
Abstract
The dual function of the immune system is to defend the host against pathogens and not to be reactive against self-antigens. The failure of these two missions is represented by primary immunodeficiencies (PID) and autoimmunity (AI), respectively. As such, PID and AI could be simplistically thought as mutually exclusive.
However, over 25% of patients with PID present one or more autoimmune disease with organ- or non-organ-specific manifestations, with an increased risk up to 120 times for autoimmune cytopenias. Defective lymphocyte maturation and activation, impaired central and peripheral tolerance, immunodysregulation and defective apoptosis are responsible of this paradoxical association.
In this chapter we briefly review autoimmune manifestations associated with PID categorized as (i) SCID/CID, (ii) LOCID, (iii) APECED, (iv) IPEX, (v) hypogammaglobulinemias, (vi) immunodysregulation disorders, and (vii) ALPS, describing each prototypical molecular defect and delving into the mechanisms that contribute to the development of AI.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Immunodeficiency
- Immunodysregulation
- Tolerance
- Autoimmunity
- Autoimmune diseases
- Cytopenias
- Hemolytic anemia
15.1 Introduction
There is no dark side in the moon really—(Eclipse, Pink Floyd 1973)
Although the Jeffrey Modell Foundation has listed ten warning signs to identify patients likely to have primary IDs [1], it would be wise to add autoimmune manifestations as the eleventh. Selective IgA deficiency (sIgAD), the most common ID in Western countries, was already known to associate with several autoimmune disorders, such as cytopenias and other organ-specific AI [2]. Further, almost 50 years ago the rarer hyper-IgM deficiency was found as able to develop autoantibodies to blood elements [3]. ID and AI have been only a curious and unexplained combination for years and considered as opposite functional poles of the immune system since recently. Now, a series of cellular and humoral IDs were found to associate with AI, and it is clear that autoimmune diseases may not merely complicate the course but can be the initial feature of an ID. Along with this, a mounting number of reviews addressing the complex dysfunction of the immune system in IDs with AI has been recently published [4,5,6].
This dark side of the moon, i.e., the apparent paradox of the immune system activated against self when failing against external offenders, is going to be revealed and as a matter of fact, it’s not all dark. Hypotheses and facts concur to explain this association.
First, monogenic IDs identified mutations for single immune regulators explaining the break of immunological tolerance. Gene mutations in SCID/CID (Omenn or DiGeorge syndrome in primis), APECED and IPEX/IPEX-like diseases undermine, respectively, central and peripheral tolerance. LRBA/CTLA4 mutations, some STAT GOF/LOF, X-linked Bruton syndrome and other hypogammaglobulinemias affect the fine mechanisms of immune regulation or B cell activation. Gene defects in ALPS and ALPS-related diseases alter apoptosis. Ataxia-telangiectasia and Wiskott-Aldrich syndrome characterized by interference in T cell activation are complicated by AI. All these pictures represent human in vivo experiments, frequently with more overt clinical manifestations than knockout animal models themselves.
Secondly, pathogens per se may contribute to the selection of the response. Pressure delivered by repeated infections may up- and dysregulate the production of proinflammatory cytokines redirecting T helper differentiation towards pathogenic (instead of protective) functional phenotypes. At the same time, the wide spectrum of colonizing pathogens may expose a mass of antigens, few of them cross-reacting with self-inciting residual responses against wrong targets (molecular mimicry) and others possibly resulting in polyclonal T cell activation (superantigens).
Finally, cell damage may produce DAMPs because of unresolved infections and unrelated (heterologous) CD8+, and CD4+ T cells might be activated following an antigen-specific, although inefficient, response (bystander activation).
Several similarities link AI to IDs and polygenic rheumatic diseases (SLE and RA) indirectly confirm shared pathomechanisms.
First, GWAS in Caucasian and Asian cohorts identified shared polymorphisms in MHC-unrelated genes in RA and some IDs as a proof of their regulatory role in tolerance [7]. Gene variants of complement proteins or GOF variants of the IFN-α signaling pathways or LOF mutations of other non-MHC genes are demonstrated in SLE analogously to the so-called interferonopathies. These findings could explain familial predisposition to AI resembling familial clustering of IDs [6].
Second, the frequent co-occurrence of autoimmune diseases within the same individual (polyautoimmunity) suggests a complex immune dysregulation as observed in some IDs where organ- and non-organ-specific AI may co-exist. Thus, the concept of the kaleidoscope could be proposed in IDs not dissimilarly from AI [8].
The prevalence of AI in IDs has been rarely addressed, and an intrinsic difficulty resides into the large variety of distribution and prevalence of IDs within different geographic areas and size of the national or international registries. The European Society for Immunodeficiencies (ESID) has recently renewed its registry, a large database collecting patients from 31 different countries, including some from Middle East, with more than 28.000 patients inserted. From the French national registry (CEREDIH), Fischer et al. [9] based the most relevant study retrospectively considering clinical pictures of dysregulation in 2183 screened ID patients aged 6 months to 92 years. One out four (26.2%) exhibited one or more AI and inflammatory manifestation, the general risk ranging 3–14 times compared to general population. Autoimmune cytopenias were the most prevalent (120-fold higher risk) with AHA stratospherically above all (830-fold). Even if gastrointestinal tract and skin were the most affected organs after the blood elements, rheumatologic disorders (i.e., RA) and large vessel vasculitides were overrepresented (at least tenfold). CVID and T cell defects were at highest risk for AI, and the overall survival time was significantly shorter when AI complicated IDs at any age (p 0.004). Prospective studies are still lacking.
If AI should be added to the ten warning signs of ID, is there any special sign which should alert in clinical practice? There is no specific and shared recommendation on this; nonetheless some suggestions might be proposed.
First, classes of immunoglobulins should be always determined as the prevalence of AI in sIgAD is globally about 36% whereas the prevalence of this immune defect is highly variable worldwide (overall prevalence 1:382 in Europe) [10].
Secondly, as cytopenias are the most frequent complication of IDs, all the patients, not only children, should be ideally screened for IDs in case of AHA, ITP or both. In the French cohort, it was estimated that given 360 pediatric cases of AHA, 15% of them (50 patients) will be ID [9]. As the risk to develop AI against blood elements persists through the ages, adult patients should not be deprived of the opportunity to be screened. Sex does not represent an adjunctive value as males are equally affected as females. Inflammatory arthritis of any kind and joint pain represent an alarm when occurring in the childhood as the risk is increased 40-fold in IDs and undoubtedly when associated with systemic autoinflammation [9]. See Table 15.1.
Finally, as AI manifestations may occur throughout the patient’s lifetime and ID may be clinically highly variable (from severe to nuanced) also in monogenic diseases, age does not represent a criterion to exclude adult patients from the screen any time.
In this chapter we provide an overview of mono- and polygenic IDs of the cellular compartment of the immune system presenting AI disorders. Other immune deficiencies such as interferonopathies, complement deficiencies and opsonization defects, or autoinflammatory disorders equally associated with manifestations of AI are beyond the purpose of this review.
15.2 Autoimmunity in Severe Combined Immunodeficiency
Severe combined immunodeficiency (SCID) is characterized by several gene mutations affecting lymphocyte maturation (IL2RG, JAK3, IL7R, etc.), VDJ recombination (RAG1, RAG2, Artemis, etc.), or purine metabolism (ADA, PNP). Clinically, they manifest in early childhood with failure to thrive and susceptibility to opportunistic infections. T lymphocytes are usually absent with variable B and/or NK cells depending on the underlying molecular defect.
15.2.1 RAG Deficiencies and AI
Recombinase genes (RAG1 and RAG2) are critical for VDJ recombination during the development of both T and B lymphocyte. Thus, biallelic RAG1 or RAG2 mutations are associated with a classical T-B-NK+ SCID phenotype. Other mutations may maintain residual enzymatic activity with a heterogeneous spectrum of manifestations, ranging from Omenn syndrome, leaky/atypical SCID, delayed-onset combined ID with granulomas and/or autoimmunity (CID-G/AI), to milder phenotypes mimicking CVID or sIgAD [11].
Omenn syndrome is paradigmatic as a SCID variant associated with hypomorphic variants in which T cells are normally present but oligoclonally expanded, skewed towards a Th2 phenotype, activated and autoreactive [12]. Clinically, eosinophilia, elevated IgE and widespread organ lymphocytic infiltration causing lymphadenopathies, hepatosplenomegaly, erythroderma, and enteropathy are present.
Whereas AI is rare in infants with typical SCID, autoimmune cytopenias and vasculitis are often observed in Omenn syndrome and leaky SCID. Interestingly, CID-G/AI is associated with multi-organ granulomatous lesions, mainly involving skin, lung and liver, in addition to organ-specific AI, including nephritis, alopecia, vitiligo, psoriasis, myasthenia gravis and Guillain-Barré syndrome [11].
The reasons why SCID patients may or may not develop AI are different and reside on the degree of oligoclonality and amount of residual recombinase activity. The lower the levels, the more restricted TCR and BCR repertoires and the early-onset severe phenotypes. Vice versa, the higher the levels, the broader repertoire and better immune response, but more autoreactive clones and AI. The reduction of the TCR repertoire is associated with a skewed V, D, and J gene segment usage and a bias towards the use of proximal TRAV/TRAJ combination [13]. Loop length and diversity of CDR3 (complementarity determining region 3) are reduced, while hydrophobic amino acids are enhanced [14]. In particular, the presence of hydrophobic residues at position 6 and 7 of CDR3β is associated with the development of autoreactive T lymphocytes [15].
Immunodysregulation may be also favored by environmental factors, i.e., chronic infections and altered microbiome interactions. The vaccine-strain rubella virus is contained inside the granulomas of patients with various T cell defects, including RAG deficiency [16]. In a mouse model of Omenn syndrome, microbiota would be a critical driver of AI, as responsible of the gut barrier impairment and mucosal expansion of self-reactive T cell clones, whereby manipulation of the microbiome with antibiotics reverted the inflammation [17].
Third, disruption of central and peripheral tolerance plays a major role in the immunodysregulation in RAG mutations. A correct thymic development requires the crosstalk between CD40+ RANK+ medullary thymic epithelial cells (mTEC) and CD40L+ RANKL+ CD4+ thymocytes. This interaction induces the expression of the autoimmune regulator (AIRE) by mTECs, which in turn increases the presentation of tissue-restricted antigens. Patients and animal models with defective recombinase activity exhibit a reduced-size thymus with a disrupted intra-thymic architecture, a lower AIRE and FoxP3 expression, and enrichment of self-reactive T cells. In atypical SCID and CID-G/AI, a disproportionate loss of Treg cells and increased self-reactive conventional T cells are indeed seen [18].
In addition, B cell tolerance is impaired, and high numbers of autoantibodies are found in RAG-deficient patients, including anti-cytokine (anti-IFN-α and anti-IFN-ω) antibodies [19]. Physiologically, RAG re-expression is critical for the B cell receptor editing in the bone marrow, and rearrangements in kappa and lambda light-chain loci reduce the frequency of self-reactive B lymphocytes. Finally, B cell-activating factor BAFF plays a cornerstone role in the survival of peripheral B cells as in physiological conditions anergic and self-reactive B cells have lower levels of BAFF receptor (BAFFR) than naïve B cells and are thus unable to survive [20]. In RAG-deficient patients, B cell lymphopenia induces high BAFF levels sustaining the survival of self-reactive B cells [21].
15.2.2 Thymic Defects and AI
Thymic development defects, similarly to RAG mutations, induce tolerance breakdown and raise of ID and AI. DiGeorge syndrome (DGS) is due to chromosome 22q11.2 deletions, with the consequence of parathyroid, thymus, cardiac and facial malformations. This ID varies from a severe SCID-like phenotype to almost normal. The prevalence of AI ranges between 10% and 33% [22, 23] with autoimmune cytopenias as the most commonly reported and possibly preceding infectious manifestations. Autoimmune polyendocrinopathies have been also observed, such as atrophic gastritis, T1D and thyroid diseases [24]. Thymus of DGS has a perturbed distribution of thymocytes, altered thymic output, and lower frequency of mature CD4+ and CD8+ T cells, in addition to reduced proportions and function of Tregs both into the thymus and peripheral blood [25]. Interestingly enough, the immunophenotypic abnormalities of the peripheral blood predict DGS patients at risk of hematologic AI and correlate with survival [26].
