
Abstract
Mitral valve prolapse (MVP) is a common disorder, with a prevalence of 2–3% in the general population. It is the most common cause of primary mitral regurgitation requiring surgery, making the underlying etiology of this condition of significant interest. As a familial pattern is observed in up to half of cases, much work has sought to determine the underlying heritable components that may contribute to this condition. In this chapter, we focus on the genetics of mitral valve prolapse, providing a broad outline of the loci known to be associated with syndromic and non-syndromic forms of MVP, a historical perspective of how some of these loci came to be identified, and a discussion of how these findings have led to novel frameworks for our understanding of at least subsets of patients with this condition. Elucidating the genetic underpinnings of MVP will further our understanding of the biological basis of this disease, and may allow earlier detection of asymptomatic individuals and prediction of disease progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
Mitral valve prolapse (MVP) is a common disorder, with a prevalence of 2–3% in the general population. It is the most common cause of primary mitral regurgitation requiring surgery [1], making the underlying etiology of this condition of significant interest. As a familial pattern is observed in up to half of cases [2, 3], much work has sought to determine the underlying heritable components that may contribute to this condition. In this chapter, we focus on the genetics of mitral valve prolapse, providing a broad outline of the loci known to be associated with syndromic and non-syndromic forms of MVP, a historical perspective of how some of these loci came to be identified, and a discussion of how these findings have led to novel frameworks for our understanding of at least subsets of patients with this condition. Elucidating the genetic underpinnings of MVP will further our understanding of the biological basis of this disease, and may allow earlier detection of asymptomatic individuals and prediction of disease progression.
Given its prevalence, MVP would be predicted to be a complex polygenic disease trait that adheres to the common disease-common variant hypothesis. Based on the latter, common heritable diseases in the population result from underlying genetic contributors (changes in coding or regulatory sequences of genes or variants) that are also common in the population [4]. According to this model, individual variants at disease-influencing genes confer a small additive or multiplicative susceptibility to the expression of the disease phenotype [4]. However, as is the case with many common diseases, a subset of MVP cases cluster tightly within families and follow a Mendelian pattern of inheritance suggesting the presence of rare variants with strong effects on disease pathogenesis [3]. Mendelian inheritance patterns relevant to non-syndromic MVP include autosomal and X-linked dominant, both of which are characterized by a single mutated copy of the causative gene is sufficient to yield the disease phenotype with the latter characterized by the absence of male-to-male transmission. Towards this end, classic genetic linkage analysis led to the earliest detection of the chromosomal locations of disease genes associated with MVP. More recent studies have leveraged next-generation sequencing technologies whose higher throughput have afforded the ability to perform genome-wide association studies (GWAS) as well as the interrogation of all coding sequences (whole-exome sequencing) in affected individuals.
Histology and Pathophysiology of MVP
MVP is characterized by a slow and progressive increase in the length and area of mitral valve tissue leading to thickening of the leaflets, their prolapse in systole beyond the mitral annulus into the left atrium, and associated mitral regurgitation. The extracellular matrix (ECM) constitutes the fibro-skeleton of a normal mitral valve. Normal mitral valve leaflets demonstrate three distinct tissue layers on histologic examination: the atrialis (composed of elastic fibers and directed towards the atrial-facing surface) provides elasticity, the spongiosa (composed of an interwoven network of glycosaminoglycans and proteoglycans within a spongy elastin network making up the middle layer) provides flexibility, and the fibrosa (composed of collagen fibers and directed towards the ventricle) provides tensile strength [5]. MVP is histologically characterized by myxomatous degeneration, a consequence of ECM dysregulation. In myxomatous valves an accumulation of proteoglycans leads to expansion of the spongiosa layer, structural alterations in all collagen components of the valve leaflets, and structurally abnormal chordae containing increased amounts of glycosaminoglycans [6,7,8].
Genetic Basis of Syndromic MVP
Syndromic MVP (Table 10.1) refers to those etiologies of disease that are thought to be secondary or associated with an identifiable disorder.
MVP as Part of Connective Tissue Disorders
The vast majority of known syndromic forms of MVP can be broadly associated with defects in the transforming growth factor β (TGF-β) superfamily signaling pathway or important components of the ECM. The TGF-β signaling pathway involves numerous cytokines acting through specific cell-surface receptors to mediate diverse cellular processes including cell proliferation, differentiation, and apoptosis via a number of intracellular signaling molecules [9] while the ECM describes highly dynamic non-cellular component of all tissues and organs that both physically scaffolds cellular constituents and initiates the biochemical and biomechanical cues necessary for appropriate tissue morphogenesis, homeostasis, and physiologic function [10]. Indeed, the ECM and TGF-β signaling pathway are themselves intimately linked, a relationship that is perhaps best exemplified in the context of Marfan syndrome, an autosomal dominant systemic connective tissue disorder that results from mutations in the ECM protein fibrillin-1 [11,12,13] resulting in cardiovascular, skeletal, and ocular manifestations in affected individuals. TGF-β isoforms are synthesized as precursor proteins that are proteolytically processed and secreted by cells in an inactive form as large latent complexes (LLCs) which are then sequestered by ECM proteins, including fibrillin-1, and rapidly released by proteases in response to physiologic or pathologic cues [14]. In Marfan syndrome, a reduction in fibrillin-1 is thought to result in deficient LLC sequestration and a concomitant excess of TGF-β signaling as has been demonstrated in mouse models of this disease [15]. MVP is frequently seen in patients with Marfan syndrome, with studies describing a widely varied prevalence from 28–80% [16,17,18,19,20]. Furthermore, Marfan syndrome mice demonstrate postnatally acquired myxomatous changes in the architecture of the mitral valve that correlate temporally with increased TGF-β signaling, a phenotype that can be rescued in vivo by TGF-β antagonism consistent with a causal mechanism [21]. Loeys-Dietz syndrome is another connective tissue disorder that shares many cardinal features of Marfan syndrome, including an increased prevalence of MVP [22, 23], and results from mutations in several TGF-β signaling pathway members including TGF-β receptors 1 and 2 (TGFBR1 and TGFBR2), the downstream TGF-β signaling pathway effector mothers against decapentaplegic homolog 3 (SMAD3), as well as the cytokines TGF-β 2 and TGF-β 3 (TGFB2 and TGFB3) [24,25,26]. Mutations in other downstream effectors of TGF-β signaling pathways are also associated with MVP including mothers against decapentaplegic homologue 4 (SMAD4) which causes juvenile polyposis [27].
