
Summary
Manganese is an essential trace metal that is a constituent of metalloenzymes and is required as an enzyme activator. Blood manganese levels are under tight homeostatic control by the liver as both manganese overload and deficiency impair neuronal function and integrity.
To date, three inherited manganese transporter defects have been identified that lead to abnormal blood manganese levels: mutations in SLC30A10 (hypermanganesaemia with dystonia 1, HMNDYT1) and SLC39A14 (hypermanganesaemia with dystonia 2, HMNDYT2) cause manganese overload, while mutations in SLC39A8 (Congenital Disorder of Glycosylation, Type IIn; CDG2N) cause manganese deficiency. SLC39A14 and SLC30A10 are required for hepatic uptake and adequate biliary excretion of manganese, respectively. Both these transporter defects are characterised by childhood-onset, progressive Parkinsonism-dystonia due to the accumulation of manganese in the basal ganglia, particularly the globus pallidus, with pathognomonic MRI brain appearances of hyperintensity on T1-weighted images. Whole blood manganese levels are highly raised. In addition to the movement disorder, SLC30A10 loss-of-function causes liver disease, polycythaemia and depletion of iron stores. Intravenous chelation with disodium calcium edetate and iron supplementation effectively lowers the manganese load and can lead to significant improvement in neurological symptoms and halt the progress of liver disease.
SLC39A8 is required for manganese uptake into the organism. Loss-of-function leads to a manganese deficiency syndrome characterised by neurodevelopmental delay, seizures, dystonia and short stature. Biochemically, SLC39A8 deficiency causes hypomanganesaemia and a characteristic dysglycosylation pattern corresponding to a type II congenital disorder of glycosylation because manganese acts as a cofactor for the β-1,4-galactosyltransferase. In addition, manganese deficiency leads to respiratory chain abnormalities and Leigh-like mitochondrial disease. Manganese supplementation can improve clinical symptoms and normalise biochemical findings.
Manganese dyshomeostasis has also been observed in a juvenile type of Parkinson’s disease associated with supranuclear gaze palsy, spasticity and dementia due to mutations in ATP13A2 (PARK9), also known as Kufor-Rakeb Syndrome. ATP13A2 has been shown to transport manganese from the cytosol to the lysosome. In addition, the phenotype can vary from neuronal ceroid lipofuscinosis type 12 (CLN12) to complicated hereditary spastic paraplegia (HSP).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
Manganese is one of the six transition metals essential for human metabolism. It is required as a cofactor for numerous enzymatic reactions including glycosylation and phosphorylation, and is involved in amino acid, lipid and carbohydrate metabolism, immune function, bone and connective tissue growth and blood clotting. As a constituent of metalloenzymes such as the manganese superoxide dismutase, it acts as a scavenger of reactive oxygen species (Chen et al. 2015).
Manganese levels are under precise homeostatic control because both excess and deficiency of manganese are deleterious for neuronal function and integrity. Excess manganese accumulates in the basal ganglia and causes a clinical syndrome known as manganism—an extrapyramidal movement disorder characterised by dystonia, bradykinesia and rigidity, accompanied by psychiatric and cognitive defects (Chen et al. 2015). Manganese overload can occur in inherited manganese transporter defects (HMNDYT1 and HMNDYT2 caused by mutations in SLC30A10 and SLC39A14, respectively) or as a result of environmental overexposure, excess manganese in parenteral nutrition or impaired hepatic excretion in patients with liver cirrhosis (Tuschl et al. 2012, 2016; Quadri et al. 2012). Manganese deficiency, on the other hand, leads to dysglycosylation and impaired mitochondrial function. Due to its ubiquitous presence in the diet, acquired manganese deficiency rarely occurs and, hence, deficiency of manganese is only observed in an inherited manganese transporter defect caused by SLC39A8 mutations (congenital disorder of glycosylation type IIn) (Park et al. 2015a; Boycott et al. 2015; Riley et al. 2017).
