
Abstract
The modification of RNA species has been well documented for decades. In mRNA, both internal and 5′ end modifications can occur. Specifically, modification of the 5′ end is known as capping. The 5′-5′ triphosphate linked N7-methyl guanosine (m7G) structure is involved in a myriad of RNA processes and was long presumed to be the only functional cap. In recent years, this view was overturned by reports that nicotinamide adenine dinucleotide (NAD+), a critical redox cofactor, can serve as an RNA cap in bacteria. Subsequently, yeast, mammalian, and plant RNA species were also found to harbor the NAD+cap. Apart from NAD+, other noncanonical nucleotide analogs, including NADH, FAD, dpCoA, UDP-Glucose, and UDP-N-acetylglucosamine, were found to be caps in endogenous RNA, suggesting that a wide repertoire of RNA caps may be present. However, the functions of noncanonical capping remain mostly unknown. This chapter describes the detection methods for noncanonical RNA caps, their mode of capping and decapping, and their potential molecular and biological functions. The discovery of noncanonical caps represents a revolution in research on RNA modifications and prompts future efforts to delve into novel epitranscriptomic processes, which may link cellular metabolism with gene expression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Noncanonical caps
- LC-MS analysis
- NAD+ captureSeq
- NAD+ tagSeq
- Capping mechanism
- Decapping enzymes
- Nudix
- DXO
- RNA stability
- Translation initiation
1 Introduction
Beyond the base genetic code provided by DNA and RNA, various kinds of chemical or “epigenetic” modifications to these structures provide another layer of information. Functionally, the addition of these chemical marks can be recognized by specific proteins, leading to versatile gene expression without changing the genetic sequence itself (Roundtree et al. 2017; Shi et al. 2019; Boo and Kim 2020). The critical need to understand RNA epigenetic modifications set off the research field known as epitranscriptomics (Fu and He 2012; Chen et al. 2020).
While both RNA and DNA can be modified, RNA modifications play a more direct role in dynamically tuning transcript output, such as by affecting stability and translatability (Chen et al. 2016). Since the first modified nucleotide in RNA was discovered in the 1960s (Cohn 1960), more than 170 different RNA modifications have been identified in coding and noncoding RNA (Boccaletto et al. 2018; Nachtergaele and He 2018). tRNA and rRNA contain the most modifications, including 2’-O-methylation and pseudouridylation (Roundtree et al. 2017). On the other hand, the known modifications on mRNA are less diverse, but contribute more to shaping the cellular transcriptome. mRNA modifications include internal modifications, such as N6-methyladenosine (m6A), N6, 2′-O-dimethyladenosine (m6Am), N1-methyladenosine (m1A), pseudouridine (Ψ), 5-methylcytidine (m5C), 5-hydroxymethylcytosine (hm5C), and N4-acetylcytidine (ac4C), as well as modifications of the 3′ end, known as the poly(A) tail and oligo(U) tail, and modification of the 5′ end, known as the caps (Boo and Kim 2020).
In particular, 5′ end capping is a critical determinant of the fate of an RNA. The 5′ cap is known to play a pivotal role in numerous RNA metabolic processes, such as polyadenylation (and possibly oligouridylation), pre-mRNA splicing, mRNA export, transcript stability, and translation initiation. Thus, this structure is mechanistically involved in every stage of the mRNA lifecycle (Ramanathan et al. 2016; Galloway and Cowling 2019). The predominant 5′ cap of mRNA is the 7-methylguanosine moiety linked via a 5′ to 5′ triphosphate chain to the first transcribed nucleotide, which is abbreviated as m7GpppN and known as cap 0. Incorporation of the m7GpppN cap is accomplished through characterized enzymatic activities (Ramanathan et al. 2016). In addition, the first and second transcribed nucleotides can be methylated on the ribose 2’-O position, resulting in m7GpppNm or m7GpppNmNm structures referred to as cap 1 and cap 2, respectively (Werner et al. 2011). In cap 1, when the first nucleotide is adenosine, another N6-methylation may also be observed at a ratio that reaches up to 20% in human cells (Mauer et al. 2017). These m7G-related cap structures, or canonical caps, have been observed at varied levels in specific tissues and cells and could be differentially regulated in specific biological processes (Wetzel and Limbach 2016; Sikorski et al. 2020). However, despite the important roles of the m7GpppN cap, this cap is found only in eukaryotes (Galloway and Cowling 2019).
Until recently, in bacteria, such as Escherichia coli (E. coli), it was assumed that the 5′ end of RNA consisted only of a 5′ triphosphate. This was overturned when nicotinamide adenine dinucleotide (NAD+) was identified as a cap in E. coli RNA (Chen et al. 2009). NAD+ is a pyridine dinucleotide and is an electron carrier involved in oxidation-reduction reactions, making it a key component of cellular signaling (Gakière et al. 2018). As NAD+ contains an adenosine moiety, it may be recognized by RNA polymerase and incorporated into the 5′ end of RNA. After the identification of NAD+-capped RNA in E. coli, yeast, mammalian, and plant RNA species were also found to harbor the noncanonical NAD+ cap (Cahová et al. 2015; Jiao et al. 2017; Walters et al. 2017; Kiledjian 2018; Julius and Yuzenkova 2019; Wang et al. 2019b; Zhang et al. 2019a). Additionally, other adenosine-containing metabolites, such as dpCoA and FAD, were also found to initiate transcription in vitro (Huang 2003).
To date, many noncanonical nucleotides have been reported to prime RNA transcription by RNA polymerases from different organisms (Fig. 1) (Wang et al. 2019a; Doamekpor et al. 2020a; Hudeček et al. 2020). In this chapter, we aim to summarize the various types of noncanonical caps in different organisms, the detection methods used to identify these structures, the mechanism of incorporation into a transcript, and the possible regulation and biological functions of noncanonical caps, with an emphasis on the NAD+ cap.

Noncanonical RNA caps discovered in different organisms. RNA cap structures that have been discovered to date are classified into adenosine-containing nucleotides and uridine-containing nucleotides in the center of the circle. In the middle ring, the structure of each cap is displayed. The outer ring indicates the organisms reported to contain each RNA cap. The cartoons of the organisms were created with BioRender.com. dpCoA, dephospho-coenzyme A; GlcNAc, N-acetylglucosamine
2 Discovery and Detection of RNA Modifications in Cells
RNA modifications can be detected, mapped, and quantified through various methods, although there are many challenges associated with the characterization of mRNA modifications in particular (Helm and Motorin 2017). For example, unlike non-coding RNAs that have relatively abundant modifications, mRNAs have low levels of modified nucleotides. The most abundant one, m6A, was estimated to only apply to 0.2% of total adenosine in cellular mRNA, equivalent to 2–3 nucleotides per transcript (Meyer et al. 2012). An additional challenge arises due to the different chemical properties of each modified residue. The need for specific detection and mapping methods for diverse RNA modifications resulted in the rise of an assortment of techniques.
The long-established method for the global detection and quantification of RNA modifications is thin layer chromatography (TLC), which relies on radioactive 32P labeling for sensitivity (Grosjean et al. 2007; Kellner et al. 2010). TLC was later supplemented by high-performance liquid chromatography (HPLC) coupled to mass-spectrometry (MS) (Thüring et al. 2016; Wetzel and Limbach 2016). In these methods, the modified nucleotides are released from mRNA by complete chemical or enzymatic digestion and identified according to their chromatographic retention times and fragmentation patterns. However, while these methods have opened the door for the detection of various RNA modifications, they do not provide information on the exact localization of the modifications.
Recently, next-generation sequencing (NGS) technologies have paved the way for mapping mRNA modifications. It was observed that some modified RNA nucleotides can naturally block primer extension or cause misincorporations during reverse transcription (RT), thus leaving a signature mark at modified sites in cDNA sequences (Ryvkin et al. 2013). However, naturally occurring abortive RT events due to RNA modifications are limited and do not apply to the majority of modifications (Helm and Motorin 2017; Schwartz and Motorin 2017). To expose further RNA modifications, mRNA can be treated with chemical reagents that react with specific modifications to change or enhance the RT signature. This type of methodology has been used to uncover Ψ, internal m7G, and m5C in specific RNAs (David et al. 2017; Zhang et al. 2019b).
Additionally, affinity-based enrichment of RNA modifications before high-throughput sequencing is highly beneficial for detection due to the low levels of modified residues in cellular RNA. Modified RNA can be selectively recognized by specific antibodies (Dominissini et al. 2012; Mishima et al. 2015; Li et al. 2016) or by clickable chemical reactions depending on the functional structure (Cahová et al. 2015). Recognition of specific, modified RNAs is followed by library preparation and sequencing, yielding information on the location and abundance of the modification. Despite the various methods to detect RNA modifications, challenges remain and information on many of these modifications is limited. Therefore, to reinforce these methods, currently used techniques to investigate RNA noncanonical capping are rapidly evolving.
2.1 Global Discovery and Detection of Noncanonical Capping in RNA
2.1.1 HPLC and MS Coupling
LC-MS has been extensively used in the detection and quantification of novel RNA modifications. In general, RNA is cleaved and fragments are subsequently analyzed, such as by fragmentation pattern or comparison to the calculated mass of the unmodified residue. This method is responsible for the discovery of NAD+-linked RNA in E. coli and Streptomyces venezuelae in 2009 (Chen et al. 2009).
In this 2009 study, a group led by David Liu employed a workflow with key treatments to detect noncanonical nucleotides in RNA (Fig. 2a). First, cellular RNA is separated through size-exclusion chromatography, and the macromolecular fraction is retained. This macromolecular fraction is further treated with the nuclease P1, an endonuclease that generates mononucleotides with a 3′ hydroxyl group and a 5′-phosphate. The treated sample is then subjected to LC-MS. Using this method, 24 and 28 unknown small molecule-RNA conjugates were significantly enriched compared to untreated samples in E. coli and S. venezuelae, respectively (Chen et al. 2009). These candidate small molecules were shown to be cleaved from cellular RNA, as the detected amount decreased if samples were pretreated with RNase.

