
Abstract
IgG4 is increasingly recognized as a pathophysiologic component of a variety of diseases. These include type 1 autoimmune pancreatitis, IgG4-related sclerosing cholangitis, retroperitoneal fibrosis, chronic sclerosing aortitis and periaortitis. Some cases of sclerosing mesenteritis, a fibrosing form of mesenteric panniculitis has been classified as IgG4-related. IgG4-related disease can affect a large number of organ systems. In most cases, more than one organ system is involved. Clinical manifestations of these conditions are dependent on the organ systems that are involved and the duration of illness. These can range from the subacute development of painless organ enlargement to severe conditions such as obstructive renal failure in patients with retroperitoneal fibrosis. Treatment consists of high doses of glucocorticoids, followed by prolonged tapering. A variety of immunosuppressive agents, (including rituximab) either alone or in combination are treatment options in patients who fail or relapse on glucocorticoid therapy. Maintenance immunosuppressive therapy is considered in patients with severe organ damage or relapsing disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- IgG4-Related Disease
- Autoimmunity
- Fibrosis
- Lymphoplasmacytic infiltration
- Plasma cells
- T cells
- Regulatory T cells
- Glucocorticoids
- Immunosuppressive agents
- Mesenteric Panniculitis
- Sclerosing Mesenteritis
- Retroperitoneal Fibrosis
- Sclerosing cholangitis
- Sclerosing aortitis
- Periaortitis
Introduction
IgG4 is one of four subcomponents of IgG, or immunoglobulin G. IgG is the most common immunoglobulin in the circulation, representing almost 75% of circulating immunoglobulins in human blood. IgG is produced in plasma cells. The role of IgG is to bind to foreign invaders such as bacteria, fungi, and viruses to prevent these organisms from producing disease states. Of the four subgroups of IgG, IgG4 represents the lowest concentration in human serum. Tissue level of IgG4 is directly related to the presence of acute inflammation. There is evidence to suggest that IgG4 has a regulatory rather than a direct inflammatory effect, and with this model, IgG4 has the physiologic function of protecting other molecules of the IgG subclasses. IgG4 produces a pathologic state when deviation from its physiologic action occurs. In this setting, rearrangement of the antibody structure of IgG4 occurs across a labile disulfide bond (Fig. 17.1). The figure demonstrates downward-facing red arrows that indicate the disulfide bonds. This is referred to as the hinge region of the molecule. Occasionally, these labile disulfide bonds enable inappropriate transformation of the IgG4 molecule, resulting in pathogenic IgG4 autoantibodies that inappropriately attack self-antigens.

Characteristic feature of Fab (Antigen-binding Fragment) arm exchange across the disulfide bond of IgG4. The hinge region, (downward-facing red arrowhead), contains the disulfide bond. These disulfide bonds are labile and thereby permit inappropriate splitting and exchange of the Fab structure found in IgG4. This is what allows IgG4 molecules to transform from regulatory to pathogenic antibodies. Reused from Kawa S. Immunoglobulin G4-related Disease: An Overview. JMA J. 2019;2(1):11–27. Attribution 4.0 International (CC BY 4.0)
These disease states are referred to as IgG4-Related Disease, or IgG4-RD. IgG4-RD are characterized by excessive inflammatory response leading to fibrosis. For this reason, IgG4-RD are referred to as a fibroinflammatory conditions. Most organs are potential targets of IgG4-RD, including the mesentery, and gastrointestinal (GI) tract organs, including the biliary tree and the pancreas.
