
Summary
Iron (Fe) is an essential element for almost every living organism. In humans and other mammals, iron homeostasis evolved to prevent iron excess, which leads to reactive and toxic oxygen species causing cell damage. This situation is attained by mechanisms for efficient regulation and internal iron recycling; however, this sophisticated control limiting iron absorption may easily promote the development of iron deficiency. Other than secondary iron overload conditions (i.e., transfusional iron overload or iron-loading anemias) and secondary iron deficiency, there are several genetically determined iron disorders. The first type of inherited iron-related disorder is “Hereditary Hemochromatosis (HH),” caused by mutations in genes maintaining Fe homeostasis. Different types of HH have been discovered; however, regardless of the mutated gene, the final outcome is an inappropriate hepcidin expression. The most common type of HH (type I) is caused by a mutation in HFE, with adult onset, and it accounts for >80% of all hemochromatosis patients, mostly Caucasian. The prevalent p.Cys282Tyr substitution leads to the inability of HFE to sense increased levels of Fe and interact with TfR1, which causes decreased hepcidin expression. Type II or juvenile HH, due to hemojuvelin (HJV) or hepcidin mutations, is a more severe disorder that affects younger individuals and causes a fast and heavy Fe overload in the liver and parenchyma. Type III HH is rare; it is similar to type 1, but is caused by mutations in the TFR2 gene. Type IV HH differs from the other ones for having an autosomal dominant transmission and for not directly affecting hepcidin expression. It is caused by mutations in the SLC40A1 gene, which encodes the Fe exporter ferroportin (Fpn), namely the hepcidin target. HH in general is not associated with anemia, whereas the conditions with iron overload associated with anemia suggest congenital atransferrinemia, hereditary aceruloplasminemia, and divalent cation transporter 1 (DMT1)-related iron overload. Finally, there are genetic defects that cause iron deficiency such as mutations occurring in TMPRSS6 (matriptase 2) responsible for an iron-refractory iron deficiency anemia (IRIDA).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Introduction
Iron (Fe) is an essential element that is involved in a variety of vital functions, including oxygen transport, DNA synthesis, metabolic energy, and cellular respiration. However, excess iron can lead to the generation of reactive oxygen species (ROS), which cause oxidative stress, lipid peroxidation, and DNA damage, compromising cell viability and promoting cell death (Coffey and Ganz 2017). Under physiologic conditions these deleterious effects are prevented by sophisticated regulatory mechanisms, which maintain systemic and cellular Fe homeostasis (Anderson and Frazer 2017). Iron homeostasis is the result of balanced cooperation between functional compartments (erythroid and proliferating cells), uptake and recycling systems (enterocytes and splenic macrophages), storage elements (hepatocytes), and mobilization processes. The intracellular iron homeostasis is maintained by a posttranscriptional mechanism based on iron responsive elements (IREs) and iron regulatory proteins (IRPs) that bind to the IREs (Muckenthaler et al. 2008). In humans, there is no regulated excretion of iron, thus the iron balance is primarily controlled at the level of intestinal absorption, which takes place in the proximal portion of the duodenum. Fe2+ iron enters enterocytes through DMT1 localized to the apical membrane and to subapical endosomes. DMT1 remains the primary transmembrane iron transporter and its expression is highly induced in iron deficiency. Once inside the intestinal epithelial cells, a portion of iron remains in the cell for use or storage and it is sloughed into the gut lumen when enterocytes become senescent; the rest is exported across the basolateral membrane of the enterocytes through the iron exporter ferroportin. Iron entry into the bloodstream is critical for systemic iron homeostasis and is negatively regulated by hepcidin, the iron regulatory hormone (Papanikolaou and Pantopoulos 2017; Ganz 2013). Hepcidin, a peptide hormone produced in the liver, is responsible for modulating iron availability to meet iron needs. Hepcidin operates by binding to ferroportin in tissue macrophages, duodenal enterocytes, and other target cells, triggering its tyrosine phosphorylation, internalization, and ubiquitin-mediated degradation in lysosomes. By removing ferroportin from the plasma membrane, hepcidin shuts off cellular iron export. The final consequence is the decrease in serum iron. Iron and inflammation are the major hepcidin inducers. Following iron intake or an increase in body iron stores, hepcidin is mainly upregulated through the activation of the Bone Morphogenetic Proteins, BMP/SMAD signaling, to prevent further dietary iron absorption. Under inflammatory conditions, hepcidin induction serves to promote hypoferremia and iron sequestration in macrophages (Nemeth et al. 2004; Ganz and Nemeth 2015). On the other hand, hepcidin expression is suppressed in iron deficiency, hypoxia, and erythropoietic expansion (stress erythropoiesis). Hepcidin inhibitors are the liver protease matriptase 2, encoded by the transmembrane serine protease 6 (TMPRSS6) gene and the erythroid-released hormone erythroferrone (ERFE). In iron deficiency, matriptase 2 inhibits hepcidin by cleaving the BMP coreceptor hemojuvelin (HJV) on the hepatocyte membrane (Silvestri et al. 2008). ERFE is an EPO target gene activated by Janus Kinase 2-Signal transducer and activator of transcription. It is widely expressed, and it is increased by EPO only in the erythropoietic (bone marrow and spleen) tissues. There are many observations that strengthen the role of ERFE in stress erythropoiesis, although TMPRSS6 has a dominant effect over ERFE (Arezes et al. 2018).