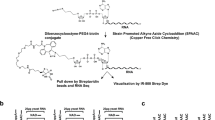
Strategies for the detection of noncanonical RNA caps in vivo. (a) Global detection and quantification of noncanonical caps. Firstly, isolated RNA is separated from the small molecular weight fraction and treated with nuclease P1 or a decapping enzyme. After a secondary size exclusion step, collected fractions are analyzed by colorimetric assays or coupled HPLC and MS. Quality or quantity is determined by comparison with a standard. (b) NAD+-capped RNA capture and sequencing technologies. The nicotinamide moiety of NAD+ is exchanged for an alkyne group by ADPRC, and the alkyne group undergoes a copper-catalyzed azide-alkyne cycloaddition reaction to link to a biotin moiety (in NAD+ captureSeq) or a tagRNA that can be hybridized with a biotinylated DNA probe (in NAD+ tagSeq). Biotinylated RNA is eluted and enriched by streptavidin beads. The profile of NAD+-capped RNAs can be analyzed by high-throughput RNA sequencing. The sequencing machine cartoons were created with BioRender.com. (c) CapZyme-Seq workflow. Noncanonically capped RNA is first processed by decapping enzymes to yield a 5′ monophosphate end and then ligated with single-stranded oligonucleotide adaptors. Finally, 5′ end sequences are analyzed by high-throughput sequencing. (d) Validation technologies for individual RNA containing noncanonical caps. A specific RNA candidate is cleaved by a DNAzyme to yield short RNA 5′ end fragments. Capped RNA is distinguished from uncapped RNA in acrylaminophenyl boronic acid electrophoresis (APB) and then hybridization with a specific probe for candidate RNA transcripts can be performed
In both species, two molecules that were found to be highly enriched after P1 digestion were NAD+ and dpCoA, alongside their derivatives. Both structures were present in shorter RNAs of lengths below ~200 nucleotides. The former showed a higher abundance at 3000 copies per cell, while the latter only displayed 100 copies per cell. Further repeated assays using isotopically labeled water revealed that NAD+ and dpCoA in cellular RNA are attached to the 5′ terminus. This novel finding showed that adenosine-based noncanonical metabolites could serve as a cap structure in bacteria. Following detection in prokaryotes, the NAD+ cap was detected using LC-MS techniques in eukaryotes such as yeast, mammalian cells, and the plant, Arabidopsis (Wang et al. 2019a; Zhang et al. 2019a).
The discovery of NADylated RNA prompted research efforts dedicated to the understanding of noncanonical RNA caps. Rapidly, more metabolites, all of which shared a nucleotide-containing structure, were found capable of being incorporated into the 5′ end of RNA by in vitro transcription. For example, FAD, a coenzyme involved in redox reactions, dinucleoside polyphosphate (NpnN), a potential alarmone, and UDP-glucose and UDP-N-acetylglucosamine (UDP-GlcNAc), cell wall precursors, could all be incorporated as cap structures in vitro (Huang 2003; Julius et al. 2018; Hudeček et al. 2020). The attachment of such a range of substrates to the 5′ end of RNA suggested that more noncanonical caps could exist in vivo. However, untargeted LC-MS analyses have not detected these caps in vivo, perhaps due to the lack of sensitivity.
To detect and quantify more of the RNA capping landscape in vivo, targeted LC-MS analyses combine off-line HPLC enrichment of cap nucleotides with triple-quadrupole mass spectrometry to enable absolute quantification of a given RNA cap structure (Wang et al. 2019a). In targeted analyses, filter parameters can be pre-set to the mass size and retention time of specific chemicals via synthesized standards to efficiently detect target molecules released from cellular RNA. In addition, highly accurate quantification can be achieved by combining isotope-labeled internal standards and a series of unlabeled external standards to generate a calibration curve. Using this technique, three novel metabolite caps (FAD, UDP-glucose, and UDP-GlcNAc) were discovered and quantified in virus, E. coli, yeast, mouse, and human cellular RNA. FAD and UDP-glucose caps accounted for <5 fmol/μg RNA (Wang et al. 2019a), but surprisingly, the UDP-GlcNAc cap was more abundant, reaching up to 28 fmol/μg RNA, higher than the NAD+ cap and consistent with the relative abundance of such cellular metabolites in cells (Yang et al. 2007; Namboori and Graham 2008; Julius et al. 2018). Using the same LC-MS-based methodology, in E. coli, dinucleoside polyphosphates (NpnN) were also detected as noncanonical caps in a short RNA fraction (Hudeček et al. 2020). The amount of NpnN caps (Ap3A, Ap3G, Ap5A) in small RNA (sRNA) was comparable to that of dpCoA (~ 75 fmol/μg sRNA) and much lower than that of NAD+ (1900 fmol/μg sRNA). LC-MS experimentation further revealed that some of the NpnN caps contained multiple methyl groups in the nucleotides (e.g. m7Gp4Gm, m6Ap3A), which maintained cap stability (Hudeček et al. 2020).
Untargeted LC-MS analysis provided an approach to discover novel RNA cap modifications but is hampered by the limited sensitivity of the MS detector and sample purity (Limbach and Paulines 2017). Conversely, targeted LC-MS analysis displays high accuracy and sensitivity, though it requires a synthesized standard and therefore can only be applied to previously identified structures. Weighing the strengths and weaknesses of each experimental approach is necessary to effectively address the desired research question.
2.1.2 CapQ Quantification
Cap detection and quantitation, known as CapQ, is another method used for RNA-cap identification that is both time-efficient and easily performed in the average laboratory (Fig. 2a). In general, the first step of CapQ is the same as LC-MS detection, where the intact 5′ end cap structure is released from RNA by enzymatic treatment, such as by nuclease P1. This step is followed by a colorimetric assay that affords measurement of the amount of released molecules based on an enzymatic cycling reaction.
Specifically, for detecting the NAD+ cap, the released NAD+ is reduced to NADH, which then reacts with a colorimetric probe to produce a colored product that can be measured at 450 nm. The intensity of the product color is proportional to the amount of NAD+ in the test sample. Using this method, the extent of NAD+ capping was determined to be ~120 fmol/μg RNA in E. coli, which is similar to previous estimates using an LC-MS approach (Chen et al. 2009; Grudzien-Nogalska et al. 2018). In other organisms, the level of NAD+ capping is lower than in E. coli (80 fmol/μg in S. cerevisiae, 20 fmol/μg in HEK293T cell, 12 fmol/μg in Arabidopsis) (Grudzien-Nogalska et al. 2018; Wang et al. 2019b). The lower NAD+ capping is reasonable as there is likely a dominant preference for the eukaryotic m7G cap.
Similar to NAD+, FAD can also be measured by a specific colorimetric assay. The recently developed FAD CapQ revealed that there is ~1 fmol FAD/μg of short RNAs in human cells. This is a comparable concentration to that measured by targeted LC-MS (Wang et al. 2019a; Doamekpor et al. 2020a).
While there are certainly benefits to the usage of CapQ methodology, this technique also has some shortcomings. For instance, so far, CapQ application is restricted to NAD+ and FAD caps and relies on commercially available colorimetric assay kits. Nonetheless, compared to LC-MS detection, the CapQ method is highly suitable for comparisons of NAD+ and FAD cap contents from different samples.
2.2 Next-Generation Sequencing Technologies for Use in the Study of Noncanonical Capping in RNA
The methods outlined above for the global quantification of noncanonical RNA caps cannot provide any information on the sequences harboring, or localizations of, these structures. Sequencing technologies have revolutionized epitranscriptomics research, affording the ability to map RNA modifications to specific transcripts and aiding in the illumination of the function of noncanonical caps.
2.2.1 NAD+ captureSeq
After the discovery that RNA potentially possessed NAD+ caps (Fig. 2b) (Chen et al. 2009), the precise transcripts that contained these caps were not profiled until 2015, when a next-generation sequencing technique known as NAD+ captureSeq was established in E. coli (Cahová et al. 2015). NAD+ captureSeq utilizes a chemoenzymatic reaction to detect and identify NAD+-capped RNA. In this reaction, adenosine diphosphate ribosylcyclase (ADPRC) removes the nicotinamide moiety from NAD+-capped RNA. This step is followed by transglycosylation with an alkyne (such as pentynol) that reacts with the remaining 5′ end of RNA and subsequently click-chemistry-mediated biotinylation (Rostovtsev et al. 2002; Cahová et al. 2015). Thus, NAD+-capped RNA is converted to biotinylated RNA, which can be captured and enriched by streptavidin beads and processed for high-throughput sequencing. Transcripts gleaned from this pipeline must be compared to a control background library without ADPRC treatment (ADPRC-) or to total RNA sequencing (RNA-seq) data. The transcripts that are significantly enriched in the ADPRC-treated sample are deemed to be NAD+-capped (Cahová et al. 2015; Kwasnik et al. 2019).
Since its development, the NAD+ captureSeq method has been widely utilized in many prokaryotes and eukaryotes, exposing new information on NAD+-capped RNA. In E. coli, it was observed that the identified NAD+-capped RNAs were mainly sRNAs involved in stress responses and mRNAs encoding enzymes involved in metabolism. The most abundantly NAD+-capped sRNA was RNAI, which had 13% of its transcripts containing an NAD+ cap (Cahová et al. 2015). Other than E. coli, the bacterium B. subtilis also exhibited NAD+-capped RNA, but at a level 14-fold less than E. coli. In B. subtilis, NAD+-capped transcripts were predominantly full-length mRNA, different from E. coli’s predisposition for NAD+-capped sRNAs (Frindert et al. 2018). Through comparing the common sequence features of identified NAD+-capped RNA, it was observed that most of the enriched RNA reads started with an adenosine, implying that NAD+ caps are incorporated into RNA during transcription initiation (Bird et al. 2016). Interestingly, neither species of bacteria displayed ribosomal RNAs or transfer RNAs that were enriched for NAD+-capped transcripts.
Following prokaryotes, NAD+ captureSeq was applied to eukaryotes. In Saccharomyces cerevisiae, 1–5% of mRNA transcripts were shown to be modified by NAD+ caps. Most of these transcripts were short RNAs involved in mitochondrial function and the translational machinery (Walters et al. 2017; Zhang et al. 2020). In human cells, NAD+-capped mRNAs were detected, and the noncoding transcripts found to be preferentially capped included small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) (Jiao et al. 2017). Finally, in plants, NAD+-capped RNAs were widespread throughout the transcriptome, except in chloroplast RNA, and these transcripts were found to be related to photosynthesis, protein synthesis, and stress responses (Wang et al. 2019b; Zhang et al. 2019a). NAD+-capped RNAs were spliced and polyadenylated in both human cells and plants.
2.2.2 NAD+ tagSeq
Based on the technique demonstrated in NAD+ captureSeq, a modified approach called NAD+ tagSeq allows for the full-length sequences of NAD+-capped transcripts to be delineated by using single-molecule RNA sequencing (Fig. 2b). Similar to NAD+ captureSeq, ADPRC removes the nicotinamide of NAD+-capped RNA, and subsequently an alkyne is introduced to the 5′ end of the RNA. However, instead of biotinylation, the copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction attaches a synthetic RNA, or tagRNA, that contains an azide group. The desired NAD+-capped RNA, now linked to this RNA tag, is isolated by a DNA probe and sequenced using Oxford Nanopore sequencing technology. Sequencing starts from the polyA tail and ends with the 5′ end of transcripts. All sequence reads containing the tagged RNA are thus NAD+-capped RNA. Through this method, features of NAD+-capped RNA can be analyzed, revealing that the 5′ end of many NAD+-capped RNAs are located around 30 to 400 bases downstream of canonical transcription start sites (TSS) in Arabidopsis. Therefore, NAD+-capped RNAs tend to have shorter 5′ UTRs than m7G-capped RNAs. NAD+ tagSeq provides more accurate and broader information about NAD+-capped RNA sequences than NAD+ captureSeq but loses the capability to analyze very short (<100 nt) RNAs due to the use of nanopore sequencing (Zhang et al. 2019a).
