
Abstract
Renal disease often crosses the boundaries of traditional medical specialties, and reduced renal function is a common consequence of diseases of the heart, liver, and gastrointestinal tract. Multiorgan transplant is a lifesaving procedure for children with multiorgan failure, yet in practice it can be challenging due to lack of clarity about the most appropriate timing and perioperative management strategies. The role of simultaneous versus sequential heart-kidney or liver-kidney transplant remains hotly debated, and governing organizations continue to work to create allocation policies that appropriately balance the needs of all stakeholders. Additionally, a child’s small size may complicate the placement of multiple allografts at one time, and the allografts often have competing needs for hydration and immunosuppression. In this chapter, we have collected and summarized the published literature on the allocation, incidence, indications, perioperative management, and outcomes of pediatric multiorgan transplants.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Multiorgan transplant
- Simultaneous heart-kidney transplant
- Combined liver-kidney transplant
- Kidney-pancreas transplant
- Multivisceral transplant
- Autosomal recessive polycystic kidney disease
- Congenital heart disease
- Primary hyperoxaluria type 1
- Wolcott-Rallison syndrome
- Diabetes
- Chronic intestinal pseudo-obstruction
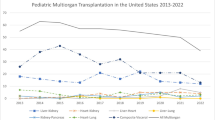
Pediatric nephrologists are familiar with the many ways in which renal-associated diseases can cross the boundaries of traditional medical subspecialties, involving the eyes, ears, heart, liver, pancreas, and intestines. In these cases, the kidney may be part of the primary syndrome or a victim of another organ disorder via hypoperfusion or toxic substances. Despite this, multiorgan transplants are uncommon in pediatrics, with 1677 reported cases in the United States since OPTN began collecting data in 1988. Incidence of pediatric multiorgan transplants rose throughout the 1990s and 2000s, peaking at 116 procedures in 2007 before declining to the current average of 55 per year [1] (Fig. 14.1).

Number of pediatric multiorgan transplants performed in the United States, by year. (Source: OPTN data [1])
Multiorgan transplantation can pose special challenges in pediatrics, especially when one of those organs is a kidney. Pediatric renal transplantation in the United States typically involves an adult- or near-adult-sized donor kidney to minimize the risk of graft thrombosis; however, heart and liver transplants involve matching the size of donor and recipient. Standard postoperative management of renal transplants involves high rates of fluid administration to maintain renal blood flow and urine output; standard management of lung and heart transplants involves limiting fluid intake to avoid pulmonary edema and heart failure. Furthermore, small abdomens may not have adequate domain to fit multiple allografts, especially if the native organs are not being removed.
Related to the relative rarity of multiorgan transplantation in children, there is a paucity of data on postoperative management and, in some cases, indications and outcomes. Here we have collected and summarized the published literature on the allocation, incidence, indications, perioperative management, and outcomes of pediatric multiorgan transplants.
Multiorgan Allocation
While the number of pediatric multiorgan transplants has stabilized over the last decade, the number among adults, excluding kidney-pancreas transplants, has more than doubled from 560 transplants in 2010 to 1074 transplants in 2019; 67% of these were combined liver-kidney transplants (CLKT) [1]. With this increase has come rising interest in the impact of multiorgan transplants on access and outcomes for single-organ recipients. The US kidney allocation system prioritizes multiorgan recipients over kidney-alone recipients, and multiorgan transplant outcomes are not included in center-specific reporting [2]. This has led to concerns about the objectivity, variability, and equitability of multiorgan allocation across the country, especially the potential impact on access to transplant for pediatric patients [2, 3]. Multiorgan transplant recipients can be listed for transplant with estimated glomerular filtration rates that would not meet the criteria for receipt of a kidney alone [4]. At the same time, kidneys allocated as part of a multiorgan transplant are not available to candidates for a kidney alone, and in 2016, 6.6% of kidney allografts were allocated with a liver or heart [2]. In the United States, the “Final Rule” governing the development of organ allocation policies requires the creation of policies “specific for each organ type or combination of organ types” [5]; however prior to 2014, there were no policies governing multiorgan transplants.
Organ allocation policies “seek to achieve the best use of donated organs” [5], requiring a balance between equity and organ utility. A multiorgan transplant may be lifesaving for one transplant candidate, but multiple single-organ transplants can save multiple lives. For example, recipients of combined heart-kidney transplant have significantly lower mortality than heart transplant recipients who remain on dialysis, but have high incidence of primary nonfunction and post-transplant dialysis [2], despite generally receiving higher quality (low kidney donor profile index) kidneys than kidney-alone recipients [3] (Fig. 14.2). Similarly, individual recipients of pediatric liver-kidney transplants have similar graft survival and lower rejection incidence compared to recipients of liver-alone transplants but worse outcomes than kidney-alone transplants, despite 49% of kidneys allocated with a liver-kidney transplant having a KDPI <35% (i.e., better quality organ) [6]. These situations result in improved outcomes for the multiorgan recipients but decrease the availability of kidney allografts [7].

Distribution of kidney donor profile index (KDPI) scores among kidney-alone and multi-organ transplant recipients. (Reprinted with permission from American Journal of Transplantation [4])
To address these concerns in the United States, the United Network for Organ Sharing (UNOS) has begun developing policies to address multiorgan transplant allocation. Kidney-pancreas allocation policies were added in 2014, establishing listing criteria that included the requirement that patients meet criteria for kidney-alone listing and specific pancreas-specific parameters including insulin use, C-peptide level, and/or body mass index threshold [8]; however, pediatric candidates are exempt from these requirements [9]. Kidney-pancreas candidates accumulate priority based on their kidney waiting time, but receive priority over kidney-alone recipients for all available local kidney-pancreas organs [10]. A 6-month evaluation of the new policy showed no change in pancreas utilization and increased regional sharing [11]. Between 2015 and 2019 there were an average of 802 kidney-pancreas transplants [1] with significant geographic variation in practice [12].
In 2017, UNOS implemented a simultaneous liver-kidney policy that established kidney eligibility criteria (either chronic kidney disease, prolonged acute kidney injury, or metabolic disease) and created a safety net giving priority for kidneys with a KDPI >20% to individuals who have continued dialysis dependency or an eGFR ≤20 ml/min/1.73m2 2–12 months after liver transplant [2]. Over the following year, the number of CLKT decreased from 740 to 676, while the number of kidney-after-liver transplants increased from 44 to 87 [2], a net decrease of 21 kidney transplants, although there may have been an initial “bolus” effect causing a transient increase in kidney-after-liver transplants immediately after policy implementation [6].
In the United States, there are currently no established national criteria for simultaneous heart-kidney or multivisceral transplant allocation, although proposals similar to the liver-kidney policy have been put forward [2].
Pediatric Heart-Kidney Transplantation
Pediatric simultaneous heart-kidney transplantation (sHKTx) is a rare procedure. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation (ISHLT), which collects data on approximately 80% of thoracic transplants worldwide, recorded only 50 pediatric heart-kidney transplants between January 1990 and June 2017 [13]. The majority of those transplants are in North America; 38 pediatric sHKTxs were performed between 1988 and April 30, 2017, in the United States [14]. The number of sHKTx overall, pediatric and adult, has risen significantly in the past ten years, likely for two main reasons. In part, heart transplant candidates are waiting longer for a transplant, developing more kidney disease related to their heart failure. Additionally, heart transplant recipients are surviving longer; 32.7% of pediatric multiorgan-heart recipients in the International Thoracic Organ Transplant Registry were re-transplants [13].
Individuals considered for heart-kidney transplantation typically have a primary cardiac disease that leads to renal failure due to multiple factors, including low cardiac output, nephrotoxic medications, and concomitant renal anomalies such as agenesis or dysplasia [14, 15]. In the ISHLT registry, 36% were repeat heart transplants with the majority of the remaining having congenital heart disease or dilated cardiomyopathy [13]. However, there is no consensus on the degree of renal dysfunction that indicates a need for renal transplantation in heart transplant candidates; 61.8% of sHKTx recipients in the ISHLT registry received dialysis prior to their transplant [13]. In a study linking Scientific Registry of Transplant Recipients (SRTR) data to the US Renal Data System, pediatric heart transplant recipients who required acute dialysis were significantly more likely to develop ESRD (14% versus 6.6%, HR 7.46 p = 0.0002) over the 25-year follow-up time, while an estimated glomerular filtration rate <60 ml/min/1.73m2 was less strongly associated with ESRD (HR 2.58, p < 0.001). The average eGFRs at transplant of patients who did and did not develop end-stage renal disease over the 25 years of the cohort were similar [16]. Complicating matters, renal dysfunction due to hypoperfusion may resolve with improved hemodynamics. In infants with renal failure at the time of cardiac surgery, mortality was high but none of the survivors required dialysis at the time of discharge [15]. Multiple studies in adult liver-kidney transplant recipients have shown that renal function, as measured by radionucleotide scans, demonstrates some level of native kidney function recovery in up to 50% of recipients [2]. These studies only included one heart-kidney recipient, whose results showed that the native kidneys were contributing 26% of the renal function while the transplant was contributing 74% [17].
