improvised by formulating a core/reference like China for facilitating trait-specific breeding in India.
On par with morphological characterization, qualitative characterization for macro- and micronutrients in foxtail millet accessions also presented a higher variability for the content of protein, carbohydrate, calcium, magnesium, iron, zinc, copper, manganese, and other minerals. Superior genotypes were characterized and identified for forwarding to bio-fortification trials (Pavani et al. 2019). This states the overall efforts in characterizing the germplasm accessions across the world for various traits in foxtail millet. These characterizations require criteria to be adopted, and these are framed by international and national organizations for germplasm conservation to avoid ambiguities. The varietal identification and germplasm documenting should be based on DUS descriptors. Such a descriptor for characterizing foxtail millet plays a major role in protecting breeder’s rights under the Protection of Plant Variety and Farmers Right Act (PPVFR Act—2001). DUS refers to the distinctiveness, uniformity, and stability of the accessions characterized. They are used to identify the uniqueness of a variety. In any admixtures or off-types, the descriptors are used as unique indicators to identify original accessions from the admixtures. The record of the observations for DUS requires some guidelines to be fulfilled. This screening should be done in two independent similar growing seasons across two locations under PPVFRA. Seeds weighing 250 g with maximum germination percentage is sent for notifying a variety by an applicant. Each observation should include the mean values of 40 plants, and the color characteristics would be recorded based on RHS (Royal Horticultural Society) color charts.
DUS characterization on 223 genotypes was performed in a reference set aiming at trait-specific improvement. These observations rendered a maximum variability for growth habit, pubescence, pigmentation, leaf attitude, inflorescence shape, grain color, and grain shape. Sixteen qualitative traits from DUS were used for documenting the variability in the accessions in India. These highly variable traits could be utilized to recognize the release of a variety in the future (Banu et al. 2018). Following this study, Amarnath et al. (2019) also utilized DUS descriptors to classify the accessions into different clusters by D2 analysis. These descriptions of an accession favor the fingerprinting of a variety to be released, and this preserves a breeders’ rights to register the nature of the varieties released. Further, this avoids the misuse of varieties by any foreign authority without any prior information, and thereby a proper documenting of the overall variability in Setaria will be done with a pedigree record.
7.4.2 Marker-Assisted Gene Introgression
Foxtail millet is a climate-resilient crop with a higher photosynthetic efficiency due to its C4 mechanism. Being a self-pollinated crop in nature, it has several constraints in exploiting its heterotic potential. Molecular markers play an essential role in characterizing and mapping genetic diversity across populations. At the same time, there are not many studies yet conducted in transferring trait-specific genes by introgression in foxtail millet. Foxtail millet naturally bestows a potential for all the favorable genes within its genome, and molecular mapping of QTLs for these traits are continuously studied from RIL populations. Recently stable QTLs for yield components and straw weight were mapped to chromosomes 3, 6, 7, and 9 of foxtail millet (Liu et al. 2020). Further, nine QTLs for drought (Qie et al. 2014), yield components related to panicle (Odonkor et al. 2018), and other QTLs for agronomic traits (Ni et al. 2017) were being explored in RIL, F2, and F7 populations.
There is not much necessity for improving the yield concerns in foxtail millet by introgression as a larger extent is being achieved from the natural variability by hybridization. Introgression could be carried out for some specific abiotic/biotic stress tolerance in epidemic zones where foxtail millet is significantly affected by stresses. This requires the identification of particular donors for various traits by genetic screening and molecular characterization. Several donors for drought tolerance, saline tolerance, flooding tolerance, and non-lodging efficiencies could be identified from comprehensive phenotyping and genotyping. These pre-breeding lines with proper background and foreground markers could be employed in marker-assisted backcross programs to improvise the genetic makeup of foxtail millet. The recurrent parent and the donors selected should also encompass a broad genetic base to avoid genetic bottlenecks in the future.
7.4.3 Gene Pyramiding
Gene pyramiding is the process of combining several desirable genes in a single cultivar to develop an elite variety. Multi-flexing in foxtail millet cultivars has not yet been initiated. Foxtail millet is a hardy crop with not much pre- and post-harvest losses in cultivation. It is almost not infested by any pests and diseases due to its spontaneous host plant resistance. The bristled inflorescence and spines in leaves are non-preferable for any insect attacks, and the higher phenol metabolites in it provide a natural tolerance against diseases. This is more yielding than other mainstream cereals with a minimum cost of cultivation and higher profit. This happens to be a primary reason for its lesser scope in gene pyramiding techniques. However, this crop is better suited as a reference for several millet species, which are now being explored.
7.4.4 Limitations and Prospects of MAS and MABCB
Marker-assisted selection (MAS) and marker-assisted backcross breeding (MABCB) are advanced techniques that brought about several elite cultivars in mainstream cereals and pulses subjected to severe yield losses. These are now utilized in biofortification trials for improving nutritional traits across crop species. In foxtail millet, these techniques have not been exploited yet. This may be because there was not much necessity in altering or introducing any trait that is absent in it. This crop is being known to possess weedy features and naturally strive in diverse conditions with a high yield. However, if this need arises in the future, these techniques could be successfully employed as the complete genomic sequence information is available in several databases like Gramene (www.gramene.org/). But coming to the backcross, effecting a crossing technique will be a limitation in this crop due to its small florets and lower seed setting capacity. This was why the breeders opted for pure line selection and mutational breeding in developing cultivars in foxtail millet. This has to be overcome with alternate strategies so that several breeding introgression libraries as that of rice could be developed in the future.
MAS has been carried out effectively across populations, followed by mapping the trait loci in chromosomes. With the advent of SNP genotyping, several advanced techniques have been used to identify suitable parental lines. But this lacks proper introgression techniques because until now, only the F2 derived progenies are used as mapping populations. Advanced techniques like GWAS and pyramiding has to be further exploited in varieties for future demands. Other than hybridization difficulties, these techniques could possibly be utilized in the future for improving foxtail millet cultivation in different agro-ecological zones.
7.5 Map-Based Cloning of Resistance/Tolerance Genes
Map-based cloning, also known as positional cloning, helps decipher the genetics of any mutant phenotype using known markers present in the genome. siago1 mutant of Foxtail millet was developed by ethyl methanesulfonate (EMS) treatment of the Yugu1 variety, which showed many developmental abnormalities like thin and curled leaf edges, dwarfed stem, and panicles, and so on. Analysis of the siago1 mutant by map-based cloning revealed that the anomalies were caused by deleting 7 bp in the C-terminus from the SiAGO1b gene and transversion (C–A). RNA-seq expression data comparison of wild type and SiAGO1b mutant showed that 1598 genes were differentially expressed, which may have a role in growth and development, abiotic stress response, cell death, and energy metabolism, etc. BiFC and Y2H assay revealed that the mutated region contained the functional motif for the interaction of SiHYL1 and SiAGO1b (Liu et al. 2016). Li et al. (2016) performed map-based cloning in yellow-green leaf mutant, viz. siygl1 that revealed SiYGL1 is responsible for the phenotype of the mutant. The mutant had a lesser accumulation of chlorophyll (Chl) with reformed ultrastructure due to change in the amino acid phenylalanine to leucine around the ATPase-conserved domain. Gene expression analysis of SiYGL1 and wild type revealed that SiYGL1 regulated genes like DEG2 (development thylakoid), LHCB1, and rbcL (photosynthesis), and SRP54CP (chloroplast signaling).