Syndromic forms of MVP are also associated with diseases that result from mutations in critical components of the ECM. Ehlers-Danlos syndrome is characterized by musculoskeletal (hypermobility of joints and muscle hypotonia), cutaneous (elastic and fragile skin), and cardiovascular (vascular aneurysms and valvular lesions) abnormalities [28]. Notably, the clinical Ehlers-Danlos syndrome results from mutations in at least 20 different genes including several different collagen types (COL1A1, COL1A2, COL3A1, COL5A1, COL5A2) as well as enzymes that process, fold, or interact with collagen (ADAMTS2, FKBP14, PLOD1, TNXB) [29]. At least two large cohort studies of over 200 patients with Ehlers-Danlos syndrome have identified an MVP prevalence of approximately 6% [30, 31]. Notably, neither of these studies classified individuals by the causative molecular lesion for their Ehlers-Danlos syndrome raising the possibility that MVP disproportionately affects a subset of these patients. There is also data suggesting a genetic association between other connective tissue diseases and MVP including pseudoxanthoma elasticum [32], Larsen-like syndrome [33], and Williams-Beuren syndrome [34]. In the case of pseudoxanthoma elasticum, a more recent report using modern imaging technologies has questioned the strength of this genetic association with MVP given its presence in only 3 of 67 patients [35].
MVP as Part of Complex Congenital Heart Disease
Several aneuploidies characterized by multiorgan system anomalies including complex congenital heart disease feature mitral valve architectures that share features common to non-syndromic forms of MVP. Trisomy 18 (Edwards syndrome), which includes micrognathia, rocker-bottom feet, biliary atresia, and profound intellectual disability, is associated with a number of cardiac lesions (atrial and ventricular septal defects, tetralogy of Fallot, double-outlet right ventricle, coarctation of the aorta, bicuspid aortic and pulmonary valves) [36]. Mitral valve abnormalities were noted to be relatively common (66%) in one study of 41 postmortem cases [37] in which the leaflets were noted to be redundant and myxomatous with absent or underdeveloped papillary muscles. Trisomy 13 (Patau syndrome) is characterized by polydactyly, cleft lip and palate, hypotelorism, and microphthalmia as well as congenital cardiac defects including atrial and ventricular septal defects, patent ductus arteriosus, and hypoplastic left heart syndrome [36]. Description of mitral valve morphology in these patients is relatively limited, with a single study of echocardiographic evaluation in 14 patients demonstrating evidence of mitral valve dysplasia and absence of a papillary muscle [38]. Klinefelter syndrome results from the presence of an additional X chromosome (47,XXY) with affected patients demonstrating a tall stature, small tetes, and delayed puberty with the most common cardiac manifestations being MVP, a patent ductus arteriosus, and atrial septal defects [36]. However, the association between Klinefelter syndrome and MVP has proven more controversial. Two early patient series describing echocardiographic analyses in Klinefelter syndrome reported MVP in approximately 55% of subjects [39, 40], However more recent and larger studies utilizing modern echocardiographic equipment have failed to reproduce these results, with no evidence of MVP identified in 94 patients with Klinefelter syndrome [41, 42]. The association of MVP with isolated congenital heart disease has also been studied. Tetralogy of Fallot, the most common cyanotic congenital heart defect [43], is associated with good long-term survival following palliation, thus allowing for assessment of MVP in adult patients. A single cohort study in 324 adult tetralogy of Fallot patients identified an 8% prevalence of systolic mitral valve abnormalities, with 4.6% demonstrating MVP [44].
Genetic Basis of Non-Syndromic MVP
Pedigree Studies
There is a long-standing appreciation that a subset of non-syndromic MVP (i.e., MVP not associated with other disorders) is inherited in a familial pattern with pedigrees demonstrating both autosomal dominant and X-linked patterns of inheritance with variable penetrance dependent upon both age and sex [45, 46]. Genetic linkage analysis, a powerful tool that allows for detection of the specific chromosomal location of disease genes, led to the identification of four loci associated with non-syndromic MVP [47]. It is important to note that within the category of non-syndromic MVP, two specific phenotypes have been described in the surgical literature [48]. Barlow’s disease is associated with diffuse myxomatous degeneration leading to prolapse of most leaflet scallops into the left atrium during systole, severe mitral annular enlargement, and elongated chordae along with histologic evidence of disrupted collagen and elastic layers. Fibroelastic deficiency, on the other hand, is associated with thinned leaflets, mild-to-moderate mitral annular dilation, and thin chordae with histologic evaluation demonstrating decreased connective tissue. Indeed, Barlow’s disease and fibroelastic deficiency are themselves associated with different natural histories with the former noted in patients presenting for surgery at young to middle-ages in which chordal rupture is rarely present and the latter in patients present at older age with chordal rupture after a shorter clinical history. These linkage analyses yielded three loci for autosomal dominant non-syndromic MVP and one for X-linked MVP, aptly named myxomatous mitral valve prolapse 1-3 (MMVP1, MMVP2, MMVP3) and X-linked myxomatous valvular dystrophy (XMVD) [47] (Table 10.1).