Tight homeostatic control of intestinal absorption and biliary excretion of manganese maintains stable tissue concentrations of the metal. This requires a group of solute carrier (SLC) transporters localised at the cell membrane. SLC39A8 and SLC39A14 facilitate uptake of manganese into the cell, while SLC30A10 mediates manganese efflux (Leyva-Illades et al. 2014; Clayton 2017). SLC39A14 appears to be the main transporter allowing for the uptake of manganese into the liver, the primary regulator of manganese homeostasis. Manganese transport also occurs at iron transporters including DMT1, transferrin/transferrin receptor complex and ferroportin, as well as the dopamine (DAT) and citrate transporters (Peres et al. 2016). Because iron competes with manganese for transport, iron supplementation can reduce manganese levels in disorders associated with excess manganese (Clayton 2017). Within the cell, manganese is transported via a number of transporters including ATP13A1 at the endoplasmic reticulum, SPCA1 at the Golgi, ATP13A2 at the lysosome and Mfrn-1 and DMT1 at the mitochondria (Chen et al. 2015; Sorensen et al. 2018; Christenson et al. 2018).
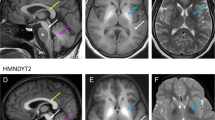
Hypermanganesaemia with dystonia 1 (HMNDYT1) caused by biallelic mutations in SLC30A10 was the first inherited manganese transporter defect described. Impaired biliary excretion leads to accumulation of manganese in the liver and brain, resulting in liver disease and generalised dystonia. Manganese deposition in the brain causes characteristic neurodegenerative features including severe neuronal loss in the globus pallidus and a vacuolated myelinopathy. MRI brain appearances are pathognomonic with hyperintensity on T1-weighted images of the globus pallidus and striatum, and the white matter of the cerebrum and cerebellum, midbrain, dorsal pons and medulla, while the ventral pons is typically spared (Fig. 38.1a, b). T2-weighted images show corresponding hypointensity of the globus pallidus and striatum (Fig. 38.1c). Activation of erythropoietin gene expression due to excess manganese leads to polycythaemia, which often precedes clinical symptoms. Whole blood manganese levels are significantly raised and usually exceed 1000 nmol/L. Another characteristic is a depletion of iron stores with increased total iron-binding capacity and low ferritin values. Some patients develop hypothyroidism, which is consistent with findings in SLC30A10 knockout mice (Hutchens et al. 2017). Disease onset is usually within the first few years of life with progressive, generalised dystonia; however, cases of adult-onset atypical Parkinson’s disease have been described. Some patients have also presented with primary hypotonia or spastic paraplegia (Gospe Jr. et al. 2000; Gulab et al. 2018; Zaki et al. 2018). Manganese chelation with intravenous disodium calcium edetate, in combination with iron supplementation to reduce the uptake of manganese, reduces manganese blood and tissue levels, improves neurological symptoms and halts liver disease progression (Tuschl et al. 2012, 1993; Quadri et al. 2012, 2015).

Characteristic MRI brain appearances due to manganese overload in SLC30A10 (HMNDYT1) and SLC39A14 (HMNDYT2) transporter defects (Tuschl et al. 2016). (a, b) T1-weighted MR imaging shows hyperintensity of the globus pallidus and striatum (blue arrow), and the white matter in the cerebrum, cerebellum, midbrain, dorsal pons (white arrows) with a pathognomonic sparing of the ventral pons (red star). (c) T2-weighted MR imaging shows corresponding hypointensity of the globus pallidus and striatum (blue arrow)
Biallelic mutations in SLC39A14 cause hypermanganesaemia with dystonia 2 (HMNDYT2) due to impaired uptake of manganese into the liver for subsequent biliary excretion. Manganese accumulates in extrahepatic tissues, causing an isolated neurological phenotype of rapidly progressive Parkinsonism-dystonia with onset in infancy or childhood. Whole blood manganese levels are highly raised and MRI brain appearances are identical to that of HMNDYT1 (Fig. 38.1). Hypointensity of the globus pallidus and striatum on T2-weighted images is often pronounced and may be mistaken as the eye of the tiger sign observed in neurodegeneration with brain iron accumulation (NBIA) disorder. Chelation therapy with disodium calcium edetate has been used with some success; however, clinical response is poor in most patients, most likely due to advanced disease progression and significant degree of neurodegeneration (Tuschl et al. 2016, 1993; Zeglam et al. 2018).