Zhang et al. (2018) isolated the sistl2 (Setaria italica stripe leaf mutant) mutant of foxtail millet. They performed map-based cloning that revealed SiSTL2 (encodes DCD: deoxycytidine monophosphate deaminase protein) to be the causal agent of the mutant phenotype. In comparison to the Yugu1 variety, sistl2 showed slowed progression in the cell cycle, leaves with stripes, dwarfed stature, and abnormal ultrastructure of the chloroplast. The SiSTL2 expression patterns, in response to low CO2, match the expression pattern of C4 genes. Silencing of SiSTL2 showed decreased 13C leaf content, and during photosynthetic carbon fixation, it increased the DEGs. In another study, Zhang et al. (2018) have performed gene expression analysis and characterized the phenotype of a yellow-green leaf mutant of foxtail millet (siygl2) variety Yugu1. The result showed that SiYGL2 is involved in the progression of leaf senescence, regulates the content of chlorophyll, and also regulates the function of PS II. Tang et al. (2019) studied the ribonucleotide reductase (RNR) in sistl1 mutant of foxtail millet that showed phenotype with striped leaf and reduced chloroplast accumulation due to the substitution of glycine to glutamate in the SiSTL1 protein. They also showed that SiSTL1, encodes the larger subunit of RNR, is essential for growth, the progression of the cell cycle, and the biogenesis of chloroplast in foxtail millet. Map-based cloning is a time-consuming method and is also tedious, but with the availability of the reference genome of the foxtail millet, this process can be made faster. Map-based cloning can also be accompanied by speed breeding method and double haploid culture to get quicker results (Watson et al. 2018).
7.6 Genomics-Aided Breeding for Resistance/Tolerance Traits
7.6.1 Details of Genome Sequencing
In 2012, two independent teams, Beijing Genome Initiative (BGI), China, and the United States Department of Energy Joint Genome Institute (USDOE-JGI), USA, sequenced the genome of foxtail millet and its wild ancestor green foxtail (Bennetzen et al. 2012; Zhang et al. 2012). The team BGI China has used the inbred foxtail millet strain “Zhang gu” and green foxtail strain “A10”. They have used the whole genome shotgun combined with next-generation sequencing for the assembly of foxtail millet genome. Insert size of 170 bp–40 kb was used to create DNA libraries followed by sequencing with Illumina second-generation sequencing. The raw data output was 63.5 Gb, which was filtered down to about 40 Gb clean reads that served as an input for SOAPdenovo for genome assembly. Generation of contigs by de Bruijn graphs and post-gap filling, the contig N50 was found to be 25.4 Kb, 90% of which was present in 16,903 contigs. Furthermore, scaffold N50 was of the size of 1.0 Mb, and 384 Mb (90% of scaffolds) was seen to be in 439 longest scaffolds. Cytogenetic methods and k-mer analysis estimated the genome size to be approximately 490 Mb and 485 Mb, respectively, with 6.6% gaps (28 Mb) wherein the scaffolds extended over about 86% of the complete genome. Transposable elements acquired ~46% of the draft genome when a complete repeat annotation was performed, which included retroelements (~133.6 Mb) like LTR (Gypsy, Copia, and others), LINEs and SINEs, and DNA transposons (~39.7 Mb) comprising tandem repeats like CACTA, hAT, Helitron, Stowaway, Tourist, etc. (Zhang et al. 2012). The team from USDOE-JGI used inbred foxtail millet strain “Yugu1” and green foxtail for sequencing with ABI3730xl capillary sequencer and Illumina Genome Analyzer II platform, respectively. Insert size of 121 Kb, covering almost 12X of the genome, was used to develop the BAC library, further used for BAC-end sequence analysis. The sequence data generated was 4 Gb that showed the genomic sequence of the “Yugu1” strain comprised of 396.7 Mb across nine chromosomes and 327 scaffolds of 4.2 Mb that covered around 80% of the genome (Bennetzen et al. 2012).
7.6.2 Gene Annotation
In foxtail millet, about 38,801 genes were found using consolidated annotation methods. Homologs of these genes with known functions were retrieved by mapping them against various protein databases like GO, InterPro, KEGG, TrEMBL, and SwissProt. Overall, 30,579 genes were annotated, while 8220 genes could not be annotated using the homolog function information. According to the gene ontology annotation, around 79% of the identified genes have homologs in public databases with well-defined functions. The transcriptome analysis of tissues from the root, spica, leaf, and stem showed expression of around 82% predicted genes in them. The average of 4.3 number of exons per gene is present. Besides, the intron’s average length was 442 bp, whereas the average length of the exon was 256 bp. The prediction of 1367 pseudogenes genes showed that they could be retrotransposed, duplicated, or be unclassified. The non-coding RNA gene prediction in the foxtail millet genome revealed that chromosomes 1, 7, 8, and 9 contained huge clusters of rRNA genes that accounted for 99 in number. In contrast, the rest non-coding RNA genes showed limited chromosomal distribution. The non-coding RNA genes contained 704 tRNA genes; 382 snRNA genes included HACA-box, CD-box, and splicing, and 159 miRNA genes (Zhang et al. 2012).
As angiosperms show a higher degree of gene conservation, Bennetzen et al. (2012) annotated the completely assembled whole-genome sequence of foxtail millet according to the reports available in other grasses and model plant—Arabidopsis. Annotation revealed 35,472 primary transcripts of protein-coding genes along with 5128 alternate transcripts and 11% of which can be putative candidates for foxtail millet study due to their novelty. Annotated genes have an average intron length of 163 bp and an average exon length of 135 bp. The protein was seen to contain an average of 329 amino acids. Foxtail millet has around 40% of TEs, which is comparatively less than the other grasses, making it a model crop. The study of C4 photosynthesis pathway genes viz. PEPC, PPDK, and MDH with maize and sorghum orthologs showed a higher conservation degree. Six clusters of drought-associated genes were found during Setaria genome analysis, with a higher number of drought-tolerant species, like Setaria and sorghum, than the drought-susceptible species like rice and maize (Bennetzen et al. 2012; Muthamilarasan and Prasad 2017). Functional studies in foxtail millet with putative candidate genes like multi-antimicrobial extrusion protein, NADH oxidase, plant lipid transfer protein, Aldo/keto reductase, AMP-dependent synthetase/ligase, and glutathione S-transferase, could aid in unraveling the genetic rationale for stress adaptation in them. The analysis of C4 photosynthetic genes, namely PEPC, PPDK, and MDH showed higher conservation with sorghum as well as maize. In contrast, the malic enzyme isoform on foxtail millet did not show any conservation. This result could be exploited to study C4 photosynthesis pathway evolution (Bennetzen et al. 2012; Muthamilarasan and Prasad 2017).