The human iron disorders are invariably disorders of iron balance or iron distribution, either in terms of iron overload or iron deficiency. Hence, understanding iron homeostasis is critical for understanding these disorders, as well as understanding genetic iron disorders (Table 1). The first type of inherited iron-related disorder is hemochromatosis (HH). This term must be reserved for iron overload of genetic origin related to hepcidin deficiency. According to the most recent classification updated in 2018 (Brissot et al. 2018, 2019), hemochromatosis encompasses the following entities:
-
1.
hemochromatosis type 1, related to mutations of the HFE gene (the C282Y mutation in the homozygous state is prevalent), which is by far the most common form, affecting mainly Caucasian populations (Allen et al. 2008)
-
2.
hemochromatosis type 2 (the so-called juvenile hemochromatosis) corresponding to mutations in the hemojuvelin (HJV) gene (type 2A hemochromatosis) or to mutations in the hepcidin gene (HAMP) (type 2B hemochromatosis) (Kong et al. 2019)
-
3.
hemochromatosis type 3 due to mutations in the transferrin receptor 2 (TFR2) gene (Kawabata 2019)
-
4.
hemochromatosis type 4 due to mutations in the ferroportin gene (SLC40A1) in rare cases where these mutations lead to a refractory state to hepcidin (“gain-of-function”). There are different mutations in the ferroportin gene that affect the subcellular localization or transporter function of ferroportin (“loss-of-function”); this condition is characterized by macrophage iron loading and preferentially should be called “ferroportin disease.” Both are autosomal dominant disorders (Pietrangelo 2017)
Thus, based on the current understanding, the molecular pathogenesis of “hemochromatosis” can be divided into three classes: first, mutations in the hepcidin gene itself (HAMP) that cause hemochromatosis by preventing the production of functional hepcidin protein; second, mutations in the genes encoding HFE (HFE), TFR2 (TFR2), and hemojuvelin (HFE2) inactivating signaling pathways that normally upregulate hepcidin expression; and finally, mutations in the gene encoding ferroportin (SLC40A1) that can cause hemochromatosis by rendering the transporter insensitive to hepcidin regulation. These different types of hemochromatosis are characterized by common signs including increased plasma iron, increased transferrin saturation, and parenchymal iron accumulation primarily into hepatocytes. The clinical expression may differ in severity among the different forms (Andrews 2008). Anemia is not a manifestation of hemochromatosis; however, there are interesting genetic conditions presenting with microcytic iron deficiency anemia associated with tissue iron overload; it is the case of atransferrinemia (Beaumont-Epinette et al. 2015), DMT1 deficiency (Iolascon et al. 2008), and aceruloplasminemia (Piperno and Alessio 2018). Congenital atransferrinemia is a rare, early onset autosomal recessive disorder caused by transferrin deficiency (<20 mg/dL) due to mutations in the transferrin-encoding TF gene on chromosome 3q22.1. The disease is also referred to as hypotransferrinemia, as the complete absence of functional transferrin is lethal. Patients exhibit very low to undetectable levels of plasma transferrin. This leads to impaired erythropoiesis, microcytic hypochromic anemia, growth retardation, and iron overload in parenchymal cells of the liver, heart, and pancreas (Beaumont-Epinette et al. 2015). Mutations in the genes encoding DMT1 (SLC11A2) are associated with autosomal recessive hypochromic, microcytic anemia (given the role of DMT1 in the uptake of iron at the apex of duodenal cells), but also have hepatic iron overload. Aceruloplasminemia is a rare autosomal recessive disorder caused by loss of ceruloplasmin function caused by mutations in the CP gene on chromosome 3q23-q24 (Kono 2013). The phenotype is quite heterogeneous but is always characterized by iron-restricted erythropoiesis leading to microcytic anemia, diabetes, and in some cases late in life to progressive retinal and neurological degeneration. An impaired iron absorption due to mutations in TMPRSS6, leading to an inability to cleave the BMP coreceptor HJV and inhibiting hepcidin, is observed in a recessive condition named IRIDA (congenital, iron-refractory, iron deficiency anemia). IRIDA patients are refractory to oral iron supplementation (Camaschella 2019).