The earliest description of familial clustering in MVP came in 1969 upon the identification of a pedigree in St. Louis, MO wherein all male members in three generations of this family exhibited congenital mitral insufficiency, suggesting a X-linked inheritance pattern [49]. Almost 30-years later, a five-generation pedigree of French origin consisting of 92 individuals in which 21 affected individuals had both MVP and a mild form of hemophilia A (a hereditary bleeding disorder that at the time had a known X-linked recessive pattern of inheritance) allowed for the rapid chromosomal mapping of XMVD to an 8 centimorgan region of the Xq28 telomeric region [50]. Detailed clinical features of this pedigree identified full-penetrance in affected males with some moderately affected female patients [51]. This region was later more finally mapped to a 2.5 megabase region with the screening of candidate genes ultimately leading to the identification of a P637Q missense mutation in filamin-A (FLNA) as the causative allele. Analysis of three other independent families identified two other missense mutations (G288R and V711D) and a 1944 kilobase in-frame genomic deletion removing exons 16–19 further supporting this conclusion [52]. FLNA encodes filamin-A, a large cytoplasmic protein that can function as a molecular tether, linking ECM-bound cell-surface integrins with the actin cytoskeleton [53, 54]. Filamin-A has also been demonstrated to regulate TGF-β signaling by associating with the known downstream TGF-β signaling effector mothers against decapentaplegic homologs 2 (SMAD2) [55]. Homozygous loss of murine Flna allele results in mid-gestational embryonic lethality with affected animals demonstrating severe cardiac malformations [56, 57]. Interestingly, immunohistochemical expression analysis of filamin-A in mice demonstrates robust expression during embryogenesis and early postnatal life with significantly reduced expression in the adult [58, 59], suggesting a potential developmental basis in this genetically distinct non-syndromic MVP phenotype. Indeed, tissue-specific deletion of Flna in the developing murine atrioventricular valves results in a myxomatous mitral valve phenotype by 2-months of age, a phenotype that likely results from a developmental error in how extracellular matrix is assembled and organized during fetal development [59].
Another family with an autosomal dominant pattern of MVP inheritance was described in 2003 upon identification of the proband as a volunteer in a course teaching echocardiographic imaging [60]. Echocardiograms and DNA were obtained for 28 individuals from this pedigree of Western European descent with 12 diagnosed with MVP, 3 with an intermediate phenotype, and the remaining 13 being unaffected. Linkage analysis revealed that the locus for autosomal dominant MVP in this family lied in a 4.3 centimorgan region on chromosome 11p15.4, which was at that time designated “MMVP2” given that it was the second such locus described. This region spanned approximately 4.45 megabases and, at the time, included several large gaps in sequence coverage and at least 46 genes [60]. The causative mutation was described 12 years later after 4 affected individuals in this pedigree underwent tiled capture and high-throughput sequencing of genomic DNA spanning 2.1 megabases within this locus yielding 4,891 single nucleotide variants and insertion/deletion polymorphisms which, upon filtering for rare variants shared among all affected members of the pedigree, yielded three heterozygous protein coding variants [61]. Two lied in DCHS1, a member of the cadherin superfamily of proteins (P197L and R2513H), and one in APBB1, the amyloid-beta A4 precursor protein-binding family B member 1 (R481H). In vivo functional assays performed in zebrafish and mice supported DCHS1 as the causative allele which was confirmed by the identification of two additional families in which MVP segregated with a novel DCHS1 protein variant (R2330C). DCHS1 is the human homologue of the drosophila gene dachsous (ds), which serves as a core component of the Wnt/planar cell polarity (PCP) signaling pathway which establishes cell orientation within the epithelial plane [62]. Mice homozygous for inactivating mutations of murine Dchs1 results in neonatal mortality with multiorgan dysfunction [63]. However, adult Dchs1 heterozygous mice exhibit mitral valve prolapse that phenocopied what was identified in the originally described proband (pronounced involvement of an elongated posterior leaflet that shifts coaptation anteriorly) with histologic analysis confirming leaflet thickening and myxomatous degeneration [60, 61] (Fig. 10.1). Evaluation valve morphology during embryonic development revealed dose-dependent alterations in the shape and anatomical patterning of the mitral valve, again consistent with a developmentally based disease that progresses over the lifespan of affected individuals.

Murine models of non-syndromic mitral valve prolapse recapitulate anatomic and histopathologic findings of human disease. Histologic examination of 6-month old mice heterozygous for a Dzip1 missense allele identified in non-syndromic familial mitral valve prolapse (MVP) demonstrate dysmorphic posterior leaflets (PL) when compared with wild type littermate controls. Similarly, immunohistochemical analyses of 9-month old mice that are haploinsufficient for Dchs1 (another allele associated with non-syndromic familial MVP) demonstrate myxomatous degeneration and expansion of proteoglycan expression when compared with wild type littermate controls. (AL = anterior leaflet; red = collagen, green = proteoglycan)
A third family of 46 individuals demonstrating MVP with an autosomal dominant pattern of inheritance was described in 2005 [64]. The proband in this third-generation pedigree of Western European descent was a dentist who self-referred for family analysis of MVP, with 43 individuals undergoing echocardiography and DNA sampling (9 of whom were affected). In this case, linkage analysis narrowed the causative locus to an 8.2 megabase candidate region on chromosome 13q31.3–q32.1 then designated “MMVP3” as it was the third such locus described in the literature. Importantly, this study was the first to demonstrate that minimal displacement or “prodromal” (no diagnostic leaflet displacement beyond the mitral annulus, but with a leaflet closure/coaptation pattern shifted anteriorly resembling that of patients with fully expressed MVP) valve morphologies that were previously classified as normal variants share the same underlying genetic substrate [64]. A subsequent study exploring the role of primary cilia in MVP assessed the linked 8.2 megabase region for candidate genes with known functions in ciliogenesis or cilia signaling, leading to the identification of DAZ-interacting protein 1 (DZIP1). Subsequent whole-exome sequencing of four affected individuals from this pedigree revealed a single missense variant in DZIP1 that affects both expressed isoforms (S70R/S24R), representing the only coding change within the linkage interval that segregated with the disease phenotype [65]. As human DZIP1 and mouse Dzip1 are highly conserved at the amino acid level, the missense variant identified in this family was introduced into the endogenous murine locus to assess the phenotypic consequences in vivo. Adult mice heterozygous for this Dzip1 missense variant develop myxomatous mitral valves and functional MVP. Further biochemical and molecular analysis on these mutant animals suggested that this Dzip1 missense variant represents a loss-of-function allele resulting in dysregulation of ECM-associated pathways [65] (Fig. 10.1).