Much of the discussion about pediatric sHKTx focuses on whether or not to perform the procedure. Simultaneous heart-kidney transplantation offers certain advantages to the recipient. Recipients of a sHKTx were less likely to have an episode of rejection within the first year post-transplant (10%) compared with recipients of a heart alone (25%) [13]. Animal models have shown that recipients of multiorgan transplants seem to develop tolerance to the donor, perhaps in part due to the increased total mass of allograft relative to body size [18]. Similarly, infant recipients of adult-sized kidneys have longer allograft survival than older children receiving similar-sized grafts [19]. Observational studies of adults have also shown decreased mortality among sHKTx recipients compared to heart transplant alone, although there is concern that these studies are subject to bias in that healthier patients were more likely to be listed for sHKTx [2]. A similar improvement in survival has not been reported in pediatric sHKTx recipients (Fig. 14.3). ISHLT data showed no difference in survival or freedom from cardiac allograft vasculopathy among recipients of a pediatric heart-kidney or heart-liver transplant [13] (Fig. 14.4).

Kaplan-Meier curve comparing survival for pediatric heart-alone transplants to that of pediatric multiorgan transplants that involved a heart, January 1990 to June 2016, 80% of which are heart-kidney transplants. (With permission from The Journal of Heart and Lung Transplantation [13])

Kaplan-Meier curve comparing time to development of cardiac allograft vasculopathy for pediatric heart-alone transplants compared to that of pediatric multiorgan transplants that involved a heart, January 1994 to June 2016, the majority of which are heart-kidney transplants. (With permission from The Journal of Heart and Lung Transplantation [13])
A sHKTx also offers potential advantages over kidney-after-heart transplantation, in that it avoids the potential immunologic risk from two donors and the additional induction therapy and surgical recovery. Recipients of a heart-alone transplant who develop ESRD often wait years to receive a kidney transplant and may have developed HLA antibodies related to their heart transplant, decreasing the pool of possible donors. The 25-year cohort studied by Choudhry et al. showed that 48% of heart transplant recipients on chronic dialysis had not received a kidney transplant at the time when the data were censored, and patients who remained on chronic dialysis had a significantly higher risk of death (HR 31.4, 95%CI 21–48.4, p < 0.0001) than those who received a kidney transplant [16].
However, sHKTx also has disadvantages for both the patient and the utility of the kidney allograft. In sHKTxs, the heart transplant is generally performed first followed by the kidney transplant [14, 20]. This results in increased cold ischemia time for the renal allograft. Heart transplant recipients may also develop vasoplegic syndrome and require ionotropic drug support, both of which compromise renal perfusion [20]. These factors likely contribute to the increased incidence of primary allograft non-function and high incidence of post-transplant dialysis (14–42%) [13, 14, 20, 21]. In the ISHLT registry, 20% of survivors of a sHKTx had severe renal dysfunction at 5 years, defined as a creatinine >2.5 g/dl or return to chronic dialysis [13]. Young children are particularly poor candidates for a simultaneous heart-kidney transplant. Heart allografts are matched to recipients based on size, but kidneys from donors less than 5 years of age have high rates of renal arterial thrombosis, delayed graft function, and early renal allograft loss [15]. Therefore, sHKTx in the smallest patients may result in worse outcomes than when kidney transplant is performed separately using an adult-sized allograft. Due to these concerns, some favor a kidney-after-heart approach, ideally using a living donor kidney [13].
Once a decision has been made to perform a sHKTx, there are several perioperative considerations. Due to the heart’s relative intolerance to ischemia, the heart transplant is performed first, followed by the kidney transplant. However, it is debated whether the kidney transplant should be performed immediately, under the same anesthetic, or whether the kidney transplant should be delayed for up to 24 hours, often called a staged procedure, to allow for recovery of the newly transplanted heart and optimization of hemodynamics and volume status [20, 22]. Immediate kidney transplant minimizes renal allograft cold ischemia time, which decreases the risk of delayed graft function and worse long-term outcomes. However, it is not clear if this benefit outweighs the risks of hypotension, post-cardiopulmonary bypass inflammatory cascade, and high doses of vasoconstrictor medications [20]. Staged sHKTx also allows for reallocation of the kidney if the heart transplant procedure does not go well.
Fluid management in sHKTx is also complex. During cardiopulmonary bypass, there is often significant ultrafiltration, causing fluid shifts that can take hours to equilibrate and further impact renal allograft perfusion [20]. Renal allografts benefit from high rates of intravenous fluids, high central venous pressures (often 10–15 mm Hg), and higher mean arterial pressures. On the other hand, cardiopulmonary bypass in heart transplant is associated with acute right ventricular failure when the donor heart is unable to adapt to higher pulmonary arterial pressures in the recipient. In this state, the heart is preload dependent but sensitive to distension; high central venous pressure should be avoided [23]. Transesophageal echocardiography can be used to closely monitor the fluid balance and ventricular function, allowing maximization of central venous pressure without overwhelming the heart [20, 23]. Several measures have also been shown to improve right ventricular function including the use of inotropes to increase contractility, provision of adequate oxygenation and ventilation, and use of inhaled pulmonary vasodilators [23].
Immediately post-transplant heart recipients often have small left ventricles with decreased compliance and increased filling pressures. The denervated heart allograft also lacks the baroreceptor reflex that compensates for hypotension due to hypovolemia or systemic vasodilation. Thus, they often require ionotropic support that can decrease renal perfusion. At this time, there is no conclusive evidence to suggest that any particular ionotrope is more protective of renal perfusion after sHKTx [20].
Post-transplant, long-term management of a sHKTx requires coordinated management by both nephrology and cardiology teams. Immunosuppression protocols for both pediatric heart and kidney transplant are center-specific, and there are no guidelines for induction or maintenance immunosuppression of a sHKTx; there is also no evidence that sHKTx recipients require more or less immunosuppression than solitary organ recipients. A review of Organ Procurement and Transplant Network data on sHKTx showed improved survival with rabbit anti-thymoglobulin induction compared to no induction or interleukin-2 receptor antibody induction, though this was no longer statistically significant in an adjusted model, and pediatric patients were excluded [24]. Both organs must be monitored for rejection individually per local protocols as they can reject simultaneously or separately [25]. Blood pressure management must also be closely coordinated.
Pediatric Liver-Kidney Transplantation
Combined liver-kidney transplant (CLKT) is the most common pediatric multiorgan transplant, with 372 performed in the United States to date [1]. Nearly half of these are in children 11–17 years of age with another 30% in children 1–5 years of age; there have been only 11 CLKTs in children less than 1 year of age and none since 2015. Worldwide, only 10–30 CLKTx are performed annually [26]. Since 2000, the incidence of CLKTx in the United States has doubled from approximately 8 per year to approximately 17 per year [1]. While adult CLKT significantly increased after the introduction of MELD score, a similar increase was not seen after the introduction of the PELD score [27], possibly because the PELD score does not give priority based on renal function.
The primary indications for CLKT in children are congenital conditions that affect both the liver and kidney. A review of CLKTs between October 1987 and February 2011 showed that 37% were performed for primary hyperoxaluria and 18% for congenital hepatic fibrosis/autosomal recessive polycystic kidney disease (ARPKD) [7]. Methylmalonic acidemia, alpha-1-antitrypsin deficiency, Alagille syndrome, atypical hemolytic uremic syndrome, and glycogen storage disease 1a are other genetic diseases affecting both organ systems for which CLKT may offer benefit. CLKT in atypical hemolytic syndrome caused by genetic mutations in liver-synthesized complement factors (complement factor H, complement factor B, and C3) is less clearly indicated in the era of terminal complement inhibition medications, but it has not been entirely excluded.