Hyperferritinemia-cataract syndrome is a dominant condition due to IRE-IRP deregulation in which mutations in the IRE of L-ferritin mRNA make L-ferritin refractory to IRP binding; as a result, the protein synthesis becomes iron-independent. Ferritin is high but total body iron is normal. L-ferritin may accumulate in the lens, leading to early onset of cataract (Tsantoula et al. 2014). A dominant rare disease named neuroferritinopathy may be due to nucleotide insertions in the C-terminus of L-ferritin leading to neurodegeneration because of increased oxidation and cell death (Kuwata et al. 2019).
Nomenclature
No. | Disorder | Alternative name | Abbreviation | Gene symbol | Chromosomal localization | Affected protein | OMIM No. |
|---|---|---|---|---|---|---|---|
Hereditary Hemochromasis type 1 | HH | HFE | 6p21.3 | Homeostatic iron regulator | 613609 | ||
Hemojuvelin deficiency | Hereditary hemochromatosis Type 2A | HJV | HFE2 | 1q21 | Hemojuvelin | 608374 | |
Hepcidin deficiency | Hereditary hemochromatosis Type 2B | HH | HAMP | 19q13 | Hepcidin | 606464 | |
Transferrin Receptor 2 deficiency | Hereditary hemochromatosis Type 3 | Tfr2 HH | TFR2 | 7q22 | Transferrin receptor 2 | 604720 | |
Ferroportin deficiency | Hemochromatosis type 4 | FPN HH | SLC40A1 | 2q32 | Ferroportin | 604653 | |
Ferritin Heavy chain dysregulation | Hereditary hemochromatosis type 5 | HH | FTH1 | 11q12 | Subunit of ferritin | 134770 | |
37.7 | Ferritin light chain deficiency | Hereditary L-ferritin deficiency | FTL | 19 | Subunit of ferritin | 134790 | |
37.8 | Ferritin light chain superactivity | Neuroferritinopathy; neurodegeneration with brain iron accumulation 3 | FTL | 19 | Subunit of ferritin | 134790 | |
Ferritin light chain dysregulation | Hyperferritinemia-cataract syndrome | FTL | 19 | Subunit of ferritin | 134790 | ||
Hereditary ceruloplasmin deficiency | Aceruloplasminemia | CP | 3q23-q24 | Ceruloplasmin | 117700 | ||
Matriptrase 2 deficiency | Iron-refractory iron deficiency anemia | IRIDA | TMPRSS6 | 22 | Matriptase 2 | 609862 | |
Hereditary transferrin deficiency | Atransferrinemia | TF | 3q22.1 | Transferrin | 190000 | ||
Transferrin receptor deficiency | Immunodeficiency type 46 | TFRC | 3q29 | Transferrin receptor | 190010 | ||
Divalent metal transporter 1 deficiency | Hypochromic microcytic anemia with iron overload type 1 | DMT1 | SLC11A2 | 12q13 | DMT1 | 600523 |
Metabolic Pathway

Brissot, P. et al. (2018) Haemochromatosis (Brissot et al. 2018) Nat. Rev. Dis. Primers. doi: https://doi.org/10.1038/nrdp.2018.16
Signs and Symptoms
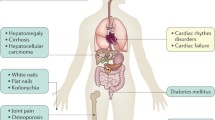
Hemochromatosis overview
Symptom | Neonatala | Infancy | Childhoodb | Adolescenceb | Adulthoodc |
|---|---|---|---|---|---|
Chronic fatigue | + | ++ | |||
Hepatomegaly | +++ | + | ++ | ++ | |
Cirrhosis | +++ | ++ | ++ | ||
Hepatocellular carcinoma | + | ||||
Join pain | + | ++ | ++ | ||
Osteoporosis | + | + | |||
Diabetes mellitus | ++ | + | |||
Melanoderma | +/− | ++ | + | ||
Skin dryness | +/− | ++ | + | ||
Hypopituitarism | + | ++ | +/− | ||
Cardiac rhythm disorder | + | + | |||
Heart failure | ++ |
Despite the high prevalence of C282Y homozygosity, only a minority of individuals will accumulate enough iron to cause organ damage. Given the autosomal recessive inheritance of C282Y, the frequency of C282Y homozygosity is similar in men and women, but the prevalence of clinical manifestations is much higher in men.