Despite the genome-level NAD+-capped RNA analysis offered by both NAD+ captureSeq and NAD+ tagSeq, there remain downfalls in using these techniques. One drawback revolves around the introduction of copper ions during the click chemistry CuAAC reaction. The introduction of copper ions is prone to causing RNA degradation, resulting in a bias toward the 5′ end (Liu et al. 2020). The density of reads at the 5′ end is increased through enrichment by streptavidin beads irrespective of the 3′ end. In addition, the alkyne moiety added during the first step seems capable of reacting with some other modified units in RNA in the absence of ADPRC, leading to nonspecific signals. For example, in Arabidopsis chloroplasts, the transcript level was comparably high in both ADPRC+ and ADPRC- samples, the latter presumably due to a false signal stemming from some other modification(s) in the RNA (Wang et al. 2019b).
2.2.3 CapZyme-Seq
NAD+ captureSeq does not provide single nucleotide resolution of 5′ ends. Although NAD+ tagSeq afforded the observation of full-length sequences, it still failed to determine the exact 5′ end sequence of NAD+-capped RNAs due to inability to call bases at the junction between the tagRNA and the 5′ end of NAD+-capped RNA (Cahová et al. 2015; Zhang et al. 2019a). Exact 5′ end high-throughput sequencing relies on adaptor ligation to RNA 5′ ends with a 5′ monophosphate. For RNA with noncanonical caps, a few decapping enzymes in various organisms were discovered that enable the removal of noncanonical caps, such as NAD+, NADH, dpCoA, or FAD, resulting in a monophosphate at the 5′ end of the RNA (Jiao et al. 2017; Doamekpor et al. 2020a). A method that takes advantage of these decapping enzymes is CapZyme-Seq, which was established to identify the exact 5′ end sequence of RNA, as well as quantify the relative amount of noncanonically capped RNA or uncapped RNA (Fig. 2c).
CapZyme-Seq combines enzymatic removal of noncanonical caps or 5′ triphosphates with high-throughput sequencing. By performing CapZyme-Seq in E. coli, it was revealed that NAD+-mediated initiation significantly preferred an adenosine at the TSS, while the capping efficiency for diverse promoter sequences varied. One sRNA with an A:T pair at the TSS position displayed a level of noncanonical capping of 22.4% compared to uncapped RNA. However, like the previous methods, this method also has limitations. For one, decapping enzymes may be unable to distinguish NAD+ caps from other noncanonical caps. In addition, the different decapping enzymes used in CapZyme-Seq may exhibit various efficiencies for specific cap types or have differing specificities toward different RNAs with the same type of noncanonical cap, which could influence the results (Vvedenskaya et al. 2018).
The methodologies described above are most useful to study NAD+-capped RNA. Unfortunately, other noncanonical caps still lack a robust sequencing technique to further explore their properties. Options for future studies on these caps could include methodologies using specific antibodies, affinity tagging through chemical reactions, or selective recognition by unique RNA or protein structures (Breaker 2012; Mishima et al. 2015).
2.3 In Vitro Research and Validation Technologies for RNA with Noncanonical Caps
Previous sections have detailed the powerful techniques used for global noncanonical cap detection in vivo. There are also simple tools available for studies of noncanonical capping in vitro. Most commonly, 32P radioactively labelled capped RNA is analyzed by TLC. This method is usually used to examine the incorporation of a cap and the efficiency of sequence extension during in vitro transcription (Julius et al. 2018). In addition, acrylaminophenyl boronic acid electrophoresis (APB) provides a visual, user-friendly technique that allows distinction of the less-mobile, capped RNA containing a vicinal-diol moiety, such as m7G, NAD+, NADH, FAD, or NpnN, from uncapped RNAs (Nübel et al. 2017; Luciano et al. 2019). For individual transcripts in vivo, APB gels also serve as a powerful validation tool for identification of capped RNAs. Combined with defined, specific oligodeoxynucleotide-mediated RNA cleavage (DNAzyme), which processes RNA to yield short 5′ end-containing fragments (Joyce 2001), APB gels can identify the noncanonical capping of RNAs, as well as distinguish between different capped species by comparing to synthetic noncanonically capped RNA standards (Fig. 2d) (Bird et al. 2018).
3 Mechanism of Noncanonical Capping
The mechanism involved in canonical m7G capping has been clearly defined. After transcription initiation, addition of the m7G cap is accomplished by a capping complex that interacts with the nascent RNA of ~20–25 nt (Shuman 2015). On the other hand, the mechanism of noncanonical capping in vivo requires further research, as current data is conflicting. After the discovery of NAD+- and dpCoA-capped RNAs in 2009, in vitro experimentation failed to incorporate these caps into RNA, suggesting that noncanonical cap addition depended on post-transcriptional processes in vivo (Chen et al. 2009; Kowtoniuk et al. 2009). In contrast, earlier research had used E. coli RNA polymerase (RNAP) to successfully synthesize short transcripts initiated with NAD+ or FAD (Malygin and Shemyakin 1979). More recently, evidence has accumulated that supports the incorporation of noncanonical caps by RNA polymerase during transcription initiation. Firstly, it has been demonstrated that eukaryotic RNAPs can use different noncanonical caps to initiate transcription (Bird et al. 2016; Julius and Yuzenkova 2017). Additionally, in vivo, NAD+-capped RNA displays similar levels of enrichment on pre-mRNAs as on mRNAs, suggesting that NAD+ is added cotranscriptionally (Walters et al. 2017; Bird et al. 2018; Sharma et al. 2020). Overall, there appear to be several mechanisms to achieve noncanonical capping of RNA in vivo, and these mechanisms can be affected by multiple factors.
3.1 RNA Polymerase
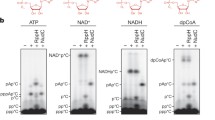
RNAPs are key enzymes in the delivery of genetic information from DNA to RNA through transcription. Usually, RNAP uses four NTPs (ATP, CTP, GTP, UTP) as substrates to initiate and extend RNA sequences. However, noncanonical substrates besides NTPs, such as coenzymes and long oligoribonucleotides, can also prime transcription by RNAP in vitro and in vivo (Fig. 3). For example, the bacteriophage T7 RNAP can use adenosine-containing NAD+, FAD, dpCoA, and NpnN to initiate transcription in vitro (Huang 2003; Hudeček et al. 2020). Structural research shows that such substrates can be accommodated in the space provided by the nucleotide-binding pocket of the T7 RNAP (Durniak et al. 2008). One exception is that T7 RNAP inefficiently incorporates NADP into transcripts, possibly owing to the additional phosphate group that causes steric hindrance in the pocket (Julius and Yuzenkova 2017).

The capping and decapping enzymes for canonical and noncanonical 5′ caps of RNA. The reported capping enzymes for each metabolite that can incorporate into RNA 5′ ends and the main decapping enzymes for each cap structure are shown. Decapping enzyme cleavage sites are displayed. RppH for NAD+ decapping is specifically from B. subtilis. NudC for NAD+ decapping is specifically from E. coli. ApaH for ApnA decapping is specifically from E. coli. Some decapping enzymes have homologs in various organisms. There are no reports of decapping enzymes for UDP-Glc and UDP-GlcNAc
The bacterial RNAP (both in E. coli and B. subtilis) and eukaryotic RNAP II can also incorporate NAD+, NADH, dpCoA, and Np4A into RNA during transcription initiation and can extend the sequence length from 2 nt up to 75 nt in vitro depending on the promoter (Bird et al. 2016; Frindert et al. 2018; Luciano and Belasco 2020). In particular, E. coli RNAP is 60 times more efficient when incorporating Np4A than NAD+, which may be due to the presence of only two bridging phosphates between the two nucleosides in NAD+. This is consistent with data showing that the incorporation efficiency of ADP only reaches up to 20% of that of ATP (Luciano and Belasco 2020). The cell wall synthesis precursors UDP-Glc and UDP-GlcNAc could also be incorporated into RNA by E. coli RNAP as pyrimidine-containing initial nucleotides, and even have higher extension efficiencies than UTP. The Km values of these noncanonical substrates (NAD+ ~ 0.36 mM; UDP-Glc and UDP-GlcNAc ~0.3 mM) during transcription initiation by E. coli RNAP are much lower than their cellular concentrations, revealing the efficiency of incorporation as an RNA cap in vivo (Julius and Yuzenkova 2017).
However, the nuclear RNAP may not be the only polymerase responsible for incorporation of noncanonical caps. Other than nuclear RNAs, up to 15% and 60% of NAD+-capped RNA in human and yeast cells, respectively, were attributed to mitochondrial transcripts (Bird et al. 2018), indicating that the mitochondrial RNAP is likely also responsible for the addition of noncanonical caps. In vitro transcription assays showed that yeast mitochondrial RNAP can use NAD+ and NADH as initial substrates, and that human mitochondrial RNAP can also use other noncanonical substrates, such as FAD and dpCoA, to initiate transcription (Bird et al. 2018; Julius et al. 2018). The efficiency of transcription when initiating with NAD+ is 40%–60% as efficient as initiating with ATP for yeast and human mitochondrial RNAP. Initiation with NAD+ by mitochondrial RNAP is about 10- to 40-fold more efficient than that by E. coli RNAP and S. cerevisiae RNAP II (Bird et al. 2018). This difference in efficiency may be due to differences in the sequences and structures of nuclear and mitochondrial RNAPs. Additionally, the mitochondrial and T7 RNAPs are single-subunit RNAPs, while the E. coli RNAP and S. cerevisiae RNAP II are multi-subunit, which may lead to quantitative differences in the efficiency of noncanonical capping (Ringel et al. 2011; Bird et al. 2018; Hillen et al. 2018).
Other polymerases that could be involved in the addition of noncanonical caps are plastid polymerases in organisms such as plants. For example, in plants, plastids contain two types of RNAPs, the nuclear-encoded single subunit RNAP (NEP) and the plastid-encoded multi-subunit RNAP (PEP) (Gray and Lang 1998). However, no study has reported their ability to initiate noncanonical caps on transcripts. The failure to detect NAD+-capped RNA in Arabidopsis chloroplasts may imply that chloroplast RNAPs are incapable of incorporating noncanonical caps (Wang et al. 2019b).
3.2 σ Factors and RNAP Structure
RNAP-dependent transcription initiation requires association with σ factors to recognize template sequences. The number of σ factors varies, altering selection of the gene targets of RNAP (Paget 2015; Barvík et al. 2017), and potentially playing a role in capping with noncanonical caps. For example, in E. coli, the RNAP holoenzyme with the σs or housekeeping σ70 factors produces most of the transcripts in the stationary or exponential phases, respectively. NAD+-capped transcript levels differ during these two phases, implying that certain RNAP factors may be involved in the specific capping of transcripts. However, no differences in capping efficiency were found in vitro between RNAP with these two σ factors when using the substrates ATP, NAD+, NADH, and FAD, suggesting that they do not have a preference for cellular substrates (Julius and Yuzenkova 2017).
One σ factor region has demonstrated some impact on the noncanonical capping of RNA. The region 3.2 of σ70 has been shown to protrude into the catalytic site of RNAP and affect nucleotide incorporation at the 5' end of transcripts (Kulbachinskiy and Mustaev 2006). Mutation of region 3.2 of σ70 did not influence the incorporation of some noncanonical caps; however, intriguingly, RNAP acquired the ability to incorporate a complex cell wall precursor, UDP-MurNAc-pentapeptide. This suggests that region 3.2 may serve as protection against the incorporation of nucleotides with long side chains (Julius and Yuzenkova 2017). Other than this example, no σ factors have effectively been demonstrated to alter the transcription initiation of RNA with noncanonical caps. Nonetheless, other alternative σ factors may have the potential to affect incorporation of noncanonical caps. For example, the E. coli gene GlmY, which produces NAD+-capped transcripts, contains the recognition sequences for σ54, implying that this σ factor may be involved in noncanonical capping (Göpel et al. 2011; Cahová et al. 2015).