15.3 AI in Late-Onset Combined Immunodeficiencies (LOCID)
Late-onset combined immunodeficiencies (LOCID) include a group of patients with severe infections for severe defects of cell-mediated immunity (e.g., opportunistic infections) and/or CD4+ lymphocyte counts <200 × 106 cells/L [27]. According to several investigations, LOCID patients tend to have a higher frequency of consanguinity and higher prevalence of lymphoproliferative disorders with hepatosplenomegaly and lymphadenopathy. Moreover, they usually present with granulomatous disease, gluten-resistant enteropathy and some features of AI. These patients might be misdiagnosed as CVID, because of frequent hypogammaglobulinemia [27]. Genetic studies, thanks to next-generation sequencing (NGS), are progressively defining new (and old) monogenic causes of LOCID [28]. Clear examples of LOCID are represented by three groups of diseases: (1) defective TCR signaling, (2) defective actin cytoskeleton, and (3) defective T cell activation/immunosenescence.
15.3.1 Defective TCR Signaling
TCR/CD3 complex engagement triggers a cascade of events resulting in T lymphocyte selection, activation, migration and effector functions, paralleled by Treg cell development and activation. The interaction between TCR and MHC-peptide complex activates Src family kinase LCK which phosphorylates CD3 ITAMs (immunoreceptor tyrosine-based activation motifs). ZAP70 (Zeta-associated protein of 70 kDa) is activated by LCK, binds to the phosphorylated ITAMs and phosphorylates downstream the adaptor molecules SLP-76 and LAT (Linker of Activated T cells). ITK (interleukin-2-inducible tyrosine kinase) upon interaction with phosphorylated SLP-76 and LAT undergoes autophosphorylation. When IDs involve proximal TCR signaling, severe abnormalities in T lymphocyte development and function with susceptibility to infections and immune dysregulation are described [29].
ZAP70 deficiency typically lacks circulating CD8+ T cells, whereas CD4+ T lymphocytes are present but do not proliferate. Hypomorphic variants with partial defects in ZAP70 signaling exhibit a peculiar immunodysregulated phenotype, similar to Omenn syndrome, with wheezing, erythroderma, lymphadenopathy, eosinophilia and high serum IgE [30]. Interestingly enough, an overwhelming AI was documented in a family with compound heterozygous ZAP70 mutations, one hypomorphic allele and one with gain of function, with bullous pemphigoid, colitis, hemophilia because of factor VIII autoantibody and nephrotic syndrome [31].
Defects in LCK present with recurrent respiratory infections, nodular skin lesions, arthritis, vasculitis, and ITP [32]. ITK deficiency clinically manifests infections, especially from herpes virus, and autoimmune cytopenias, lymphadenopathy, hepatosplenomegaly and EBV-associated lymphoproliferative disease, usually localized to the lung [33].
After the proximal TCR engagement (ZAP70, SLP-76, LAT, ITK, LAT), phospholipase C-γ1 (PLC-γ1) is activated, allowing hydrolysis of phosphatidylinositol (4,5) diphosphate (PIP2) to inositol (1,4,5) trisphosphate (IP3) and diacylglycerol (DAG), release of Ca2+ from endoplasmic reticulum (ER) stores, Ca2+ influx, activation of Erk, reorganization of the actin cytoskeleton and activation of transcriptional program.
Ca2+ release from ER stores is called SOCE (store-operated Ca2+ entry). It is activated by CRAC (Ca2+ release-activated Ca2+) channels which are composed by forming ion-conducting pore on the plasma membrane ORAI1 and STIM1 and 2 (stromal interaction molecule) located in the ER. LOF mutations of the CRAC complex proteins , ORAI1 and STIM1, are characterized by a SCID-like disease with AI, muscular hypotonia, and ectodermal dysplasia, with defects in sweat gland function and dental enamel formation [34].
Caspase recruitment domain (CARD) proteins, B cell CLL/lymphoma 10 (BCL10) and MALT1 paracaspase (MALT1) form adaptor complexes (CBM) critical in downstream signaling of numerous membrane receptors, including innate immunity pattern-recognition receptors (PRR) and GPCRs in non-hematopoietic cells. TCR and BCR signaling is mediated by the CBM complex expressing CARD11, which finely regulates cell activation, proliferation, metabolism in addition to survival pathways such as NF-kB, JNK and mTOR signaling. Numerous phenotypes associated with defects in the CBM complex have been recently described [35]. Mutations of CARD11 manifest with different phenotypes. Biallelic LOF associates with a SCID/CID-like phenotype [36]. Monoallelic germline or somatic GOF variants cause a polyclonal B cell expansion with NF-kB and T cell anergy (BENTA) [37]. Heterozygous dominant-negative mutations are responsible for CARD11-associated atopy with dominant interference of NF-kB signaling (CADINS) . CADINS is characterized by variable degrees of ID, including viral and bacterial infections in skin and airways, respectively, low to normal Ig and prominent atopic features, namely, asthma and atopic dermatitis, followed by food allergy, eosinophilic esophagitis and rhino-conjunctivitis. AI is found in 20% of the patients, most commonly alopecia, ITP, neutropenia and bullous pemphigoid [38].
15.3.2 Defective Actin Cytoskeleton
Defects in the assembly and dynamics of the actin cytoskeleton are an expanding group of IDs. Polymerization of actin plays an active role in multiple cellular processes, such as proliferation, endo- and exocytosis, intracellular trafficking and migration. Some of those defects are associated with significant inflammation and AI [39].
Wiskott-Aldrich syndrome protein (WASP) and WASP-interacting protein (WIP) are nucleation-promoting factors that promote F-actin branching. Defects in these genes are associated with Wiskott-Aldrich syndrome (WAS), which is the prototype of actin assembly defects. WAS is characterized by a triad: ID, micro-thrombocytopenia and eczema. Classical WASP defects are X-linked, while WIP defects are associated with autosomal recessive phenocopy. Hematological malignancies and AI are common complications of WAS and are often associated with the most severe forms. Reported autoimmune diseases include AHA, neutropenia, small and large vessels vasculitis, IBD and glomerulonephritis. A broad spectrum of autoantibodies has been observed [40]. Numerous mechanisms have been implicated in the WAS-associated AI: Tregs dysfunction, loss of tolerance and expansion of autoreactive B cells, defective Fas-mediated apoptosis and phagocytosis of apoptotic cells [41].
Arp2/3 complex (actin-related protein 2/3 complex) is required for assembly and branching of actin filaments. ARPC1B acts as a bridge between the actin filaments and the Arp2/3 complex. Biallelic mutations in ARPC1B have been reported in patients with a WAS-like ID, in which AI is prominent. Patients typically presents with lung and skin infections, bleeding, failure to thrive, early-onset eczema and atopic features. Cutaneous small vessel vasculitis, IBD and arthritis are between the most common presentations [42].
DOCK8 (dedicator of cytokinesis 8) is a guanine nucleotide exchange factor that activates Rho GTPases such as CDC42. DOCK8 activation is regulated by MST1 (mammalian sterile 20-like 1), also known as STK4 (serine/threonine kinase 4). DOCK8 deficiency causes autosomal recessive hyper-IgE syndrome, associated with a profound combined ID. Patients suffer from severe bacterial, viral (severe warts, CMV, EBV, HPV, HSV, VZV) and fungal (mucocutaneous candidiasis) infections, food allergies and malignancies. They have decreased and dysfunctional Tregs and produce autoantibodies [43]. Described autoimmune manifestations include AHA, colitis, sclerosing colitis and vasculitis. However, overt AI is uncommon, and it has been speculated that the profound T effector function deficiency might protect DOCK8-deficient patients from AI [44].
Disassembly of actin filaments is also crucial in cytoskeletal dynamics. Depolymerization is mediated by the recruitment of coronin, cofilin, and Aip1 (actin-interacting protein 1), encoded by WDR1 (WD repeat-containing protein 1). WDR1 deficiency is embryonic lethal; however hypomorphic mutations have been recently discovered being the cause of the lazy leukocyte syndrome described in the 1970s. Affected patients suffer from severe pyogenic infections, defective wound healing, chronic stomatitis leading to oral stenosis, neutropenia, thrombocytopenia and autoinflammation with periodic fever [45].
15.3.3 Defective T Cell Activation/Immunosenescence
mTOR (serine/threonine kinase mechanistic/mammalian target of rapamycin) is a sensor detecting clues coming from the microenvironment and controlling cell growth, proliferation and terminal cell differentiation. mTOR is part of a complex pathway including phosphoinoside3-kinase (PI3K), AKT and S6 kinase [46]. Hyperactivation of this pathway switches cells towards an anabolic state and impairs T cell function. Accordingly, naïve CD4+ repertoire is extremely limited and counterbalanced by an excess of terminally differentiated senescent T CD8+ cells. mTOR hyperactivation might be the consequence of heterozygous GOF mutations of two PI3Kdelta subunits, p110delta catalytic and p85alfa regulatory subunit. These mutations are responsible for the activated PI3Kdelta syndrome (APDS) 1 and 2 , respectively. APDS1 was formerly named PASLI (p110delta activating mutation causing senescent T cells, lymphadenopathy and immunodeficiency) [47].
The clinical picture is usually characterized by the presence of both ID and immunodysregulation. Classically, all the patients present recurrent respiratory tract infections, commonly pneumonia and bronchiectasis, these latter higher in the APDS1 cohort. Bacterial pathogens are more frequently capsulated (Streptococcus pneumoniae, Haemophilus influenzae). Half of the patients show persistent/recurrent severe herpes virus infections, with EBV viremia detected in 50% of them. More than three quarter of the patients show features of non-infective immune complications, the most common being non-neoplastic lymphoproliferation, clinically evidenced by diffuse chronic lymphadenopathy, splenomegaly and/or hepatomegaly. Evolution to lymphoma and other malignancies are more common in APDS2 patients (25%). Over than 1/3 of the patients present with some autoimmune features. AI is common in APDS1 with organ- or non-organ-specific manifestations: cytopenias (e.g., Coombs-positive AHA), GN, thyroiditis, insufficiency of exocrine pancreas, seronegative arthritis and sclerosing cholangitis. Recurrent pericarditis and enteropathy are also possible. In APDS2 AI manifestations are similar although less frequent (AHA, ITP and arthritis) in addition to organ-specific diseases (T1D, autoimmune hepatitis). Growth retardation is a prerogative of APDS2. Although most of the patients have been misdiagnosed as CVID, Ig deficiency is not a typical feature. Pneumococcal vaccine response is reduced in 90% of the patients, but IgG and IgA are low in less than half of the patients, whereas IgM levels are high and APDS1 and 2 need to be differentially diagnosed from hyper-IgM syndromes [48, 49].
Despite the differences between APDS1 and 2 cohorts, there is no genotype-phenotype correlation. Similarly, no different treatments between the two cohorts have been suggested [50]. Antimicrobial prophylaxis, immunoglobulin replacement and HSCT are used. Autoimmune manifestations have been treated with steroids and rituximab, with good clinical response, although followed by sustained B cell lymphopenia. Sirolimus has been used in non-lymphomatous lymphoproliferation but with less benefits on AI and cytopenias. Leniolisib, a potent oral p110delta subunit inhibitor, seems promising to control lymphoproliferation with no significant adverse reactions so far [50].
15.4 AI in Primary Defects of Central Tolerance
The paradigmatic link between IDs and AI is represented by the autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy (APECED) also called autoimmune polyendocrine syndrome (APS)-1 . APS-1 is a complex disorder with predominant organ-specific manifestations as parathyroids, thyroid, adrenal glands and gonad involvement. As a ID, 80% of patients are also affected by chronic mucocutaneus candidiasis [51].
Characteristically, the production of organ- and non-organ-specific autoantibodies in APECED is strictly linked to the failure of central tolerance [52], that is, physical elimination or functional anergy of harmful self-reactive lymphocytes before their maturation [53]. The expression of self-antigens within the thymus is tightly regulated by a transcriptional regulating and chromatin remodeling complex called AIRE. AIRE mutations have a knockout effect on the development of regulatory T (Treg) cells which account for the clinical features of the disease [53]. Indeed, the different clinical phenotypes described so far might be explained by the large amount of mutations described, the most common being R257* in exon 6, the so-called Finnish mutation [54].