Overview of hematological signs of iron deficiency anemia with tissue iron overload and IRIDA
Atransferrinemia | DMT1 deficiency | Aceruloplasminemia | IRIDA | |
|---|---|---|---|---|
Hb | ↓ | ↓ | ↓ | ↓ |
MCV | ↓ | ↓ | Normal | ↓ |
Fe | ↑ | ↑ | ↓ | ↓ |
Transferrin | ↓ Undetectable | ↓ | ↑ | ↑ |
Transferrin saturation | ↑ | ↑ | ↓ | ↓ |
S. Ferritin | ↑ | ↑ | ↑ | Normal |
S. Hepcidin | ↓ | ↓ Normal | ↓ | ↑ |
Reference and Pathological Values
Serum | Hb (g/L) ± 2SD | Iron (μmol/L) | Ferritin (μg/L) | Transferrin (g/L; range) |
|---|---|---|---|---|
Newborn | 185 ± 30 | 6.4–33.0 | 110–503 | 1.8 (1.42.29) |
3–6 months | 115 ± 20 | 6.4–33.0 | 4–405 | 2.03 (1.58–2.57) |
6–12 months | 120 ± 15 | 6.4–33.0 | 4–405 | – |
2–6 years | 125 ± 10 | 6.4–33.0 | 4–405 | 2.39 (1.86–3.03) |
6–12 years | 135 ± 20 | 6.4–33.0 | 4–405 | 2.17 (1.97–3.19) |
12–18 years (w) | 140 ± 20 | 6.4–33.0 | 9–79 | 2.17 (1.97–3.19) |
12–18 years (m) | 145 ± 15 | 6.4–33.0 | 9–59 | 2.17 (1.97–3.19) |
>18 years (w) | 140 ± 20 | 6.6–26.0 | 6–81 | 2.0–3.4 |
>18 years (m) | 155 ± 20 | 10.6–28.0 | 30–233 | – |
CSF | – | 0.4 (0.2–0.6) | – | 14.4 mg/L |
HFE | >30 | >300 (up to 5000) | >70 |
Diagnostic Flowchart

Brissot, P. et al. (2018) Haemochromatosis (Brissot et al. 2018) Nat. Rev. Dis. Primers. doi: https://doi.org/10.1038/nrdp.2018.16
In genetic conditions characterized by iron overload, transferrin saturation and ferritin levels are the key parameters to be assessed. However, increased ferritin levels (>300 mcg/L for men and >200 mcg/L for women) need rigorous interpretation before they are assigned to iron overload. Several conditions can be associated with increased ferritin levels independent of substantial iron overload such as metabolic syndrome (which is the most frequent cause), alcoholism, inflammation, and marked cytolysis. Despite these limitations, increased ferritin levels are critical for the diagnosis of hemochromatosis. Any acquired iron overload situation must be excluded (i.e., blood transfusions, dyserythropoiesis, or parenteral iron supplementation) by clinical history; family history could be helpful in some cases. Ethnicity is important considering the fact that HFE-associated hemochromatosis is observed almost exclusively in Caucasians and more frequently in men because the phenotypic expression of hemochromatosis is usually less pronounced in women. Age of onset is also important as HFE-associated (type 1) and TFR2-associated (type 3) hemochromatosis are generally observed in individuals >30 years of age, whereas clinical expression in younger individuals is typical of HJV-related (type 2A) or HAMP-related (type 2B) hemochromatosis. The non-HFE hemochromatosis diseases are very rare, in contrast to HFE-associated hemochromatosis.
Treatment
Phlebotomy (weekly) remains the key of treatment for hemochromatosis. The goal of phlebotomy is to reach iron depletion to prevent tissue damage. After achieving such iron balance, maintenance phlebotomy (1–4 yearly) is advisable lifelong. In the most severe cases with decompensated cirrhosis or heart failure (for example, individuals with severe juvenile hemochromatosis) that badly tolerate phlebotomy, adjunctive oral chelation can be used. Phlebotomies are also efficient for treatment of patients with loss-of-function ferroportin disease but should be carried out on a less intensive schedule given the risk of anemia (Kowdley et al. 2019).