7.6.3 Impact on Germplasm Characterization and Gene Discovery
The release of the draft genome sequence of foxtail millet paved the way for several studies that identified genes responsible for molecular and physiological processes. These genes may play a pivotal role in the growth and development of the crop and protect the plant from various environmental stresses. Characterization of genes also threw light on the regulation of few genes. The foxtail millet specific genes might also play a major function in making it a climate-resilient crop. In foxtail millet, 586 genes were observed by Zhang et al. (2012) associated with “response to water” that could be putative candidates in studying dehydration and drought stress machinery. Many genes were identified and characterized at a genome-wide level that showed differential expression when exposed to various abiotic stress viz. NAC (Puranik et al. 2013), AP2/ERF (Lata et al. 2014), WD40 (Mishra et al. 2014), MYB (Muthamilarasan et al. 2014b), C2H2 type zinc finger (Muthamilarasan et al. 2014a), Nuclear factor Y (Feng et al. 2015), WRKY (Muthamilarasan et al. 2015), RDR, AGO, and DCL (Yadav et al. 2015), 14-3-3 proteins (Kumar et al. 2015), Heat shock protein (Singh et al. 2016), autophagy associated protein (Li et al. 2016), SET (Yadav et al. 2016a, b), DOF (Zhang et al. 2017), HD-Zip (Chai et al. 2018), CDPK genes (Yu et al. 2018), LIM genes (Yang et al. 2019) and C4 photosynthetic genes (Muthamilarasan et al. 2020). Regulation of root development by SiMYB3 showed auxin biosynthesis regulation in low nitrogen conditions (Ge et al. 2019).
7.6.4 Structural and Functional Genomic Resources Developed
With the foxtail millet genome sequence release, there was a sudden increase in the structural and functional genomic resources. After scanning the plant’s genome, 28,342 microsatellite motifs were identified with an average coverage of 69 microsatellites/Mb of foxtail millet genome sequence. Out of 28,342 microsatellites, primer pairs for 21,294 were developed, and a physical map was constructed, and non-uniform distribution was observed. Chromosome 9 showed a maximum density of 46.4 per Mb and the highest average marker frequency of about 17%. Whereas chromosome number 8 showed a minimum density of about 30/Mb and the lowest average marker frequency of about 8%. The physical gap size between the SSR markers was seen to be 24 kb. Forty percent polymorphism was seen in these markers, with 89% cross-transferability among other bioenergy grasses, millets, and cereals. Guinea grass showed a maximum percentage in transferability, with an average of 98.2% and wheat, with a minimum of 71.2% (Pandey et al. 2013). In foxtail millet, 24,828 unigenes were generated by assembling 66,027 ESTs reported in NCBI dbEST to develop 534 eSSRs (Kumari et al. 2013). The development of 5,123 ILP markers in foxtail millet was done by using EST database. These ILP markers could be used in marker-assisted breeding, comparative genome mapping, evolutionary studies, generating high-density genetic linkage maps, and genes and QTL mapping for beneficial agronomic traits (Muthamilarasan et al. 2014a; Muthamilarasan and Prasad 2015). Around 176 miRNA-based markers were developed by analyzing the genome-wide miRNAs reported in the foxtail millet and its related species and showed about 55% polymorphism and about 70% cross-transferability. These markers also showed good reproducibility, high stability, and efficiency (Yadav et al. 2014). The plant genome contains a substantial amount of transposable elements and is uniformly distributed in the genome, making it easier to develop TE-based markers. In foxtail millet, 30,706 TEs were found that were further divided into two classes with 6314 class I retrotransposons and 24,392 class II DNA retrotransposons. Repeat junction markers were developed (20,278), grouped in six types namely RMAP (57), IRAP (3,239), RJM (4,451), RBIP (4,801), ISBP (7,401) and RJJM (329) (Yadav et al. 2015).
The available genome sequence and annotation have eased identifying and studying gene and gene families across the foxtail millet genome using computational methods. This also helps in learning the role of the genes in plant growth and their fight against stress. For breeding in foxtail millet, allele-specific markers were developed from the DREB2 locus that contained a dehydration tolerance linked SNP (Lata et al. 2013). NAC TFs coding 147 genes were seen in foxtail millet, out of these 50 were used for analyzing the differential gene expression against hormone and stress conditions and reported that SiNAC128 could be used in stress associated research (Puranik et al. 2013) Genome-wide analysis of several other transcription factors were performed to find genes encoding them, like, 110 genes encoding WRKY, 171 genes encoding AP2/ERF, 124 genes encoding C2H2 zinc finger and 209 genes encoding MYB (Lata et al. 2013; Muthamilarasan et al. 2014b, 2015). RNA silencing-related genes were studied by Yadav et al. (2015) and found putative candidates like SiDCL06, SiRDR07, and SiAG008. A study on ADP-ribosylation factors of rice and foxtail millet were reported to have 23 ARF proteins in rice and 25 in foxtail millet. The presence of cis-regulatory elements in their promoter might have a role in the regulation of stress and could be studied further.
7.6.5 Application of Structural and Functional Genomics in Genomics-Assisted Breeding
Structural and functional genomics of foxtail millet is being studied extensively to establish genetic and genomic resources that could further help know the physiological and molecular basis of tolerance to several stress factors like heat, drought, and salinity. The genomics data helps develop molecular markers, QTLs, and platforms for genotyping, which can be used in genomics-assisted breeding (GAB) for the rapid development of elite varieties of foxtail millet. Stress-responsive genetic determinants, viz. alleles, genes, and QTLs in contrasting cultivars, are identified, followed by NGS-based GAB to provide stress tolerance. Prerequisites of GAB is to perform association mapping, QTL mapping as well as recurrent selection screening.
7.7 Gene Editing Strategies Developed in Foxtail Millet
The efficiency and precision of the gene-editing tools help in the rapid development of elite varieties of crops. The proven and most effective tools of genome editing are zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), programmed homing nuclease (Meganucleases), transposons, recombinant Adeno-associated virus (rAAV), and CRISPR/Cas9. CRISPR/Cas9, owing to its simple, efficient, and robust characteristic, has become very popular. Nevertheless, optimization in the construction of vector, tissue culture, gene expression, and transformation protocol are prerequisites before applying it in a new crop (Yin et al. 2017). CRISPR/Cas9 is also capable of multiplexing. In foxtail millet, CRISPR/Cas9 has been applied to mutate the PDS gene via protoplast transfection wherein the transfection efficiency was found to be 51%, and mutagenesis efficiency was 10.2% (Lin et al. 2018). In 2019, Huang et al. used the CRISPR/Cas9 strategy to knockout the SvLES1 gene from S. viridis (green foxtail, wild ancestor of foxtail millet ; Huang et al. 2019). Domesticated foxtail millet has an insertion of a retrotransposon in the gene, Less Shattering 1, responsible for seed shattering in S. viridis. In 2020, Weiss’ group has developed and optimized protoplast-based multiplexed CRISPR/Cas9_Trex2 tool for genome editing in green foxtail (Weiss et al. 2020). In the study, they have targeted and knocked out Drm1a and Drm1b genes that are highly linked (domain rearranged methylase). In CRISPR/Cas9, still an efficient delivery system is needed. The genome editing of foxtail millet is still in its infancy. Nevertheless, multiple types of research (as mentioned above) are going on to develop and optimize genome editing protocols in foxtail millet. More research is done in the wild ancestor of foxtail millet, i.e., S. viridis, and hence an efficient CRISPR/Cas9 protocol is still lacking in S. italica.