Chronic mucocutaneous candidiasis (CMC) is usually the first manifestation of the disease, appearing during the first 5 years of age and caused by the presence of anti-interleukin (IL)-17 and anti-IL-22 autoantibodies [55]. Chronic hypoparathyroidism and adrenal insufficiency that constitute the classical triad of APS-1 might appear up to the fourth decade of life [51]. Chronic hypoparathyroidism is usually the first endocrine organ hit by AI with a vague clinical presentation (muscle cramps and mild paresthesia), as hypocalcemia seems to be slowly progressive. Fever may be the precipitating factor, leading to seizure and, eventually, to diagnosis [56]. Relevant autoantigens have not been discovered yet. Vice versa, autoantibodies against a recognized target (anti-12-hydroxilase) can be detected many years before the onset of adrenal insufficiency, being fatigue, weight loss and increased pigmentation the most common (and non-specific) symptoms [57]. Hypergonadotropic hypogonadism, T1D, thyroid diseases and pituitary defects are other endocrine features [57]. Gastrointestinal AI (i.e., autoimmune gastritis and hepatitis) is also common. Malabsorption and steatorrhea may be explained by pancreatic failure or activity of anti- intestinal endocrine cell autoantibodies [58]. Cutaneous vasculitis, Sjögren’s syndrome, and celiac disease have also been described. Autoimmune encephalitis may occur but is not common [59]. Hormone replacement therapy is necessary for the endocrine failure, and immunosuppressive therapy, including rituximab, is used to control clinical symptoms and the autoimmune manifestations [60]. Finally, ectodermal abnormalities are also common and appear early during the disease course. Although autoimmune mechanisms might explain the clinical picture, no specific autoantibodies have been reported so far. Dental enamel hypoplasia, pitted nail dystrophy, alopecia, and vitiligo are the most common manifestations, and an urticarial rash associated with fever might appear at first [61].
15.5 AI in Primary Defects of Peripheral Tolerance
Peripheral tolerance is the general mechanism of inactivation of mature lymphocytes that escaped the central tolerance but recognizing self antigens. It may occur by re-exposure to the antigen (anergy), death by apoptosis or active suppression by Tregs [62]. CD4+ regulatory T cells express high levels of CD25 (IL-2 receptor alpha chain), CD127 (IL-7 receptor) and the transcription factor FOXP3 (forkhead box P3) [63]. FOXP3 is, indeed, critical for the development and function of Treg cells [63].
15.5.1 IPEX
Hemizygous mutations of FOXP3 gene located on the X-chromosome are responsible for the immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome associated with a deficiency of Treg cells, and the subsequent failure of peripheral tolerance [62].
Even though the typical presentation of the disease is characterized by severe enteropathy, T1D and severe eczema during the first months of life, diagnosis can be difficult as it may manifest later in childhood with isolated features and failure to thrive [64]. No correlation genotype-phenotype has been described so far and the cause of this heterogeneity remains obscure [63].
Enteropathy occurs usually early in life (within 7 months of age), occurs in >90% of the patients and causes watery diarrhea and malabsorption that eventually brings to failure to thrive. In one half of these patients, histology demonstrates small bowel villous atrophy, probably due to anti-harmonin and anti-villin autoantibodies [64]. Skin manifestations are common with eczema and/or exfoliative dermatitis [63]. Severe food allergy typically associates with elevated IgE levels with overall normal immunoglobulins [64]. It is probably caused by inappropriate skewing towards a type-2 immunity due to defective Treg cells and reduced T cell receptor repertoire [65].
T1D usually follows enteritis and skin disease and is marked by anti-glutamic acid decarboxylase (GAD) or anti-islet cell antibodies. Besides this classic triad, other common manifestations of AI include thyroid dysfunction, cytopenias (anemia, neutropenia, thrombocytopenia), renal involvement (GN and interstitial nephritis), autoimmune hepatitis and interstitial lung disease. The response to common immunosuppressant such as corticosteroids or cyclosporine A is usually transient and weak. Thus, early hematopoietic stem cell transplantation (HSCT) is the only curative approach with a long-term survival (over 30 years) in 50% [64].
15.5.2 IPEX-Like Syndromes
IL-2 is pivotal for the survival and proliferation of all T lymphocytes [66]. Treg cells are characterized by the isolate presence of the low-affinity IL-2 receptor CD25 (IL-2 receptor alpha chain) able to equally bind IL-2, differently from the other T cell populations expressing the high-affinity receptor for IL-2, constituted by beta (CD122) and gamma (CD132) chains in addition to CD25. In this way, Treg cells compete and deprive other T cells from IL-2, with the consequence of reduced differentiation and proliferation and, ultimately, suppression of the immune response [63]. The IL-2 high-affinity receptor signals through the STAT5b transduction pathway, allowing FOXP3 transcription. However, transcription factor STAT5b is shared by other signaling pathways and is pivotal for growth hormone (GH) [67]. For this reason, mutations of CD25 and STAT5b show a clinical picture similar but not identical to IPEX (so-called IPEX-like syndromes), where AI and ID are present at the same time and, in case of STAT5b, together with GH insensitivity and dwarfism [63]. At variance with IPEX, however, patients with IPEX-like syndromes less likely suffer from T1D and food allergies which generally develop later in life [64].
CD25 deficiency is an IPEX-like syndrome characterized by severe enteropathy, eczema, AI (such as T1D), lymphoproliferation, systemic lymphadenopathy and susceptibility to chronic infections, especially towards herpes family viruses [68].
STAT5b deficiency is a rare autosomal recessive disorder which combines GH insensitivity syndrome (impaired postnatal growth in the presence of normal-to-high GH levels) and features of an IPEX-like syndrome with infections. Dominant-negative STAT5b mutations have been recently described with predominant growth defects and none-to-mild immune dysregulation. This latter is evidenced by eczema, celiac disease and autoimmune thyroiditis [69].
Ten-year survival is significantly higher in IPEX-like than IPEX patients (81.5% vs 65%), but 30-year survival is poor in both groups, and in both cohorts, HSCT improves the survival rate, supporting its early implementation [64].
15.6 AI in Immunoglobulin Defects
Activation, proliferation and survival of B cells, correct somatic hypermutation of immunoglobulin (Ig) genes and antibody class switch recombination (CSR) are critical points for a successful (and regulated) humoral response. Thus, any perturbation of these checkpoints alters the quantity of circulating immunoglobulins and, only apparently, paradoxical AI development. The main clinical syndromes discussed in this chapter are agammaglobulinemia, selective IgA deficiency, class-switch recombination defects and common variable immunodeficiency (CVID).
Bruton agammaglobulinemia is a rare X-linked ID with agammaglobulinemia (XLA) characterized by the absence of mature B cells and Ig-secreting plasma cells, severe antibody deficiency and recurrent infections. More than 500 different mutations (nonsense, splice defects, deletions, insertions) of the BTK gene located on the long arm of the X-chromosome have been described. Mutated BTK (Bruton tyrosine kinase) impairs B cell development from pro-B to pre-B lymphocytes as being involved downstream into signal transduction after a successful immunoglobulin heavy-chain rearrangement [70]. Severe recurrent infections are the mainstay of this ID. This may account for reactive manifestations resembling clear AI (arthritis secondary to Mycoplasma infections, GNs, IBDs due to Campylobacter or Enteroviruses), but XLA is characterized per se by AI although much less frequently than CVID or other Ig defects. Fewer than one third of the patients may develop painful joints, but frank formal arthritis is only occasionally observed. Thrombocytopenia, anemia and leukopenia affect less than 10% of patients, and neutropenia seems to be due to infective agents rather than autoantibodies [71].
Selective IgA deficiency (SIgAD) is the most common ID in both adults and children with a highly variable prevalence also in populations of similar descent [72]. Similarly, SIgAD is highly heterogeneous in its immunological defects: a) defects of B cells (altered class switch and Ig rearrangement, increased B cell apoptosis, reduced survival of IgA+ plasma cells or reduced terminal differentiation of IgA plasmablasts), b) unbalanced production of cytokines crucial for class switch or maturation of B lymphocytes (TGF-beta, IL-10 or IL-21) or c) defective T cell help [73]. Patients with sIgAD may carry several chromosomal abnormalities and cytogenetic defects (monosomy, trisomy, translocation or deletions) [74], but low IgA levels can be found in a series of inborn errors of immunity. Beyond possible infectious susceptibility, allergic diseases, gastrointestinal disorders and malignancies, sIgAD characteristically associate with AI with 5–30% prevalence [73, 75]. Individuals with symptomatic or asymptomatic SIgAD indeed show an increased ability to produce autoantibodies not necessarily associated with overt AI and anti-thyroid autoantibodies (anti-thyroglobulin or -microsomal antigens), Coomb’s positivity, anti-nuclear and anti-cardiolipin antibodies, can be detected. When autoimmunity clinically develops, cytopenias (ITP, AHA), thyroid diseases (Graves’ disease, Hashimoto’s thyroiditis), T1D, celiac disease, RA or SLE can occur. Almost one fifth of SIgAD affected adult patients develops at least one of the autoimmune diseases [76], whereas in children celiac disease is the most prevalent (about 15%) followed by thyroid disease and then vitiligo, psoriasis, T1D, and alopecia as seen in one recent cohort undergoing a long follow-up [77].
Class-switch recombination (CSR) is an intrachromosomal deletional recombination event leading to the production of all the isotypes of immunoglobulins, but IgM, in mature B cells. Thus, CSR-ID are characterized by normal/elevated circulating IgM in the absence/low levels of IgG, IgA and IgE. They are thus also known as hyper-IgM syndromes (HIGM) . CSR-ID/HIGM are rare, far less common than classic CVID (at most 1/1,000,000), and are classified in five different disorders (HIGM1–5 and X-linked hyper-IgM syndrome [XHIM]) because different genes are involved, namely, CD40L and CD40, AICDA, UNG, and IKBKG [78].
CD40 is codified on 20q12-q13.2 and plays an essential role for T cell-dependent immunoglobulin class switching, memory B cell development, and germinal center formation [75, 79]. Only in 2001 absence of CD40 expression was related to the rare autosomal recessive HIGM3.
CD40L encoding gene (CD40LG) is located in the X chromosome and its defects are responsible for the most frequent XHIM. It codifies for the activated T cell receptor CD40L (CD154) which engages the B-cell costimulatory receptor CD40 to induce the production of antibodies of correct affinity. In mice CD40L hyperexpression associates with accelerated lymphocyte apoptosis, high titers of autoantibodies and SLE-like [80]. In humans, CD40LG microduplications associate with AI (cytopenias, arthralgias, thyroid disease) [81].
In general, every perturbation of CD40-CD40L interaction correlates with AI and XHIGM and HIGM3 patients are similarly affected by autoimmune manifestations. Autoimmune cytopenias (neutropenia, AHA, ITP), hypothyroidism, IBD, and kidney disease are described in addition to infections. HIGM2 is an autosomal recessive (mostly) or autosomal dominant (rarely) inherited defect of AICDA. The gene encodes the DNA editing enzymatic protein AID (activation-induced cytidine deaminase) involved in somatic hypermutation and Ig gene conversion [82]. Cytopenias, hepatitis, IBD, and arthritis are the most common manifestations occurring in about one fifth of the patients, but also SLE and uveitis have been described and autoreactive antibodies detected into the circulation [83, 84]. The recognition of the genetic defects of HIGM4 still lacks, and the syndrome has a mild course with involvement of blood series, uvea and joints [85]. So far, no manifestations of AI have been recognized, although expected, in HIGM5 deficiency related to UNG (uracil N-glycosylase) [86].
Other diseases including IDs exhibit increased levels of IgM both occasionally (RAG-2 deficiencies) and regularly (APDS, PMS2 gene mutations, A-T or NBS, CVID). Both PMS2 (postmeiotic segregation increased 2) gene mutations and NBS (Nijmegen breakage syndrome) are constitutional mismatch repair deficiencies primarily characterized by malignancies in the absence (PMS2) or presence (NBS) of ID. AI occurs only in the latter, mainly described in individuals of Slavic descent, with single descriptions of AHA and juvenile idiopathic arthritis (JIA)-like polyarthritis [87]. Revisions of AI in selected IDs with hyper IgM are available [78, 88].