Such patients should be evaluated on a case-by-case basis, including assessment of the risks of liver transplant and access to complement inhibition therapy [28]. Approximately 18% of pediatric CLKT recipients reported primary liver disease, including TPN-induced liver disease, biliary atresia, and familial and neonatal cholestasis with a second kidney disease [27]. Hepatorenal syndrome, which typically resolves with liver transplantation, is not generally considered an indication for CLKT. However, a subset of patients who require dialysis for greater than 6–8 weeks prior to liver transplantation may not recover, and CLKT has been used for adults with this indication [27].
Combined liver-kidney transplantation in children is a technically complex procedure that is mostly performed using deceased donor organs [29]. Living donation of both organs from one donor is technically possible [30] but is riskier for the donor; living donation is more commonly done as two sequential procedures. The liver is transplanted first followed by the kidney using standard surgical technique. In patients with anuric renal failure pre-transplant, such as ARPKD, fluid management during the liver transplant phase can be difficult, and continuous renal replacement therapy (CRRT) may be helpful [29]. Intraoperative CRRT can be safely performed, but its use is complicated by the patient’s changing coagulation status; systemic heparin increases the risk of bleeding, citrate anticoagulation may result in citrate excess toxicity due to impaired liver clearance, and anticoagulation-free dialysis is associated with risk of clotting [31]. In one adult cohort that largely avoided heparin or citrate anticoagulation, 40% of filters clotted during the procedure [32]. Patients with ARPKD may also require native nephrectomy to create sufficient intrabdominal space for CLKT due to the large size of the native kidneys. In this situation, the risk and benefits of performing a third procedure during the CLKT should be weighed against those of performing nephrectomy prior to transplant [26, 29].
Immediately after transplant, CLKT recipients require close monitoring for bleeding and vascular complications. Liver transplant recipients can have significant blood loss and disturbances of coagulation during transplantation, especially if intra-abdominal varices or hypersplenism was present. Overcorrection with excessive fresh frozen plasma, platelets, cryoprecipitate, and/or fibrinogen can result in vascular thrombosis after organ reperfusion. Management is therefore a matter of maintaining balance [26]. In one single-center cohort, 8 out of 18 pediatric CLKT recipients had bleeding complications [33], while another cohort reported bleeding in 6 of 12 children, half of whom required operative revision and vascular complications in two children [34]. For comparison, among pediatric liver-alone transplants, the incidence of bleeding is 5% and the incidence of vascular complications is 18% [26]. Frequent Doppler ultrasound examinations are necessary to monitor for complications.
The need for postoperative hemodialysis is also high among pediatric CLKT recipients, although these numbers are somewhat confounded by the routine use of hemodialysis after transplant in recipients with primary hyperoxaluria type 1. Harps et al. reported a cohort of 16 pediatric CLKT recipients, of whom 9 required continuous renal replacement therapy post-transplant; 8 of these had primary hyperoxaluria type 1 [35]. Similarly, Büscher et al. reported a need for dialysis in 5 of 11 children with primary hyperoxaluria type 1 and 1 of 10 patients with other indications for transplant [36]. In a review of SRTR data, Calinescu et al. reported an overall incidence of delayed graft function of only 22.4% [7], underscoring the idea that the need for dialysis is not likely related to kidney function.
Combined liver-kidney transplant may be particularly challenging in younger children due to their size. Harps et al. reported that increasing donor to recipient weight ratio and donor to recipient age ratio were strongly associated with longer intensive care unit (ICU) stay, with a receiver operating curve suggesting an age ratio of 5.34 and weight ratio of 3.4 as cutoffs for a good ICU outcome [35]. However, the use of kidneys from donors less than 5 years of age is associated with worse renal allograft outcomes due to increased vascular complications [15]. Therefore, the kidneys of young liver donors may have worse outcomes. There are reports of successful CLKTs in children under 10 kg [35, 37], but a staged procedure, with isolated liver transplant followed by kidney transplant once the patient has grown, may be necessary [36, 37]. Smaller patients also typically require split-liver transplants, which have higher rates of bleeding and thrombosis [36].
There are no guidelines regarding immunosuppression management for CLKT recipients, and induction and maintenance immunosuppression protocols are generally center-specific [29]. There are data suggesting that transplantation of a liver and kidney from the same donor is associated with a lower incidence of acute rejection and improved renal graft survival [29], and there are cases of a pre-transplant-positive lymphocytotoxic crossmatch becoming negative after liver transplant [27]. Multiple reasons for this have been theorized, including neutralization of circulating alloantibodies by soluble class I HLA antigens produced by the liver allograft, inhibition of natural killer and cytotoxic T cells by liver-produced HLA-G antigen, and liver clearance of circulating class 1 HLA antibodies [27, 29]. Adult observational studies have shown decreased kidney graft loss to chronic rejection among CLKT recipients (2%) compared to kidney-alone recipients (8%) [38]. However, a similar benefit was not seen among pediatric CLKT recipients in the European Society for Pediatric Nephrology/European Renal Association-European Dialysis and Transplant Registry [39]. In this cohort of 202 pediatric patients with ARPKD, there was no difference in 5-year death-censored kidney allograft survival between recipients of a CLKT and recipients of a kidney alone (92.1% vs 85.9%, p = 0.4), though age- and sex-adjusted risk for death was 6.7 times higher among the CLKT recipients [39]. Three of the four deaths within 1 month post-transplant were among the CLKT recipients; causes of death included cardiovascular disease, infection, and “other/unknown” factors [39].
A 2014 study evaluated outcomes for 152 children in the United States who had received CLKT. Patient survival for CLKT was 86.8% at 1 year, 82.1% at 5 years, and 78.9% at 10 years. A total of 12 of the 32 deaths occurred within 30 days post-operation and 17 had primary hyperoxaluria. The primary causes of death were infectious and cardiovascular complications. In comparison, patient survival after isolated liver transplant over a comparable period was 86.7% at 1 year, 81.2% at 5 years, and 77.4% at 10 years. Patient survival after isolated kidney transplant was 98.2% at 1 year, 95.4% at 5 years, and 90% at 10 years [7] (Fig. 14.5).

Patient survival following pediatric combined liver-kidney transplantation (CLKT) compared to patient survival in isolated kidney transplantation (KT) and isolated liver transplantation (LT). (Reprinted with permission from American Journal of Transplantation [7])
Liver graft survival was 81.9% at 1 year, 76.5% at 5 years, and 72.6% at 10 years. Liver graft survival was significantly worse among those with primary hyperoxaluria (p = 0.01), possibly due to the complications of systemic oxalosis, and the most common causes of liver graft failure were venous thrombosis (37.5%) and infection (25%). Kidney graft survival was 83.4% at 1 year, 76.5% at 5 years, and 66.8% at 10 years. Primary hyperoxaluria was significantly associated with reduced renal allograft survival (p = 0.01), and the most common causes of renal graft loss were chronic rejection (24%), infection (24%), and venous thrombosis (12%). Fourteen children (9.2%) had failure of both the liver and kidney, with kidney allograft failure preceding liver allograft failure in 57.1%. The time between failure of the two allografts was less than 40 days in 64.2% of children [7].
Primary Hyperoxaluria
Special consideration must be given to the question of CLKT in patients with primary hyperoxaluria. Primary hyperoxaluria type 1 is a defect in the AXGT gene, resulting in defective production or trafficking of alanine-glyoxylate aminotransferase in the liver [27]. This deficiency leads to overproduction of oxalate and excessive urinary excretion of calcium oxalate that causes a decline in renal function. As the estimated glomerular filtration rate falls below 30–50 ml/min/1.73 m, renal oxalate excretion can no longer keep pace with production, and oxalate accumulates in the bones, vessels, heart, joints, and retina. Oxalate is poorly cleared by hemodialysis , with levels only decreasing by about 40% and returning to 95% of pre-dialysis levels within 48 hours; peritoneal dialysis clearance is worse. Children with primary hyperoxaluria often require daily hemodialysis with or without nightly peritoneal dialysis in order to keep pace with the continuous excess production of oxalate by the liver [40]. End-stage renal disease develops in 50% of children with primary hyperoxaluria type 1 by 15 years of age and 80% by age 30 years [27], though the course can be highly variable, even in siblings with the same genetic mutation [41].