Additionally, the Rif pocket of RNAP is an important structural determinant for noncanonical capping, as the nascent transcripts both make contact with and pass through the Rif pocket. Crystal structures of the E. coli RNAP complex show the nicotinamide moiety of the NAD+ nucleotide interacting with residues D516 and H1237 of this pocket (Bird et al. 2016). Mutation of D516 indeed strongly decreased the NAD+ utilization efficiency (Julius and Yuzenkova 2017). However, in B. subtilis, no altered efficiency of NAD+ capping was observed when the E. coli homologous site for the Rif pocket was mutated (Frindert et al. 2018). The cell wall precursors, UDP-Glc and UDP-GlcNAc, were also not affected by the amino acid substitutions in the Rifampicin binding pocket. This could be because they may not make specific contact with the amino acids of the Rif pocket (Julius and Yuzenkova 2017). Finally, while the addition of Rifampicin to the transcription reaction inhibited the extension of ATP-initiated transcripts due to its ability to block transcription elongation (Campbell et al. 2001), NAD+-capped short RNAs were not affected, suggesting that the 5′ NAD+ prevents Rifampicin binding to RNAP and thus stabilizes these short transcripts (Julius and Yuzenkova 2017). Collectively, the influence of the Rifampicin pocket as a determinant for capping might depend on different RNAPs in various organisms, as well as the noncanonical substrates themselves.
3.3 Promoter Sequence
Another determinant of incorporation of a noncanonical cap is the promoter sequence. Experimentation using in vitro transcription suggested that noncanonical cap initiation only occurs from template DNA containing A:T at the transcription start site (+1) (Bird et al. 2016). In the case of E. coli, the RNA polymerase selects a position not far downstream (ranges from 7 to 10 nt) of the promoter −10 element as TSS. Normally, TSS selection for NAD+-mediated initiation differs from that of NTPs due to this strong preference for an A:T base pair at the TSS position (Vvedenskaya et al. 2018). To put the selection preference for NAD+-mediated initiation into perspective, half of the TSS selected by bacterial and eukaryotic RNAPs are +1A, whereas all of the TSS selected by yeast and mitochondrial RNAPs are +1A (Tsuchihara et al. 2009; Thomason et al. 2015; Bird et al. 2018). This preference for TSS further demonstrated that noncanonical capping is accomplished via transcription initiation, rather than post-transcriptional mechanisms.
Besides the TSS, the promoter sequence close to the TSS strongly affects the efficiency of capping. Bird et al. (2016) demonstrated that NAD+ capping with the E. coli RNAI and gadY promoters exhibits higher efficiencies than with the PN25 and PT7A1 promoters. This is consistent with the relative extent of NAD+-capped transcripts attributed to each after detection in vivo (Cahová et al. 2015). Further analysis revealed that the identity of the base −1 upstream of the TSS plays a particularly important role in NAD+ capping efficiency. This may be due to the nicotinamide moiety of NAD+ interacting with the −1 position, thus leading to different efficiencies depending on the identity of the −1-position base, with G facilitating NAD+ capping and C repressing it (in the coding sequence) (Bird et al. 2016; Vvedenskaya et al. 2018). This trend was also observed in the B. subtilis veg promoter, where a T to C transition (in the coding strand) decreased the amount of NAD+ capping by around 40% both in vitro and in vivo. Only 9% of the promoters of all NAD+-capped RNAs contain a C at the −1 position (Frindert et al. 2018). Additionally, in Staphylococcus aureus, the efficiency of NAD+ capping in RNAIII transcripts depends on the −1 position of the P3 promoter, further supporting this view (Morales-Filloy et al. 2020). All of these alterations in efficiency could be explained by the nicotinamide moiety experiencing severe steric hindrance with the template strand A or G at the −1 position (Vvedenskaya et al. 2018). However, later studies argued that the preference of NAD+ at the −1 position is not specifically due to pairing of the nicotinamide moiety with the −1 base because the same trend was also observed for ATP (Julius and Yuzenkova 2017).
The efficiency of NAD+ capping also depends on the identity of the nucleotides −3 and −2 upstream and + 2, +3, and + 4 downstream of the TSS. In particular, the +2 base has a large, 6–8-fold effect on the efficiency of noncanonical capping, which makes it the second strongest determinant of capping with a noncanonical initiating nucleotide. Through the CapZyme-seq method, a consensus promoter sequence for the highest efficiency of NAD+ capping was determined as HRRASWW (H, ATC; R, GA; S, GC; W, AT), where A is the +1 base in E. coli. Replacing the bases with their anti-consensus sequence, GYYAWSS (Y, TC), leads to a 40-fold decrease in NAD+ capping efficiency (Vvedenskaya et al. 2018). Differing from E. coli, in yeast, the highly conserved promoter motif, YAAG, is associated with efficient NAD+ incorporation and is more likely to be recognized by the yeast RNAP II (Zhang et al. 2020).
Comparable to NAD+, capping by Np4A in E. coli also depends on the identity of the base pair at position −1. The levels of capping are higher when the −1 base on the coding strand is a purine rather than a pyrimidine, whereas the −2 and − 3 positions only modestly affect Np4A incorporation (Luciano and Belasco 2020). Taken together, it appears that the promoter sequence strongly affects the incorporation efficiency of noncanonical caps.
3.4 Cellular Metabolite Concentration
The intracellular concentration of NTPs and other noncanonical substrates utilized by RNAP for transcription plays a central role in regulating noncanonical cap initiation. Higher concentrations of NTPs lead to a greater chance of penetration into the active site of RNAP to initiate transcription (Haugen et al. 2008). RNAPs therefore seem to serve as both sensors to and actuators for the level of cellular metabolites, adjusting the transcriptional yield accordingly (Bird et al. 2018). For example, when high mitochondrial NAD(H) levels were changed to low levels, the levels of NAD(H)-capped mitochondrial RNAs changed from 15% to 0% (Bird et al. 2018).
Further support for the notion that cellular metabolite concentration influences the incorporation of noncanonical caps has been demonstrated in bacteria. In E. coli, the average cellular ATP concentration is about 1.54 mM, and the cellular NAD+ concentration is about 0.6 mM, while the NADH concentration is up to 10 times lower than that of NAD+. This predicts the probability of incorporation of each nucleotide, which would have an order of ATP > NAD+ > NADH (Lin and Guarente 2003; Zhou et al. 2011; Yaginuma et al. 2014). The concentrations of other noncanonical substrates, such as dpCoA and FAD, are only around 10 μM to 600 μM, lower than that of NAD+ (Takamura and Nomura 1988; Louie et al. 2003). These cellular concentrations are consistent with that of the respective, noncanonically capped RNA transcript levels detected in vivo (Chen et al. 2009; Kowtoniuk et al. 2009; Wang et al. 2019a). As for dinucleoside polyphosphates (Np4N) in E. coli, concentrations are even lower than those of FAD, but this concentration elevates during oxidative stress. Thus, E. coli mRNA and sRNA can only acquire Np4N caps under disulfide stress conditions that increase Np4N cellular concentrations (Luciano et al. 2019; Luciano and Belasco 2020). Only one substrate’s concentration, UDP-GlcNAc, is comparable with that of NAD+. UDP-GlcNAc is the most abundant noncanonical cap in vivo, consistent with the relative level of the cellular metabolite (>1 mM) in E. coli and human cells (Mao et al. 2006; Namboori and Graham 2008; Wang et al. 2019a). These studies indicate that the cellular concentration of noncanonical substrates is an important factor for transcript capping. Conversely, negative regulation by high NTP levels also leads to nascent transcription abortion (Turnbough and Switzer 2008). However, this negative regulation has not yet been reported during noncanonical capping.
3.5 Post-Transcription
Based on the limited research available, biosynthesis of noncanonically capped RNA by RNA polymerases during transcription initiation is the most common route. However, other mechanisms could also occur. For example, in mammalian cells, snoRNAs and small Cajal body RNAs (scaRNAs), some of which are produced from introns via splicing, also contain NAD+ caps, particularly after the removal of the decapping enzyme, DXO, in cells. This observation led to the proposal that an alternate, post-transcriptional NAD+ capping mechanism exists (Jiao et al. 2017).
There are capping mechanisms independent of those of RNAPs. In E. coli, some aminoacyl-tRNA synthetases, such as LysU, enable catalysis during the reaction of aminoacyl-adenylates with not only the 5′ triphosphate of mononucleotides but also with the triphosphorylated 5′ end of polynucleotides. This reaction produces Ap4A capped yeiP RNA (Luciano et al. 2019). Additionally, in vitro, ribozymes that are able to incorporate NAD+, FAD, and dpCoA into the 5′ terminal of RNA may also represent a potential method of capping in the in vivo synthesis of capped RNA (Huang 2003). Furthermore, m7G-capped RNAs can undergo m7G cap removal under specific conditions, and re-capping by NAD+ may also be possible (Zhang et al. 2019a). Collectively, these studies suggest that alternative post-transcriptional noncanonical capping mechanisms may exist and need to be looked into.
In summary, RNA polymerases, initiation σ factors, Rif pockets, promoter sequences, and cellular metabolite concentration all influence the profile of NAD+-capped RNA in organisms. However, the steady-state level of noncanonically capped RNAs may not only depend on such determinants. This level is also dynamically regulated by decapping mechanisms.
4 Decapping Enzymes of Noncanonical RNA Caps
Equally important to understanding the mechanisms involved in the modification of the 5′ end of RNA is understanding how noncanonical caps may be removed in a process referred to as decapping. While research in the decapping of noncanonical caps is only recently budding, decapping of the canonical eukaryotic m7G structure and conversion of the bacterial triphosphate 5′ end to a monophosphate have been studied extensively over the past several decades.
In eukaryotes, decapping of the m7G structure is tied to the regulation of gene expression and is recognized to play a role in mRNA turnover. The degradation of mRNA can be accomplished through various mechanisms, where decapping is a critical step for 5′-to-3′ decay in particular. This decapping occurs in a deadenylation-dependent or deadenylation-independent manner. During deadenylation-dependent decay, deadenylases and associated proteins encourage decapping after poly(A) tail shortening, whereas during deadenylation-independent decay, decapping is triggered through mechanisms such as mRNA uridylation or endonucleolytic cleavage. With successful decapping, an exoribonuclease, such as the mammalian XRN1 or the plant XRN4, degrades RNA containing a 5′ end monophosphate (Łabno et al. 2016).