Inherited manganese deficiency with low blood manganese levels is caused by biallelic mutations in SLC39A8 and leads to a type II congenital disorder of glycosylation (Type IIn; CDG2N). Dysglycosylation occurs because manganese is a cofactor for the β-1,4-galactosyltransferase, essential for galactosylation of glycan chains. Affected individuals present as early as infancy with neurodevelopmental delay, hypotonia, seizures, dystonia, ataxia, vision and hearing impairment, dysmorphism and short stature/dwarfism (Park et al. 2015a; Boycott et al. 2015). This disorder may also manifest with Leigh-like mitochondrial disease including features such as raised CSF lactate, respiratory chain abnormalities, bilateral basal ganglia hyperintensities on T2-weighted imaging and cerebellar atrophy (Riley et al. 2017). Treatment has been attempted with oral manganese sulphate with remarkable clinical and biochemical improvement. Upon manganese supplementation, the glycosylation pattern and blood manganese levels normalised. Clinically, this was associated with cessation of seizures and improvement in hearing, vision and motor abilities (Park et al. 2017).
There is evidence that manganese dyshomeostasis also plays a role in individuals with mutations in ATP13A2. ATP13A2 facilitates manganese transport into the lysosome and thereby protects cells from manganese toxicity. Biallelic mutations in ATP13A2 have been reported in three neurodegenerative disorders with parkinsonism as a shared clinical feature: (1) Juvenile-onset Parkinson’s disease (PARK9), also known as Kufor-Rakeb-Syndrome, which is accompanied by supranuclear gaze palsy, dementia and generalised brain atrophy (Racette et al. 2012). (2) Neuronal ceroid lipofuscinosis (CLN12) with typical accumulation of autofluorescent lipopigment, which presents with learning difficulties, parkinsonism, spinocerebellar ataxia, bulbar syndrome and pyramidal involvement (Bras et al. 2012). (3) Complex hereditary spastic paraplegia (SPG78), which is accompanied by ataxia and neuropathy, parkinsonism and cognitive decline (Estrada-Cuzcano et al. 2017). The parkinsonian features tend to respond well to treatment with levodopa; however, dyskinesias develop early. In addition, heterozygous mutations in ATP13A2 pose a genetic risk factor for early-onset Parkinson’s disease (Park et al. 2015b). Despite the fact that ATP13A2 is involved in manganese transport, blood manganese levels are normal in affected individuals. T2-weighted MR images show iron accumulation in the basal ganglia, and, hence, ATP13A2 mutations are also classified as an NBIA disorder (Schneider et al. 2010) (Fig. 38.2).
Nomenclature
No. | Disorder | Alternative name | Abbreviation | Gene symbol | Chromosomal location | Mode of Inheritance | Affected protein | OMIM |
|---|---|---|---|---|---|---|---|---|
Hypermanganesemia with Dystonia 1 | SLC30A10 deficiency; Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia | HMNDYT1 | SLC30A10 | 1q41 | AR | SLC30A10 (ZNT10) | 613280 | |
Hypermanganesemia with Dystonia 2 | SLC39A14 deficiency | HMNDYT2 | SLC39A14 | 8p21.3 | AR | SLC39A14 (ZIP14) | 617013 | |
Congenital Disorder of Glycosylation, Type IIn | SLC39A8 deficiency | CDG2N | SLC39A8 | 4q24 | AR | SLC39A8 (ZIP8) | 616721 | |
Parkinson disease 9 | Kufor-Rakeb Syndrome; Neuronal ceroid lipofuscinosis 12; Complex hereditary spastic paraplegia 78 | PARK9, NCL12, SPG78 | ATP13A2 | 1p36 | AR | ATP13A2 | 606693 |
Metabolic Pathways

Our current understanding of how manganese homeostasis (a) is maintained under physiological conditions and (b–d) is impaired in manganese transporter defects (Tuschl et al. 2016). (a) In healthy individuals, manganese from the diet is taken up in the small intestine via SLC39A8 and enters the liver via SLC39A14. The liver regulates blood manganese levels via biliary excretion through SLC30A10. (b) In SLC30A10 deficiency (HMNDYT1), biliary excretion of manganese is impaired leading to accumulation of manganese in the liver (causing liver cirrhosis and polycythaemia), the blood (hypermanganesaemia) and the brain where it causes neurotoxicity within the globus pallidus resulting in dystonia. (c) In SLC39A14 deficiency (HMNDYT2), manganese uptake into the liver is reduced and manganese accumulates in extrahepatic tissues. (d) In SLC39A8 deficiency (CDG2N), uptake of manganese is impaired resulting in systemic manganese deficiency
Signs and Symptoms
Reference Values
Reference value | ||
|---|---|---|
Mn (B) | 73–325 nmol/L | |
Zn (S) | 8–20 μmol/L | |
Hb (B) | Prepubertal | |
0–6 days | 145–220 g/L | |
7 days | 140–186 g/L | |
8 days–3 months | 95–125 g/L | |
3 months–4 years | 110–140 g/L | |
5–12 years | 115–140 g/L | |
Postpubertal | ||
Female | 131–175 g/L | |
Male | 130–175 g/L | |
TIBC (S) | 50–90 μmol/L | |
Ferritin (S) | 7–150 μg/L |
Pathological Values
Mn (B) | Zn (S) | Hb (B) | TIBC (S) | Ferritin (S) | |
|---|---|---|---|---|---|
HMNDYT1 (SLC30A10 deficiency) | ↑ | N | ↑ | ↑ | ↓ |
HMNDYT2 (SLC39A14 deficiency) | ↑ | N | N | N | N |
CDG2N (SLC39A8 deficiency) | ↓ | ↓/N | N | N | N |
Diagnostic Flowchart
HMNDYT1 and HMNDYT2

In any individuals with childhood-onset, progressive Parkinsonism-dystonia, whole blood manganese levels should be determined as well as brain MR imaging obtained. If features of manganese accumulation are identified, the above diagnostic algorithm should be followed.