The first step in considering CLKT for primary hyperoxaluria is genetic testing or liver biopsy to confirm alanine-glyoxylate aminotransferase deficiency [33]. Outcomes for kidney-alone transplant in primary hyperoxaluria type 1 are dismal, with a 5-year graft survival of 14% in children, primarily due to ongoing overproduction of oxalate in the liver that damages the allograft. However, the indications for liver transplantation in primary hyperoxaluria secondary to causes other than AXGT mutation remain unclear, with sources reporting both good outcomes and graft losses due to oxalate deposition among recipients of kidney-alone transplant in primary hyperoxaluria type 2, a milder form of the disease associated with mutations in glyoxylate reductase/hydroxypyruvate reductase and decreased risk of progression to ESRD. Another important step is determining if the patient is pyridoxine sensitive, as approximately 25–30% of patients with primary hyperoxaluria type 1 will have reduced oxalate excretion with pharmacologic doses of pyridoxine (5–10 mg/kg/dose twice a day). Soliman et al. reported that 8 of 26 patients on pyridoxine therapy were able to maintain normal renal function after 2 years of follow-up [42]. There are case reports suggesting that pyridoxine-responsive patients may have successful kidney-alone transplants, but the overall results of this practice remain unclear [33]. Similarly, there are multiple case reports of primary hyperoxaluria diagnosed only after kidney-alone transplantation; most cases resulted in early graft dysfunction (often within days to months of transplant) and early graft loss, but there also are reports of renal function stabilizing with aggressive fluid intake [43].
Among patients with confirmed primary hyperoxaluria type 1, there remains great debate about the appropriate timing of kidney transplantation [44]. After liver transplantation corrects the underlying genetic defect, systemically deposited oxalate is progressively mobilized into the blood to be filtered by, and damage, the kidneys. One management option is sequential liver and kidney transplant. The liver transplant occurs first, allowing for immediate correction of the oxalate overproduction, followed by a period of intensive dialysis to clear the mobilized systemic oxalate. Once plasma oxalate levels are lowered or normalized, the kidney transplant occurs, typically in a range of 51 days to 9 months post-liver transplant [45]. Advantages of the sequential strategy include protection of the renal allograft from systemic oxalosis and stabilization of liver function and coagulation prior to proceeding to kidney transplantation. Sequential transplant may also be more appropriate for children with the infantile form of primary hyperoxaluria type 1, for whom it is difficult to find appropriately size-matched liver and kidney allografts from the same donor. There are also multiple reports of successful sequential liver and kidney transplants using the same living donor [44, 46, 47]. However, if a patient requires a deceased donor for both organs, the wait time between liver and kidney transplants on intensive dialysis can be long [48].
In combined liver-kidney transplant, the metabolic defect and renal failure are corrected immediately, allowing earlier discontinuation of dialysis. Plasma oxalate levels drop significantly after CLKT, from >60–100 μmol/L to <20 μmol/L, but they can remain elevated for months or years as systemically deposited oxalate is mobilized [45, 49] (Fig. 14.6). Post-transplant management with high fluid intake, urine crystallization inhibitors, such as citrate, and pyridoxine (if patient is pyridoxine-responsive) is necessary to protect the renal allograft until oxalate levels are normal [33, 45]. The value of dialysis immediately after CLKT, to clear oxalate and prevent early oxalate deposition in the new renal allograft, remains unclear [33], as the anticoagulation needed for dialysis increases the risk of bleeding in a patient whose synthetic liver function is still recovering. CLKT also allows the patient to benefit from the lower rejection rates seen in CLKT [45]. In a recent case series reported by Horoub et al., 24 patients with primary hyperoxaluria type 1 underwent transplantation between 2011 and 2018, 13 of whom were less than 18 years of age. Thirteen patients received sequential liver and kidney transplant and eight patients received CLKT. The authors reported no differences in mortality at 3 years, need for hemodialysis after transplant, acute cellular rejection, or estimated glomerular filtration rate between the sequential and combined transplant strategies [50].

Plasma oxalate levels declined rapidly following successful combined kidney-liver transplantation but remained above normal in most patients during the first year after transplant. The normal range for plasma oxalate levels is <1.8 μmol/L, shown in the gray-shaded area. (Reprinted with permission from American Journal of Transplantation [49])
Pre-emptive liver transplant for patients with primary hyperoxaluria type 1 remains controversial. It may be an option for patients who are diagnosed prior to significant decline in renal function, correcting the metabolic defect before systemic oxalosis develops and preventing the need for a renal transplant. “Late” pre-emptive liver transplant, in patients with a glomerular filtration rate <30 mL/min/1.73m2, may also delay the need for kidney transplant [48]. In young children, preemptive liver transplant avoids the technical and anatomic issues of CLKT in a small abdomen [48]. However, primary hyperoxaluria has a heterogeneous course; it can present clinically in infancy with rapid progression, in adolescence with recurrent nephrolithiasis, or even in adulthood [51]. Factors predicting the onset of renal failure are unclear but may include a higher urinary oxalate level and nephrocalcinosis [52, 53]. Family history or genetic mutation is not necessarily predictive [41].
Given this uncertainty, and liver transplant graft survival outcomes of 85% at 5 years, 70% at 10 years, and 50% at 20 years [51], and the risks of immunosuppression, the difference between “preemptive” and “premature” liver transplant remains debated. There have been 24 published cases of preemptive liver transplantation for pediatric primary hyperoxaluria type 1, of which 20 patients were free of end-stage renal disease at follow-up of 0.7–16 years [48]. Ongoing research in hepatocyte transplantation [48] and the recent US Food and Drug Administration approval of lumasiran, an RNA interference-based treatment that decreases oxalate production by reducing levels of glycolate oxidase, [29] may change management of primary hyperoxaluria type 1, further complicating considerations for preemptive liver transplantation.
Pediatric Kidney-Pancreas Transplantation
Pediatric kidney-pancreas transplantation is a rare procedure, with only 69 reported cases in the US Organ Procurement and Transplantation Network [1]. The number of kidney-pancreas transplants in children has been declining in recent years, from a median of 4 per year 2006–2010 to a median of 2 per year 2016–2020. Pancreas-alone transplant is more common in children, with 703 performed since 1988. Of all pediatric kidney-pancreas transplants performed in the United States, 8 (11.5%) were in children less than 1 year old, 22 (31.9%) were in children 1–5 years old, 15 (21.7%) were in children 6–10 years old, and 22 (31.9%) were in children 11–17 years old [1].
Nearly 50 (72.4%) of the 69 pediatric kidney-pancreas recipients did not have diabetes [1]; their indications for transplant are not clearly documented in published OPTN data but may include cystic fibrosis and chronic pancreatitis [1]. Seven (10.1%) pediatric kidney-pancreas recipients reported type I diabetes, while the remainder are “unknown.” Forty-three (62.3%) recipients reported “other” as their indication for renal transplant. As of writing, there were three pediatric patients waitlisted for kidney-pancreas transplant, two of whom had congenital/metabolic disorders [1].
In 1996, Bendel-Stenzel et al. published two cases of children who received simultaneous pancreas-kidney transplants. Both patients had a history of diarrhea-associated hemolytic uremic syndrome that resulted in pancreatic insufficiency requiring insulin and renal failure. One patient received a deceased donor transplant and one received a living-related simultaneous kidney and segmental pancreas transplant. Induction was with antithymocyte globulin in both patients. Both patients received tacrolimus and prednisone maintenance immunosuppression; in addition, one patient received azathioprine and one patient received mycophenolate mofetil. The need for insulin in both patients resolved within 6 hours of post-transplant; one patient continued to require pancreatic enzyme supplements. One patient had multiple episodes of rejection, while the other had none. Both patients had functioning allografts at 1-year follow-up. The authors further reported on a total of eight pediatric simultaneous kidney-pancreas transplants in the International Pancreas Transplant Registry, six of whom had function of both grafts at follow-up [54].
Due to the rarity of the procedure, published data on simultaneous kidney-pancreas transplant are largely limited to the adult population, where the primary indication is type 1 diabetes with concurrent diabetic nephropathy and patient comorbidities may be substantially different. The pancreas transplant is completed first to limit ischemic time [55]. The pancreas is transplanted heterotopically, but the exact location is a matter of surgeon and center preference. Common anastomosis sites include the pelvis, with anastomoses to the common or external iliac artery and vein, or the small bowel mesentery, with anastomoses to the iliac artery or aorta and the portal vein or superior mesenteric vein. Venous anastomosis to the systemic circulation is technically easier but carriers a higher risk of hyperinsulinemia; venous anastomosis to the portal circulation is theoretically more physiologic but has not been shown to improve graft survival [55]. The pancreas is a technically challenging organ on which to operate. Technical complications are common and include pancreatic pseudocyst and thrombosis (5–10%) [55, 56]. Abdominal infections are responsible for 9.2–15% of technical failures and remain a major cause of mortality in pancreatic transplantation [57].