CVID is the most common symptomatic hypogammaglobulinemia in adults. It is characterized by reduced immunoglobulins, impaired production of specific antibodies, autoimmunity and lymphoproliferation. However, CVID is an extremely heterogeneous syndrome. In fact, it is extensively used as a broad umbrella diagnosis. Monogenic forms account for a variable percentage of the patients, depending on selection criteria and sequencing technique. Genes that have been implicated in monogenic CVID include ICOS, TACI, BAFF-R, TWEAK, CD19, CD81, CD21, CD20, CD27, IL-21, IL21R, LRBA, CTLA4, PRKCD, PLCG2, NF-KB1, NF-KB2, PIK3CD, PIK3R1, VAV1, RAC2, BLK, IKZF1 and IRF2BP2 [89]. Moreover, the discovery of the molecular defects underlying some CVID patients greatly helped understating the pathogenesis of the apparently paradoxical autoimmune and lymphoproliferative complications. Some of the most complex clinical pictures characterized by overwhelming autoimmune phenomena have been re-classified as “immunodysregulation disorders” [90] and are discussed separately in this manuscript, despite the clinical presentation often mimics CVID.
B cell-activating factor (BAFF) regulates antibody response through the binding of its three receptors: (i) BAFF-R (BAFF-receptor) , promoting survival and maturation of peripheral B cells; (ii) BCMA (calcium modulator and B cell maturation antigen), promoting the survival of long-lived bone marrow plasma cells; and (iii) TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor) , promoting differentiation and survival of plasma cells, inhibition of B cell expansion and induction of CSR towards IgG and IgA [91, 92]. In mice, deletions in the BAFF-encoding gene or BAFFR deficiency interrupt maturation of transitional B cells causing ID. In humans the homozygous deletion within the exon 2 of the TNFRSF13C gene produces an altered transmembrane region of BAFFR with late-onset IgG and IgM hypogammaglobulinemia, severe lymphopenia and possibly infections [93]. Altered extracellular or intracellular chain of BAFFR due to missense mutations is more common and may be found in CVID, but its contribution remains elusive [94]. Several genetic defects (biallelic and monoallelic loss-of-function variants) in TNFRSF13B TACI-encoding gene have been found in almost 10% of the patients with CVID, mostly heterozygous C104R and A181E point mutations, with associated variable clinical phenotypes [95]. Variants have also been detected in sIgAD, IgG subclass deficiency, asymptomatic relatives of CVID and 1–2% general population; thus the combination with other defect(s) is likely involved into overt ID [89, 96]. More interestingly, BAFF and its receptors are highly involved in AI. TACI−/− or BAFF transgenic mice develop a SLE-like disease. In humans BAFF/APRIL concentrations are increased in RA and SLE and correlate with disease activity and autoantibody production. High levels of BAFF maintain the pool of transitional B cells in which high numbers of autoreactive lymphocytes are included [91, 92, 97]. In CVID, levels of BAFF are indeed increased [98]. In TACI as well as in BAFFR variants, cytopenias, rheumatic diseases (sacroiliitis) and IBD may develop [89].
ICOS (inducible T cell costimulator) is a T cell-associated receptor belonging to the CD28/CTLA-4 family. ICOS-ICOS-L interaction is crucial for antibody production, effector T cell responses and tolerance [99]. In ICOS mutations (missense mutations, homozygous deletions, compound heterozygous mutations), clinical AI with cytopenias, rheumatic disease, and early onset of Crohn’s-like colitis are found together with altered B cell numbers, hypogammaglobulinemia, impaired CTLA-4 expression, abnormal cytokine production and TH1/TH2 unbalance, repeated respiratory and/or viral infections [100, 101] without any clear genotype-phenotype correlation.
NF-κB (nuclear factor “kappa-light-chain-enhancer” of activated B cells) represent a family of transcription factors (NF-κB1, NF-κB2, RelA, RelB, c-Rel) crucial for cell activation [102]. Deficiency of NF-κB1 (heterozygous LOF variants) and NF-κB2 (germline dominant-negative heterozygous mutations) is responsible for CVID with early onset (NF-κB2) or ranging from infants to aged individuals (NF-κB1). ID varies from mild hypogammaglobulinemias to severe forms with only NF-κB1 mutations associated with organ-specific AI (cytopenias, alopecia areata or universalis, vitiligo, and Hashimoto thyroiditis) [103].
15.7 Diseases of Immunodysregulation
15.7.1 Checkpoints Defects
Lymphocyte deactivation after antigen recognition requires negative signals which are provided by a series of surface receptors and/or regulatory cells. CTLA-4, ICOS, OX40, and PD1 exert downregulation activities and control lymphocyte proliferation and differentiation. Thus, mutations of genes coding for these checkpoint molecules are followed by lymphoproliferation and abnormal antigen recognition including self in the context of different degrees of ID.
Cytotoxic T lymphocyte antigen 4 (CTLA-4) (CD152) is a homodimer coded by four exons acting as negative regulator of immunity [104]. CTLA4 gene is located on 2q33.3 where CD28 and ICOS are also placed [105]. CTLA-4 is constitutively expressed on Tregs [106] where it represents a sine-qua-non molecule for the suppressive function being CTLA4, a target gene of FOXP3 [106, 107]. Functionally, it binds CD80 and CD86 with greater (500–2500-fold) affinity and avidity than its homolog CD28, thus competing with this latter and terminating cell activation. Deprivation of IL-2 is one of the mechanisms of cell deactivation by CTLA-4 [108]. Actually, homozygous CTLA4−/− mice succumb for fatal AI and lymphoproliferation [109]. In humans, several observations link the CTLA4 gene with thyroid autoimmunity (hypothyroidism, Graves’), dysregulated IgE production, insulin-dependent diabetes and malignancies [110, 111]. Conversely, the fusion protein with agonist properties abatacept is a useful therapeutic tool in autoimmune diseases including RA [112].
Heterozygous germline mutations in CTLA4 have been described for the first time in 2014 [113, 114]. Since then, more than 50 heterozygous CTLA4 germline mutations have been identified (missense, insertions or deletions, nonsense) all placed in exons 1–2-3 [115]. Asymptomatic or mildly symptomatic carriers of CTLA4 mutation have been found in the same families of the affected individuals.
The largest known cohort of CTLA4 mutation carriers including 133 subjects and 28 novel mutations has clarified the complex spectrum of the clinical manifestations in heterozygous CTLA-4 insufficiency [116]. Three out four of the patients had CVID and respiratory tract infections or sepsis. AI is eventually represented by cytopenias (30%) frequently bi- or tri-lineage (ITP, AHA, pure red cell aplasia or autoimmune neutropenia). Autoimmune endocrinopathies (thyroid diseases and T1D, Addison disease), psoriatic arthritis, alopecia, autoimmune encephalitis, atrophic gastritis, celiac disease and primary biliary cholangitis may also occur. Immune dysregulation finally manifests as non-malignant or malignant lymphoproliferation and solid organ neoplasms. This kaleidoscopic picture is completed by other clinical manifestations as atopic dermatitis (about 60%), neurological features (about 30%), mental and/or growth retardation. Phenotypically, lymphopenia with reduced CD19+ CD27+ IgD- and relative increase of CD21low are found [116]. A complete review on CTLA-4 in relation to immunodeficiency has been recently provided [115].
In 2012 mutations in chromosomal region 4q31 encoding LRBA (lipopolysaccharide-responsive beige-like anchor protein) , already suspected as an ID candidate gene [117], were described [118]. Homozygous individuals exhibited a deleterious ID with hypogammaglobulinemia and repeated severe infections. AI was almost invariably the onset of the disease (ITP followed by AHA) plus autoimmune enteropathy resembling Crohn’s disease, atrophic gastritis with autoantibodies against intrinsic factor, hypothyroidism, myasthenia gravis, and arthritis. Disturbed B cell development and reduced plasmablast formation were initially described, and, in 2015, marked depletion of Tregs with defective levels of canonical markers (FOXP3, CD25, Helios and CTLA-4) was added [119]. LBRA is codified into the BEACH (BEige and Chediak-Higashi syndrome) family of cytoplasmic proteins that regulate intracellular vesicle trafficking and exocytosis [120]. CTLA4 and LRBA co-localize within recycling endosomes and the TGN, but the tail of CTLA-4 needs to interact with the pleckstrin homology (PH)-like and the BEACH domains of LRBA to be maintained intracellularly stored [121]. In LRBA-deficient individuals CTLA4 mRNA levels are thus normal and LRBA controls the expression of CTLA-4 at post-translational level [121].
Recessive-inherited mutations in LRBA deficiency vary (complete absence of the protein, truncated protein with altered expression of PH, BEACH or WD40 domains, truncated protein devoid of the BEACH and WD40 domains, normal expression of a not-functional protein) [122]. The clinical course is close to CTLA-4 deficiency: hypogammaglobulinemia, repeated infections, interstitial lung disease, neurologic disease (cerebral or cerebellar atrophy, myasthenia gravis, space-occupying lesions usually of granulomatous origin), skin diseases resembling atopic dermatitis, and chronic diarrhea. About AI, besides ITP and AIHA, autoimmune thyroid disease and autoimmune enteropathy are the most common, but, also, vitiligo, celiac or sprue-like disease with villous atrophy, T1D and juvenile idiopathic arthritis may occur [122].
One more mutated protein associated with decreased ability to recycle CTLA-4 is DEF6 (Differentially Expressed in FDCP6 homolog) also named as IRF4 binding protein (IBP) or SWAP-70-like adaptor of T cells (SLAT) [123, 124]. It activates downstream of TCR and promotes Ca2+ signaling, NFAT1 activation and T cell adhesion [125]. In AI it was invoked in early-onset large vessel vasculitis and other systemic autoimmune manifestations [126]. Very recently, three patients from two families from Iraqi and Pakistan were described as carrying homozygous or compound heterozygous mutations with reduced or complete loss of expression of DEF6 protein. In addition to increased susceptibility to viral and bacterial infections, mixed autoimmune/autoinflammatory pictures were present: IBD and atrophic gastritis due to lymphocytic infiltration with severe diarrhea, AHA, ITP, high levels of anti-neutrophil cytoplasmic antibodies (ANCA) and anti-phospholipid autoantibodies, successful treatment with abatacept. Cycling CTLA-4 was reduced, CTLA-4 did not effectively reach surfaces and CD80 capture and transendocytosis were impaired. A defective interaction between DEF6 and the small GTPase RAB11 necessary for correct recycling endosomes is responsible for this novel ID [125].
The interaction between tumor necrosis factor receptor OX40 (CD134) and its cognate ligand OX40L (CD134L, CD252) has been long supposed as a possible therapeutic target in AI. OX40 is encoded by the TNFRSF4 gene placed on chromosome 1. Lack of OX40 is characterized by reduced numbers of Tregs and reduced IFN-ɣ production [127]. However, only one healthy female carrying an autosomal recessive OX40 deficiency has been described [128]. More recently, a single case with a homozygous nonsense mutation of the RC3H1 gene, affecting OX40, ICOS, and TNF expression, has been described, but the clinical phenotype was hyperinflammation and presentation as relapsing HLH instead of ID and AI [129].
15.7.2 Defects of JAK/STAT Signaling
Maturation, survival, and differentiation of B cells and Ig class switch are also dependent by a correct signaling after the engagement of the cell surface receptors for cytokines, chemokines, growth factors or hormones. The activation of different combinations of JAKs (Janus kinase signal transducer) is responsible for the recruitment of various STATs (activator of transcription signaling pathway), depending on the type (I or II) of the cytokine receptor. This chain of events is involved in host defense but also in AI as redirecting T cell differentiation, and JAK inhibitors are successful therapeutic arms for increasing numbers of autoimmune diseases [130, 131].