Specimen Collection
Test | Precondition | Material | Handling | Pitfalls |
|---|---|---|---|---|
Mn (B) | – | Blood | Ambient | False elevation due to contamination. Secondary causes of hypermanganesaemia due to liver disease, environmental manganese exposure, parenteral nutrition. |
Ideally, blood for metal determination should be collected in trace-metal-free EDTA blood containers. Alternatively, if such containers are unavailable, common EDTA blood containers can be used and an empty blood container analysed in parallel to rule out background contamination of the container.
Prenatal Diagnosis
Prenatal diagnosis can be performed by molecular analysis of all four disorders of manganese transport provided the mutation of each biological parent is known. DNA can be isolated from cells directly or by culture after amniocentesis, from chorionic villous samples, from maternal blood, or at the preimplantation stage.
DNA Testing
Genetic diagnosis can be made by standard molecular diagnostic procedures including gene sequencing of SLC30A10, SLC39A14, SLC39A8 and ATP13A2, whole exome sequencing, and deletion/duplication analysis using genomic DNA of any source.
Treatment Summary
HMNDYT1 and HMNDYT2
The recommended treatment for disorders of manganese overload including HMNDYT1 and HMNDYT2 is the regular chelation of manganese with intravenous disodium calcium edetate in combination with iron supplementation to maintain iron parameters (TIBC, ferritin) within the high normal range. Individuals with HMNDYT1 deficiency seem to respond well with improvement of Parkinsonism-dystonia and liver disease, normalization of haemoglobin, reduction in blood manganese levels and manganese accumulation on brain MRI. However, individuals with HMNDYT2 have a less favourable response, probably due to advanced disease progression and different disease pathogenesis. Chelation treatment can lead to a deficiency of calcium, zinc, copper and selenium, which need frequent monitoring and supplementation as required (Tuschl et al. 2016, 1993).
CDG2N
High-dose manganese supplementation in individuals with CDG2N can improve neurological symptoms and normalise glycosylation and blood manganese levels. Therapy requires close monitoring of glycosylation assays and blood manganese to prevent manganese toxicity (Park et al. 2017). In addition, galactose and uridine supplementation can normalise glycosylation patterns (Park et al. 2015a; Riley et al. 2017).
PARK9
Initially, the parkinsonian features in individuals with ATP13A2 mutations tend to respond well to the standard treatment of Parkinson’s disease with levodopa. However, early development of dyskinesias and visual hallucinations complicates treatment (Schneider et al. 2010; Di Fonzo et al. 2007).
Standard Treatment
Therapy | Application | Dose | Duration | |
|---|---|---|---|---|
HMNDYT1 | Disodium calcium edetate | iv | 20 mg/kg/dose (max 1 g) twice daily for 5–8 days every 4 weeks | Lifelong |
Ferrous sulphate/fumarate | po | To keep iron parameters within the normal range (TIBC low normal, ferritin high normal) | Lifelong | |
HMNDYT2 | Disodium calcium edetate | iv | 20 mg/kg/dose (max 1 g) twice daily for 5–8 days every 4 weeks | Lifelong |
Ferrous sulphate/fumarate | po | To keep iron parameters within the normal range (TIBC low normal, ferritin high normal) | Lifelong | |
CDG2N | Manganese sulphate | po | 15–20 mg/kg to keep blood manganese within the normal range | Lifelong |
PARK9 | Carbidopa/levodopa (and standard Parkinson’s disease treatment) | po | 150–1000 mg daily in 3–4 divided doses | Lifelong |
References
Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, et al. Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am J Hum Genet. 2015;97(6):886–93.
Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. 2012;21(12):2646–50.
Chen P, Chakraborty S, Mukhopadhyay S, Lee E, Paoliello MM, Bowman AB, et al. Manganese homeostasis in the nervous system. J Neurochem. 2015;134(4):601–10.
Christenson ET, Gallegos AS, Banerjee A. In vitro reconstitution, functional dissection, and mutational analysis of metal ion transport by mitoferrin-1. J Biol Chem. 2018;293(10):3819–28.
Clayton PT. Inherited disorders of transition metal metabolism: an update. J Inherit Metab Dis. 2017;40(4):519–29.
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68(19):1557–62.
Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T, et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain. 2017;140(2):287–305.
Gospe SM Jr, Caruso RD, Clegg MS, Keen CL, Pimstone NR, Ducore JM, et al. Paraparesis, hypermanganesaemia, and polycythaemia: a novel presentation of cirrhosis. Arch Dis Child. 2000;83(5):439–42.
Gulab S, Kayyali HR, Al-Said Y. Atypical neurologic phenotype and novel SLC30A10 mutation in two brothers with hereditary hypermanganesemia. Neuropediatrics. 2018;49(1):72–5.
Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, et al. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem. 2017;292(23):9760–73.
Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, et al. SLC30A10 is a cell surface-localized manganese efflux transporter, and Parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci. 2014;34(42):14079–95.
Park JH, Hogrebe M, Gruneberg M, DuChesne I, von der Heiden AL, Reunert J, et al. SLC39A8 deficiency: a disorder of manganese transport and glycosylation. Am J Hum Genet. 2015a;97(6):894–903.
Park JS, Blair NF, Sue CM. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov Disord. 2015b;30(6):770–9.
Park JH, Hogrebe M, Fobker M, Brackmann R, Fiedler B, Reunert J, et al. SLC39A8 deficiency: biochemical correction and major clinical improvement by manganese therapy. Genet Med. 2017;20(2):259–68.
Peres TV, Schettinger MR, Chen P, Carvalho F, Avila DS, Bowman AB, et al. Manganese-induced neurotoxicity: a review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol Toxicol. 2016;17(1):57.
Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90(3):467–77.
Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, et al. Manganese transport disorder: Novel SLC30A10 mutations and early phenotypes. Mov Disord. 2015;30(7):996–1001.
Racette BA, Aschner M, Guilarte TR, Dydak U, Criswell SR, Zheng W. Pathophysiology of manganese-associated neurotoxicity. Neurotoxicology. 2012;33(4):881–6.
Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40(2):261–9.
Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord. 2010;25(8):979–84.
Sorensen DM, Holemans T, van Veen S, Martin S, Arslan T, Haagendahl IW, et al. Parkinson disease related ATP13A2 evolved early in animal evolution. uPLoS One. 2018;13(3):e0193228.
Tuschl K, Clayton PT, Gospe SM, Mills PB. Dystonia/parkinsonism, hypermanganesemia, polycythemia, and chronic liver disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. SourceGeneReviews®. Seattle, WA: University of Washington, Seattle; 1993–2019.
Tuschl K, Gregory A, Meyer E, Clayton PT, Hayflick SJ, Mills PB, et al. SLC39A14 deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. SourceGeneReviews®. Seattle, WA: University of Washington, Seattle; 1993–2019.
Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90(3):457–66.
Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601.
Zaki MS, Issa MY, Elbendary HM, El-Karaksy H, Hosny H, Ghobrial C, et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: Six novel SLC30A10 mutations and further phenotype delineation. Clin Genet. 2018;93(4):905–12.
Zeglam A, Abugrara A, Kabuka M. Autosomal-recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene SLC39A14. Acta Neurol Belg. 2018; https://doi.org/10.1007/s13760-018-1024-7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tuschl, K., Mills, P.B., Clayton, P.T. (2022). Disorders of Manganese Metabolism. In: Blau, N., Dionisi Vici, C., Ferreira, C.R., Vianey-Saban, C., van Karnebeek, C.D.M. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-67727-5_38
Download citation
DOI: https://doi.org/10.1007/978-3-030-67727-5_38
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67726-8
Online ISBN: 978-3-030-67727-5
eBook Packages: MedicineMedicine (R0)