7.8 Achievements of Transgenics
In foxtail millet, the transgenics were developed using the biolistic or Agrobacterium tumefaciens mediated methods. Gene transfer into immature inflorescence derived embryogenic callus was performed using biolistic methods (Diao et al. 1999). In another study, the bombardment of foxtail millet explants from pollen, and inflorescence was done to develop transgenic, but the efficiency was abysmal (Dong and Duan 1999, 2000). Later, embryogenic calli derived from floret was bombarded to generate overexpression as well as RNAi lines of SiPf40. The study shed light on auxin homeostasis and suggested that SiPf40 played a role in tillering (Liu et al. 2009). (Liu et al. 2005) were the first to report Agrobacterium-mediated transformation in foxtail millet with 6.6% efficiency. A modified version of Liu et al. protocol was used for transformation using Agrobacterium in calli derived from panicle and immature inflorescence (Qin et al. 2008; Wang et al. 2011). This modified version was exploited by Wang et al. to overexpress SiLEA14 (LEA proteins’ homolog; LEA: late embryogenesis abundant), the transgenic lines displayed increased tolerance against salt and drought stress (Wang et al. 2014). SiARDP (ABA-responsive DRE-binding protein: ARDB), along with SiASR4 uses the ABA-dependent pathway in transgenic foxtail millet to provide drought tolerance (Li et al. 2014, 2017). In 2016, Pan et al. developed SiLTP (LTP: Lipid transfer protein) overexpressed and RNAi lines of foxtail millet and elucidated the function of SiLTP against salt and drought stress. For conferring abiotic stress tolerance following the ABA-dependent signaling pathway, SiLTP could be a probable candidate for SiARDP. An efficient transformation protocol is a prerequisite to developing transgenics, but due to genetic transformation recalcitrance in foxtail millet, not many transgenics could be developed. In foxtail millet, Santos et al. (2020) has developed an Agrobacterium-mediated transformation protocol with an average efficiency of 19% using seed as explant. Sood et al. (2020) has developed an efficient protocol for gene expression using Agrobacterium transformation in foxtail millet seeds with a transformation efficiency of 27%. Recently, Yang et al. (2020) have developed a miniature mutant of foxtail millet named Xiaomi with a tiny life cycle like Arabidopsis. An efficient transformation protocol has been developed for it and the availability of transcriptomics and genomics resources to ease research in foxtail millet. Xiaomi could act as a model to study the molecular functions of C4 plants.
7.9 Brief Account on the Role of Bioinformatics as a Tool
7.9.1 Gene and Genome Databases
It was the Joint Genome Institute (JGI) that, for the first time, introduced the foxtail millet genome sequence in Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html) wherein the genome assembly of foxtail millet genome was around 405.7 Mb present across 336 scaffolds wherein, with approximately 1.2% gap, 400.9 Mb was present across 6791 contigs representation of around 90 data is done in nine pseudomolecule. Protein coding transcripts were present in 34,584 loci, and also 43,001 protein-coding transcripts are reported (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Sitalica). Zhang et al. (2012) hosted the foxtail millet sequence in the Foxtail millet database (http://foxtailmilletmillet.genomics.org.cn) to avail the genome sequence to the researchers. PlantGDB also hosted the foxtail millet sequence (http://www.plantgdb.org/SiGDB/). KEGG database (https://www.genome.jp/kegg/) was used to map the proteins of foxtail millet. Gramene database is an open resource with integrated data containing the genome data of foxtail millet (http://www.gramene.org/Setaria_italica/Info/Index) (Tello Ruiz et al. 2016). The databases are an open resource and contain tools that could help in sequence assembly, bowser for datasets, special datasets, and more. The Foxtail millet Marker Database (FmMDb) was created in 2013 as an open-source where the large-scale datasets of markers could be retrieved, visualized, and managed in order to develop elite cultivars of the crop (http://www.nipgr.res.in/foxtailmillet.html). Several other browsers like Ensembl and UCSC Genome Browser can also be used for gene and genome search.
7.9.2 Comparative Genome Databases
Databases like Gramene, PlantGDB, Phytozome v12.1, and NCBI are used for comparative genome analysis of foxtail millet. These databases contain the integrated and updated data of the crop species. They also provide several tools that could ease the data search process wherein data can be downloaded, analyzed, or developed. They include various search functions like BLAST, pBLAST, gene viewer, and so on. Gradually with time, many application-based databases were created. Setaria italica functional genome database (SIFGD) is a platform created in lieu to integrate sequences of the genome, transcript, protein, miRNA, and RNA of foxtail millet from different public databases like BGI, NCBI, Phytozome, and so on (http://structuralbiology.cau.edu.cn/SIFGD/index.html). MiRNA database for foxtail millet (FmMiRNADb) contains the data for 355 miRNAs of foxtail millet and 123 molecular markers based on miRNA that would aid in molecular breeding also genotyping of cereals and millets. Foxtail millet transposable elements-based marker database (FmTEMDb) was developed with TEs data of approximately 30,000 foxtail millet with markers of six types that could be applied for a large-scale genotyping (http://59.163.192.83/ltrdb/index.html).
7.9.3 Gene Expression Databases
The Foxtail millet Transcription Factor Database (FmTFDb) was created in the year 2014. It was an open-access platform and contained 2295 transcription factors across 55 families. The transcription factor related information like sequence, phylogeny, and gene ontology could be looked into using several tools like BLAST search and set of annotation query interfaces (http://59.163.192.91/FmTFDb/index.html). NCBI (https://www.ncbi.nlm.nih.gov/guide/genes-expression/) contains data of foxtail millet gene expression along with tools to analyze the gene expression data. Also, Phytozome can be used to study the gene expression data of foxtail millet (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Sitalica). These databases provide functional annotation of several tissue-specific expression data and other corresponding information.
7.9.4 Protein or Metabolome Databases
To map the proteins of foxtail millet, SwissProt/TrEMBL (https://www.uniprot.org/statistics/TrEMBL) is extensively used as it gives easy access to the sequence of the protein and their annotation. It contains several tools like sequence BLAST, alignment, retrieval and ID mapping, and peptide search. Protein function analysis could be done by InterPro (https://www.ebi.ac.uk/interpro/). It could help predict protein domains and classify them in protein families using predictive signature models available from other member databases. Plant metabolic pathway databases (PMN) help generate a framework to mingle different plant metabolism data sources. These encompass the metabolic pathways of various plant species and corresponding enzymes, genes, and substrate (https://pmn.plantcyc.org/SETARIA). The plant metabolome database (PMDB) contains the data of plant metabolites and small molecules that are functionally and structurally annotated. For quick access, the web interface has several tools where a query can be searched, followed by data retrieval and analysis.