However, bacterial mRNA degradation occurs through different pathways, as bacterial RNAs do not contain the m7G cap. Bacterial RNA largely contains a triphosphate at the 5′ end and a stabilizing hairpin structure at the 3′ end. Broadly, in bacteria, RNA degradation occurs through two pathways referred to as “direct access” and “5′-end-dependant” degradation (Hui et al. 2014; Kramer and McLennan 2019). Direct access degradation begins with cleavage by an endonuclease, such as RNase E in E. coli, and subsequently proceeds through 3′-to-5′ or 5′-to-3′ decay by exonucleases. On the other hand, 5′-end-dependant degradation initiates through the hydrolysis of the triphosphorylated 5′ end to a monophosphate by an enzyme, such as RppH, which makes the RNA susceptible to endonucleases and exonucleases, such as RNase E and RNase J, respectively.
As for noncanonical caps, the decapping process has been less extensively examined. Enzymes that are responsible for this decapping largely fall into two protein families: Nudix and DXO (Fig. 3). These two families are also involved in the hydrolysis of the canonical eukaryotic m7G cap and the bacterial triphosphate 5′ end. Similar to canonical decappers, decapping proteins for noncanonical caps generally encourage the conversion of the 5′ end to a monophosphate, which subjects the RNA to further degradation.
4.1 Nudix Enzymes Involved in Decapping of Noncanonical Caps
The Nudix superfamily consists mainly of pyrophosphohydrolases that were initially classified for demonstrating activity on various nucleoside diphosphates linked to moiety X, although this family also includes proteins of other functionalities (Srouji et al. 2017). Nudix proteins are ancient, widespread, and evolutionarily conserved between all three branches of life, as well as viruses, with 13, 7, 22, and 28 Nudix genes found in E. coli, Saccharomyces cerevisiae, humans, and Arabidopsis thaliana, respectively (McLennan 2006; Yoshimura and Shigeoka 2015; Carreras-Puigvert et al. 2017). Many proteins in this family can be defined by a conserved region termed the Nudix motif, GX5EX7REUXEEXGU, where U is a hydrophobic amino acid and X is any amino acid. This motif is critical for catalytic activity and the binding of divalent cations like Mg2+ and Mn2+, which function as cofactors for the pyrophosphohydrolase activity (Mildvan et al. 2005; McLennan 2006). In particular, Nudix hydrolases have diverse functions and substrates, and were originally described as “housecleaning enzymes” that act to rid cells of toxic materials and reduce the accumulation of metabolites and intermediates (McLennan 2006). However, Nudix hydrolases have also recently been demonstrated to be proficient decappers of noncanonical RNA caps (Fig. 3).
4.1.1 Canonical Decappers: Dcp2, Nudt16, and Nudt3
The function of Nudix hydrolases in decapping has been recognized since the identification of the decapping abilities of Dcp2 (Wang et al. 2002). Dcp2 is conserved in eukaryotes and functions in the hydrolysis of the canonical m7G cap. However, Dcp2 isn’t the only enzyme responsible for the decapping of the m7G structure. In vitro studies suggest that several other Nudix enzymes, such as human Nudt16 and Nudt3, could be involved in m7G decapping (Song et al. 2013). Of the various Nudix enzymes that demonstrate m7G decapping activity, only Nudt16 has recently shown potential in the hydrolysis of NAD+-, FAD-, and dpCoA-capped RNA in vitro and (for NAD+-capped RNA and FAD-capped RNA) in cells (Sharma et al. 2020).
4.1.2 NudC
NudC was the first member of the Nudix superfamily recognized to have activity on noncanonically capped RNA. In E.coli, NudC can hydrolyze NAD+-capped RNA in the presence of Mg2+ to release nicotinamide mononucleotide (NMN) and monophosphorylated RNA, which is susceptible to further degradation by RNase E (Cahová et al. 2015; Bird et al. 2016; Kiledjian 2018). Following NudC deletion, NAD+-capped RNA levels rise, supporting that NudC functions in cells as a regulator of NAD+-capped RNA. Additionally, NudC can hydrolyze NAD+ and NADH at lower efficiency compared to NAD+-capped RNA but displays no significant activity against 5′ triphosphorylated RNA, indicating it may primarily serve to remove NAD+ caps (Cahová et al. 2015; Höfer et al. 2016; Abele et al. 2020). In vitro, NudC also exhibits activity on RNA capped with NADH and dpCoA (Bird et al. 2016).
NudC prefers single-stranded substrates with three or more unpaired bases at the 5′ end and a purine as the first base of the RNA. In terms of RNA lengths, NudC can hydrolyze the NAD+ cap of both longer, complex RNA and short RNA (Höfer et al. 2016). Structurally, NudC functions as a symmetric homodimer, where both monomers bind to an individual NAD+. This dimerization is essential for substrate recognition and binding, as the catalytic pocket containing the Nudix motif is comprised of residues from each monomer (Höfer et al. 2016; Zhang et al. 2016).
Recently, several close NudC homologs have been characterized. In mammals, Nudt12 was demonstrated to hydrolyze cytosolic NAD+-capped RNA (Grudzien-Nogalska et al. 2019; Wu et al. 2019). Like NudC, loss of Nudt12 increases the levels of NAD+-capped RNA, indicating that Nudt12 regulates the stability of a subset of NAD+-capped RNA in cells. Specifically, Nudt12 may regulate transcripts involved in metabolism, as NAD+-capped transcripts that increased after nutrient stress were responsive to Nudt12 decapping and included nuclear-encoded mitochondrial protein mRNAs. Nudt12 may also have a role in the regulation of circadian clock transcripts. Structurally similar to NudC, Nudt12 functions as a homodimer, with most of the structural differences occurring in the N-terminal domain instead of the C-terminal domain, which contains the conserved Nudix motif (Grudzien-Nogalska et al. 2019; Wu et al. 2019). Nudt12 interacts with bleomycin hydrolase (BLMH), forming a dodecamer that likely contains a BLMH hexamer and three Nudt12 dimers. The interaction between Nudt12 and BLMH is necessary to localize Nudt12 to cytoplasmic granules that are distinct from P-bodies. This sequestration of Nudt12 to cytoplasmic granules may be beneficial to regulate Nudt12 activity on m7G and unmethylated caps, since Nudt12 can hydrolyze these structures to release m7GMP and GMP/GDP, respectively. Finally, Nudt12 shows activity on NAD+ and NADH, but prefers NADH (Grudzien-Nogalska et al. 2019).
Other close NudC homologs have been identified. Recently, in yeast, Npy1 was demonstrated to hydrolyze NAD+-capped RNA in the cytosol (Zhang et al. 2020). Additionally, in vitro, Nudt19 in Oryza sativa contained NAD+-decapping capabilities (Zhang et al. 2016).
4.1.3 RppH
A second bacterial protein involved in decapping is RppH (Deana et al. 2008). RppH is an RNA pyrophosphohydrolase with two differing prototypes, one from E. coli (EcRppH) and the other from B. subtilis (BsRppH). Orthologs of the EcRppH prototype are found within many classes of proteobacteria and flowering plants, while those of BsRppH are mainly restricted to the order Bacillales (Foley et al. 2015; Bischler et al. 2016). Both prototypes are involved in the hydrolysis of the 5′ triphosphate present in bacterial RNA, but key sequence and structural differences result in unique substrate specificity and function. EcRppH and BsRppH share only 23% identity, with much of the sequences outside of the Nudix motif differing significantly (Richards et al. 2011; Foley et al. 2015), leading to crucial differences between the two.
Recently, both BsRppH and EcRppH have been implicated in the removal of noncanonical caps. It has been demonstrated in vitro that BsRppH can decap NAD+-capped RNA, resulting in monophosphorylated RNA and NMN (Frindert et al. 2018). This removal of the NAD+ cap is enhanced by Mn2+ ions and the presence of guanosine at the second base position, but is inhibited by double-stranded structures present at the 5′ end. However, loss of BsRppH did not significantly affect NAD+-capped RNA levels, suggesting that NAD+ cap removal may not be the primary function of this enzyme in vivo (Frindert et al. 2018). Similarly, EcRppH has been shown to decap NAD+-capped RNA in vitro in some studies (Frindert et al. 2018; Grudzien-Nogalska et al. 2019), although this finding remains debatable due to contrasting studies that demonstrate that EcRppH has little efficiency on NAD+-capped RNA (Cahová et al. 2015; Bird et al. 2016; Abele et al. 2020). Widely, it is instead theorized that the NAD+ cap may serve to protect transcripts from EcRppH-dependent degradation (Cahová et al. 2015; Bird et al. 2016; Abele et al. 2020). Other than NAD+-capped transcripts, EcRppH has been demonstrated to hydrolyze NpnN caps to a 5′ monophosphate, although methylation of the NpnN cap structure can inhibit this activity (Luciano et al. 2019; Hudeček et al. 2020).
4.2 DXO Enzymes Involved in Noncanonical Decapping
A second family of proteins that is recognized for having activity on a variety of RNA caps is the DXO family of proteins (Fig. 3). This protein family shares an active site with six conserved motifs, which function in cleavage, RNA binding, and the coordination of divalent cations. Outside of this active site, there is little conservation between proteins in this family (Xiang et al. 2009; Chang et al. 2012; Wang et al. 2015). An important difference between the DXO family of proteins and the Nudix superfamily of proteins is that the two cleave noncanonical caps at different locations, with the DXO family removing the entire cap structure (Fig. 3). In this section, three prototypes in the DXO family will be discussed: Rai1, Dxo1, and DXO.
4.2.1 Rai1
The fungal Rai1 is present in the nucleus and was initially found to be a pyrophosphohydrolase with activity on 5′ triphosphorylated RNA, releasing diphosphate and RNA with a monophosphorylated 5′ end (Xiang et al. 2009). Association of Rai1 with the 5′-to-3′ exoribonuclease Rat1 affords degradation of the remaining 5′ monophosphorylated RNA product and stimulates both cleavage by Rai1 and 5′ to 3′ exonuclease activity by Rat1 (Xiang et al. 2009; Jiao et al. 2010). Subsequent to the revelation that Rai1 functions on 5′ triphosphorylated RNA, it was demonstrated that Rai1 could remove the canonical m7G cap, but was most efficient in removing unmethylated caps, releasing the entire cap structure, GpppN (Jiao et al. 2010). In addition, Rai1 homologs can have triphosphonucleotide hydrolase activity, releasing pppN (Wang et al. 2015). These functions indicated that the primary role of Rai1 was surveillance against aberrantly capped RNA. Recently, this role has expanded to include decapping of noncanonical caps. Rai1 cleaves NAD+-capped RNA to release NAD+ and also has activity on RNA capped with dpCoA and FAD in vitro (Jiao et al. 2017; Vvedenskaya et al. 2018; Doamekpor et al. 2020a). Finally, the complex formed by Rai1 and Rat1 can also degrade 5’ OH RNA (Doamekpor et al. 2020b).
4.2.2 Dxo1
Dxo1 works together with Rai1 in some yeast species to monitor aberrantly capped RNA, but is present in both the cytoplasm and the nucleus, indicating that there may be a hierarchical order to this surveillance (Zhang et al. 2020). Unlike Rai1, Dxo1 displays no pyrophosphohydrolase activity on 5′ triphosphorylated RNA; however, Dxo1 is highly efficient at removal of unmethylated GpppN cap structures (Chang et al. 2012). Additionally, this protein can remove the canonical m7G cap more efficiently than Rai1, although it prefers unmethylated caps (Chang et al. 2012). Unlike Rai1, which generally depends on Rat1 for exonuclease activity, Dxo1 contains 5′ to 3′ exonuclease activities of its own, though it is prone to stalling at secondary structures (Chang et al. 2012). Finally, Dxo1 has activity on NAD+-, dpCoA-, and FAD-capped RNA in vitro (Jiao et al. 2017; Doamekpor et al. 2020a).