As most adult simultaneous kidney-pancreas transplants are for type 1 diabetes, the patient has intact exocrine function of their native pancreas; therefore, the exocrine duct of the pancreatic allograft is typically diverted to either the bladder or jejunum. Anastomosis with the bladder was more common historically, but this can result in chronic metabolic acidosis due to loss of bicarbonate-rich fluid in the urine, infections, and damage to the urethra by pancreatic enzymes [58]. A duodenal or jejunal anastomosis of the exocrine duct is now more commonly used [55, 56], though this may result in malabsorption and diarrhea [58]. In pediatric patients with exocrine dysfunction, the location of the duct anastomosis may affect their need for pancreatic enzyme supplementation.
Post-transplant, close monitoring of blood glucose level is critical as it reflects graft function; failure to achieve normoglycemia quickly after transplant is a sign of graft dysfunction, rejection, pancreatitis, and/or an allograft that is too small for the patient [56]. Pancreatic allograft rejection in adult simultaneous kidney-pancreas recipients is concordant with kidney graft rejection in 60% of cases [55]. Elevated C-peptide and lipase are suggestive of dysfunction. Biopsy of the pancreatic graft is technically challenging and may be nondiagnostic in 12% of cases due to sampling error [55].
Data in adults suggest that simultaneous kidney-pancreas transplant has increased graft survival (>90% at 1 year) compared to isolated pancreas (80%) or pancreas-after-kidney (82%) transplantation [56]. Simultaneous kidney-pancreas recipients also have longer graft survival (72% at 8 years) than deceased donor kidney-alone recipients (55% at 8 years) [55]. However, wait times for simultaneous kidney-pancreas transplant are substantially longer than for kidney or pancreas alone. For example, between 2011 and 2014, the median wait time to receive a simultaneous kidney-pancreas transplant in the United States for an 11- to 17-year-old was 1033 days, compared to 680 days for a kidney alone and 758 days for a pancreas alone [1]. Therefore, the improved graft survival must be balanced with the longer wait time.
Liver-Kidney-Pancreas Transplant
There have been two pediatric liver-kidney-pancreas multiorgan transplants performed in the United States [1], which correspond to two case reports of such transplants for Wolcott-Rallison syndrome. Wolcott-Rallison syndrome is a rare genetic disorder that causes neonatal-onset insulin-dependent diabetes, skeletal dysplasia (mostly spondyloepiphyseal dysplasia), short stature, and hepatic dysfunction with recurrent acute liver failure [59]. The disease is autosomal recessive and is caused by mutations in EIF2AK3, which encodes pancreatic endoplasmic reticulum kinase (PERK). In the absence of PERK, the endoplasmic reticulum cannot respond to stress from accumulated unfolded proteins [60]. Infection, medications, or anesthesia can trigger episodic acute liver failure, often accompanied by acute renal failure [61]; the first “aggravation” is fatal in approximately 50% of cases [60].
Rivera et al. reported an 8-year-old girl with genetic-confirmed Wolcott-Rallison syndrome who presented with a dry cough, low-grade fever, and abdominal pain. She quickly developed multisystem organ failure, including a need for continuous renal replacement therapy. Nine days later, she underwent en bloc liver, pancreas, and kidney transplant from an 8-year-old ABO-compatible donor with thymoglobulin induction. Abdominal wall closure was completed on postoperative day 2; she was extubated and began eating on postoperative day 4. Post-transplant course was complicated by acute rejection of the liver and pancreas on day 45 and Enterococcus urosepsis at 6 months. She was in good health with good graft function at 18 months of follow-up [62].
Tzakis et al. reported a 6-year-old girl who presented with acute hepatic failure and was confirmed to have Wolcott-Rallison syndrome by genetic testing. She required mechanical ventilation, dialysis, and plasmapheresis for 6 weeks before recovering. Once she had been discharged, she was evaluated and listed for liver, pancreas, and kidney transplant. She received en bloc transplant with both kidneys; the native kidneys were not removed. The abdominal wall was closed on postoperative day 6. Post-transplant course was complicated by severe rejection of all three organs with acute respiratory distress syndrome, from which she recovered after 2 months of hospitalization. She was in good health with good graft function at 18 months of follow-up [60].
Kidney-Intestinal and Multivisceral Transplants
Composite visceral transplants are any transplant including the intestine and at least one other abdominal organ; multivisceral transplants are intestinal transplants that also include the stomach, duodenum, and pancreas with or without the liver and kidney [63]. Per OPTN data, 55 pediatric composite visceral transplants that include a kidney have been performed in the United States to date; 50 of these are liver-kidney-intestinal-pancreas transplants. Of the remainder, two were kidney-intestinal transplants, two were kidney-intestinal-pancreas transplants, and one was a liver-kidney- intestinal transplant. A total of 21 (42%) of the 50 transplants were performed in children aged 6–10 years. The incidence of multivisceral transplantation peaked in 2008–2010 before declining. In the past 5 years, multivisceral transplantation has been limited exclusively to liver-kidney-intestinal-pancreas transplants performed in children of ages 1–10 years at a rate of 1–3 transplants per year [1].
Intestinal and multivisceral transplant is the standard of care for patients with irreversible intestinal failure who can no longer be maintained on parental nutrition [63]. The primary causes of intestinal failure are short bowel syndrome, congenital enteropathies, and intestinal motility disorders. Primary treatment for intestinal failure is parental nutrition, with a goal of intestinal rehabilitation and return to full enteral nutrition, but maintenance of parental nutrition may be limited by severe cholestatic liver disease, recurrent catheter-related infections, and/or loss of vascular access [64]. Intestinal failure-associated liver disease (IFALD) occurs in 40–60% of children, and as many as 85% of neonates, who depend on parenteral nutrition for prolonged periods; approximately 15% of these will progress to end-stage liver disease [65]. IFALD is multifactorial and related to prematurity, recurrent infections, and parenteral lipid intake, especially the soybean oil routinely used in the United States [64, 65]. In infants with IFALD who are considered to have a high likelihood of intestinal adaptation and return to enteral nutrition, liver transplant alone may be considered, as liver disease has been shown to interfere with intestinal adaptation [65]. However, this can be difficult to predict. IFALD often recurs in the liver allograft, and the immunosuppressive medications may increase the risks of sepsis in children who still require parenteral nutrition [65]. Intestinal failure is less commonly associated with kidney disease; in the OPTN/SRTR 2016 Annual Data Report, only 3.7% of intestinal recipients required simultaneous kidney transplant [66]. Of note, many multivisceral transplant recipients do not require a pancreas transplant, but the pancreas is often included for technical reasons, as it eliminates the need for biliary reconstruction (and associated risk of bile leaks), simplifies backtable preparation, and allows for procurement of longer superior mesenteric artery and vein vessels [67].
The intestinal transplant procedure is complex and individualized to the patient, depending on the organs being transplanted and the abdominal anatomy of the patient. In the pre-transplant phase, a full assessment of upper and lower vascular patency is key as many patients will have vascular thrombosis related to their parental nutrition dependence that can complicate or even preclude the procedure. The kidney may be transplanted en bloc with the intestine or separately [63]. A recent case series by Kunzler de Oliveira Maia et al. reported using infant en bloc kidneys with a bladder segment, using the bladder patch technique in three children receiving multivisceral transplants [68]. All three patients had good vascular flow; one developed ureteral stenosis. One patient died of sepsis, while the other two were alive with graft function at 2 and 5 years of post-transplant [68]. Small patients may not have adequate abdominal domain to place an en bloc multivisceral transplant with abdominal wall closure, but abdominal wall closure at the time of surgery is preferred to decrease the risk of infection. There are several reports of using the abdominal rectus as fascia to allow tension-free abdominal closure with good results [69, 70]. Postoperatively, multivisceral transplant recipients typically continue parental nutrition with gradual, stepwise introduction of enteral feeds [63]. Acute kidney injury is common (25% incidence in adults), and is likely related to erratic intestinal absorption of tacrolimus, which can lead to markedly elevated tacrolimus levels and calcineurin inhibitor-related renal artery vasoconstriction [71].