Pathogenesis
Pathogenetic development of IgG4-RD can ultimately be summarized as lymphoplasmacytic infiltration of IgG4+ plasma cells. The term lymphoplasmacytic indicates a concentration of lymphoid cells and plasma cells. Interestingly, IgG4-containing plasma cells may be confined to affected tissues and frequently are not circulating in the blood; therefore, IgG4 levels may not be elevated when measured in the peripheral blood. When local inflammation is present, lymphoid cells are generally recruited along with the plasma cells. Specifically, regulatory T (Treg) cells are most commonly associated with IgG4-RD as well as other autoimmune diseases. Treg cells are responsible for secreting specific cytokines in normal immune physiology. In IgG4-RD, dysregulation of Treg cells occurs with associated inappropriate cytokines secretion, (see Fig. 11.4 in Chap. 11 Immunologic Function of the Mesentery). The most notable cytokines secreted by Treg cells are IL-4, IL-5, IL-10, IL-13, and TGF-beta. These cytokines play an important role in the production and recruitment of eosinophils. This ultimately leads to eosinophilic deposition in target tissues and organs. Peripheral eosinophilia and elevated serum IgE levels also occur. IL-10 was found to have the most intimate relationship with IgG4 production and this ultimately leads to local fibrosis via TGF-beta expression. Local fibrosis can best be explained by the fact that TGF-beta is primarily responsible for cell growth, proliferation, and apoptosis (cell death). Therefore, if any of these three roles of TGF-beta are dysregulated, inappropriate cell growth and proliferation and inadequate cell death follow.
Storiform fibrosis is a key histologic feature of IgG4-RD and is defined as “swirling fibrotic tissue encasing cellular infiltrate in collagen fibrils.” When seen on tissue biopsy, storiform fibrosis is highly suggestive, although not pathognomonic, of IgG4-RD. Storiform fibrosis is commonly seen in Type I Autoimmune Pancreatitis, an IgG4-RD affecting the pancreas.
Autoimmune pancreatitis (AIP) is the most well studied IgG4-RD of the GI tract. AIP can present as an acute pancreatitis, but more often, it manifests as a more insidious onset. It is estimated that AIP accounts for approximately 5% of all cases of chronic pancreatitis. IgG4-related sclerosing cholangitis, has attracted recent investigation, as have studies of the pathogenesis of retroperitoneal fibrosis and other mesenteric manifestations of IgG4-RD. Human leukocyte antigen (HLA) genes have been implicated in the development of IgG4-RD. Specifically, HLA subtypes DQB1_0401 and DRB1_0405 were found to be highly expressed in patients with AIP as compared to healthy patients. Furthermore, AIP patients also have higher levels of DQB1_0401 and DRB1_0405 expression compared to other patients with chronic pancreatitis.
Non-HLA genes that are associated with AIP consist of single nucleotide polymorphisms (SNPs). SNPs have been increasingly found to have clinical significance as it relates to the development, response to treatment, and prognosis of a variety of diseases. The SNPs associated with AIP are CTLA-4_49A, Cytotoxic T-Lymphocyte Antigen-4, and TNF-alpha promoter_863, a Tumor Necrosis Factor-alpha promoter gene. Studies performed in Chinese and Japanese patients have found these SNPS to be more highly expressed in AIP patients than in patients without known disease. In terms of clinical significance, CTLA-4_49A was a marker for a higher likelihood of relapse after seemingly adequate initial treatment of AIP.
Clinical Manifestations
IgG4-RD has been identified in virtually all organ systems. In most cases (60–90%) it affects more than one system simultaneously by causing subacute painless enlargement of the affected organs. As expected, clinical manifestations vary depending on the organ system involved and commonly include complications secondary to compression of nearby structures. Thus, IgG4-RD should be considered in patients presenting with unexplained enlargement of one or more organs. At the time of diagnosis, the patients are usually well and have no systemic symptoms, especially fever. However, significant weight loss in the months preceding diagnosis is not uncommon. Due to an increased awareness of the IgG4-RD, these more commonly being diagnosed as an incidental finding seen on imaging or histopathology in otherwise asymptomatic patients.
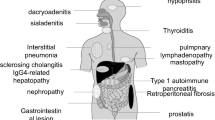
IgG4-RD include type 1 autoimmune pancreatitis (AIP)*, sclerosing cholangitis*, sclerosing mesenteritis*, Mikulicz disease, sclerosing sialadenitis, Inflammatory orbital pseudotumor, chronic sclerosing dacryoadenitis, idiopathic retroperitoneal fibrosis*, Chronic sclerosing aortitis and periaortitis*, and Riedel’s thyroiditis.
Because diseases with an (*) are closely related to the mesentery, they will be discussed in more detail.