STAT1 is principally activated upon binding of type I (α and β) and II (ɣ) interferons in addition to IL-2, IL-10, IL-21 and IL-27. While LOF mutations on STAT1 gene are included into the group of MSMD (Mendelian Susceptibility to Mycobacterial Disease) as deficient in the IL-12/IFN-ɣ axis, GOF mutations associate with CMC (chronic mucocutaneous candidiasis) [132, 133]. Heterozygous GOF STAT1 mutations are characterized by ID (hypogammaglobulinemia, CMC, disseminated fungal infections, viral and Staphylococcus aureus infections) and AI in more than 30% of the patients, seldom as the prevalent feature. AI is typically represented by hypothyroidism, T1D, autoimmune cytopenias, and SLE, but other manifestations can occur such as IBD, autoimmune hepatitis, arthritis, multiple sclerosis, as well as cerebral vascular aneurysms, even if infrequently [133]. A single case of fatal Takayasu arteritis has been described [134].
STAT3 is activated downstream after type I, II and III interferons, IL-6, IL-10, IL-17, IL-21, IL-22 and IL-23 signaling. Autosomal dominant-negative STAT3 LOF variants are the cause of hyper-IgE syndrome, also known as Job’s syndrome. Those patients display defective TH17 lymphocyte polarization and IL-17 production. They present clinically with a wide array of infectious diseases (candidiasis, S. aureus) and typical phenotypical features (facies, pneumatoceles, connective tissue malformations). They often present with symptoms suggestive of AI diseases such as arthritis and Raynaud’s, skin, and mucosal ulcerations, but seldomly meet diagnostic criteria. Between 4.8% and 14.5% have a possible AI connective tissue disease. Organ-specific diseases like T1D or thyroiditis are rare [135].
STAT3 GOF germline mutations (more than 28 described, autosomal dominant trait inheritance) enhance transcriptional activity of STAT3, STAT3 DNA-binding activity, or delay STAT3 dephosphorylation. At the same time, STAT1 and STAT5 phosphorylation decreases and STAT5 is crucial for growth hormone signaling. STAT3 hyperactivation favors TH17 differentiation and suppresses Treg function. In fact, STAT3 polymorphisms associate with increased predisposition to psoriasis and multiple sclerosis [136]. Patients with STAT3 heterozygous GOF mutations exhibit multiple different clinical features as short stature, inconstant recurrent infections proper of ID (disseminated non-tuberculous mycobacteria, viruses and fungi) with low IgG levels and reduced class-switched memory B lymphocytes, non-malignant lymphoproliferation (lymphadenopathy, splenomegaly) and AI [137]. Autoimmune disorders range from cytopenias (about 50% of the patients), to T1D with infancy onset (25%), enteropathies (50%), primary hypothyroidism (25%), and interstitial lung disease (about one third) [138].
15.7.3 Miscellaneous Disorders
Adenosine deaminase 2 (ADA2) is an extracellular enzyme, secreted by monocytes and dendritic cells involved into the catabolism of purines [139]. It also regulates the balance between M1 and M2 macrophages. M1 macrophages induce TNF-α production and enhance inflammation, characterized by vascular injury and upregulation of neutrophil-expressed proinflammatory genes. Thus, anti-TNF-α drugs have shown efficacy in the treatment of DADA2 vasculitis and prevention of strokes [139]. Homozygous mutations in ADA2 (DADA2) exhibit different clinical phenotypes, with hematological, immunological and vascular involvement. Although it is generally accepted that the lower the activity of ADA2, the worse is the clinical phenotype, no clear genotype-phenotype correlation exists. Clinically, livedo reticularis and erythema nodosum are classical signs. Cutaneous vasculitis can be severe, with skin ulcers, and systemic vasculitis mimicking panarteritis nodosa (PAN), with juvenile onset. Both central and peripheral nervous systems (ischemic and hemorrhagic strokes) can occur. Strokes can recur in approximately one half of the patients [139]. Peripheral neuropathy is common. Arthritis is a frequent finding. The clinical picture also includes the hematological involvement: pure red cell aplasia (sometimes mimicking Diamond-Blackfan anemia) and bone marrow failure signs (including neutropenia and pancytopenia). Finally, hepatosplenomegaly, diffuse lymphadenopathy and hematological malignancies are observed. Homozygous ADA2 mutations have been recently found in patients with hypogammaglobulinemia and recurrent infections (CVID-like DADA2) [140].
Ikaros is a zinc-finger transcription factor encoded by IKZF1 gene, strongly expressed in lymphoid and hematopoietic progenitors. Ikaros homodimers bind to DNA at the pericentromeric heterochromatin regions, acting mainly as a gene repressor [141]. In humans Ikaros regulates B cell development, commitment and activation, Ig gene recombination together with EBF1 and PAX5 [142] but also the differentiation of dendritic cells. In IKZF1 haploinsufficiency plasmacytoid dendritic cells are absent, but conventional dendritic cells are normal or expanded [143]. In Ikaros deficiency very low B cell counts, raised CD8+ T lymphocytes counts, and reduction of two or more Ig classes are observed. Autoimmune manifestations include arthritis, SLE, and ITP [144]. Combined ID with myeloid abnormalities (neutropenia, eosinopenia and reduced myeloid dendritic cells) and increased susceptibility to hematological malignancies, especially B cell and T cell acute lymphoblastic leukemia, are seen [141].
GATA2 is a member of a family of zinc finger transcription factors critical for generation and function of stem cells, progenitor cells and the following cell lineages [145]. Heterozygous mutations into GATA2 gene (deletions, mutations in exons or intronic regulatory regions) associate with a spectrum of human diseases, which follows an autosomal dominant pattern of inheritance, due to haploinsufficiency: hematological abnormalities, recurrent infections, alveolar proteinosis and pulmonary dysfunction, lymphedema, thrombotic events, congenital deafness and hematological malignancies. AI manifests with panniculitis (up to 50% of the patients), erythema nodosum, Sweet syndrome, arthritis and SLE-like, hypothyroidism, autoimmune hepatitis or primary biliary cholangitis [146, 147].
15.8 Autoimmune Lymphoproliferative Syndrome (ALPS)
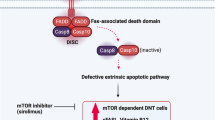
Peripheral tolerance is crucial for a healthy immune system, because the deletion of every autoreactive T cell is impossible in central lymphoid organs. Peripheral tolerance thus prevents dangerous expansion and activation of autoreactive T cells in secondary lymphoid organs and peripheral tissues. Among the several effector mechanisms responsible for peripheral tolerance, the Fas-Fas ligand (FasL) system plays an important role in eliminating autoreactive lymphocytes. Fas (CD95) is a member of the TNF superfamily expressed in T, B and antigen-presenting cells, including dendritic cells. It is a homotrimer with three extracellular cysteine-rich domains, one transmembrane domain and a functional intracellular domain or Death Domain (DD). Its ligand (Fas-L) equally belongs to the TNF superfamily and is a homotrimer. Fas-DD allows the interaction between Fas and a group of cytoplasmic DD-containing proteins, named FADD (Fas-associated DD). FADD also contains a Death Effector Domain (DED), recruiting other cytoplasmic DED-containing proteins such as procaspase-8 and procaspase-10. This cascade promotes the assembly of the Death Inducing Signalling Complex (DISC). Procaspase-8 and procaspase-10 are recruited to the DISC, converted into their active forms, thus activating other procaspase with their proteolytic action. This process culminates in cellular apoptosis. The DISC is negatively regulated by the enzymatically inactive caspase homolog FLIP (Flice-Inhibitory Protein), which inhibits procaspase activation [148].
At rest, Fas and FasL are not expressed on the cell surface, as appearing within 24 hours after cell activation. Initially, due to a relative abundance of FLIP, T cells are resistant to apoptosis, becoming more susceptible one week after the activation. Fas-FasL system is crucial for the downregulation of the normal immune response, as preventing persistent immune activation and tissue damage. Fas is also involved in AICD (activation-induced cell death), preventing AI. Dendritic cells continuously patrol peripheral tissues, sampling and presenting antigens to naïve T cells. Repeated TCR stimulation by autoantigens in the absence of costimulatory signals triggers the expression of Fas and FasL in activated T cells with deletion of activated T cells and dendritic cells. In this way autoreactive cells are removed in the peripheral tissues.
Mutations of Fas-FasL system demonstrate the importance of these molecules. Autoimmune lymphoproliferative syndrome (ALPS) depends on several different gene mutations of Fas, FasL and CASP10. De novo or inherited Fas mutations arise in hematopoietic progenitors. Mutations are mostly autosomal dominant and less frequently recessive and not fully penetrant [149].
Mutated lymphocytes display a survival advantage which explains the clinical hallmark of ALPS, i.e., lymphoproliferation. Lymphocytes infiltrate secondary lymphoid organs (SLO), such as spleen and lymph nodes, explaining the usual clinical presentation of ALPS with splenomegaly and lymphadenopathy. Lymphoproliferation is mediated by an abnormal CD4-CD8-TCRαβ+ subset, namely, double negative cells (DNC). Spleen infiltration is responsible for marginal zone-like B cell deficiency and poor pneumococcal polysaccharide response, a critical risk factor for pneumococcal sepsis. Loss of Fas signaling impairs deletion of autoreactive lymphocytes, resulting in the clinical picture of autoimmune cytopenias which commonly occur in ALPS. AHA, ITP, and autoimmune neutropenia can be severe, and due to autoantibodies. Other autoimmune diseases, such as glomerulonephritis and uveitis, are much rarer and probably coincidental. It is supposed that ALPS accelerates organ-specific AI that would have happened later in the life. The survival advantage of lymphocytes in ALPS is also a risk factor for malignant lymphoproliferation, in particular Hodgkin lymphoma (50-fold) and non-Hodgkin lymphoma (14-fold) [150].
ALPS clinical phenotype is not limited to Fas, FasL or caspase-10 mutations. The same phenotype is shared by other primary IDs, collectively called ALPS-like syndromes . Here, many other mechanisms are involved, but all with disturbed apoptosis. Defects in Fas signaling involving FADD and caspase-8 have been found, but the clinical picture is consistent with combined IDs, given the role of these proteins in lymphocyte proliferation [149].
Other IDs share disturbed apoptosis. STAT3 can downregulate the expression of Fas while enhancing many anti-apoptotic proteins of the BCL-2 family. Thus, STAT3 GOF mutation , previously discussed in this manuscript, may exhibit an ALPS-like phenotypes [151].
Somatic mutations of KRAS and NRAS are responsible for constitutive activation of the RAS pathway followed by BIM expression defect. This is the molecular pathogenesis of a block of activated cell autonomous death, i.e., cell death after growth factor deprivation. Lymphocytes of these patients are resistant to IL-2 starvation in vitro. From a clinical point of view, lymphoproliferation and autoimmune cytopenias are the hallmarks of this syndrome, often associated with monocytosis without any DNT expansion [148].
Finally, immune checkpoint defects, such as CTLA-4 haploinsufficiency and LRBA deficiency , can mimic ALPS. The removal of the inhibitory effect of CTLA-4 on lymphocytes is a risk factor for lymphoproliferation and infiltration of SLO and peripheral tissues. Consistently, these patients often display AI manifestations as described above [148].
15.9 Therapeutic Perspectives in IDs with Autoimmune Complications
The growing knowledge of the molecular defects in immune dysregulation syndromes has paved the way to targeted therapy. See Fig. 15.1.

Molecular pathways and possible targeted immunomodulatory drugs for the treatment of autoimmune manifestations associated with monogenic primary immunodeficiencies
APDS due to GOF mutations in PIK3CD or PIK3R1 genes is characterized by PI3K hyperactivity with many and complex effects on the immune system, including AI and lymphoproliferation. A downstream effector of PI3K is mTOR (mammalian target of rapamycin), a conserved serine/threonine kinase that participates in two complexes, mTORC1 and 2. APDS, where high mTOR activity is present, is indeed included in the so-called immune TOR-opathies [46]. For this reason, the use of sirolimus , an antibiotic produced by Streptomyces hygroscopicus and inhibiting mTOR, is reasonable. Sirolimus has been used in APDS as effective in reversing hepatosplenomegaly and lymphadenopathy, even if less efficient in cytopenias and gastrointestinal disease [50]. Selective p110δ inhibitors are currently under evaluation as possible more targeted and safe options. The most promising molecule, leniolisib, was nicely able to revert lymphoproliferation (hepatosplenomegaly, lymphadenopathy) and fatigue and normalize immunological parameters (transitional B cells, senescent T cells, naive B cells) [50].