Compared to other organs, the intestinal allograft includes significantly more lymphoid tissue and is highly antigenic; both rejection and graft versus host disease can and do occur. Acute cellular rejection has been reported in 30–60% of intestinal transplant recipients by 3 months of post-transplant [69, 71]. The rejection is typically isolated to the intestine with other transplanted organs relatively spared [71]. Intestinal rejection can present with fever, diarrhea, abdominal pain, dissension, and bacteremia due to translocation of bacteria. Tacrolimus levels may appear elevated during rejection due to impaired enterocyte tacrolimus metabolism [71]. To monitor for rejection, an ileostomy is commonly created to allow for surveillance biopsies [63] as often as twice a week immediately after transplant [71]. Depending on which other organs are transplanted, routine monitoring of hepatic enzymes, pancreatic enzymes, and serum creatinine is also necessary. Immunomodulatory strategies, including allograft radiation and bone marrow augmentation, have also been explored with some improvement in outcomes [63, 72].
Simultaneously, the patient must be monitored for graft versus host disease (GvHD), which occurs in 4–30% of recipients [71, 73]. The risk is highest in patients with an immunodeficiency (such as in familial multiple intestinal atresia or trichoheptoenteric syndrome) or who are under the age of 5 years [73, 74]. Unlike GvHD of after hematopoietic stem cell transplant, GvHD after multivisceral transplant nearly always involves the skin (often a maculopapular rash that starts on the palms and soles) [73], though intestine, lungs, and bone marrow involvement may occur concurrently [75]. The diagnosis is confirmed by the presence of donor leukocytes in the recipient’s peripheral blood or organs [63]; management involves steroids and reduction in other immunosuppression. Mortality among multivisceral transplant recipients has been reported to be as high as 60–70% in one small case series [75, 76].
Intestinal transplant recipients have the highest rates of Epstein-Barr virus (EBV) infection and post-transplant lymphoproliferative disease (PTLD), with an incidence ranging from 13 to 33%, likely related to the comparatively younger age of recipients, more frequent use of T-cell depleting induction therapy, and larger amount of donor lymphoid tissue present in the intestine [77, 78]. One case series also reported two cases of intra-abdominal EBV-associated smooth muscle tumor, an overall incidence of 14% in the pediatric multivisceral population [78]. Both PTLD- and EBV-associated smooth muscle tumors are treated with reduction in immunosuppression, but case reports suggest that resection of smooth muscle tumors may improve survival [79].
Related to these complications, outcomes for multivisceral transplants remain significantly worse than other solid organ transplants. They are also worse than outcomes for individuals with intestinal failure who can be maintained on chronic parenteral nutrition [80]. Data on kidney-inclusive multivisceral transplants are not available, but current 5-year patient survival among all pediatric multivisceral transplant recipients is approximately 50–60% [69, 80, 81]. Younger age at transplant, receipt of a liver transplant from the same donor, and use of rapamycin as maintenance immunosuppression have all been associated with improved graft and patient outcomes [81]. However, Ramisch et al. reported that 93% of graft recipients were taking full enteral nutrition within 1 month of post-transplant [69], and quality of life is reportedly good after intestinal transplantation, though lower than the general population, especially in areas of school functioning [82, 83]. With improvements in intestinal rehabilitation and prevention of IFALD, the role of multivisceral transplant may be limited to that of a “final option” therapy until outcomes improve.
Future Directions
Despite growth in multiorgan transplantation over the last two decades, current information on management and outcomes, especially among pediatric patients, remains limited. Existing data supporting or refuting the role of multiorgan transplantation versus single-organ or sequential transplantation are often subject to confounding by indication and is difficult to interpret. National and international consensus on indications for multiorgan transplant is lacking. Peri- and post-operative management remains largely center-specific, driven by expert opinion rather than data. National and international registries often combine pediatric and adult data in their reporting, making it challenging to apply the results to children. Centers that frequently perform multiorgan transplants should be encouraged to publish their experience (especially in the field of pediatric kidney-pancreas transplantation), and existing registries should be encouraged to publish pediatric-specific outcomes from their databases to continue to improve the care we provide to this small but vulnerable population.
Questions
-
1.
Which of the following statements is true regarding multiorgan allocation?
-
A.
Incidence of multiorgan allocation to pediatric patients has risen over the last 5 years.
-
B.
There are no policies surrounding allocation of organs for multiorgan transplantation.
-
C.
Current allocation policies result in multiorgan transplant recipients receiving kidneys with a lower KDPI than kidney-alone transplant recipients.
-
D.
There are simple and straightforward ways to balance equity and utility in multiorgan transplant allocation.
C. Because multiorgan transplant takes priority over solo pediatric kidney, recipients of multiorgan transplants often receive better quality kidneys (lower KDPI) than recipients of kidney alone.
-
A.
-
2.
Outcomes among pediatric simultaneous heart-kidney transplant recipients:
-
A.
Show increased rejection within the first-year post-transplant compared to heart-alone transplantation
-
B.
Show increased rates of primary allograft nonfunction compared to kidney-alone transplantation
-
C.
Are likely to be better for infants <10 kg than for larger children
-
D.
Show increased mortality compared to heart-alone transplantation
B. Recipients of combined heart-kidney transplant have significantly lower mortality than heart transplant recipients who remain on dialysis but have high incidence of primary nonfunction and post-transplant dialysis, despite generally receiving higher quality (low KDPI) kidneys than kidney-alone recipients.
-
A.
-
3.
Perioperative management of combined liver-kidney transplant recipients involves:
-
A.
Transplanting the kidney first, followed by the liver
-
B.
Routine use of CRRT with heparin anticoagulation in all patients
-
C.
Careful balancing of bleeding and thrombosis risks
-
D.
Rare use of postoperative dialysis
C. Immediately after transplant, CLKT recipients require close monitoring for bleeding and vascular complications. Liver transplant recipients can have significant blood loss and disturbances of coagulation during transplantation, especially if intra-abdominal varices or hypersplenism was present. Overcorrection with excessive fresh frozen plasma, platelets, cryoprecipitate, and/or fibrinogen can result in vascular thrombosis after organ reperfusion.
-
A.
-
4.
Compared to other solid-organ transplant recipients, multivisceral transplant recipients are at higher risk for:
-
A.
Graft versus host disease
-
B.
Rejection
-
C.
Post-transplant lymphoproliferative disease
-
D.
All of the above
D. Multivisceral transplant recipients are at higher risk of all of the listed complications and have to be monitored carefully.
-
A.
References
National Data - OPTN. https://optn.transplant.hrsa.gov/data/view-data-reports/national-data/#. Accessed 11 Oct 2020.
Cheng XS, Khush KK, Wiseman A, Teuteberg J, Tan JC. To kidney or not to kidney: applying lessons learned from the simultaneous liver-kidney transplant policy to simultaneous heart-kidney transplantation. Clin Transpl. 2020. https://doi.org/10.1111/ctr.13878.
OPTN/UNOS Ethics Committee. Ethical implications of multi-organ transplants. June 2019.
Reese PP, Veatch RM, Abt PL, Amaral S. Revisiting multi-organ transplantation in the setting of scarcity. Am J Transplant. 2014;14:21–6.
About the final rule - OPTN. https://optn.transplant.hrsa.gov/governance/about-the-optn/final-rule/. Accessed 11 Oct 2020.
Lum EL, Cárdenas A, Martin P, Bunnapradist S. Current status of simultaneous liver-kidney transplantation in the United States. Liver Transpl. 2019;25:797–806.
Calinescu AM, Wildhaber BE, Poncet A, Toso C, McLin VA. Outcomes of combined liver-kidney transplantation in children: analysis of the scientific registry of transplant recipients. Am J Transplant. 2014;14:2861–8.
Kidney-pancreas allocation system frequently asked questions - OPTN. https://optn.transplant.hrsa.gov/resources/guidance/kidney-pancreas-allocation-system-frequently-asked-questions/. Accessed 11 Oct 2020.
Andrews WS, Kane BJ, Hendrickson RJ. Organ allocation and utilization in pediatric transplantation. Semin Pediatr Surg. 2017;26:186–92.
Optn OPTN policies effective as of June 18 2020 [Eliminate the use of regions in VCA distribution].