The first genetic locus identified to segregate in an autosomal dominant pattern with MVP (“MMVP1”) paradoxically remains the only locus with an unidentified causal gene to date (Table 10.1). The study describing MMVP1 systematically screened first-degree relatives of 17 patients who underwent MV repair yielding 4 pedigrees of varied ethnicities (Ashkenazi Jewish, western France, and eastern France) with a total of 79 subjects undergoing echocardiography and blood sampling, 25 of whom were affected [66]. Genetic linkage studies narrowed the locus to a 5 centimorgan region of chromosome 16 (16p11.2–16p12.1). The causative allele in this setting remains to be identified. The 5 centimorgan region defined by the sequence tag sites D16S3068 and D16S420 identified from these pedigrees span a region of approximately 1.3 megabases [66]. This region contains at least eight protein coding genes along with several long intergenic non-coding RNAs.
Genome-Wide Association Studies
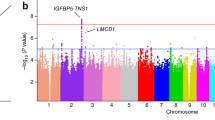
Genome-wide association studies (GWAS) test genetic variants across the genomes of many individuals to identify genotype-phenotype associations and have revolutionized the fields of complex disease genetics, leading to the identification of novel disease susceptibility genes and biological pathways [67]. As genetic variants are present along a spectrum of allele frequencies and effect sizes, GWAS is best suited for the identification of common variants with small effects, intermediate frequency variants with moderate effects, or highly penetrant rare mutations. Rare variants with small effect sizes are difficult to detect by GWAS while common variants with large effects are not commonly seen in complex disease traits [67]. GWAS in the context of MVP, consisting of a meta-analysis including 2,864 cases and 9,218 controls, identified 6 risk loci that are robustly associated with MVP [68]. These 6 loci implicated four intergenic (IGFBP5-TNS1, SETD4-CBR1, PITBNB-MN1, and PCNX-SIPA1L1) and two intronic regions (LMCD1 and SMG6). These candidates were narrowed from an initial list of 53 genes that were located within 500 kilobases to 1 megabase from the lead associated single nucleotide polymorphism (SNP) by taking into account proximity to the SNP, level of cardiac expression, proximity to previously identified GWAS signals for cardiovascular traits, and a biological link with mitral valve or cardiac development [68]. This study highlighted functional data for two of these genes: LMCD1 (a transcription factor known to repress the activity of the critical cardiac transcription factor GATA6) knockdown in zebrafish embryos caused regurgitation of the single atrioventricular valve of the two-chamber heart in these animals while Tensin 1 (a protein that localizes to focal adhesions and interacts with actin) showed sustained expression by immunohistochemistry during murine valve morphogenesis with adult Tns1-/- mice demonstrating enlarged mitral valves with evidence of myxomatous degeneration. A follow-up study sought to utilize the same data to identify additional loci that despite not having met the stringent GWAS statistical thresholds (P < 5 × 108) may still have biological relevance for MVP by applying computational pathway enrichment tools [69]. This led to the identification of GLIS family zinc finger 1 (GLIS1), a Kruppel-like zinc finger protein containing transactivation and repressor functions as a potential susceptibility gene for MVP, supported by its expression of murine Glis1 in developing valvular endothelial and interstitial cells [69].
Mitral Valve Prolapse: An Unrecognized Ciliopathy?