7.9.5 Integration of Different Data
Biological databases have turned out to be one of the crucial resources for researchers worldwide and are used daily. All the databases mentioned above may differ in functions, but they share a similar framework and show the interests and expertise of people who manage those databases. Each database has its own specialty and role, wherein it becomes difficult to answer questions related to other databases. The integration of these databases can hamper the information resource as the process will require several unwanted compromises. For instance, maintaining the exact name of the biological samples may vary across the databases. The task of continuously updating the integrated databases would also be problematic as the biological databases always change. A single database for all the queries might make our work more comfortable, but it does not seem possible shortly. Many databases have formed a consortium so that they can exchange the information by cross-database search of queries.
7.10 Conclusions and Future Perspectives
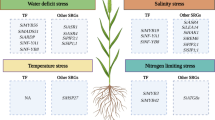
Compared to other major cereal crops, foxtail millet is higher in nutritional compounds and can be grown in adverse climatic conditions. Lodging is one of the significant drawbacks in foxtail millet yield loss and poor grain quality, but cultivars like Longgu 28 and Nenxian 13 developed by China are lodging resistant (Dwivedi et al. 2012). Waterlogging is also one of the constraints in foxtail millet cultivation. Lugu No. 7, a foxtail millet cultivar resistant to waterlogging, was developed by Chen and Qi (1993). Studies are conducted to identify QTLs associated with climate resilient traits (Fig. 7.1).

Strategies involved in identification of quantitative trait loci controlling climate-resilient traits in foxtail millet
Cereal grain cultivation is affected by the changing climatic conditions and decreasing fertility and area of agricultural land. The population worldwide is increasing, and so is food scarcity, and therefore, food production across the globe calls for an urgent solution. Speeding up crop improvement through genomic assisted breeding could help identify abiotic stress-responsive genes with better agronomic characters. Integration of data from multi-omics technology (genomics, transcriptomics, proteomics and metabolomics), statistical analysis, and computational biology in breeding would fasten up the crop improvement process and produce elite variety that can withstand adverse weather and give higher yield (Fig. 7.2). After the release of the reference genome, molecular maps were constructed for the tolerant and susceptible cultivars using QTLs, molecular markers, and many other candidate genes. These maps are brought into use in MAS. Speed breeding strategy can be integrated with breeding approaches like genomic assisted breeding and breeding with haplotypes to fast track the release of elite varieties. Until now, breeding was the sole source of foxtail millet improvement. With the availability of the reference genome sequence, genome editing tools, and the efficient transformation protocols discussed in the previous section can also be implied for rapid gene discovery manipulation of traits with better agronomic characteristics. These researches are majorly limited to multinational companies, and hence collaboration and coordination of the public sector are required to accelerate the release of elite varieties.

Flowchart showing the strategies involving multi-omics tools to develop abiotic stress tolerant cultivars of foxtail millet
References
Austin D (2006) Fox-tail millets (Setaria: Poaceae)—abandoned food in two hemispheres. Econ Bot 60:143–158
Banu H, Gowda J, Gowda MC (2018) Characterization and identification of DUS traits in reference set of foxtail millet germplasm (Setaria italica (L.) Beauv). Intl J Curr Microb Appl Sci 7(12):175–183
Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J et al (2012) Reference genome sequence of the model plant Setaria. Nat Biotechnol 30(6):555–561
Brunda SM, Kamatar MY, Hundekar R, Naveenkumar KL (2015) Studies on correlation and path analysis in foxtail millet [Setaria italica (L.) P. B.]. Intl J Appl Agric Hortic Sci 6(5):966–969
Ceccarelli S, Grando S (1996) Drought as a challenge for the plant breeder. Plant Growth Regul 20:149–155. https://doi.org/10.1007/BF00024011
Chai W, Si W, Ji W, Qin Q, Zhao M et al (2018) Genome-wide investigation and expression profiling of HD-Zip transcription factors in foxtail millet (Setaria italica L.). BioMed Res Intl 2018(8457614):18. https://doi.org/10.1155/2018/8457614
Chander S, Bhat KV, Kumari R, Sen S, Gaikwad AB et al (2017) Analysis of spatial distribution of genetic diversity and validation of Indian foxtail millet core collection. Physiol Biol Plants 23(3):663–673
Chen J, Qi Y (1993) Recent developments in foxtail millet cultivation and research in China. In: Riley KW, Gupta SC, Seetharam A, Mushonga JN (eds) Advances in small millets. Oxford and IBH Publishing Co, New Delhi, India, pp 101–107
Deaton A, Dreze J (2009) Food and nutrition in India: facts and interpretations. Available at http://www.princeton.edu/deaton/downloads/FoodandNutritioninIndiaFactsandInterpretations.pdf
Devos KM, Wang ZM, Beales CJ, Sasaki T, Gale MD (1998) Comparative genetic maps of foxtail millet (Setaria Italica) and rice (Oryza sativa). Theor Appl Genet 96:63–68
Diao X, Jia G (2017) Foxtail millet breeding in China. In: Doust A, Diao X (eds) Genetics and genomics of setaria. Springer International Publishing, Cham, Switzerland, pp 93–113
Diao X, Chen Z, Duan S, Liu Y, Zhao L et al (1999) Factors influencing foxtail millet embryogenic calli transformation by particle bombardment. Acta Agric Boreali Sin 14(3):31–36
Diao X, Schnable J, Bennetzen JL, Li J (2014) Initiation of Setaria as a model plant. Front Agric Sci Eng 1:16. https://doi.org/10.15302/J-FASE-2014011
Dong Y, Duan S (1999) Establishment of embryogenic cell suspension culture and plant regeneration of millet and gene transfer. J Basic Sci Eng 7(1):34–40
Dong Y, Duan S (2000) Production of transgenic millet plants via particle bombardment. Acta Bot Boreal-Occident Sin 20(2):175–178
Doust AN, Devos KM, Gadberry MD, Gale MD, Kellogg EAl (2004) Genetic control of branching in foxtail millet. Proc Natl Acad Sci USA 101:9045–9050