Carrico R, Fridell J, Odorico J, Stewart Z. How did the new kidney and pancreas allocation systems affect pancreas utilization in the first six-months? [abstract]. Am J Transplant. 2016;16:1689–99.
Axelrod DA, Sung RS, Meyer KH, Wolfe RA, Kaufman DB. Systematic evaluation of pancreas allograft quality, outcomes and geographic variation in utilization. Am J Transplant. 2010;10:837–45.
Rossano JW, Cherikh WS, Chambers DC, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: twenty-first pediatric heart transplantation report—2018; focus theme: multiorgan transplantation. J Heart Lung Transplant. 2018;37:1184–95.
Weng PL, Alejos JC, Halnon N, Zhang Q, Reed EF, Tsai Chambers E. Long-term outcomes of simultaneous heart and kidney transplantation in pediatric recipients. Pediatr Transplant. 2017. https://doi.org/10.1111/petr.13023.
Sahney S, Chinnock R. Management of infants and young children with combined heart and kidney failure. Pediatr Transplant. 2006;10:408–12.
Choudhry S, Dharnidharka VR, Castleberry CD, Goss CW, Simpson KE, Schechtman KB, Canter CE. End-stage renal disease after pediatric heart transplantation: a 25-year national cohort study. J Heart Lung Transplant. 2018;37:217–24.
Mosconi G, Panicali L, Persici E, Conte D, Cappuccilli ML, Cuna V, Capelli I, Todeschini P, D’Arcangelo GL, Stefoni S. Native kidney function after renal transplantation combined with other solid organs in preemptive patients. Transplant Proc. 2010;42:1017–20.
Madsen JC, Yamada K, Allan JS, Choo JK, Erhorn AE, Pins MR, Vesga L, Slisz JK, Sachs DH. Transplantation tolerance prevents cardiac allograft vasculopathy in major histocompatibility complex class I-disparate miniature swine. Transplantation. 1998;65:304–13.
Sarwal MM, Cecka JM, Millan MT, Salvatierra O. Adult-size kidneys without acute tubular necrosis provide exceedingly superior long-term graft outcomes for infants and small children. Transplantation. 2000;70:1728–36.
Mc Loughlin S, Bianco JC, Marenchino RG. Anesthetic and perioperative considerations for combined heart-kidney transplantation. J Cardiothorac Vasc Anesth. 2018;32:44–9.
Riaz A, Adebiyi O, Mishler D, Yaqub M, Taber T, Sharfuddin A. Kidney graft outcomes in combined heart-kidney transplantation: a UNOS database analysis 2000–2015 [abstract].
Ruzza A, Czer LSC, Trento A, Esmailian F. Combined heart and kidney transplantation: what is the appropriate surgical sequence? Interact Cardiovasc Thorac Surg. 2013;17:416–8.
Chacon MM, Roberts EK. Dilemmas for the cardiac anesthesiologist: managing conflicting fluid management strategies during combined heart-kidney transplantation. J Cardiothorac Vasc Anesth. 2018;32:50–2.
Ariyamuthu VK, Amin AA, Drazner MH, et al. Induction regimen and survival in simultaneous heart-kidney transplant recipients. J Heart Lung Transplant. 2018;37:587–95.
Fritzsche SD, McCabe JL, Chinnock RE. Rare combined heart and kidney transplant in a pediatric patient: a case study. J Transpl Coord. 1999;9:145–8.
Jalanko H, Pakarinen M. Combined liver and kidney transplantation in children. Pediatr Nephrol. 2014;29:805–14.
Chava SP, Singh B, Pal S, Dhawan A, Heaton ND. Indications for combined liver and kidney transplantation in children. Pediatr Transplant. 2009;13:661–9.
Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31:15–39.
Grenda R, Kaliciński P. Combined and sequential liver–kidney transplantation in children. Pediatr Nephrol. 2018;33:2227–37.
Sakamoto S, Kasahara M, Fukuda A, Tanaka H, Kakiuchi T, Karaki C, Kanazawa H, Kamei K, Ito S, Nakazawa A. Pediatric liver-kidney transplantation for hepatorenal fibrocystic disease from a living donor. Pediatr Transplant. 2012;16:99–102.
Matuszkiewicz-Rowińska J, Małyszko J, Wieliczko M. Renal support during liver transplantation: when to consider it? Transplant Proc. 2013;45:3157–62.
Townsend DR, Bagshaw SM, Jacka MJ, Bigam D, Cave D, Gibney RTN. Intraoperative renal support during liver transplantation. Liver Transpl. 2009;15:73–8.
Bacchetta J, Mekahli D, Rivet C, Demède D, Leclerc AL. Pediatric combined liver-kidney transplantation: a 2015 update. Curr Opin Organ Transplant. 2015;20:543–9.
Herden U, Kemper M, Ganschow R, Klaassen I, Grabhorn E, Brinkert F, Nashan B, Fischer L. Surgical aspects and outcome of combined liver and kidney transplantation in children. Transpl Int. 2011;24:805–11.
Harps E, Brinkert F, Ganschow R, Briem-Richter A, Van Husen M, Schmidtke S, Herden U, Nashan B, Fischer L, Kemper MJ. Immediate postoperative intensive care treatment of pediatric combined liver-kidney transplantation: outcome and prognostic factors. Transplantation. 2011;91:1127–31.
Büscher R, Büscher AK, Cetiner M, Treckmann JW, Paul A, Vester U, Hoyer PF. Combined liver and kidney transplantation and kidney after liver transplantation in children: indication, postoperative outcome, and long-term results. Pediatr Transplant. 2015;19:858–65.
Sutherland SM, Alexander SR, Sarwal MM, Berquist WE, Concepcion W. Combined liver-kidney transplantation in children: indications and outcome. Pediatr Transplant. 2008;12:835–46.
Fong TL, Bunnapradist S, Jordan SC, Selby RR, Cho YW. Analysis of the United Network for Organ Sharing database comparing renal allografts and patient survival in combined liver-kidney transplantation with the contralateral allografts in kidney alone or kidney-pancreas transplantation. Transplantation. 2003;76:348–53.
Mekahli D, van Stralen KJ, Bonthuis M, et al. Kidney versus combined kidney and liver transplantation in young people with autosomal recessive polycystic kidney disease: data from the European Society for Pediatric Nephrology/European Renal Association−European Dialysis and Transplant (ESPN/ERA-EDTA) registry. Am J Kidney Dis. 2016;68:782–8.
Nair P, Al-Otaibi T, Nampoory N, Al-Qabandi W, Said T, Halim MA, Gheith O. Combined liver and kidney transplantation in primary hyperoxaluria: a report of three cases and review of the literature. Saudi J Kidney Dis Transpl. 2013;24:969–75.
Eytan Mor MD, Weismann I. Editorial: current treatment for primary hyperoxaluria type 1: when should liver/kidney transplantation be considered. Pediatr Transplant. 2009;13:805–7.
Soliman NA, Nabhan MM, Abdelrahman SM, et al. Clinical spectrum of primary hyperoxaluria type 1: experience of a tertiary center. Nephrol Ther. 2017;13:176–82.
Cai R, Lin M, Chen Z, et al. Primary hyperoxaluria diagnosed after kidney transplantation failure: lesson from 3 case reports and literature review. BMC Nephrol. 2019. https://doi.org/10.1186/s12882-019-1402-2.
Ozer A, Aktas H, Bulum B, Emiroglu R. The experience of combined and sequential liver and kidney transplantation from a single living donor in patients with primary hyperoxaluria type 1. Pediatr Transplant. 2019. https://doi.org/10.1111/petr.13406.
Kemper MJ. Concurrent or sequential liver and kidney transplantation in children with primary hyperoxaluria type 1? Pediatr Transplant. 2005;9:693–6.
Astarcioglu I, Karademir S, Gülay H, Bora S, Astarcioglu H, Kavukcu S, Türkmen M, Soylu A. Primary hyperoxaluria: simultaneous combined liver and kidney transplantation from a living related donor. Liver Transpl. 2003;9:433–6.
Angelico R, Guzzo I, Pellicciaro M, et al. Same donor laparoscopic liver and kidney procurement for sequential living donor liver-kidney transplantation in primary hyperoxaluria type I. J Laparoendosc Adv Surg Tech. 2019;29:1616–22.