Primary cilia are microtubule-based solitary organelles that extend from the surface of most vertebrate cell types to receive and process molecular and mechanical signaling cues [70]. Long regarded as an “evolutionary vestige,” the primary cilium has emerged an essential signaling hub that coordinates a variety of signaling pathways including those regulated by Hedgehog, G protein-coupled receptors, Wnt, TGF-β, and receptor tyrosine kinases to control developmental processes, tissue plasticity, and organ function [71]. Primary ciliary dysfunction underlies a pleiotropic group of disease and syndromic disorders termed ciliopathies which may affect a number of different organs in the body (e.g., Bardet-Biedl syndrome, Leber’s congenital amaurosis, and polycystic kidney disease). Indeed, an association between MVP has long been appreciated in the context of autosomal dominant polycystic kidney disease (ADPKD) [72]. ADPKD patients have a 10-fold increase in rates of MVP compared with the general population [73, 74]. Furthermore, unbiased forward genetics approaches in mice have revealed a critical role for the cilium and cilia transduced signaling pathways in the pathogenesis of congenital heart disease [75]. As our collective understanding of the biology associated with causative genes for MVP increases, there is a growing body of evidence to support that some forms of non-syndromic MVP are ciliopathies. All three of the known causative genes identified from large linkage analyses for non-syndromic MVP (DCHS1, DZIP1, and FLNA) are intimately involved in ciliogenesis or ciliary function (Fig. 10.2). DCHS1 (the causative gene identified at the MMVP2 locus) encodes Dachsous-1, a component of the primary cilium. In the context of the MMVP3 locus, the determination that DZIP1 was the causative allele emanated from a series of experiments exploring the hypothesis that primary cilia may contribute to valvulogenesis [65]. These studies found that primary cilia are robustly expressed on mesenchymal cells within valve primordia with their length changing over the course of development concomitant with the type of ECM produced within the mitral valves. In fact, the way that DZIP1 was identified as the causative allele amongst the 16 genes in the MMVP3 locus was through its known role in ciliogenesis and cilia signaling [65]. At the time of its discovery as the causative allele at the XMVD locus, FLNA was known to associate with downstream effectors of the TGF-β signaling pathway, overactivity of which was thought to contribute to the increased incidence of MVP in Marfan syndrome patients. Subsequent studies have since shown that filamin-A directly interacts with meckelin, a transmembrane receptor that localizes to the primary cilium and basal body (the protein structure at the base of the cilium) [76]. MKS3, the gene that encodes meckelin, is mutated in Meckel-Gruber syndrome, a ciliopathy characterized by renal cystic dysplasia, hepatic developmental defects, and severe neurodevelopmental anomalies. Interestingly, studies exploring this filamin-a/meckelin interaction have demonstrated that loss of filamin-A leads to defects in basal body positioning and ciliogenesis [76]. GLIS1, the aforementioned gene identified through additional bioinformatic analyses of previously published GWAS data for MVP, has also been suggested to have a potential role in ciliogenesis [77]. A mechanistic basis for how defective ciliary function may lead to MVP was best illustrated by tissue-specific knockout of the ciliogenic gene intraflagellar transport protein 88 (ift88) from endothelial-derived mesenchymal cells in mice in vivo. Ift88-loss led to the loss of primary cilia, valve enlargement, and a molecular signal demonstrating robust activation of ECM gene pathways consistent with early stages of myxomatous degeneration [65].

Mitral valve prolapse as a ciliopathy. Primary cilia play a critical role during normal cardiac valvular morphogenesis. Three of the four loci identified as being causative for familial non-syndromic mitral valve prolapse (DCHS1, DZIP1, and FLNA) are all involved in ciliogenesis or ciliary function
Future Perspectives
Technological Advances
While significant progress has been made in our understanding of the genetic basis for MVP, there remains much to be discovered. Technological advances have played an important role in our ability to accelerate efforts to identify the molecular underpinnings of these diseases which, to date, have yielded important insights into the genes and pathways that ultimately result in an MVP phenotype and a growing recognition that this disease results from primary pathologic insults that occur during development. These findings further underscore that MVP is not a single disease, but instead exists along a phenotypic spectrum wherein a degree of dosage sensitivity conferred by individual risk alleles collectively drive alterations in highly stereotyped and regulated morphogenetic events during cardiac development, likely through a core group of signaling pathways, to yield a mature phenotype that may meet the diagnostic criteria for MVP or instead yield a “prodromal” intermediate [64].
Indeed, technological innovations have changed enormously our approach to understanding the genetic basis of complex disease. Complex and iterative linkage analyses are no longer required as the cost, throughput, and availability of DNA sequencing has significantly reduced the barriers to the application of these cutting-edge technologies. One group recently utilized an exome slice methodology wherein 101 probands with MVP underwent sequencing of the coding regions of 522 genes associated with cardiac development and/or disease [78]. This study found 1 individual with a likely pathogenic mutation in DCHS1 along with an additional 10 probands with likely pathogenic mutations in six different genes that have been associated with cardiomyopathies (DSP, HCN4, MYH6, TMEM67, TRPS1, and TTN). Increasing the yield of studies of this nature necessitates that unbiased interrogations of coding or non-coding DNA sequences in affected and unaffected populations be linked with very careful clinical phenotyping of each individual. Towards that end, the specificity of echocardiographic diagnosis for MVP was increased significantly upon an improvement of our understanding of the three-dimensional shape of the mitral valve [79, 80]. As echocardiographic imaging technologies continue to improve yielding higher resolution images and the ability to render multi-dimensional projections of cardiac anatomy, we may be able to better categorize MVP into subtypes that will yield a greater signal-to-noise ratio in subsequent unbiased genomic interrogations. Whole-exome sequencing, a methodology wherein the entire protein coding DNA sequence of an individual genome is defined, in carefully phenotyped pedigrees has the ability to reveal novel modes of inheritance for MVP. Oligogenic patterns of inheritance have been suggested in hypertrophic cardiomyopathy [81] and recently shown in the context of left ventricular noncompaction cardiomyopathy [82]. It is also interesting to note that, even within families with non-syndromic MVP who share the same underlying causative genetic lesion, the phenotypic spectrum of disease presentation is quite varied suggesting the existence of unlinked enhancer and/or suppressor loci. Our ability to systematically identify and define these modifier loci will have important prognostic implications for MVP patients with the potential to inform clinical care decisions.
Genetic Discoveries Outside of Coding Regions
A critical challenge in understanding the genetic underpinnings of all inherited disease lies in the assessment of genetic variation outside of coding regions of the genome. As outlined, the most significant loci identified in the GWAS performed for MVP were within intergenic regions of the genome. While intergenic variants from GWAS can lead to incredible discoveries regarding gene regulation and disease pathogenesis [83, 84], examples of this nature are exceedingly rare. Although sequencing the entire genome of an individual has become a relatively straightforward undertaking both in terms of cost and availability, we remain in the nascent stages of our ability to interpret the consequences of variants in non-coding sequences.