Type 1 Autoimmune Pancreatitis (AIP)
Type 1 AIP was the first identified form of IgG4-RD and is considered to be the prototypical form. It accounts for approximately 5% of patients with chronic pancreatitis. Almost all patients are older than 45 at the time of diagnosis. Type 1 AIP often presents with a pancreatic mass or painless jaundice and can therefore be mistaken for pancreatic cancer. It has been estimated that 3–9% of patients who undergo pancreatic resection are discovered to have had Type 1 AIP. Patients with Type 1 AIP develop diabetes mellitus due to destruction of endocrine portions of the pancreas. It is commonly associated with other IgG4-related conditions. Radiologic features of AIP include a “sausage-shaped” pancreas with a surrounding halo of edema around the organ. It is very difficult to distinguish AIP from pancreatic adenocarcinoma on imaging. Some authors have suggested that diffusion-weighted magnetic resonance (MR) imaging findings maybe more useful than CT in making such a distinction.
IgG4-Related Sclerosing Cholangitis (SC)
IgG4-related SC is the second most common manifestation of IgG4-RD. Although IgG4-related SC affects 60–80% of patients with type 1 AIP, it can occasionally occur as an isolated manifestation. Because it leads to focal or diffuse bile duct wall thickening IgG4-related SC tends to present as obstructive jaundice. The most commonly affected segment is the intra-pancreatic portion of the common bile duct. IgG4-related SC and primary sclerosing cholangitis have similar magnetic resonance cholangiopancreatographic (MRCP) and endoscopic retrograde cholangiopancreatographic (ERCP) findings. Primary sclerosing cholangitis is usually a disease of young and middle age males while IgG4-related SC typically affects middle aged and elderly me. IgG4-related SC exhibits a good response to glucocorticoid therapy.
Sclerosing mesenteritis (SM), mesenteric lipodystrophy and mesenteric panniculitis are terms that have been used interchangeably in the literature (Table 17.1). There have been some case reports relating SM to IgG4-RD. However, to this date, there is not enough evidence to determine this association (see Chap. 20 Mesenteric Panniculitis).
SM likely represents a specific form of mesenteric panniculitis that is predominated by fibrosis. As in other IgG4-RD, SM may present with weight loss fever and malaise. In these cases, an abdominal mass can be palpated on examination, and occurs in one-third to one-half of patients. Abdominal tenderness, distension, signs of peritoneal irritation and ascites are less common. Some patients present with bowel obstruction, obstructive uropathy or renal failure, chylous ascites, and vascular occlusion. Laboratory findings include elevated erythrocyte sedimentation rate and C-reactive protein. The natural history of SM associated with IgG4-RD has not been fully established in the literature.
Retroperitoneal Fibrosis, Chronic Sclerosing Aortitis and Periaortitis
Retroperitoneal fibrosis (RF), chronic sclerosing aortitis and periaortitis are other forms of IgG4D. It has been suggested that previously described “idiopathic” RF is actually an IgG4-mediated disease. RF typically affects the infrarenal portion of the abdominal aorta with the simultaneous involvement of the iliac arteries. Progressive fibro-inflammatory changes in the distal aorta lead to obstructive uropathy. On imaging, IgG4-related RF is seen as a soft-tissue mass encasing the abdominal aorta and its branches. Hydronephrosis and hydroureter are not uncommon. As with AIP, RP is responsive to glucocorticoid therapy. IgG4-related periaortitis, may lead to aneurysmal formation and possible rupture.
Diagnosis of IgG4-RD
The diagnostic process of IgG4-RD begins by clinical recognition of the symptoms and physical exam findings suggestive of the disease. Alternatively, these conditions are suggested by incidental findings found on imaging studies. Biopsy findings are the cornerstone of diagnosing IgG4-RD. These findings include lymphoplasmacytic tissue infiltration of IgG4-positive plasma cells and lymphocytes in association with storiform fibrosis. These findings are often accompanied by obliterative phlebitis and tissue eosinophilia. IgG4 lymphoplasmacytic infiltrates are not specific to IgG4-RD. They can be found in malignancies and autoimmune disorders. It is important to rule out those disorders first as they are much more common than IgG4-RD. After the diagnosis of IgG4-RD has been established, the extent of the disease should be determined. Imaging of the chest, abdomen and pelvis via CT or MRI is highly effective in achieving that purpose. Characteristic findings include diffuse and focal organ infiltration and encasement by inflammatory and fibrotic tissue.