Sirolimus was also clinically efficient in ALPS, as reducing lymphocytes’ survival and removing the stop to apoptosis. In some studies, sirolimus also reduced lymphoproliferation and DNC numbers and improved autoimmune cytopenias [152]. It decreases effector T cell proliferation with little impairment of Treg function. IPEX syndrome takes advantage of this activity of sirolimus [152] exhibiting fewer side effects than calcineurin inhibitors , usually representing the first-line therapy.
Other IDs might be susceptible of targeted therapy, i.e., CTLA-4 haploinsufficiency and LRBA deficiency. Abatacept , a fusion protein joining the extracellular domain of CTLA-4 to the Fc region of IgG1, has been successfully used in RA to dampen joint inflammation, but it is also able to inhibit lymphocyte activation. The absence of the natural CTLA-4 protein in these disorders can be replaced by abatacept. Although used into a small number of patients, a significant improvement in lymphadenopathy, colitis, autoimmune cytopenias and lymphocytic interstitial lung disease was observed [153]. Notably, abatacept seems to restore immunological balance, as increasing naive T cells and partially restoring vaccine response [153] without any long-term complications.
GOF STAT1 mutations increase STAT1 expression or phosphorylation or impair STAT1 dephosphorylation. STAT1 is placed downstream several cytokine receptors which transmit their signals by JAK1, JAK2 and JAK3. Therefore, inhibition of JAKs is an appealing approach to reverse immune dysregulation in these patients. Selective inhibitors of JAK (JAKi) have been recently developed to cure RA and psoriatic arthritis. Five JAK inhibitors are now on the market: ruxolitinib (JAK1/JAK2 inhibitor), tofacitinib (JAK1/JAK3 inhibitor), baricitinib (JAK1/JAK2 inhibitor), filgotinib (JAK1 selective inhibitor), and decernotinib (JAK3 selective inhibitor). JAKi have been used in limited case series with good clinical efficacy in cytopenias, interstitial lung diseases and enteropathy. JAKi also improved the control of infections and ameliorated immunological parameters such as NK cell cytotoxicity and proportion of TH1, TH17, and Tfh cells [133, 153].
STAT3 GOF mutations enhance transcriptional activity of STAT3 or delay STAT3 dephosphorylation. Here, two targeted approaches have been tried. Given the important role of STAT3 in IL-6 signaling, the anti-IL-6 receptor monoclonal antibody tocilizumab has been administered to few patients with only a partial effect. JAKi alone or together with anti-IL-6 strategies have been tried and the combination of the two seems to be particularly effective [153].
Besides targeted therapies, hematopoietic stem cell transplantation (HSCT) offers the fascinating prospective of a complete cure of IDs, as providing a new immune system. This therapy is now safe, reaching high survival percentage (90%).
When approaching HSCT, the type of ID must be first considered. HSCT is suitable for defects stemming from hematopoietic compartment but not for thymic stromal or other extra-hematopoietic defects. Secondly, timing of HSCT needs to be considered. Best results are obtained in patients undergoing HSCT early, before anatomical injuries or severe infections develop. In young adults, HSCT must be wisely chosen, weighting accurately benefits versus risks. HSCT is lifesaving and urgent when SCID is diagnosed. This therapeutic option is otherwise challenging in immune dysregulation syndromes, although likely to be useful as avoiding long-term disease and treatment-related injuries. However few data have been indeed published. As targeted therapy is available, the opportunity of HSCT should be individualized and HSCT still remains a controversial option in humoral IDs, such as CVID [154].
New strategies have been developed to improve the access to HSCT. Matched-related donor (MRD), which is the gold standard for HSCT, may be difficult, because of the high frequency of consanguinity in patients with IDs. Matched unrelated donor (MUD) may be the answer, but patients often belong to ethnic minorities, underrepresented in donor registries. Haplo-identical transplant with selective ex vivo depletion of αβ T lymphocytes (as responsible for graft-versus-host disease, GVHD) and B lymphocytes (possibly harboring EBV), as well as HSCT with genetically modified αβ T lymphocytes with an added caspase suicide gene, allowed remarkable survival in ID cohorts. In the latter, the activator of the caspase suicide gene rimiducid is administered if GVHD arises, to selectively remove donor’s T lymphocytes [154]. Further, less toxic conditioning regimens and T lymphocytes specifically directed against viral epitopes from donor banks have been used to control post-HSCT infections with remarkably improved outcomes.
Gene therapy is the last and more promising therapy for IDs as aimed to directly correct the responsible gene defect(s) by modification(s) of host genome. Retroviruses have been extensively used as vectors for DNA modification, as able to integrate into host DNA. Gamma-retroviruses, simple retroviruses without additional regulatory genes, have been used in the past. However, they were not so efficient and raised safety concerns, because of significant rate of insertional mutagenesis with possible development of leukemia. Thus, lentiviruses are now used as devoid of insertional mutagenesis and more efficient in transducing non-dividing cells. Gene therapy has been used in ADA-SCID, X-linked SCID, WAS and CGD, with good results in children. Also, adult patients poorly responding to HSCT seem to benefit, but fewer data are available. Gene therapy for ADA-SCID is now available on the market [154].
Two more advanced approaches to gene therapy are autologous gene-corrected T cell therapy and gene editing . In the former gene therapy is restricted to T cells only or to a specific T cell subset only. The latter aims to correct the gene defect. Specific endonucleases introduce breaks into the host DNA and exploit the normal DNA repairing systems to restore gene function. These approaches are currently under evaluation on preclinical animal models and clinical studies are eagerly awaited [154].
References
Modell V, Orange JS, Quinn J, Modell F (2018) Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res 66:367–380. https://doi.org/10.1007/s12026-018-8996-5
Liblau RS, Bach J-F (1992) Selective IgA deficiency and autoimmunity. Int Arch Allergy Immunol 99:16–27. https://doi.org/10.1159/000236330
Rosen FS, Kevy SV, Merler E, Janeway CA, Gitlin D (1961) Dysgammaglobulinæmia and recurrent bacterial infection. Lancet 277:700. https://doi.org/10.1016/s0140-6736(61)91725-1
Amaya-Uribe L, Rojas M, Azizi G, Anaya J-M, Gershwin ME (2019) Primary immunodeficiency and autoimmunity: a comprehensive review. J Autoimmun 99:52–72. https://doi.org/10.1016/j.jaut.2019.01.011
Walter JE, Ayala IA, Milojevic D (2019) Autoimmunity as a continuum in primary immunodeficiency. Curr Opin Pediatr 31:851–862. https://doi.org/10.1097/mop.0000000000000833
Schmidt RE, Grimbacher B, Witte T (2017) Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol 14:7–18. https://doi.org/10.1038/nrrheum.2017.198
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K et al (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506:376–381. https://doi.org/10.1038/nature12873
Shoenfeld Y (1993) The kaleidoscope of autoimmunity. Autoimmunity 15:245–252. https://doi.org/10.3109/08916939309019934
Fischer A, Provot J, Jais J-P, Alcais A, Mahlaoui N, Adoue D et al (2017) Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol 140:1388–1393.e8. https://doi.org/10.1016/j.jaci.2016.12.978
Singh K, Chang C, Gershwin ME (2014) IgA deficiency and autoimmunity. Autoimmun Rev 13:163–177. https://doi.org/10.1016/j.autrev.2013.10.005
Delmonte OM, Schuetz C, Notarangelo LD (2018) RAG deficiency: two genes, many diseases. J Clin Immunol 38:646–655. https://doi.org/10.1007/s10875-018-0537-4
Milner JD, Fasth A, Etzioni A (2008) Autoimmunity in Severe Combined Immunodeficiency (SCID): lessons from patients and experimental models. J Clin Immunol 28:29–33. https://doi.org/10.1007/s10875-007-9159-y
Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA et al (2016) Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci Immunol 1:eaah6109. https://doi.org/10.1126/sciimmunol.aah6109
Daley SR, Koay H-F, Dobbs K, Bosticardo M, Wirasinha RC, Pala F et al (2019) Cysteine and hydrophobic residues in CDR3 serve as distinct T-cell self-reactivity indices. J Allergy Clin Immunol 144:333–336. https://doi.org/10.1016/j.jaci.2019.03.022
Stadinski BD, Shekhar K, Gómez-Touriño I, Jung J, Sasaki K, Sewell AK et al (2016) Hydrophobic CDR3 residues promote the development of self-reactive T cells. Nat Immunol 17:946–955. https://doi.org/10.1038/ni.3491
Buchbinder D, Hauck F, Albert MH, Rack A, Bakhtiar S, Shcherbina A et al (2019) Rubella virus-associated cutaneous granulomatous disease: a unique complication in immune-deficient patients, not limited to DNA repair disorders. J Clin Immunol 39:81–89. https://doi.org/10.1007/s10875-018-0581-0.