4.2.3 Mammalian DXO
The predominately nuclear mammalian homolog, DXO, is a pyrophosphohydrolase, RNA-specific 5′ to 3′ exonuclease, and decapper of canonical and noncanonical RNA caps. DXO may have a preference for activity on pre-mRNA (Jiao et al. 2013) and can release diphosphate from 5′ triphosphorylated RNA, GpppG from RNA with unmethylated caps, and NAD+, dpCoA, and FAD from RNA with noncanonical caps (Jiao et al. 2013, 2017; Doamekpor et al. 2020a). Additionally, DXO is efficient against methylated caps (Jiao et al. 2013) and can remove a 5′-OH dinucleotide before degrading 5′ OH RNA, making it a hydroxyl dinucleotide hydrolase (Doamekpor et al. 2020b). In cells, NAD+-capped RNA and FAD-capped RNA levels rise when DXO activity is absent (Jiao et al. 2017; Doamekpor et al. 2020a). DXO likely functions on distinct subsets of these RNAs with a potential tie to RNA involved in environmental stress, such as heat shock (Grudzien-Nogalska et al. 2019). Due to the high activity of DXO on a variety of cap structures, this protein must be highly regulated. For example, cap binding proteins such as CBP20 and eIF4E can inhibit DXO activity, effectively protecting properly capped RNA (Jiao et al. 2013). The 2′-O-methylated cap structure also protects RNA from degradation by DXO (Picard-Jean et al. 2018).
4.2.4 Plant DXO1
In plants, the only DXO homolog present is the nuclear and cytoplasmic DXO1, which was also demonstrated to have deNADding, exoribonuclease, and hydroxyl dinucleotide hydrolase activity (Kwasnik et al. 2019; Doamekpor et al. 2020b; Pan et al. 2020). However, this protein does contain a plant-specific modification of the active site that hampers its 5′ to 3′ exonuclease activity and its activity on other 5′ RNA modifications. Despite this modification and independent of its role as a potent deNADding enzyme, Arabidopsis DXO1 has likely evolved to have a role in chloroplast-, development-, and immunity-related processes. For example, the N-terminal extension of the protein may promote chloroplast functions, potentially serving as a connection between nuclear and plastid signaling (Kwasnik et al. 2019; Pan et al. 2020).
4.3 Other Enzymes Involved in the Decapping of Noncanonical RNA Caps
Other enzymes outside of these two protein families may also be capable of decapping noncanonical RNA caps. For example, a bis (5′-nucleosyl)-tetraphosphatase (ApaH) was demonstrated to be able to efficiently remove NpnN caps (Hudeček et al. 2020). Additionally, CD38, a human glycohydrolase, can process NAD+-capped RNA in vitro (Abele et al. 2020). Diverse other enzymes could be involved in the decapping of RNA with noncanonical caps, and further research is required to delve into these possibilities.
5 Potential Molecular and Biological Functions of Noncanonical Capping
5.1 Does Noncanonical Capping Promote RNA Stability or Decay?
The 5′ terminal structure can affect the stability of an RNA. In E. coli, 5′ triphosphate RNA generally has a longer half-life than 5′ monophosphate RNA, while in eukaryotes, the m7G cap plays a central role in mRNA stability. However, whether noncanonical RNA caps also regulate mRNA stability remains somewhat controversial (Fig. 4). In E. coli, the 5′ end of triphosphorylated RNA can be hydrolyzed by the Nudix protein, RppH, to yield a 5′ monophosphorylated RNA, thereby triggering RNase-E-mediated decay (Deana et al. 2008). In vitro experiments showed that 5′ end modification with NAD+ strongly decelerates processing by RppH, thus heightening stability against RNase E (Cahová et al. 2015). NAD+ capping also resulted in a three to fourfold increase in RNA stability in vivo (Bird et al. 2016). Similarly, NAD+ capping in B. subtilis stabilized mRNA against exonucleolytic decay by RNase J1, which prefers degrading 5′ monophosphorylated RNA (Frindert et al. 2018).

Model of the potential molecular functions of the NAD+ cap in RNA in E. coli and eukaryotes. NAD+-capped RNA is altered dynamically in vivo through regulation by capping and decapping enzymes. In E. coli, pppRNA undergoes 5′ to 3′ decay enabled by RppH pyrophosphohydrolase and RNase E endonuclease activity, while the NAD+ cap promotes RNA stability against RppH and RNase E. In eukaryotes, the m7G cap protects mRNA against decay, while the NAD+ cap promotes 5′ to 3′ decay through recruitment of deNADding enzymes. CBC (cap binding complex) binds to the m7G cap to mediate splicing, polyadenylation, and nuclear export, though these steps remain unclear for the NAD+ cap. m7G-capped RNA recruits the eIF4F complex to initiate translation, while NAD+-capped RNA does exist on plant ribosomes but does not support translation in vitro or in transfected human cells. NAD+-capped RNA can be regulated by environmental stimuli and growth conditions, but the exact molecular and biological functions need to be further investigated
Contrary to observations in bacteria, the 5′ NAD+ cap promoted decay of RNAs in eukaryotes. In human cells, transfected NAD+-capped and polyadenylated luciferase mRNA was less stable and decayed via deNADding followed by 5′-3′ decay by DXO. The observed opposite response to NAD+ capping of prokaryotic and eukaryotic cells is perhaps due to the different features between these organisms, as well as differences in experimental methods. In E. coli, the main machinery for RNA degradation is a complex of the endoribonuclease, RNase E, and an exoribonuclease. Therefore, the inhibition of this complex by NAD+ capping could stabilize the RNA transcripts. However, in eukaryotes, most of the RNA transcripts are under the protection of the m7G cap at the 5′ end, while less than 10% of RNA transcripts are NAD+-capped. Thus, the NAD+ cap is more likely to be a 5′ end mark to recruit DXO and mediate decay of RNA that is unneeded or non-functional, save for special conditions (Jiao et al. 2017). Knockdown of mDXO or AtDXO in human or plant cells, respectively, causes the enrichment of NAD+-capped RNA (Jiao et al. 2017; Pan et al. 2020). In particular, most enriched NAD+ RNA in human cells are sno/scaRNAs, which are highly resistant to exonucleolytic degradation. This suggests that NAD+ capping for sno/scaRNAs probably triggers DXO-mediated decay (Filipowicz and Pogacić 2002; Jiao et al. 2017).
In addition, noncanonical capping may mediate RNA stability indirectly via a 5′-independent mechanism. For example, most of the NAD+-capped RNA revealed by NAD+ captureSeq in E. coli are short fragments, which might imply that RNA degradation also occurs without removing the 5′ cap (Cahová et al. 2015). A direct entry and attack mechanism by RNase E might not need a 5′ monophosphate end and perhaps could be induced by noncanonical capping (Bouvier and Carpousis 2011). In plants, sRNA biogenesis is an alternative way to degrade NAD+-capped RNA when there is a loss of the decapping enzyme DXO (Pan et al. 2020). Conversely, noncanonical capping might promote RNA stability by blocking the polyadenylation process that initiates degradation in E. coli, as its poly(A) polymerase prefers monophosphorylated substrates (Kushner 2004). However, how RNA stability is altered by noncanonical capping remains largely undefined and still requires further experimental support.
5.2 Is Noncanonical Capping of RNA Involved in Translation Regulation?
The initiation step of translation is critical to protein production. It requires the delivery of the ribosomal subunit to an mRNA, usually at the 5′ end. In eukaryotes, translation initiation is primarily achieved by the 5′ mRNA m7G cap through binding with the eIF4F complex, which recruits the ribosomal subunit pre-bound to a complex of initiation factors (Mitchell and Parker 2015). Caps other than m7G may not be recognized by this translation complex (Issur et al. 2013). Therefore, whether noncanonically capped RNA possesses the ability to be translated remains uncertain (Fig. 4).
In vitro translation experiments for yeast nuclear NAD+-capped transcripts suggest that NAD+-capped RNAs are unable to be translated, producing even less protein than triphosphorylated RNA and monophosphorylated RNA (Zhang et al. 2020). NAD+-capped and polyadenylated luciferase mRNA transfected into human cells displayed a translation signal no greater than that for uncapped RNA, similarly suggesting that NAD+-capped RNA is unable to initiate translation (Jiao et al. 2017). However, this study was performed using artificial, exogenous NAD+-capped RNA, which may not reflect the natural conditions in vivo. An alternate study in plants demonstrated that NAD+-capped mRNAs are enriched in the polysome fraction with translating ribosomes and therefore can probably be translated (Wang et al. 2019b). So far, there are no studies that report NAD+ capping mediating translation initiation or observations of the translation initiation complex binding with NAD+-capped RNA.
In eukaryotes, there exist other translation mechanisms that are independent of the 5′ end cap. Some mRNAs contain specific internal ribosome entry sites (IRES) to recruit ribosomal subunits, and m6A modification in the 5′ UTR can promote the translation of a transcript (Mitchell and Parker 2015). It is possible that NAD+-capped RNAs enriched in the polysome fraction might undergo translation through a cap-independent mechanism involving internal ribosome entry. Alternatively, additional modifications could promote the translation of NAD+-capped RNA. For example, the presence of m6Am modification on the second nucleotide next to the m7G cap increases translation initiation (Meyer et al. 2015). It is unclear if NAD+-capped 5′ ends contain these m6Am modified nucleotides. A recent report shows that the m6Am next to the m7G cap can be specifically demethylated by fat mass and obesity-associated protein (FTO), whose activity is enhanced by binding with NADP (Mauer et al. 2017; Wang et al. 2020). Thus, the possibility exists that NAD+ capping can recruit a protein factor to promote or inhibit translation initiation. In pathogens, NAD+ capping in RNAIII impairs the translation of its target gene, hla. This is perhaps due to the pseudo-base pairing between the nicotinamide of NAD+ and the target RNA (Morales-Filloy et al. 2020).
5.3 The Relationship Between Noncanonical Capping and Cellular Metabolism
NAD+, which is one of the most common organic cofactors, plays a critical role in cellular metabolism. Genes involved in the NAD+-NADP synthesis pathway, or encoding NAD+-NADP utilizing enzymes, were observed to produce NAD+-capped RNAs in different organisms (Morales-Filloy et al. 2020). For example, L-threonine 3 dehydrogenase (tdh) catalyzes an NAD+-dependent oxidation reaction in B. subtilis. NAD+-capped tdh mRNA may directly provide a regulatory feedback mechanism for the synthesis of this protein (Frindert et al. 2018). Another gene involved in NAD+ synthesis, nadA, is usually regulated by the nadA motif in the 5′ UTR that binds ligands, and might also be modulated by NAD+ RNA capping (Malkowski et al. 2019). These findings imply that NAD+ RNA capping may substitute for direct feedback regulation by the cofactor NAD+ to regulate NAD+ synthesis.