Doust AN, Devos KM, Gadberry MD, Gale MD, Kellogg EA (2005) The genetic basis for inflorescence variation between foxtail and green millet (Poaceae). Genetics 169(3):1659–1672
Dvořáková Z, Čepková PH, Janovská D, Viehmannová I, Svobodová E et al (2015) Comparative analysis of genetic diversity of 8 millet genera revealed by ISSR markers. Emir J Food Agri 617–628
Dwivedi S, Upadhyaya H, Senthilvel S, Hash C, Fukunaga K et al (2012) Millets: genetic and genomic resources. In: Janick J (ed) Plant breeding reviews, vol 35. Wiley, USA, pp 247–375
Fang X, Dong K, Wang X, Liu T, He J et al (2016) A high density genetic map and QTL for agronomic and yield traits in Foxtail millet [Setaria italica (L) P Beauv]. BMC Genom 17(1):336
Feng ZJ, He GH, Zheng WJ, Lu PP, Chen M et al (2015) Foxtail Millet NF-Y Families: Genome-wide survey and evolution analyses identified two functional genes important in abiotic stresses. Front Plant Sci 6:1142
Fukunaga K, Domon E, Kawase M (1997) Ribosomal DNA variation in foxtail millet, Setaria italica (L) P Beauv, and a survey of variation from Europe and Asia. Theor Appl Genet 95(5–6):751–756
Fukunaga K, Wang Z, Kato K, Kawase M (2002) Geographical variation of nucleargenome RFLPs and genetic differentiation in foxtail millet, Setaria italica (L) P Beauv. Genet Resour Crop Evol 49(1):95–101
Fukunaga K, Kato K (2003) Mitochondrial DNA variation in foxtail millet, Setaria italica (L.) P. Beauv. Euphytica 129:7–13
Ge L, Dou Y, Li M, Qu P, He Z et al (2019) SiMYB3 in foxtail millet (Setaria italica) confers tolerance to low-nitrogen stress by regulating root growth in transgenic plants. Intl J Mol Sci 20. https://doi.org/10.3390/ijms20225741
Hirschi KD (2009) Nutrient biofortification of food crops. Annu Rev Nutr 29:401–421
Huang P, Mamidi S, Healey A, Grimwood J, Jenkins J et al (2019) The Setaria viridis genome and diversity panel enables discovery of a novel domestication gene. bioRxiv 2019:744557
Huang Z, Zhao L, Chen D, Liang M, Liu Z et al (2013) Salt stress encourages proline accumulation by regulating proline biosynthesis and degradation in Jerusalem Artichoke Plantlets. PLoS ONE 8(4):e62085
Jaiswal V, Gupta S, Gahlaut V, Muthamilarasan M, Bandyopadhyay T et al (2019) Genome-wide association study of major agronomic traits in foxtail millet (Setaria italica L) using ddRAD sequencing. Sci Rep 9(1):1–11
Jia X, Zhang Z, Liu Y, Zhang C, Shi Y et al (2009) Development and genetic mapping of SSR markers in foxtail millet [Setaria italica (L) P Beauv]. Theor Appl Genet 118(4):821–829
Jia X, Zhang Z, Liu Y, Zhang C, Shi Y, et al (2009) Development and genetic mapping of SSR markers in foxtail millet [Setaria italica (L) P Beauv]. Theor Applied Genet 118(4):821-829
Kole C, Muthamilarasan M, Henry R, Edwards D, Sharma R et al (2015) Application of genomics-assisted breeding for generation of climate resilient crops: progress and prospects. Front Plant Sci 6:563
Kumar K, Muthamilarasan M, Bonthala VS, Roy R, Prasad M (2015) Unraveling 14-3-3 proteins in C4 panicoids with emphasis on model plant Setaria italica reveals phosphorylation-dependent subcellular localization of RS splicing factor. PLoS ONE 10:
Kumari K, Muthamilarasan M, Misra G, Gupta S, Subramanian A et al (2013) Development of eSSR-markers in Setaria italica and their applicability in studying genetic diversity, cross-transferability and comparative mapping in millet and non-millet species. PLoS ONE 8(6):
Lata C, Gupta S, Prasad M (2013) Foxtail millet: a model crop for genetic and genomic studies in bioenergy grasses. Crit Rev Biotechnol 33:328–343
Lata C, Mishra AK, Muthamilarasan M, Bonthala VS, Khan Y et al (2014) Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L). PLoS ONE 9(11):
Liu X, Tang S, Jia G, Schnable JC, Su H, et al (2016) The C-terminal motif of SiAGO1b is required for the regulation of growth, development and stress responses in foxtail millet (Setaria italica (L.) P. Beauv). J Exp Bot 67:3237–3249
Li Y-M (1997) Breeding for foxtail millet drought tolerant cultivars (in Chinese). In: Li Y (ed) Foxtail millet breeding. Chinese Agriculture Press, Beijing, China, pp 421–446
Li Y, Wu SZ (1996) Traditional maintenance and multiplication of foxtail millet (Setaria italica (L.) P. Beauv) landraces in China. Euphytica 87:33–38
Li C, Yue J, Wu X, Xu C, Yu J (2014) ABA-responsive DRE-binding protein gene from Setaria italica, SiARDP, the target gene of SiAREB, plays a critical role under drought stress. J Exp Bot 65:5415–5427
Li W, Chen M, Wang E, Hu L, Hawkesford MJ et al (2016) Genome-wide analysis of autophagy-associated genes in foxtail millet (Setaria italica L.) and characterization of the function of SiATG8a in conferring tolerance to nitrogen starvation in rice. BMC Genomics 17:797
Li J, Dong Y, Li C, Pan Y, Yu J (2017) SiASR4, the target gene of SiARDP from Setaria italica, improves abiotic stress adaption in plants. Front Plant Sci 7:2053. https://doi.org/10.3389/fpls.2016.02053
Lin CS, Hsu CT, Yang LH, Lee LY, Fu JY et al (2018) Application of protoplast technology to CRISPR/Cas9 mutagenesis: from single-cell mutation detection to mutant plant regeneration. Plant Biotechnol J 16:1295–1310. https://doi.org/10.1111/pbi.12870
Liu Y, Yu J, Zhao Q, Zhu D, Ao G (2005) Genetic transformation of millet (Setaria italica) by Agrobacterium-mediated. Chin J Agric Biotechnol 13:32–37
Liu Y, Feng X, Xu Y, Yu J, Ao G et al (2009) Overexpression of millet ZIP-like gene (SiPf40) affects lateral bud outgrowth in tobacco and millet. Plant Physiol Biochem 47:1051–1060
Liu Z, Bai G, Zhang D, Zhu C, Xia X et al (2011) Genetic diversity and population structure of elite foxtail millet [Setaria italica (L) P Beauv] germplasm in China. Crop Sci 51(4):1655–1663
Liu T, He J, Dong K, Wang X, Wenwen W et al (2020) QTL mapping of yield component traits on bin map generated from resequencing a RIL population of foxtail millet (Setaria italica). BMC Genom 21(1):141
Lu H, Zhang J, Liu KB, Wu N, Li Y et al (2009) Earliest domestication of common millet (Panicum miliaceum) in East Asia extended to 10,000 years ago. Proc Natl Acad Sci USA 106:7367–7372
Mishra AK, Muthamilarasan M, Khan Y, Parida SK, Prasad M et al (2014) Genome-wide investigation and expression analyses of WD40 protein family in the model plant foxtail millet (Setaria italica L). PLoS ONE 9(1):