Rigoni R, Fontana E, Guglielmetti S, Fosso B, D’Erchia AM, Maina V et al (2016) Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med 213:355–375. https://doi.org/10.1084/jem.20151116
Rowe JH, Stadinski BD, Henderson LA, Ott de Bruin L, Delmonte O, Lee YN et al (2017) Abnormalities of T-cell receptor repertoire in CD4 + regulatory and conventional T cells in patients with RAG mutations: Implications for autoimmunity. J Allergy Clin Immunol 140:1739–1743.e7. https://doi.org/10.1016/j.jaci.2017.08.001
Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M et al (2015) Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest 125:4135–4148. https://doi.org/10.1172/JCI80477
Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu H-B et al (2004) Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20:441–453. https://doi.org/10.1016/S1074-7613(04)00079-2
Walter JE, Rucci F, Patrizi L, Recher M, Regenass S, Paganini T et al (2010) Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med 207:1541–1554. https://doi.org/10.1084/jem.20091927
Lima K, Abrahamsen TG, Wolff AB, Husebye E, Alimohammadi M, Kämpe O et al (2011) Hypoparathyroidism and autoimmunity in the 22q11.2 deletion syndrome. Eur J Endocrinol 165:345–352. https://doi.org/10.1530/EJE-10-1206
Gennery AR (2002) Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch Dis Child 86:422–425. https://doi.org/10.1136/adc.86.6.422
McLean-Tooke A, Spickett GP, Gennery AR (2007) Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand J Immunol 66:1–7. https://doi.org/10.1111/j.1365-3083.2007.01949.x
Marcovecchio GE, Bortolomai I, Ferrua F, Fontana E, Imberti L, Conforti E et al (2019) Thymic epithelium abnormalities in DiGeorge and down syndrome patients contribute to dysregulation in T cell development. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.00447
Montin D, Marolda A, Licciardi F, Robasto F, Di Cesare S, Ricotti E et al (2019) Immunophenotype anomalies predict the development of autoimmune cytopenia in 22q11.2 deletion syndrome. J Allergy Clin Immunol Pract 7:2369–2376. https://doi.org/10.1016/j.jaip.2019.03.014
Malphettes M, Gérard L, Carmagnat M, Mouillot G, Vince N, Boutboul D et al (2009) Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis Off Publ Infect Dis Soc Am 49:1329–1338. https://doi.org/10.1086/606059
Ameratunga R, Lehnert K, Woon S-T, Gillis D, Bryant VL, Slade CA et al (2018) Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol 54:261–268. https://doi.org/10.1007/s12016-017-8645-0
Notarangelo LD (2014) Immunodeficiency and immune dysregulation associated with proximal defects of T cell receptor signaling. Curr Opin Immunol 31:97–101. https://doi.org/10.1016/j.coi.2014.10.003
Au-Yeung BB, Shah NH, Shen L, Weiss A (2018) ZAP-70 in signaling, biology, and disease. Annu Rev Immunol 36:127–156. https://doi.org/10.1146/annurev-immunol-042617-053335
Chan AY, Punwani D, Kadlecek TA, Cowan MJ, Olson JL, Mathes EF et al (2016) A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. J Exp Med 213:155–165. https://doi.org/10.1084/jem.20150888
Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A et al (2012) Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol 130:1144–1152.e11. https://doi.org/10.1016/j.jaci.2012.07.029
Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K et al (2011) IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica 96:472–476. https://doi.org/10.3324/haematol.2010.033910
Lacruz RS, Feske S (2015) Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 1356:45–79. https://doi.org/10.1111/nyas.12938
Lu HY, Biggs CM, Blanchard-Rohner G, Fung S-Y, Sharma M, Turvey SE (2019) Germline CBM-opathies: from immunodeficiency to atopy. J Allergy Clin Immunol 143:1661–1673. https://doi.org/10.1016/j.jaci.2019.03.009
Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I et al (2013) Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol 131:1376–1383.e3. https://doi.org/10.1016/j.jaci.2013.02.012
Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G et al (2015) Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol 35:32–46. https://doi.org/10.1007/s10875-014-0106-4
Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM et al (2019) Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. J Allergy Clin Immunol 143:1482–1495. https://doi.org/10.1016/j.jaci.2018.08.013
Tangye SG, Bucciol G, Casas-Martin J, Pillay B, Ma CS, Moens L et al (2019) Human inborn errors of the actin cytoskeleton affecting immunity: way beyond WAS and WIP. Immunol Cell Biol 97:389–402. https://doi.org/10.1111/imcb.12243
Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA (1994) A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 125:876–885. https://doi.org/10.1016/S0022-3476(05)82002-5
Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C et al (2007) The Wiskott-Aldrich syndrome protein is required for the function of CD4+CD25+Foxp3+ regulatory T cells. J Exp Med 204:381–391. https://doi.org/10.1084/jem.20061338
Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M et al (2019) A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol 143:2296–2299. https://doi.org/10.1016/j.jaci.2019.02.003
Biggs CM, Keles S, Chatila TA (2017) DOCK8 deficiency: insights into pathophysiology, clinical features and management. Clin Immunol 181:75–82. https://doi.org/10.1016/j.clim.2017.06.003
Janssen E, Kumari S, Tohme M, Ullas S, Barrera V, Tas JMJ et al (2017) DOCK8 enforces immunological tolerance by promoting IL-2 signaling and immune synapse formation in Tregs. JCI Insight 2:e94298. https://doi.org/10.1172/jci.insight.94298
Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, Priel DL et al (2016) Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood 128:2135–2143. https://doi.org/10.1182/blood-2016-03-706028
Jung S, Gámez-Díaz L, Proietti M, Grimbacher B (2018) “Immune TOR-opathies,” a novel disease entity in clinical immunology. Front Immunol 9:966. https://doi.org/10.3389/fimmu.2018.00966
Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K (2016) PI3Kδ and primary immunodeficiencies. Nat Rev Immunol 16:702–714. https://doi.org/10.1038/nri.2016.93
Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N et al (2017) Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol 139:597–606.e4. https://doi.org/10.1016/j.jaci.2016.06.021
Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L et al (2016) Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: a cohort study. J Allergy Clin Immunol 138:210–218.e9. https://doi.org/10.1016/j.jaci.2016.03.022
Coulter TI, Cant AJ (2018) The treatment of activated PI3Kδ syndrome. Front Immunol 9:2043–2043. https://doi.org/10.3389/fimmu.2018.02043
Aaltonen J, Björses P, Perheentupa J, Horelli-Kuitunen N, Palotie A, Peltonen L et al (1997) An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 17:399–403. https://doi.org/10.1038/ng1297-399
Hinterberger M, Aichinger M, Prazeres da Costa O, Voehringer D, Hoffmann R, Klein L (2010) Autonomous role of medullary thymic epithelial cells in central CD4+ T cell tolerance. Nat Immunol 11:512–519. https://doi.org/10.1038/ni.1874
Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D (2005) The cellular mechanism of aire control of T cell tolerance. Immunity 23:227–239. https://doi.org/10.1016/j.immuni.2005.07.005
Stolarski B, Pronicka E, Korniszewski L, Pollak A, Kostrzewa G, Rowińska E et al (2006) Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: novel AIRE mutations and an estimate of disease prevalence. Clin Genet 70:348–354. https://doi.org/10.1111/j.1399-0004.2006.00690.x
Sarkadi AK, Taskó S, Csorba G, Tóth B, Erdős M, Maródi L (2014) Autoantibodies to IL-17A may be correlated with the severity of mucocutaneous candidiasis in APECED patients. J Clin Immunol 34:181–193. https://doi.org/10.1007/s10875-014-9987-5
Alimohammadi M, Björklund P, Hallgren Å, Pöntynen N, Szinnai G, Shikama N et al (2008) Autoimmune polyendocrine syndrome Type 1 and NALP5, a parathyroid autoantigen. N Engl J Med 358:1018–1028. https://doi.org/10.1056/nejmoa0706487
Ferre EMN, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE et al (2016) Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 1. https://doi.org/10.1172/jci.insight.88782
Gianani R, Eisenbarth GS (2003) Autoimmunity to gastrointestinal endocrine cells in autoimmune polyendocrine syndrome Type I. J Clin Endocrinol Metab 88:1442–1444. https://doi.org/10.1210/jc.2003-030247
Mazza C, Buzi F, Ortolani F, Vitali A, Notarangelo LD, Weber G et al (2011) Clinical heterogeneity and diagnostic delay of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome. Clin Immunol 139:6–11. https://doi.org/10.1016/j.clim.2010.12.021
Popler J, Alimohammadi M, Kämpe O, Dalin F, Dishop MK, Barker JM et al (2012) Autoimmune polyendocrine syndrome type 1: utility of KCNRG autoantibodies as a marker of active pulmonary disease and successful treatment with rituximab. Pediatr Pulmonol 47:84–87. https://doi.org/10.1002/ppul.21520
Perheentupa J (2006) Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab 91:2843–2850. https://doi.org/10.1210/jc.2005-2611
Georgiev P, Charbonnier L-M, Chatila TA (2019) Regulatory T cells: the many faces of Foxp3. J Clin Immunol 39:623–640. https://doi.org/10.1007/s10875-019-00684-7
Cepika A-M, Sato Y, Liu JM-H, Uyeda MJ, Bacchetta R, Roncarolo MG (2018) Tregopathies: monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol 142:1679–1695. https://doi.org/10.1016/j.jaci.2018.10.026
Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S et al (2018) Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-Linked (IPEX) syndrome. Front Immunol 9:2411. https://doi.org/10.3389/fimmu.2018.02411
Santoni de Sio FR, Passerini L, Restelli S, Valente MM, Pramov A, Maccari ME et al (2018) Role of human forkhead box P3 in early thymic maturation and peripheral T-cell homeostasis. J Allergy Clin Immunol 142:1909–1921.e9. https://doi.org/10.1016/j.jaci.2018.03.015
Mitra S, Leonard WJ (2018) Biology of IL-2 and its therapeutic modulation: mechanisms and strategies. J Leukoc Biol 103:643–655. https://doi.org/10.1002/JLB.2RI0717-278R
Kanai T, Jenks J, Nadeau KC (2012) The STAT5b pathway defect and autoimmunity. Front Immunol 3. https://doi.org/10.3389/fimmu.2012.00234
Vignoli M, Ciullini Mannurita S, Fioravanti A, Tumino M, Grassi A, Guariso G et al (2019) CD25 deficiency: a new conformational mutation prevents the receptor expression on cell surface. Clin Immunol 201:15–19. https://doi.org/10.1016/j.clim.2019.02.003
Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D et al (2018) Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun 9:2105. https://doi.org/10.1038/s41467-018-04521-0
Vetrie D (1993) The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 364:362–362. https://doi.org/10.1038/364362a0
Hernandez-Trujillo VP, Scalchunes C, Cunningham-Rundles C, Ochs HD, Bonilla FA, Paris K et al (2014) Autoimmunity and inflammation in X-linked agammaglobulinemia. J Clin Immunol 34:627–632. https://doi.org/10.1007/s10875-014-0056-x
Yel L (2010) Selective IgA deficiency. J Clin Immunol 30:10–16. https://doi.org/10.1007/s10875-009-9357-x
Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A (2017) Selective IgA deficiency: epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Scand J Immunol 85:3–12. https://doi.org/10.1111/sji.12499
Abolhassani H, Aghamohammadi A, Hammarström L (2016) Monogenic mutations associated with IgA deficiency. Expert Rev Clin Immunol 12:1321–1335. https://doi.org/10.1080/1744666x.2016.1198696
Castigli E, Geha RS (2008) TACI, isotype switching, CVID, and IgAD. Natl Inst Allergy Infect Dis NIH:343–348. https://doi.org/10.1007/978-1-59745-569-5_38
Nechvatalova J, Pikulova Z, Stikarovska D, Pesak S, Vlkova M, Litzman J (2012) B-lymphocyte subpopulations in patients with selective IgA deficiency. J Clin Immunol 32:441–448. https://doi.org/10.1007/s10875-012-9655-6
Lougaris V, Sorlini A, Monfredini C, Ingrasciotta G, Caravaggio A, Lorenzini T et al (2019) Clinical and laboratory features of 184 Italian pediatric patients affected with selective IgA deficiency (SIgAD): a Longitudinal Single-Center Study. J Clin Immunol 39:470–475. https://doi.org/10.1007/s10875-019-00647-y
Davies EG, Thrasher AJ (2010) Update on the hyper immunoglobulin M syndromes. Br J Haematol 149:167–180. https://doi.org/10.1111/j.1365-2141.2010.08077.x
Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ (2009) Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229:152–172. https://doi.org/10.1111/j.1600-065x.2009.00782.x
Kishi Y, Aiba Y, Higuchi T, Furukawa K, Tokuhisa T, Takemori T et al (2010) Augmented antibody response with premature germinal center regression in CD40L transgenic mice. J Immunol 185:211–219. https://doi.org/10.4049/jimmunol.0901694
Le Coz C, Trofa M, Syrett CM, Martin A, Jyonouchi H, Jyonouchi S et al (2018) CD40LG duplication-associated autoimmune disease is silenced by nonrandom X-chromosome inactivation. J Allergy Clin Immunol 141:2308–2311.e7. https://doi.org/10.1016/j.jaci.2018.02.010
Shinkura R, Ito S, Begum NA, Nagaoka H, Muramatsu M, Kinoshita K et al (2004) Separate domains of AID are required for somatic hypermutation and class-switch recombination. Nat Immunol 5:707–712. https://doi.org/10.1038/ni1086
Quartier P, Bustamante J, Sanal O, Plebani A, Debré M, Deville A et al (2004) Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol 110:22–29. https://doi.org/10.1016/j.clim.2003.10.007
Durandy A, Cantaert T, Kracker S, Meffre E (2013) Potential roles of activation-induced cytidine deaminase in promotion or prevention of autoimmunity in humans. Autoimmunity 46:148–156. https://doi.org/10.3109/08916934.2012.750299
Imai K, Catalan N, Plebani A, Maródi L, Sanal Ö, Kumaki S et al (2003) Hyper-IgM syndrome type 4 with a B lymphocyte–intrinsic selective deficiency in Ig class-switch recombination. J Clin Invest 112:136–142. https://doi.org/10.1172/jci18161