Genetic Basis of Sudden Cardiac Death Risk in MVP
Another important finding associated with MVP whose genetic etiology requires further evaluation is the increased risk for sudden cardiac death, which is roughly twice that observed in the general population [85, 86]. Although much of this risk can be attributed to left ventricular dysfunction secondary to severe mitral regurgitation [87], life-threatening ventricular arrhythmias can be seen in MVP patients with even trivial to mild mitral regurgitation [88]. A recent case report described familial segregation of a truncating variant in FLNC, which encodes filamin-C (An actin-binding protein necessary for the structural integrity of the sarcomere in cardiac and skeletal muscle cells) with both MVP and ventricular arrhythmias [89]. FLNC mutations have been associated with dilated cardiomyopathy [90, 91], hypertrophic cardiomyopathy [92], and restrictive cardiomyopathy [93,94,95], but not with valvular abnormalities. Whether both the MVP and ventricular arrhythmia phenotypes result from a single genetic lesion as is suggested by this report, or instead results from cosegregation of an independent risk modifier remains to be determined. Uncovering the genetic predisposition to ventricular arrhythmias and sudden cardiac death in the setting of MVP is an important area for future investigation.
Conclusions
MVP is a common, highly heritable phenotype and is associated with important clinical sequelae including severe mitral regurgitation and sudden cardiac death. Multiple genes have been identified from pedigree investigations and GWAS studies of individuals with MVP and have been linked to abnormal valve embryogenesis and cell differentiation, ultimately leading to ECM dysregulation and myxomatous degeneration of the mitral valve. As our collective understanding of the biology associated with causative genes for MVP increases, there is a growing body of evidence to support that some forms of non-syndromic MVP are ciliopathies. Indeed, all three of the known causative genes identified from large linkage analyses for non-syndromic MVP (DCHS1, DZIP1, and FLNA) are intimately involved in ciliogenesis or ciliary function. Genetic discoveries overall have exponentially accelerated our understanding of MVP. However, many questions remain unanswered in relation to MVP pathophysiology, clinical progression, and therapeutic options. Future studies are needed to address these important issues.
References
Dziadzko V, Clavel M-A, Dziadzko M, Medina-Inojosa JR, Michelena H, Maalouf J, Nkomo V, Thapa P, Enriquez-Sarano M. Outcome and undertreatment of mitral regurgitation: a community cohort study. Lancet. 2018;391:960–9.
Delling FN, Rong J, Larson MG, Lehman B, Osypiuk E, Stantchev P, Slaugenhaupt SA, Benjamin EJ, Levine RA, Vasan RS. Familial clustering of mitral valve prolapse in the community. Circulation. 2015;131:263–8.
Delling FN, Li X, Li S, et al. Heritability of mitral regurgitation: observations from the framingham heart study and Swedish population. Circ Cardiovasc Genet. 2017; https://doi.org/10.1161/CIRCGENETICS.117.001736.
Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–9.
Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008;118:1864–80.
Tamura K, Fukuda Y, Ishizaki M, Masuda Y, Yamanaka N, Ferrans VJ. Abnormalities in elastic fibers and other connective-tissue components of floppy mitral valve. Am Heart J. 1995;129:1149–58.
Grande-Allen KJ, Griffin BP, Calabro A, Ratliff NB, Cosgrove DM 3rd, Vesely I. Myxomatous mitral valve chordae. II: Selective elevation of glycosaminoglycan content. J Heart Valve Dis. 2001;10:325–32. discussion 332–3
Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–32.
Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–8.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–200.
Kainulainen K, Pulkkinen L, Savolainen A, Kaitila I, Peltonen L. Location on chromosome 15 of the gene defect causing Marfan syndrome. N Engl J Med. 1990;323:935–9.
Dietz HC, Cutting GR, Pyeritz RE, et al. Defects in the fibrillin gene cause the Marfan syndrome; linkage evidence and identification of a missense mutation. Nature. 1991;352:337–9.
Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366:1965–76.
ten Dijke P, Arthur HM. Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–69.
Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–11.
Pyeritz RE, Wappel MA. Mitral valve dysfunction in the Marfan syndrome. Clinical and echocardiographic study of prevalence and natural history. Am J Med. 1983;74:797–807.
Hirata K, Triposkiadis F, Sparks E, Bowen J, Boudoulas H, Wooley CF. The Marfan syndrome: cardiovascular physical findings and diagnostic correlates. Am Heart J. 1992;123:743–52.
van Karnebeek CD, Naeff MS, Mulder BJ, Hennekam RC, Offringa M. Natural history of cardiovascular manifestations in Marfan syndrome. Arch Dis Child. 2001;84:129–37.
Porciani MC, Attanasio M, Lepri V, Lapini I, Demarchi G, Padeletti L, Pepe G, Abbate R, Gensini GF. Prevalence of cardiovascular manifestations in Marfan syndrome. Ital Heart J Suppl. 2004;5:647–52.
Taub CC, Stoler JM, Perez-Sanz T, Chu J, Isselbacher EM, Picard MH, Weyman AE. Mitral valve prolapse in Marfan syndrome: an old topic revisited. Echocardiography. 2009;26:357–64.
Ng CM, Cheng A, Myers LA, et al. TGF-β–dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–92.
van der Linde D. Aggressive cardiovascular phenotype of aneurysms-osteoarthritis syndrome caused by pathogenic SMAD3 variants. J Am Coll Cardiol. 2012;60(5):397–403.
Attias D, Stheneur C, Roy C, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in marfan syndrome and related disorders. Circulation. 2009;120:2541–9.
Rienhoff HY Jr, Yeo C-Y, Morissette R, et al. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys-Dietz syndrome. Am J Med Genet A. 2013;161A:2040–6.
MacCarrick G, Black JH, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, Sponseller PD, Loeys B, Dietz HC. Loeys–Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16:576–87.
Kuechler A, Altmüller J, Nürnberg P, Kotthoff S, Kubisch C, Borck G. Exome sequencing identifies a novel heterozygous TGFB3 mutation in a disorder overlapping with Marfan and Loeys-Dietz syndrome. Mol Cell Probes. 2015;29:330–4.
Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A. 2011;155A:1165–9.
Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med. 2010;12:597–605.
Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C: Semin Med Genet. 2017;175:8–26.
Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr. 2011;158:826–830.e1.
Asher SB, Chen R, Kallish S. Mitral valve prolapse and aortic root dilation in adults with hypermobile Ehlers-Danlos syndrome and related disorders. Am J Med Genet A. 2018;176:1838–44.
Lebwohl MG, Distefano D, Prioleau PG, Uram M, Yannuzzi LA, Fleischmajer R. Pseudoxanthoma elasticum and mitral-valve prolapse. N Engl J Med. 1982;307:228–31.
Baasanjav S, Al-Gazali L, Hashiguchi T, et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89:15–27.
Collins RT, Thomas Collins R. Cardiovascular disease in Williams syndrome. Circulation. 2013;127:2125–34.
Prunier F, Terrien G, Le Corre Y, et al. Pseudoxanthoma elasticum: cardiac findings in patients and Abcc6-deficient mouse model. PLoS One. 2013;8:e68700.
Pierpont ME, Basson CT, Benson DW Jr, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–38.
Van Praagh S, Truman T, Firpo A, Bano-Rodrigo A, Fried R, McManus B, Engle MA, Van Praagh R. Cardiac malformations in trisomy-18: a study of 41 postmortem cases. J Am Coll Cardiol. 1989;13:1586–97.
Musewe NN, Alexander DJ, Teshima I, Smallhorn JF, Freedom RM. Echocardiographic evaluation of the spectrum of cardiac anomalies associated with trisomy 13 and trisomy 18. J Am Coll Cardiol. 1990;15:673–7.
Fricke GR, Mattern HJ, Schweikert HU. Mitral valve prolapse in Klinefelter syndrome. Lancet. 1981;2:1414.
Fricke GR, Mattern HJ, Schweikert HU, Schwanitz G. Klinefelter’s syndrome and mitral valve prolapse. an echocardiographic study in twenty-two patients. Biomed Pharmacother. 1984;38:88–97.
Andersen NH, Bojesen A, Kristensen K, Birkebaek NH, Fedder J, Bennett P, Christiansen JS, Gravholt CH. Left ventricular dysfunction in Klinefelter syndrome is associated to insulin resistance, abdominal adiposity and hypogonadism. Clin Endocrinol. 2008;69:785–91.
Pasquali D, Arcopinto M, Renzullo A, et al. Cardiovascular abnormalities in Klinefelter syndrome. Int J Cardiol. 2013;168:754–9.
van der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–7.
Agarwal A, Harris IS, Mahadevan VS, Foster E. Coexistence of abnormal systolic motion of mitral valve in a consecutive group of 324 adult Tetralogy of Fallot patients assessed with echocardiography. Open Heart. 2016;3:e000518.
Devereux RB. Inheritance of mitral valve prolapse: effect of age and sex on gene expression. Ann Intern Med. 1982;97:826.
Zuppiroli A, Roman MJ, O’Grady M, Devereux RB. A family study of anterior mitral leaflet thickness and mitral valve prolapse. Am J Cardiol. 1998;82(823–6):A10.
Delling FN, Vasan RS. Epidemiology and pathophysiology of mitral valve prolapse: new insights into disease progression, genetics, and molecular basis. Circulation. 2014;129:2158–70.
Fornes P, Heudes D, Fuzellier JF, Tixier D, Bruneval P, Carpentier A. Correlation between clinical and histologic patterns of degenerative mitral valve insufficiency: a histomorphometric study of 130 excised segments. Cardiovasc Pathol. 1999;8:81–92.
Monteleone PL, Fagan LF. Possible X-linked congenital heart disease. Circulation. 1969;39:611–4.
Kyndt F, Schott JJ, Trochu JN, et al. Mapping of X-linked myxomatous valvular dystrophy to chromosome Xq28. Am J Hum Genet. 1998;62:627–32.
Trochu JN, Kyndt F, Schott JJ, Gueffet JP, Probst V, Bénichou B, Le Marec H. Clinical characteristics of a familial inherited myxomatous valvular dystrophy mapped to Xq28. J Am Coll Cardiol. 2000;35:1890–7.
Kyndt F, Gueffet J-P, Probst V, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–9.
Shizuta Y, Shizuta H, Gallo M, Davies P, Pastan I. Purification and properties of filamin, and actin binding protein from chicken gizzard. J Biol Chem. 1976;251:6562–7.
Kim H, Sengupta A, Glogauer M, McCulloch CA. Filamin A regulates cell spreading and survival via beta1 integrins. Exp Cell Res. 2008;314:834–46.
Sasaki A, Masuda Y, Ohta Y, Ikeda K, Watanabe K. Filamin Associates with Smads and Regulates Transforming Growth Factor-β Signaling. J Biol Chem. 2001;276:17871–7.
Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci U S A. 2006;103:19836–41.
Hart AW, Morgan JE, Schneider J, West K, McKie L, Bhattacharya S, Jackson IJ, Cross SH. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum Mol Genet. 2006;15:2457–67.
Norris RA, Moreno-Rodriguez R, Wessels A, et al. Expression of the familial cardiac valvular dystrophy gene, filamin-A, during heart morphogenesis. Dev Dyn. 2010;239:2118–27.
Sauls K, de Vlaming A, Harris BS, et al. Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc Res. 2012;96:109–19.
Freed LA, Acierno JS Jr, Dai D, Leyne M, Marshall JE, Nesta F, Levine RA, Slaugenhaupt SA. A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am J Hum Genet. 2003;72:1551–9.
Durst R, Sauls K, Peal DS, et al. Mutations in DCHS1 cause mitral valve prolapse. Nature. 2015;525:109–13.