Muthamilarasan M, Prasad M (2015) Advances in Setaria genomics for genetic improvement of cereals and bioenergy grasses. Theor Appl Genet 128:1–14
Muthamilarasan M, Prasad M (2017) Genetic determinants of drought stress tolerance in Setaria. In: Doust A, Diao X (eds) Genetics and genomics of setaria. Springer, Berlin, pp 267–289
Muthamilarasan M, Bonthala VS, Mishra AK, Khandelwal R, Khan Y et al (2014a) C2H2-type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct Integr Genom 14:531–543
Muthamilarasan M, Khandelwal R, Yadav CB, Bonthala VS, Khan Y et al (2014b) Identification and molecular characterization of MYB transcription factor superfamily in C4 model plant foxtail millet (Setaria italica L.). PLoS One 9:e109920
Muthamilarasan M, Venkata Suresh B, Pandey G, Kumari K, Parida S et al (2014c) Development of 5123 intron-length polymorphic markers for large-scale genotyping applications in foxtail millet. DNA Res 21:41–52
Muthamilarasan M, Bonthala VS, Khandelwal R, Jaishankar J, Shweta S et al (2015) Global analysis of WRKY transcription factor superfamily in Setaria identifies potential candidates involved in abiotic stress signaling. Front Plant Sci 6:910
Muthamilarasan M, Dhaka A, Yadav R, Prasad M (2016) Exploration of millet models for developing nutrient rich graminaceous crops. Plant Sci 242:89–97
Muthamilarasan M, Singh RK, Suresh BV, Rana S, Dulani P et al (2020) Genomic dissection and expression analysis of stress-responsive genes in C4 panicoid models, Setaria italica and Setaria viridis. J Biotechnol 318:57–67. https://doi.org/10.1016/j.jbiotec.2020.05.007
Nandini C, Sujata B, Tippeswamy V (2018) Characterization of foxtail millet (Sateria italica (L) Beauv) germplasm for qualitative and quantitative traits to enhance its utilization. Acad J Agric Res 6(5):121–129
Ni X, Xia Q, Zhang H, Cheng S, Li H et al (2016) Gene mapping of nine agronomic traits and genome assembly by resequencing a foxtail millet RIL population bioRxiv: 069625
Ni X, Xia Q, Zhang H, Cheng S, Li H et al (2017) Updated foxtail millet genome assembly and gene mapping of nine key agronomic traits by resequencing a RIL population. GigaScience 6(2):giw005
Nadeem F, Ahmad Z, Wang R, Han J, Shen, Q, et al (2018) Foxtail Millet [Setaria italica (L.) Beauv.] Grown under low nitrogen shows a smaller root system, enhanced biomass accumulation, and nitrate transporter expression. Front Plant Sci 9:205
Nirmalakumari A, Vetriventhan M (2010) Characterization of foxtail millet germplasm collections for yield contributing traits. Elect J Plant Breed 1(2):140–147
Odonkor S, Choi S, Chakraborty D, Bello LM, Wang X et al (2018) QTL mapping combined with comparative analyses identified candidate genes for reduced shattering in Setaria italica. Front Plant Sci 9:918
Pan Y, Li J, Jiao L, Li C, Zhu D et al (2016) A non-specific Setaria italica lipid transfer protein gene plays a critical role under abiotic stress. Front Plant Sci 7:1752. https://doi.org/10.3389/fpls.2016.01752
Pandey G, Misra G, Kumari K, Gupta S, Parida SK et al (2013) Genome-wide development and use of microsatellite markers for large–scale genotyping applications in foxtail millet [Setaria italica (L.)]. DNA Res 20:197–207
Pavani A, Ratna Babu D, Kumar V, Ramesh D (2019) Characterisation of quality traits in foxtail millet germplasm (Setaria italica (L) Beau). Intl J Chem Studies 7(6):769–772
Pramitha JL, Joel AJ, Srinivas S, Sreeja R, Hossain R et al (2020) Enumerating the phytic acid content in maize germplasm and formulation of reference set to enhance the breeding for low phytic acid. Physiol Mol Biol Plants 26(2):353–365
Puranik S, Sahu PP, Mandal SN, Suresh VB, Parida SK et al (2013) Comprehensive genome-wide survey, genomic constitution and expression profiling of the NAC transcription factor family in Foxtail Millet (Setaria italica L.). PLoS One 8:e64594
Qie L, Jia G, Zhang W, Schnable J, Shang Z et al (2014) Mapping of quantitative trait locus (QTLs) that contribute to germination and early seedling drought tolerance in the interspecific cross Setaria italica Setaria viridis. PLoS ONE 9(7): https://doi.org/10.1371/journal.pone.0101868
Qin F, Zhao Q, Ao G, Yu JJ (2008) Co-suppression of Si401 a maize pollen specific Zm401 homologous gene, results in aberrant anther development in foxtail millet. Euphytica 163(1):103–111
Radha BN, Channakeshava BC, Bhanuprakash K (2014) Biochemical landmarks for Identification of foxtail millet genotypes. Intl J Appl 9(1):9–26
Reddy VG, Upadhyaya HD, Gowda CLL (2006) Current status of sorghum genetic resources at ICRISAT: their sharing and impacts. Intl Sorghum Millets Newsl 47:9–13
Santos CM, Romeiro D, Silva JP, Basso MF, Molinari HBC et al (2020) An improved protocol for efficient transformation and regeneration of Setaria italica. Plant Cell Rep 39:501–510. https://doi.org/10.1007/s00299-019-02505-y
Sato K, Mukainari Y, Naito K, Fukunaga K, Mukainari Y et al (2013) Construction of a foxtail millet linkage map and mapping of spikelet-tipped bristles1 (stb1) by using transposon display markers and simple sequence repeat markers with genome sequence information. Mol Breed 31(3):675–684
Schontz D, Rether B (1998) Genetic variability in foxtail millet, Setaria italica (L) P Beauv-RFLP using a heterologous rDNA probe. Plant Breed 117(3):231–234
Singh RK, Jaishankar J, Muthamilarasan M et al (2016) Genome-wide analysis of heat shock proteins in C 4 model, foxtail millet identifies potential candidates for crop improvement under abiotic stress. Sci Rep 6(1):1–14
Sood P, Singh RK, Prasad M (2020) An efficient Agrobacterium-mediated genetic transformation method for foxtail millet (Setaria italica L.). Plant Cell Rep 39:511–525. https://doi.org/10.1007/s00299-019-02507-w
Sun J, Luu NS, Chen Z, Chen B, Cui X et al (2019) Generation and characterization of a Foxtail Millet (Setaria italica) mutant library. Front Plant Sci 10:369
Tang C, Tang S, Zhang S, Luo M, Jia G et al (2019) SiSTL1, encoding a large subunit of ribonucleotide reductase, is crucial for plant growth, chloroplast biogenesis, and cell cycle progression in Setaria italica. J Exp Bot 70:1167–1182. https://doi.org/10.1093/jxb/ery429
Tello-Ruiz MK, Stein J, Wei S, Preece J, Olson A et al (2016) Comparative plant genomics and pathway resources. Nucleic Acids Res 2016 Jan 4;44(D1):D1133-40. https://doi.org/10.1093/nar/gkv1179. Epub 2015 Nov 8. PMID: 26553803; PMCID: PMC4702844