Treatment
The location, severity and duration of patient symptomology and clinical findings determine the treatment regimen for IgG4-RD. Gastrointestinal disease (e.g. mesentery, biliary tree, and pancreatic) often presents acutely and requires urgent treatment. Treatment principles focus on reducing inflammation and host immune response. The initial treatment focuses on glucocorticoid therapy, often administered at high doses followed by a slow taper. However, some patients may not be able to tolerate corticosteroids or may have ongoing symptoms and disease activity despite corticosteroids. In these patients, other immunosuppressive agents are administered and these treatments may be used in combination with glucocorticoids. Rituximab monotherapy is has shown to eliminate relapses within a six-month follow up period in over 75% of patients with a variety of IgG4-RD. Other immunosuppressive agents that are commonly used (sometimes in combination with glucocorticoids) include mycophenolate, azathioprine, 6-mercaptopurine methotrexate, and cyclophosphamide. If a patient enters disease remission, maintenance therapy is considered. The decision for use of maintenance therapy focuses on prior history of relapsing disease and whether the patient’s initial presentation resulted in severe organ damage. In Japan, AIP maintenance therapy consists of low-dose glucocorticoids. If relapses occur, other immunosuppressive agents are added to the treatment regimen.
Suggested Reading
Crescioli S, Correa I, Karagiannis P, Davies AM, Sutton BJ, Nestle FO, Karagiannis SN. IgG4 Characteristics and Functions in Cancer Immunity. Current Allergy Asthma Rep. 2016;16(1):7.
Van der Neut KM, Schuurman J, Losen M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317(5844):1554–7.
Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51.
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th ed. New York: Garland Science; 2001.
Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–738.
Nishimori I, Tamakoshi A, Otsuki M, Research committee on intractable diseases of the pancreas, Ministry of Health, Labour, and Welfare of Japan. Prevalence of autoimmune pancreatitis in Japan from a nationwide survey in 2002. J Gastroenterol. 2007;42(18):6.
Zen Y, Nakanuma Y. Pathogenesis of IgG4-related disease. Curr Opin Rheumatol. 2011;23(12):114–118.
Stone JH. IgG4-Related Disease: Harrison’s Principles of Internal Medicine, 19e. 2017 November; Part 14: Section 391e.
Khosroshahi A, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheum. 2015;67:1688–99.
Carruthers MN, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74:1171–7.
Kubo K, Yamamoto K. IgG4-related disease. Int J Rheum Dis. 2016;19:747–62. https://doi.org/10.1111/1756-185X.12586.
Yadlapati S, Verheyen E, Efthimiou P. IgG4-related disease: a complex under-diagnosed clinical entity. Rheumatol Int. 2018;38(2):169–77. https://doi.org/10.1007/s00296-017-3765-7.
Anxo Martínez-de-Alegría mailto:anxomartinezdealegria@gmail.com, Sandra Baleato-González, Roberto García-Figueiras, Anaberta Bermúdez-Naveira, Ihab Abdulkader-Nallib, José A. Díaz-Peromingo, Carmen Villalba-Martín: IgG4-related Disease from Head to Toe. RadioGraphics 2015;35:2007–2025.
Danford CJ, Lin SC, Wolf JL. Sclerosing mesenteritis. Am J Gastroenterol. 2019;114(6):867–73.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Khattab, A., Kruchko, D.H., Ehrenpreis, E.D. (2021). IgG4-Related Diseases and the Mesentery. In: Ehrenpreis, E.D., Alverdy, J.C., Wexner, S.D. (eds) The Mesenteric Organ in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-71963-0_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-71963-0_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-71962-3
Online ISBN: 978-3-030-71963-0
eBook Packages: MedicineMedicine (R0)