Yousif AS, Stanlie A, Begum NA, Honjo T (2014) Opinion: uracil DNA glycosylase (UNG) plays distinct and non-canonical roles in somatic hypermutation and class switch recombination. Int Immunol 26:575–578. https://doi.org/10.1093/intimm/dxu071
Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M (2012) Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis 7:13. https://doi.org/10.1186/1750-1172-7-13
Giardino G, Gallo V, Prencipe R, Gaudino G, Romano R, De Cataldis M et al (2016) Unbalanced immune system: immunodeficiencies and autoimmunity. Front Pediatr 4. https://doi.org/10.3389/fped.2016.00107
Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F (2016) Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet 53:575–590. https://doi.org/10.1136/jmedgenet-2015-103690
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A et al (2020) Human inborn errors of immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 40:24–64. https://doi.org/10.1007/s10875-019-00737-x
Zhang Y, Li J, Zhang Y-M, Zhang X-M, Tao J (2015) Effect of TACI signaling on humoral immunity and autoimmune diseases. J Immunol Res 2015:1–12. https://doi.org/10.1155/2015/247426
Smulski CR, Kury P, Seidel LM, Staiger HS, Edinger AK, Willen L et al (2017) BAFF- and TACI-Dependent processing of BAFFR by ADAM proteases regulates the survival of B cells. Cell Rep 18:2189–2202. https://doi.org/10.1016/j.celrep.2017.02.005
Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J et al (2009) B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci 106:13945–13950. https://doi.org/10.1073/pnas.0903543106
Smulski CR, Eibel H (2018) BAFF and BAFF-receptor in B cell selection and survival. Front Immunol 9. https://doi.org/10.3389/fimmu.2018.02285
Salzer U, Chapel HM, Webster ADB, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M et al (2005) Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet 37:820–828. https://doi.org/10.1038/ng1600
Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L et al (2005) TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet 37:829–834. https://doi.org/10.1038/ng1601
Vincent FB, Morand EF, Schneider P, Mackay F (2014) The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol 10:365–373. https://doi.org/10.1038/nrrheum.2014.33
Knight AK, Radigan L, Marron T, Langs A, Zhang L, Cunningham-Rundles C (2007) High serum levels of BAFF, APRIL, and TACI in common variable immunodeficiency. Clin Immunol 124:182–189. https://doi.org/10.1016/j.clim.2007.04.012
Sharpe AH, Freeman GJ (2002) The B7–CD28 superfamily. Nat Rev Immunol 2:116–126. https://doi.org/10.1038/nri727
Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R et al (2003) Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol 4:261–268. https://doi.org/10.1038/ni902
Abolhassani H, El-Sherbiny YM, Arumugakani G, Carter C, Richards S, Lawless D et al (2019) Expanding clinical phenotype and novel insights into the pathogenesis of ICOS deficiency. J Clin Immunol. https://doi.org/10.1007/s10875-019-00735-z
BEINKE S, LEY SC (2004) Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J 382:393–409. https://doi.org/10.1042/bj20040544
Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E et al (2018) Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol 142:1285–1296. https://doi.org/10.1016/j.jaci.2018.01.039
Schwartz J-CD, Zhang X, Fedorov AA, Nathenson SG, Almo SC (2001) Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 410:604–608. https://doi.org/10.1038/35069112
Ling V, Wu PW, Finnerty HF, Agostino MJ, Graham JR, Chen S et al (2001) Assembly and annotation of human chromosome 2q33 sequence containing the CD28, CTLA4, and ICOS gene cluster: analysis by computational, comparative, and microarray approaches. Genomics 78:155–168. https://doi.org/10.1006/geno.2001.6655
Jago CB, Yates J, Câmara NOS, Lechler RI, Lombardi G (2004) Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol 136:463–471. https://doi.org/10.1111/j.1365-2249.2004.02478.x
Hori S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057–1061. https://doi.org/10.1126/science.1079490
Walker LSK, Sansom DM (2011) The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 11:852–863. https://doi.org/10.1038/nri3108
Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP et al (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270:985–988. https://doi.org/10.1126/science.270.5238.985
Heward JM, Allahabadia A, Armitage M, Hattersley A, Dodson PM, Macleod K et al (1999) The development of graves’ disease and the CTLA-4 gene on chromosome 2q331. J Clin Endocrinol Metab 84:2398–2401. https://doi.org/10.1210/jcem.84.7.5820
Jury EC, Flores-Borja F, Kalsi HS, Lazarus M, Isenberg DA, Mauri C et al (2010) Abnormal CTLA-4 function in T cells from patients with systemic lupus erythematosus. Eur J Immunol 40:569–578. https://doi.org/10.1002/eji.200939781
Moreland L, Bate G, Kirkpatrick P (2006) Abatacept. Nat Rev Drug Discov 5:185–186. https://doi.org/10.1038/nrd1989
Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A et al (2014) Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 20:1410–1416. https://doi.org/10.1038/nm.3746
Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT et al (2014) Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345:1623–1627. https://doi.org/10.1126/science.1255904
Mitsuiki N, Schwab C, Grimbacher B (2018) What did we learn from CTLA-4 insufficiency on the human immune system? Immunol Rev 287:33–49. https://doi.org/10.1111/imr.12721
Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D et al (2018) Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 142:1932–1946. https://doi.org/10.1016/j.jaci.2018.02.055
Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A et al (2012) LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol 130:481–488.e2. https://doi.org/10.1016/j.jaci.2012.05.043
Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K et al (2012) Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 90:986–1001. https://doi.org/10.1016/j.ajhg.2012.04.015
Charbonnier L-M, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT et al (2015) Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol 135:217–227. https://doi.org/10.1016/j.jaci.2014.10.019
Cullinane AR, Schäffer AA, Huizing M (2013) The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic Cph Den 14:749–766. https://doi.org/10.1111/tra.12069
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C et al (2015) Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349:436–440. https://doi.org/10.1126/science.aaa1663
Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z et al (2015) Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol 36:33–45. https://doi.org/10.1007/s10875-015-0224-7
Gupta S, Lee A, Hu C, Fanzo J, Goldberg I, Cattoretti G et al (2003) Molecular cloning of IBP, a SWAP-70 homologous GEF, which is highly expressed in the immune system. Hum Immunol 64:389–401. https://doi.org/10.1016/s0198-8859(03)00024-7
Tanaka Y, Bi K, Kitamura R, Hong S, Altman Y, Matsumoto A et al (2003) SWAP-70-like adapter of T cells, an adapter protein that regulates early TCR-initiated signaling in Th2 lineage cells. Immunity 18:403–414. https://doi.org/10.1016/s1074-7613(03)00054-2
Serwas NK, Hoeger B, Ardy RC, Stulz SV, Sui Z, Memaran N et al (2019) Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis. Nat Commun 10:3106. https://doi.org/10.1038/s41467-019-10812-x
Canonigo-Balancio AJ, Fos C, Prod’homme T, Bécart S, Altman A (2009) SLAT/Def6 plays a critical role in the development of Th17 cell-mediated experimental autoimmune encephalomyelitis. J Immunol Baltim Md 1950 183:7259–7267. https://doi.org/10.4049/jimmunol.0902573
Webb GJ, Hirschfield GM, Lane PJL (2015) OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 50:312–332. https://doi.org/10.1007/s12016-015-8498-3
Byun M, Ma CS, Akçay A, Pedergnana V, Palendira U, Myoung J et al (2013) Inherited human OX40 deficiency underlying classic Kaposi sarcoma of childhood. J Exp Med 210:1743–1759. https://doi.org/10.1084/jem.20130592
Tavernier SJ, Athanasopoulos V, Verloo P, Behrens G, Staal J, Bogaert DJ et al (2019) A human immune dysregulation syndrome characterized by severe hyperinflammation with a homozygous nonsense Roquin-1 mutation. Nat Commun 10:4779–4779. https://doi.org/10.1038/s41467-019-12704-6
O’Shea JJ, Plenge R (2012) JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36:542–550. https://doi.org/10.1016/j.immuni.2012.03.014
Villarino AV, Kanno Y, O’Shea JJ (2017) Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat Immunol 18:374–384. https://doi.org/10.1038/ni.3691
Lorenzini T, Dotta L, Giacomelli M, Vairo D, Badolato R (2016) STAT mutations as program switchers: turning primary immunodeficiencies into autoimmune diseases. J Leukoc Biol 101:29–38. https://doi.org/10.1189/jlb.5ri0516-237rr
Notarangelo LD, Fleisher TA (2017) Targeted strategies directed at the molecular defect: toward precision medicine for select primary immunodeficiency disorders. J Allergy Clin Immunol 139:715–723. https://doi.org/10.1016/j.jaci.2017.01.004
Maeshima K, Ishii K, Shibata H (2018) An adult fatal case with a STAT1 Gain-of-function mutation associated with multiple autoimmune diseases. J Rheumatol 46:325–327. https://doi.org/10.3899/jrheum.180210
Heimall JR, Collins KR, Milner JD, Holland SM, Freeman AF, Siegel R (2010) Autoimmune phenotypes in patients with autosomal dominant hyper-IgE/Job’s syndrome. J Allergy Clin Immunol 125:AB9. https://doi.org/10.1016/j.jaci.2009.12.066
Hillmer EJ, Zhang H, Li HS, Watowich SS (2016) STAT3 signaling in immunity. Cytokine Growth Factor Rev 31:1–15. https://doi.org/10.1016/j.cytogfr.2016.05.001
Vogel TP, Milner JD, Cooper MA (2015) The Ying and Yang of STAT3 in human disease. J Clin Immunol 35:615–623. https://doi.org/10.1007/s10875-015-0187-8
Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich P-S et al (2019) Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J Allergy Clin Immunol Pract 7:1958–1969.e9. https://doi.org/10.1016/j.jaip.2019.02.018
Sahin S, Adrovic A, Kasapcopur O (2020) A monogenic autoinflammatory disease with fatal vasculitis. Curr Opin Rheumatol 32:3–14. https://doi.org/10.1097/bor.0000000000000669
Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W et al (2020) Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2). J Allergy Clin Immunol. https://doi.org/10.1016/j.jaci.2019.12.908
Boutboul D, Kuehn HS, Van de Wyngaert Z, Niemela JE, Callebaut I, Stoddard J et al (2018) Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest 128:3071–3087. https://doi.org/10.1172/JCI98164
Eskandarian Z, Fliegauf M, Bulashevska A, Proietti M, Hague R, Smulski CR et al (2019) Assessing the functional relevance of variants in the IKAROS family Zinc Finger Protein 1 (IKZF1) in a Cohort of patients with primary immunodeficiency. Front Immunol 10:568–568. https://doi.org/10.3389/fimmu.2019.00568
Bigley V, Cytlak U, Collin M (2019) Human dendritic cell immunodeficiencies. Semin Cell Dev Biol 86:50–61. https://doi.org/10.1016/j.semcdb.2018.02.020
Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H et al (2017) Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol 140:223–231. https://doi.org/10.1016/j.jaci.2016.09.029
Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR et al (2014) GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123:809–821. https://doi.org/10.1182/blood-2013-07-515528
McReynolds LJ, Calvo KR, Holland SM (2018) Germline GATA2 mutation and bone marrow failure. Hematol Oncol Clin North Am 32:713–728. https://doi.org/10.1016/j.hoc.2018.04.004
Collin M, Dickinson R, Bigley V (2015) Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169:173–187. https://doi.org/10.1111/bjh.13317
Rieux-Laucat F (2017) What’s up in the ALPS. Curr Opin Immunol 49:79–86. https://doi.org/10.1016/j.coi.2017.10.001
Rieux-Laucat F, Magérus-Chatinet A, Neven B (2018) The autoimmune lymphoproliferative syndrome with defective FAS or FAS-ligand functions. J Clin Immunol 38:558–568. https://doi.org/10.1007/s10875-018-0523-x
Straus SE, Jaffe ES, Puck JM et al (2001) The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98(1):194–200. Blood 2017;130:232–232. https://doi.org/10.1182/blood-2017-05-787341
Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T et al (2017) STAT3 gain-of-function mutations associated with autoimmune lymphoproliferative syndrome like disease deregulate lymphocyte apoptosis and can be targeted by BH3 mimetic compounds. Clin Immunol 181:32–42. https://doi.org/10.1016/j.clim.2017.05.021
Vignesh P, Rawat A, Singh S (2016) An update on the use of immunomodulators in primary immunodeficiencies. Clin Rev Allergy Immunol 52:287–303. https://doi.org/10.1007/s12016-016-8591-2
Delmonte OM, Castagnoli R, Calzoni E, Notarangelo LD (2019) Inborn errors of immunity with immune dysregulation: from bench to bedside. Front Pediatr 7:353–353. https://doi.org/10.3389/fped.2019.00353
Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD (2019) Hematopoietic stem cell transplantation in primary immunodeficiency diseases: current status and future perspectives. Front Pediatr 7:295–295. https://doi.org/10.3389/fped.2019.00295
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Palterer, B., Vitiello, G., Vivarelli, E., Parronchi, P. (2021). Autoimmunity in Cellular Immunodeficiencies. In: D'Elios, M.M., Baldari, C.T., Annunziato, F. (eds) Cellular Primary Immunodeficiencies. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-030-70107-9_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-70107-9_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-70106-2
Online ISBN: 978-3-030-70107-9
eBook Packages: MedicineMedicine (R0)