Khorsandi SE, Samyn M, Hassan A, et al. An institutional experience of pre-emptive liver transplantation for pediatric primary hyperoxaluria type 1. Pediatr Transplant. 2016;20:523–9.
Bergstralh EJ, Monico CG, Lieske JC, Herges RM, Langman CB, Hoppe B, Milliner DS. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10:2493–501.
Horoub R, Shamsaeefar A, Dehghani M, Nikoopour H, Entezari M, Moradi A, Kazemi K, Eshraghian A, Nikeghbalian S, Malek-Hosseini SA. Liver transplant for primary hyperoxaluria type 1: results of sequential, combined liver and kidney, and preemptive liver transplant. Exp Clin Transplant. 2019. https://doi.org/10.6002/ect.2019.0150.
Squires J, Nguyen C. Complexity of pre-emptive liver transplantation in children with primary hyperoxaluria type 1. Pediatr Transplant. 2016;20:604–6.
Zhao F, Bergstralh EJ, Mehta RA, Vaughan LE, Olson JB, Seide BM, Meek AM, Cogal AG, Lieske JC, Milliner DS. Predictors of incident ESRD among patients with primary hyperoxaluria presenting prior to kidney failure. Clin J Am Soc Nephrol. 2016;11:119–26.
Tang X, Bergstralh EJ, Mehta RA, Vrtiska TJ, Milliner DS, Lieske JC. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int. 2015;87:623–31.
Bendel-Stenzel MR, Kashtan CE, Sutherland DER, Chavers BM. Simultaneous pancreas-kidney transplant in two children with hemolytic-uremic syndrome. Pediatr Nephrol. 1997;11:485–7.
Samoylova ML, Borle D, Ravindra KV. Pancreas transplantation: indications, techniques, and outcomes. Surg Clin North Am. 2019;99:87–101.
Mittel AM, Wagener G. Anesthesia for kidney and pancreas transplantation. Anesthesiol Clin. 2017;35:439–52.
Haidar G, Green M. Intra-abdominal infections in solid organ transplant recipients: guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin Transpl. 2019. https://doi.org/10.1111/ctr.13595.
Bottino R, Balamurugan AN, Giannoukakis N, Trucco M. Islet/pancreas transplantation: challenges for pediatrics. Pediatr Diabetes. 2002;3:210–23.
Rubio-Cabezas O, Patch AM, Minton JAL, et al. Wolcott-Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metab. 2009;94:4162–70.
Tzakis AG, Nunnelley MJ, Tekin A, Buccini LD, Garcia J, Uchida K, Neville HL, Nares MA, Ruiz P, Bodamer O. Liver, pancreas and kidney transplantation for the treatment of Wolcott-Rallison syndrome. Am J Transplant. 2015;15:565–7.
Julier C, Nicolino M. Wolcott-Rallison syndrome. Orphanet J Rare Dis. 2010. https://doi.org/10.1186/1750-1172-5-29.
Rivera E, Gupta S, Chavers B, Quinones L, Berger MR, Schwarzenberg SJ, Pruett T, Verghese P, Chinnakotla S. En bloc multiorgan transplant (liver, pancreas, and kidney) for acute liver and renal failure in a patient with Wolcott-Rallison syndrome. Liver Transpl. 2016;22:371–4.
Costa G, Parekh N, Osman M, Armanyous S, Fujiki M, Abu-Elmagd K. Composite and multivisceral transplantation nomenclature, surgical techniques, current practice, and long-term outcome. https://doi.org/10.1016/j.suc.2018.09.010.
D’Antiga L, Goulet O. Intestinal failure in children: the European view. J Pediatr Gastroenterol Nutr. 2013;56:118–26.
Nehra D, Fallon EM, Puder M. The prevention and treatment of intestinal failure-associated liver disease in neonates and children. Surg Clin North Am. 2011;91:543–63.
Smith JM, Weaver T, Skeans MA, Horslen SP, Harper AM, Snyder JJ, Israni AK, Kasiske BL. OPTN/SRTR 2016 annual data report: intestine. Am J Transplant. 2018;18:254–90.
Mercer DF, Brady MJ, Jain G. Should the pancreas be routinely included with an intestinal graft? Curr Transplant Rep. 2015;2:159–63.
Kunzler de Oliveira Maia F, Tekin A, Nicolau-Raducu R, Beduschi T, Selvaggi G, Vianna R, Ammar Al Nuss M, González J, Gaynor JJ, Ciancio G. Use of pediatric donor en bloc kidneys along with bladder segment in pediatric liver-kidney and multivisceral-kidney transplantation. Pediatr Transplant. 2020. https://doi.org/10.1111/petr.13596.
Ramisch D, Rumbo C, Echevarria C, et al. Long-term outcomes of intestinal and multivisceral transplantation at a single center in Argentina. Transplant Proc. 2016;48:457–62.
Caso Maestro O, Abradelo De Usera M, Justo Alonso I, Calvo Pulido J, Manrique Municio A, Cambra Molero F, García Sesma A, Loinaz Segurola C, Moreno González E, Jiménez Romero C. Porcine acellular dermal matrix for delayed abdominal wall closure after pediatric liver transplantation. Pediatr Transplant. 2014;18:594–8.
Hopfner R, Tran TT, Island ER, McLaughlin GE. Nonsurgical care of intestinal and multivisceral transplant recipients: a review for the intensivist. J Intensive Care Med. 2013;28:215–29.
Abu-Elmagd K, Reyes J, Bond G, et al. Clinical intestinal transplantation: a decade of experience at a single center. Ann Surg. 2001;234:404–17.
Rambhia PH, Hanna R, Bergfeld WF. Graft versus host disease in a pediatric multiple organ transplant recipient with trichohepatoenteric syndrome - a unique case report. Int J Dermatol. 2018;57:89–91.
Fischer RT, Friend B, Talmon GA, Grant WJ, Quiros-Tejeira RE, Langnas AN, Coccia PF. Intestinal transplantation in children with multiple intestinal atresias and immunodeficiency. Pediatr Transplant. 2014;18:190–6.
Andres AM, Santamaría ML, Ramos E, Sarriá J, Molina M, Hernandez F, Encinas JL, Larrauri J, Prieto G, Tovar JA. Graft-vs-host disease after small bowel transplantation in children. J Pediatr Surg. 2010;45:330–6.
Kato T, Tzakis AG, Selvaggi G, et al. Intestinal and multivisceral transplantation in children. Ann Surg. 2006;243:756–64.
Dharnidharka VR, Tejani AH, Ho PL, Harmon WE. Post-transplant lymphoproliferative disorder in the United States: young Caucasian males are at highest risk. Am J Transplant. 2002;2:993–8.
Hryhorczuk AL, Kim HB, Harris MH, Vargas SO, Zurakowski D, Lee EY. Imaging findings in children with proliferative disorders following multivisceral transplantation. Pediatr Radiol. 2015;45:1138–45.
Boudjemaa S, Boman F, Guigonis V, Boccon-Gibod L. Brain involvement in multicentric Epstein-Barr virus-associated smooth muscle tumours in a child after kidney transplantation. Virchows Arch. 2004;444:387–91.
van Stralen KJ, Borzych-Dużalka D, Hataya H, et al. Survival and clinical outcomes of children starting renal replacement therapy in the neonatal period. Kidney Int. 2014;86:168–74.
Grant D, Abu-Elmagd K, Mazariegos G, Vianna R, Langnas A, Mangus R, Farmer DG, Lacaille F, Iyer K, Fishbein T. Intestinal transplant registry report: global activity and trends. Am J Transplant. 2015;15:210–9.
Andres AM, Alameda A, Mayoral O, Hernandez F, Dominguez E, Martinez Ojinaga E, Ramos E, Prieto G, Lopez Santamaría M, Tovar JA. Health-related quality of life in pediatric intestinal transplantation. Pediatr Transplant. 2014;18:746–56.
Ngo KD, Farmer DG, McDiarmid SV, et al. Pediatric health-related quality of life after intestinal transplantation. Pediatr Transplant. 2011;15:849–54.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Engen, R.M., Verghese, P. (2021). Multiorgan Transplantation Challenges. In: Twombley, K.E. (eds) Challenges in Pediatric Kidney Transplantation . Springer, Cham. https://doi.org/10.1007/978-3-030-74783-1_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-74783-1_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-74782-4
Online ISBN: 978-3-030-74783-1
eBook Packages: MedicineMedicine (R0)