5.4 Regulation of Noncanonical Capping by Developmental and Environmental Stimuli
Cellular NAD+ plays a vital role in metabolism and acts as a factor linking cellular metabolism, transcript level, and environmental stimulus (Gakière et al. 2018). Perhaps due to the roles of NAD+ in the cell, NAD+-capped RNA is affected by developmental stage and environmental condition. For instance, NAD+-capped transcripts in the stationary phase of E. coli are twofold higher than in the exponential phase (Bird et al. 2016), and yeast cultures in synthetic media result in more NAD+-capped transcripts compared to those in rich media (Frindert et al. 2018). These results demonstrate that NAD+ capping could be modulated in response to environmental changes.
Additionally, it was found that NAD+-capped RNA significantly increased when human cells were exposed to either heat shock or glucose deprivation, while cellular NAD+ levels did not consistently demonstrate the same response. This suggests that NAD+ capping can be directly modulated under stress and isn’t only altered though sensing the cellular NAD+ level by RNAP. Moreover, the target NAD+-capped transcripts of DXO or Nudt12 were altered, further indicating that distinct regulation of NAD+-capped RNA is undertaken following different stresses (Grudzien-Nogalska et al. 2019). Likewise, Np4N in bacteria is thought to act as alarmones through receptor mediated signaling in environmental stress response. However, the generation of Np4A-capped RNAs under disulfide stress implies that the physiological responses previously attributed to Np4A signaling might be due to an Np4A RNA capping mechanism (Luciano et al. 2019).
6 Conclusion and Outlook
For a long time, the hallmark for mRNA capping in eukaryotes was the traditional m7G cap. After about 50 years of research, the molecular and biological function of m7G RNA capping in different organisms has been uncovered. In recent years, the new discovery of the NAD+ cap on mRNA opened a novel and exciting research field for RNA biology. With the present detection strategies, NAD+-capped RNAs appear widespread in various prokaryotes and eukaryotes. NAD+ capping occurs mainly on mRNA but also on noncoding RNAs. Additionally, NAD+-capped transcripts encode proteins involved in a range of biological processes, particularly cellular metabolism and stress responses.
The mechanism of incorporation of noncanonical caps like NAD+ continues to be elucidated. At present, NAD+ is only known to be introduced into the 5′ end of RNA by RNA polymerases during transcription initiation. However, many questions remain. Do RNA polymerases deposit NAD+ differently at unique genes? At one gene, are the transcription initiation sites different when NAD+ vs. ATP is used as the initiating nucleotide? Besides this capping mechanism, are there alternative mechanisms of NAD+ capping? After capping by RNA polymerases, how are NAD+-capped transcripts exported out of the nucleus? Are there any “readers” that recognize such transcripts? Finally, can these transcripts be translated by m7G-cap-independent mechanisms? A plethora of questions remain unanswered concerning the mechanisms surrounding RNA with noncanonical caps.
Decapping enzymes are involved in maintaining the steady-state levels of noncanonically capped transcripts in vivo. The Nudix and DXO families of proteins, which have long been known as hydrolases for various cellular metabolites (Ogawa et al. 2008), have been demonstrated to possess potent decapping activities that target different noncanonically capped transcripts. How decapping enzymes specifically regulate noncanonical capping and perform uncharacterized biological functions highlights a major bottleneck to obtaining a full understanding of RNA capping by NAD+ and other metabolites.
Besides capping by NAD+, other noncanonical substrates (FAD, dpCoA, UDP-Glc, UDP-GlcNAc, NpnN) have been identified in RNA in some organisms. Unfortunately, we still await robust sequencing technologies for such noncanonically capped RNA, which will pave the way to understanding their profiles in various transcriptomes. However, so far, no phenotypic changes were observed upon increasing or decreasing these noncanonically capped RNA in vivo. This brings up another critical inquiry: What are the functions of RNA noncanonical capping? How did noncanonical capping come to exist in evolution? Was it an accidental or specific event? We trust that further work on RNA with noncanonical caps will shed light on various questions in the epitranscriptomics field and will afford more practical applications.
References
Abele F, Höfer K, Bernhard P et al (2020) A novel NAD-RNA decapping pathway discovered by synthetic light-up NAD-RNAs. Biomol Ther 10:513
Barvík I, Rejman D, Panova N et al (2017) Non-canonical transcription initiation: the expanding universe of transcription initiating substrates. FEMS Microbiol Rev 2:131–138
Bird JG, Basu U, Kuster D et al (2018) Highly efficient 5′ capping of mitochondrial RNA with NAD+ and NADH by yeast and human mitochondrial RNA polymerase. elife 7:e42179
Bird JG, Zhang Y, Tian Y et al (2016) The mechanism of RNA 5′ capping with NAD+, NADH and desphospho-CoA. Nature 535:444–447
Bischler T, Hsieh P, Resch M et al (2016) Identification of the RNA pyrophosphohydrolase RppH of helicobacter pylori and global analysis of its RNA targets. J Biol Chem 292:1934–1950
Boccaletto P, Machnicka MA, Purta E et al (2018) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 46:D303–D307
Boo SH, Kim YK (2020) The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med 52:400–408
Bouvier M, Carpousis AJ (2011) A tale of two mRNA degradation pathways mediated by RNase E. Mol Microbiol 82:1305–1310
Breaker RR (2012) Riboswitches and the RNA world. Cold Spring Harb Perspect Biol 4:a003566
Cahová H, Winz ML, Höfer K (2015) NAD capture Seq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 519:374–377
Campbell EA, Korzheva N, Mustaev A et al (2001) Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912
Carreras-Puigvert J, Zitnik M, Jemth AS et al (2017) A comprehensive structural, biochemical and biological profiling of the human NUDIX hydrolase family. Nat Commun 8:1541
Chang J, Jiao X, Chiba K et al (2012) Dxo1 is a novel eukaryotic enzyme with both decapping and 5′-3′ exoribonuclease activity. Nat Struct Mol Biol 19:1011–1017
Chen YG, Kowtoniuk WE, Agarwal I et al (2009) LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat Chem Biol 5:879–881
Chen K, Zhao BS, He C (2016) Nucleic acid modifications in regulation of gene expression. Cell Chem Biol 23:74–85
Chen LQ, Zhao WS, Luo GZ (2020) Mapping and editing of nucleic acid modifications. Comput Struct Biotechnol J 18:661–667
Cohn WE (1960) Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: isolation, structure, and chemical characteristics. J Biol Chem 235:1488–1498
David R, Burgess A, Parker B et al (2017) Transcriptome-wide mapping of RNA 5-methylcytosine in Arabidopsis mRNAs and noncoding RNAs. Plant Cell 29:445–460
Deana A, Celesnik H, Belasco JG (2008) The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451:355–358
Doamekpor SK, Gozdek A, Kwasnik A et al (2020b) A novel 5′-hydroxyl dinucleotide hydrolase activity for the DXO/Rai1 family of enzymes. Nucleic Acids Res 48:349–358
Doamekpor SK, Grudzien-Nogalska E, Mlynarska-Cieslak A et al (2020a) DXO/Rai1 enzymes remove 5′-end FAD and dephospho-CoA caps on RNAs. Nucleic Acids Res 48:6136–6148
Dominissini D, Moshitch-Moshkovitz S, Schwartz S et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206
Durniak KJ, Bailey S, Steitz TA (2008) The structure of a transcribing T7 RNA polymerase in transition from initiation to elongation. Science 322:553–557
Filipowicz W, Pogacić V (2002) Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol 14:319–327
Foley PL, Hsieh PK, Luciano DJ et al (2015) Specificity and evolutionary conservation of the Escherichia coli RNA pyrophosphohydrolase RppH. J Biol Chem 290:9478–9486
Frindert J, Zhang Y, Nübel G et al (2018) Identification, biosynthesis, and decapping of NAD-capped RNAs in B. subtilis. Cell Rep 24:1890–1901.e8
Fu Y, He C (2012) Nucleic acid modifications with epigenetic significance. Curr Opin Chem Biol 16:516–524
Gakière B, de Bont L, Fernie AR et al (2018) NAD+ biosynthesis and signaling in plants. Crit Rev Plant Sci 37:259–307
Galloway A, Cowling VH (2019) mRNA cap regulation in mammalian cell function and fate. Biochim Biophys Acta Gene Regul Mech 1862:270–279
Göpel Y, Lüttmann D, Heroven AK et al (2011) Common and divergent features in transcriptional control of the homologous small RNAs GlmY and GlmZ in Enterobacteriaceae. Nucleic Acids Res 39:1294–1309
Gray MW, Lang BF (1998) Transcription in chloroplasts and mitochondria: a tale of two polymerases. Trends Microbiol 6:1–3
Grosjean H, Droogmans L, Roovers M et al (2007) Detection of enzymatic activity of transfer RNA modification enzymes using radiolabeled tRNA substrates. Methods Enzymol 425:55–101
Grudzien-Nogalska E, Bird JG, Nickels BE et al (2018) "NAD-capQ" detection and quantitation of NAD caps. RNA 24:1418–1425
Grudzien-Nogalska E, Wu Y, Jiao X et al (2019) Structural and mechanistic basis of mammalian Nudt12 RNA deNADding. Nat Chem Biol 15:575–582
Haugen SP, Ross W, Gourse RL (2008) Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol 6:507–519
Helm M, Motorin Y (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18:275–291
Hillen HS, Temiakov D, Cramer P (2018) Structural basis of mitochondrial transcription. Nat Struct Mol Biol 25:754–765
Höfer K, Li S, Abele F et al (2016) Structure and function of the bacterial decapping enzyme NudC. Nat Chem Biol 12:730–734
Huang F (2003) Efficient incorporation of CoA, NAD and FAD into RNA by in vitro transcription. Nucleic Acids Res 31:e8
Hudeček O, Benoni R, Reyes-Gutierrez PE et al (2020) Dinucleoside polyphosphates act as 5'-RNA caps in bacteria. Nat Commun 11:1052
Hui MP, Foley PL, Belasco JG (2014) Messenger RNA degradation in bacterial cells. Annu Rev Genet 48:537–559
Issur M, Bougie I, Despins S et al (2013) Enzymatic synthesis of RNAs capped with nucleotide analogues reveals the molecular basis for substrate selectivity of RNA capping enzyme: impacts on RNA metabolism. PLoS One 8:e75310
Jiao X, Chang JH, Kilic T et al (2013) A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell 50:104–115
Jiao X, Doamekpor SK, Bird JG et al (2017) 5′ end nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding. Cell 168:1015–1027.e10
Jiao X, Xiang S, Oh C et al (2010) Identification of a quality-control mechanism for mRNA 5′-end capping. Nature 467:608–611