Matis M, Axelrod JD. Regulation of PCP by the Fat signaling pathway. Genes Dev. 2013;27:2207–20.
Mao Y, Mulvaney J, Zakaria S, Yu T, Morgan KM, Allen S, Basson MA, Francis-West P, Irvine KD. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development. 2011;138:947–57.
Nesta F, Leyne M, Yosefy C, Simpson C, Dai D, Marshall JE, Hung J, Slaugenhaupt SA, Levine RA. New locus for autosomal dominant mitral valve prolapse on chromosome 13: clinical insights from genetic studies. Circulation. 2005;112:2022–30.
Toomer KA, Yu M, Fulmer D, et al. Primary cilia defects causing mitral valve prolapse. Sci Transl Med. 2019; https://doi.org/10.1126/scitranslmed.aax0290.
Disse S, Abergel E, Berrebi A, Houot AM, Le Heuzey JY, Diebold B, Guize L, Carpentier A, Corvol P, Jeunemaitre X. Mapping of a first locus for autosomal dominant myxomatous mitral-valve prolapse to chromosome 16p11.2–p12.1. Am J Hum Genet. 1999;65:1242–51.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20:467–84.
Dina C, Bouatia-Naji N, Tucker N, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet. 2015;47:1206–11.
Yu M, Georges A, Tucker NR, et al. Genome-wide association study–driven gene-set analyses, genetic, and functional follow-up suggest GLIS1 as a susceptibility gene for mitral valve prolapse. Circ Genom Precis Med. 2019; https://doi.org/10.1161/circgen.119.002497.
Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123:499–503.
Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol. 2019;15:199–219.
Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med. 1988;319:907–12.
Ivy DD, Shaffer EM, Johnson AM, Kimberling WJ, Dobin A, Gabow PA. Cardiovascular abnormalities in children with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;5:2032–6.
Lumiaho A, Ikäheimo R, Miettinen R, Niemitukia L, Laitinen T, Rantala A, Lampainen E, Laakso M, Hartikainen J. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis. 2001;38:1208–16.
Li Y, Klena NT, Gabriel GC, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520–4.
Adams M, Simms RJ, Abdelhamed Z, et al. A meckelin-filamin A interaction mediates ciliogenesis. Hum Mol Genet. 2012;21:1272–86.
Yasuoka Y, Matsumoto M, Yagi K, Okazaki Y. Evolutionary history of GLIS genes illuminates their roles in cell reprogramming and ciliogenesis. Mol Biol Evol. 2020;37:100–9.
van Wijngaarden AL, Hiemstra YL, Koopmann TT, Ruivenkamp CAL, Aten E, Schalij MJ, Bax JJ, Delgado V, Barge-Schaapveld DQCM, Ajmone Marsan N. Identification of known and unknown genes associated with mitral valve prolapse using an exome slice methodology. J Med Genet. 2020; https://doi.org/10.1136/jmedgenet-2019-106715.
Levine RA, Handschumacher MD, Sanfilippo AJ, Hagege AA, Harrigan P, Marshall JE, Weyman AE. Three-dimensional echocardiographic reconstruction of the mitral valve, with implications for the diagnosis of mitral valve prolapse. Circulation. 1989;80:589–98.
Perloff JK, Child JS. Mitral valve prolapse. Evolution and refinement of diagnostic techniques. Circulation. 1989;80:710–1.
Li L, Bainbridge MN, Tan Y, Willerson JT, Marian AJ. A potential oligogenic etiology of hypertrophic cardiomyopathy. Circ Res. 2017;120:1084–90.
Gifford CA, Ranade SS, Samarakoon R, et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science. 2019;364:865–70.
Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–9.
Gupta RM, Hadaya J, Trehan A, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 2017;170:522–33.
Nishimura RA, McGoon MD, Shub C, Miller FA, Ilstrup DM, Jamil Tajik A. Echocardiographically documented mitral-valve prolapse. N Engl J Med. 1985;313:1305–9.
Zipes DP, Wellens HJJ. Sudden cardiac death. Circulation. 1998;98:2334–51.
Kligfield P, Levy D, Devereux RB, Savage DD. Arrhythmias and sudden death in mitral valve prolapse. Am Heart J. 1987;113:1298–307.
Vohra J, Sathe S, Warren R, Tatoulis J, Hunt D. Malignant ventricular arrhythmias in patients with mitral valve prolapse and mild mitral regurgitation. Pacing Clin Electrophysiol. 1993;16:387–93.
Bains S, Tester DJ, Asirvatham SJ, Noseworthy PA, Ackerman MJ, Giudicessi JR. A novel truncating variant in FLNC-encoded filamin C may serve as a proarrhythmic genetic substrate for arrhythmogenic bileaflet mitral valve prolapse syndrome. Mayo Clin Proc. 2019;94:906–13.
Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–51.
Begay RL, Graw SL, Sinagra G, et al. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell–cell adhesion structures. J Am Coll Cardiol EP. 2018;4:504–14.
Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014; https://doi.org/10.1038/ncomms6326.
Brodehl A, Ferrier RA, Hamilton SJ, et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat. 2016;37:269–79.
Tucker NR, McLellan MA, Hu D, et al. Novel mutation in FLNC (Filamin C) causes familial restrictive cardiomyopathy. Circ Cardiovasc Genet. 2017; https://doi.org/10.1161/CIRCGENETICS.117.001780.
Kiselev A, Vaz R, Knyazeva A, et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum Mutat. 2018;39:1161–72.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Padmanabhan, A., Delling, F.N. (2021). Genetics of Mitral Valve Disease. In: Wells, F.C., Anderson, R.H. (eds) Mitral Valve Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-67947-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-67947-7_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67946-0
Online ISBN: 978-3-030-67947-7
eBook Packages: MedicineMedicine (R0)