Upadhyaya HD, Gowda CLL, Sastry (2008) Plant genetic resources management: collection, characterization, conservation and utilization. J SAT Agric Res 6:16
Upadhyaya HD, Pundir RPS, Dwivedi SL, Gowda CLL, Reddy VG et al (2009a) Developing a mini core collection of sorghum for diversified utilization of germplasm. Crop Sci 49(5):1769–1780
Upadhyaya HD, Pundir RPS, Gowda CLL, Reddy VG, Sl Singh (2009b) Establishing a core collection of foxtail millet to enhance the utilization of germplasm of an underutilized crop. Plant Genet Resour 7(2):177–184
Veeranagamallaiah G, Sudhakar C (2017) Determination of polyamines by dansylation, benzoylation, and capillary electrophoresis. In: Plant stress tolerance, pp 313–323. Humana Press, New York, NY
Wang J, Wang Z, Du X, Yang H, Han F, et al (2017) A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L) P Beauv] using RAD-seq. PLoS ONE 12(6):e0179717
Wang ZM, Devos KM, Liu CJ, Wang RQ, Gale MD (1998) Construction of RFLP-based maps of foxtail millet, Setaria italica (L) P Beauv. Theor Appl Genet 96(1):31–36
Wang M, Pan Y, Li C, Pan Y, Jiang X et al (2011) Culturing of immature inflorescences and Agrobacterium-mediated transformation of foxtail millet (Setaria italica). Afr J Biotechnol 10:16466–16479. https://doi.org/10.5897/ajb10.2330
Wang M, Li P, Li C, Pan Y, Jiang X et al (2014) SiLEA14, a novel atypical LEA protein, confers abiotic stress resistance in foxtail millet. BMC Plant Biol 14(1):290. https://doi.org/10.1186/s12870-014-0290-7
Wang Z, Wang J, Peng J, Du X, Jiang M et al (2019) QTL mapping for 11 agronomic traits based on a genome-wide Bin-map in a large F 2 population of foxtail millet (Setaria italica (L) P Beauv). Mol Breed 39(2):18
Watson A, Ghosh S, Williams MJ, Cuddy WS, Simmonds J et al (2018) Speed breeding is a powerful tool to accelerate crop research and breeding. Nat Plants 4:23–29. https://doi.org/10.1038/s41477-017-0083-8
Weiss T, Wang C, Kang X, Zhao H, Gamo ME et al (2020) Optimization of multiplexed CRISPR/Cas9 system for highly efficient genome editing in Setaria viridis. Plant J 0–2. https://doi.org/10.1111/tpj.14949
Wen Q-F, Wang L, Wang XY (2005) The foxtail millet germplasm resources and screening and utilization of drought resistance germplasm in Shanxi (In Chinese with English abstract) J Shanxi Agric Sci 33:32–33
Willweber-kishiomoto E (1962) Interspecific relationships in the genus setaria. Contributions from the biological laboratory. Kyoto University 14:1–41 http://hdlhandlenet/2433/155927
Yadav CB, Muthamilarasan M, Pandey G, Khan Y, Prasad M (2014) Development of novel microRNA-based genetic markers in foxtail millet for genotyping applications in related grass species. Mol Breed. https://doi.org/10.1007/s11032-014-0137-9
Yadav CB, Muthamilarasan M, Pandey G, Khan Y, Prasad M (2015) Identification, characterization and expression profiling of Dicer-like, Argonaute and RNA dependent RNA polymerase gene families in foxtail millet. Plant Mol Biol Rep 33:43–55
Yadav CB, Muthamilarasan M, Dangi A, Shweta S, Prasad M (2016a) Comprehensive analysis of SET domain gene family in foxtail millet identifies the putative role of SiSET14 in abiotic stress tolerance. Sci Rep 6:32621
Yadav CB, Muthamilarasan M, Dangi A, Shweta S, Prasad M (2016b) Comprehensive analysis of 890 SET domain gene family in foxtail millet identifies the putative role of SiSET14 in abiotic 891 stress tolerance. Sci Rep 6:32621
Yang R, Chen M, Sun J, Yue Y, Min DH et al. (2019) Genome-wide analysis of LIM family genes in foxtail millet (Setaria italica L.) and characterization of the Role of SiWLIM2b in drought tolerance. Intl J Mol Sci 20:1303
Yang Z, Zhang H, Li X, Shen H, Gao J et al (2020) A mini foxtail millet with an Arabidopsis-like life cycle as a C4 model system. Nat Plants. https://doi.org/10.1038/s41477-020-0747-7
Yin K, Gao C, Qiu JL (2017) Progress and prospects in plant genome editing. Nat Plants 3:17107. https://doi.org/10.1038/nplants.2017.107 PMID: 28758991
Yoshitsu Y, Takakusagi M, Abe A, Takagi H, Uemeura A et al (2017) QTL-seq analysis identifies two genomic regions determining the heading date of foxtail millet, Setaria italica (L) P Beauv. Breed Sci 67(5):518–527
Yu T-F, Zhao W-Y, Fu J-D, Lei Y-W, Chen M et al (2018) Genome-wide analysis of CDPK family in foxtail millet and determination of SiCDPK24 functions in drought stress. Front Plant Sci 9:651. https://doi.org/10.3389/fpls.2018.00651
Zhang K, Fan G, Zhang X, Zhao F, Wei W, et al (2017) Identification of QTLs for 14 agronomically important traits in Setaria italica based on SNPs generated from high-throughput sequencing. G3 (Bethesda) 7(5):1587–1594
Zhang HZ (1980), Study on the natural hybridization rate and the acquisition of sexual hybrids of millet. Shanxi Agric Sci 12–13
Zhang J, Liu T, Fu J, Zhu Y, Jia J et al (2007) Construction and application of EST library from Setaria italica in response to dehydration stress. Genomics 90:121–131
Zhang X, Xia J, Li YE, Blanca-figureoa BE, Gao XZ et al (2012) Genome-wide analysis of plant nat-siRNAs reveals insights into their distribution, biogenesis and function. Genome Biol 13:R20
Zhang S, Tang S, Tang C, Luo M, Jia G et al (2018) SiSTL2 is required for cell cycle, leaf organ development, chloroplast biogenesis, and has effects on C4 photosynthesis in setaria italica (L.) P, Beauv. Front Plant Sci 9:1–16. https://doi.org/10.3389/fpls.2018.01103
Acknowledgements
The authors’ research in millet genetics and genomics is supported by the DST INSPIRE Grant (File No. DST/INSPIRE/04/2016/002341) and DST-SERB ECRA Grant (File No. ECR/2017/001526) awarded by the Department of Science and Technology, Govt. of India, and Science and Engineering Research Board, Govt. of India.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rana, S., Pramitha, L., Muthamilarasan, M. (2021). Genomic Designing for Abiotic Stress Tolerance in Foxtail Millet (Setaria Italica L.). In: Kole, C. (eds) Genomic Designing for Abiotic Stress Resistant Cereal Crops. Springer, Cham. https://doi.org/10.1007/978-3-030-75875-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-75875-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-75874-5
Online ISBN: 978-3-030-75875-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)