Joyce GF (2001) RNA cleavage by the 10-23 DNA enzyme. Methods Enzymol 341:503–517
Julius C, Riaz-Bradley A, Yuzenkova Y (2018) RNA capping by mitochondrial and multi-subunit RNA polymerases. Transcription 9:292–297
Julius C, Yuzenkova Y (2017) Bacterial RNA polymerase caps RNA with various cofactors and cell wall precursors. Nucleic Acids Res 45:8282–8290
Julius C, Yuzenkova Y (2019) Noncanonical RNA-capping: discovery, mechanism, and physiological role debate. Wiley Interdiscip Rev RNA 10:e1512
Kellner S, Burhenne J, Helm M (2010) Detection of RNA modifications. RNA Biol 7:237–247
Kiledjian M (2018) Eukaryotic RNA 5′-end NAD. Trends Cell Biol 28:454–464
Kowtoniuk WE, Shen Y, Heemstra JM et al (2009) A chemical screen for biological small molecule-RNA conjugates reveals CoA-linked RNA. Proc Natl Acad Sci U S A 106:7768–7773
Kramer S, McLennan AG (2019) The complex enzymology of mRNA decapping: enzymes of four classes cleave pyrophosphate bonds. Wiley Interdiscip Rev RNA 10:e1511
Kulbachinskiy A, Mustaev A (2006) Region 3.2 of the sigma subunit contributes to the binding of the 3′-initiating nucleotide in the RNA polymerase active center and facilitates promoter clearance during initiation. J Biol Chem 281:18273–18276
Kushner SR (2004) mRNA decay in prokaryotes and eukaryotes: different approaches to a similar problem. IUBMB Life 56:585–594
Kwasnik A, Wang VY, Krzyszton M et al (2019) Arabidopsis DXO1 links RNA turnover and chloroplast function independently of its enzymatic activity. Nucleic Acids Res 47:4751–4764
Łabno A, Tomecki R, Dziembowski A (2016) Cytoplasmic RNA decay pathways - enzymes and mechanisms. Biochim Biophys Acta 1863:3125–3147
Li X, Xiong X, Wang K et al (2016) Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol 12:311–316
Limbach PA, Paulines MJ (2017) Going global: the new era of mapping modifications in RNA. Wiley Interdiscip Rev RNA 8
Lin SJ, Guarente L (2003) Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol 15:241–246
Liu K, Lat PK, Yu HZ et al (2020) CLICK-17, a DNA enzyme that harnesses ultra-low concentrations of either cu+ or Cu2+ to catalyze the azide-alkyne 'click' reaction in water. Nucleic Acids Res 48:7356–7370
Louie TM, Xie XS, Xun LY (2003) Coordinated production and utilization of FADH(2) by NAD(P)H-flavin oxidoreductase and 4 hydroxyphenylacetate 3-monooxygenase. Biochemistry 42:7509–7517
Luciano DJ, Belasco JG (2020) Np4A alarmones function in bacteria as precursors to RNA caps. Proc Natl Acad Sci U S A 117:3560–3567
Luciano DJ, Levenson-Palmer R, Belasco JG (2019) Stresses that raise Np4A levels induce protective nucleoside tetraphosphate capping of bacterial RNA. Mol Cell 75:957–966.e8
Malkowski SN, Spencer TCJ, Breaker RR (2019) Evidence that the nadA motif is a bacterial riboswitch for the ubiquitous enzyme cofactor NAD+. RNA 25:1616–1627
Malygin AG, Shemyakin MF (1979) Adenosine, NAD and FAD can initiate template-dependent RNA synthesis catalyzed by Escherichia coli RNA polymerase. FEBS Lett 102:51–54
Mao Z, Shin HD, Chen RR (2006) Engineering the E. coli UDP-glucose synthesis pathway for oligosaccharide synthesis. Biotechnol Prog 22:369–374
Mauer J, Luo X, Blanjoie A et al (2017) Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541:371–375
McLennan AG (2006) The Nudix hydrolase superfamily. Cell Mol Life Sci 63:123–143
Meyer KD, Patil DP, Zhou J et al (2015) 5' UTR m(6)a promotes cap-independent translation. Cell 163:999–1010
Meyer KD, Saletore Y, Zumbo P et al (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149:1635–1646
Mildvan AS, Xia Z, Azurmendi HF et al (2005) Structures and mechanisms of Nudix hydrolases. Arch Biochem Biophys 433:129–143
Mishima E, Jinno D, Akiyama Y et al (2015) Immuno-northern blotting: detection of RNA modifications by using antibodies against modified nucleosides. PLoS One 10:e0143756
Mitchell SF, Parker R (2015) Modifications on translation initiation. Cell 163:796–798
Morales-Filloy HG, Zhang Y, Nübel G et al (2020) The 5' NAD cap of RNAIII modulates toxin production in Staphylococcus aureus isolates. J Bacteriol 202:e00591–e00519
Nachtergaele S, He C (2018) Chemical modifications in the life of an mRNA transcript. Annu Rev Genet 52:349–372
Namboori SC, Graham DE (2008) Enzymatic analysis of uridine diphosphate N-acetyl-D-glucosamine. Anal Biochem 381:94–100
Nübel G, Sorgenfrei FA, Jäschke A (2017) Boronate affinity electrophoresis for the purification and analysis of cofactor-modified RNAs. Methods 117:14–20
Ogawa T, Yoshimura K, Miyake H et al (2008) Molecular characterization of organelle-type Nudix hydrolases in Arabidopsis. Plant Physiol 3:1412–1424
Paget MS (2015) Bacterial sigma factors and anti-sigma factors: structure, function and distribution. Biomol Ther 5:1245–1265
Pan S, Kai-en L, Huang W et al (2020) Arabidopsis DXO1 possesses deNADding and exonuclease activities and its mutation affects defense-related and photosynthetic gene expression. J Integr Plant Biol 62:967
Picard-Jean F, Brand C, Tremblay-Létourneau M et al (2018) 2'-O-methylation of the mRNA cap protects RNAs from decapping and degradation by DXO. PLoS One 13:e0193804
Ramanathan A, Robb GB, Chan SH (2016) mRNA capping: biological functions and applications. Nucleic Acids Res 44:7511–7526
Richards J, Liu Q, Pellegrini O et al (2011) An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol Cell 43:940–949
Ringel R, Sologub M, Morozov YI et al (2011) Structure of human mitochondrial RNA polymerase. Nature 478:269–273
Rostovtsev VV, Green LG, Fokin VV et al (2002) A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem Int Ed Engl 41:2596–2599
Roundtree IA, Evans ME, Pan T et al (2017) Dynamic RNA modifications in gene expression regulation. Cell 169:1187–1200
Ryvkin P, Leung YY, Silverman IM et al (2013) HAMR: high-throughput annotation of modified ribonucleotides. RNA 19:1684–1692
Schwartz S, Motorin Y (2017) Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol 14:1124–1137
Sharma S, Grudzien-Nogalska E, Hamilton K et al (2020) Mammalian Nudix proteins cleave nucleotide metabolite caps on RNAs. Nucleic Acids Res 48:6788–6798
Shi H, Wei J, He C (2019) Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74:640–650
Shuman S (2015) RNA capping: progress and prospects. RNA 21:735–737
Sikorski PJ, Warminski M, Kubacka D et al (2020) The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res 48:1607–1626
Song MG, Bail S, Kiledjian M (2013) Multiple Nudix family proteins possess mRNA decapping activity. RNA 19:390–399
Srouji JR, Xu A, Park A et al (2017) The evolution of function within the Nudix homology clan. Proteins 85:775–811
Takamura T, Nomura G (1988) Changes in the intracellular concentration of acetyl-CoA and Malonyl-CoA in relation to the carbon and energy-metabolism of Escherichia-Coli-K12. J Gen Microbiol 134:2249–2253
Thomason MK, Bischler T, Eisenbart SK et al (2015) Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197:18–28
Thüring K, Schmid K, Keller P et al (2016) Analysis of RNA modifications by liquid chromatography-tandem mass spectrometry. Methods 107:48–56
Tsuchihara K, Suzuki Y, Wakaguri H et al (2009) Massive transcriptional start site analysis of human genes in hypoxia cells. Nucleic Acids Res 37:2249–2263
Turnbough CL, Switzer RL (2008) Regulation of pyrimidine biosynthetic gene expression in bacteria: repression without repressors. Microbiol Mol Biol Rev 72:266–300
Vvedenskaya IO, Bird JG, Zhang Y et al (2018) CapZyme-seq comprehensively defines promoter-sequence determinants for RNA 5′ capping with NAD. Mol Cell 70:553–564.e9
Walters RW, Matheny T, Mizoue LS et al (2017) Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 114:480–485
Wang J, Alvin Chew BL, Lai Y et al (2019a) Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res 47:e130
Wang Z, Jiao X, Carr-Schmid A et al (2002) The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A 99:12663–12668
Wang L, Song C, Wang N et al (2020) NADP modulates RNA m6A methylation and adipogenesis via enhancing FTO activity. Nat Chem Biol
Wang VY, Jiao X, Kiledjian M et al (2015) Structural and biochemical studies of the distinct activity profiles of Rai1 enzymes. Nucleic Acids Res 43:6596–6606
Wang Y, Li S, Zhao Y et al (2019b) NAD +-capped RNAs are widespread in the Arabidopsis transcriptome and can probably be translated. Proc Natl Acad Sci U S A 116:12094–12102
Werner M, Purta E, Kaminska KH et al (2011) 2'-O-ribose methylation of cap2 in human: function and evolution in a horizontally mobile family. Nucleic Acids Res 39:4756–4768
Wetzel C, Limbach PA (2016) Mass spectrometry of modified RNAs: recent developments. Analyst 141:16–23
Wu H, Li L, Chen KM et al (2019) Decapping enzyme NUDT12 partners with BLMH for cytoplasmic surveillance of NAD-capped RNAs. Cell Rep 29:4422–4434.e13
Xiang S, Cooper-Morgan A, Jiao X et al (2009) Structure and function of the 5′>3′ exoribonuclease Rat1 and its activating partner Rai1. Nature 458:784–788
Yaginuma H, Kawai S, Tabata KV et al (2014) Hiromi Imamura diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci Rep 6:6522
Yang H, Yang T, Baur JA et al (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130:1095–1107
Yoshimura K, Shigeoka S (2015) Versatile physiological functions of the Nudix hydrolase family in Arabidopsis. Biosci Biotechnol Biochem 79:354–366
Zhang D, Liu Y, Wang Q et al (2016) Structural basis of prokaryotic NAD-RNA decapping by NudC. Cell Res 26:1062–1066
Zhang H, Zhong H, Zhang S et al (2019a) NAD tagSeq reveals that NAD+-capped RNAs are mostly produced from a large number of protein-coding genes in Arabidopsis. Proc Natl Acad Sci U S A 116:12072–12077
Zhang LS, Liu C, Ma H et al (2019b) Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol Cell 74:1304–1316.e8
Zhang YQ, Kuster D, Schmidt T et al (2020) Extensive 5′-surveillance guards against non-canonical NAD-caps of nuclear mRNAs in yeast. Nat Commun 11:5508
Zhou Y, Wang L, Yang F et al (2011) Determining the extremes of the cellular NAD(H) level by using an Escherichia coli NAD(+)-auxotrophic mutant. Appl Environ Microbiol 77:6133–6140
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hu, H., Flynn, N., Chen, X. (2021). Discovery, Processing, and Potential Role of Noncanonical Caps in RNA. In: Jurga, S., Barciszewski, J. (eds) Epitranscriptomics. RNA Technologies, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-030-71612-7_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-71612-7_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-71611-0
Online ISBN: 978-3-030-71612-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)