Although randomized clinical trials are missing, a sufficient body of data has suggested that phlebotomy therapy can improve chronic fatigue and cardiac function, stabilize liver disease, reverse hepatic fibrosis, and reduce skin pigmentation in patients with hemochromatosis (Adams and Barton 2010). The effectiveness of phlebotomy is much better if it starts before the development of severe organ damage such as cirrhosis. An alternative to phlebotomy could be erythrocytapheresis; this procedure could be useful in patients suffering from hypoproteinemia or thrombocytopenia (Rombout-Sestrienkova et al. 2016). A phase I/II clinical trial with Deferasirox in non-cirrhotic HFE hemochromatosis patients has been conducted, showing a dose-dependent ferritin reduction.
IRIDA, differently than classical iron deficiency anemia where hepcidin levels are low or even undetectable, has normal or high hepcidin levels, and is resistant to oral iron and only partially responsive to intravenous iron, which still remains the advisable treatment.
Future Treatments
Although phlebotomy is inexpensive, safe, and effective in reversing many complications of iron overload, it is not well tolerated by a minority of patients. Moreover, phlebotomy is not feasible in iron-loading anemias because the patients become even more anemic. For these reasons, there is a consensus that novel therapeutic approaches are needed for all iron overload diseases. As hepcidin represents the iron homeostasis controller, the use of hepcidin agonists or antagonists could be beneficial, depending on the specific disorder (Katsarou and Pantopoulos 2018).
-
(a)
Hepcidin agonists include compounds that mimic the activity of hepcidin and agents that increase the production of hepcidin by targeting hepcidin-regulatory molecules. The potential of these future drugs includes the improvement in erythropoiesis as shown in thalassemia mouse models and in phase I/II clinical trial.
-
(b)
Hepcidin antagonists may be beneficial in IRIDA or in anemias associated with a variety of inflammatory disorders and malignancies, and in chronic renal disease with or without inflammatory etiology.
References
Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317–25.
Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221–30.
Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr. 2017;106:1559S–66S.
Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112:219–30.
Arezes J, Foy N, McHugh K, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132:1473–7.
Beaumont-Epinette MP, Delobel JB, Ropert M, et al. Hereditary hypotransferrinemia can lead to elevated transferrin saturation and when associated to HFE or HAMP mutations to iron overload. Blood Cells Mol Dis. 2015;54:151–4.
Brissot P, Pietrangelo A, Adams PC. Hemochromatosis. Nat Rev Dis Primers. 2018; article 18016.
Brissot P, Troadec MB, Loréal O, Brissot E. Pathophysiology and classification of iron overload diseases; update 2018. Transfus Clin Biol. 2019;26:80–8.
Camaschella C. Iron deficiency. Blood. 2019;133:30–9.
Coffey R, Ganz T. Iron Homestasis: an anthropocentric perspective. J Biol Chem. 2017;292(31):12727–34.
Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93:1721–41.
Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500–10.
Iolascon A, Camaschella C, Pospisilova D, et al. Natural history of recessive inheritance of DMT1 mutations. J Pediatr. 2008;152:136–9.
Katsarou A, Pantopoulos K. Hepcidin therapeutics. Pharmaceuticals. 2018;11:E127.
Kawabata H. Transferrin and transferrin receptors update. Free Radic Biol Med. 2019;133:46–54.
Kong X, Xie L, Zhu H, et al. Genotypic and phenotypic spectra of hemojuvelin mutations in primary hemochromatosis patients: a systematic review. Orphanet J Rare Dis. 2019;14:171.
Kono S. Aceruloplasminemia: an update. Int Rev Neurobiol. 2013;110:125–51.
Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG clinical guideline: hereditary hemochromatosis. Am J Gastroenterol. 2019;114:1202–18.
Kuwata T, Okada Y, Yamamoto T, et al. Structure, function, folding and aggregation of a Neuroferritinopathy-related ferritin variant. Biochemistry. 2019;58:2318–25.
Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28:197–213.
Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3.
Papanikolaou G, Pantopoulos K. Systemic iron homeostasis and erythropoiesis. IUBMB Life. 2017;69:399–413.
Pietrangelo A. Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica. 2017;102(12):1972–84.
Piperno A, Alessio M. Aceruloplasminemia: waiting for an efficient therapy. Front Neurosci. 2018;12:903.
Rombout-Sestrienkova E, van Kraij MG, Koek GH. How we manage patients with hereditary hemochromatosis. Br J Haematol. 2016;175:759–70.
Silvestri L, Pagani A, Nai A, et al. The serine pro- tease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502–11.
Tsantoula F, Kioumi A, Germenis AE, Speletas M. Hereditary hyperferritinemia cataract syndrome as a cause of childhood hyperferritinemia. J Pediatr Hematol Oncol. 2014;36:304.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cappellini, M.D. (2022). Disorders of Iron Metabolism. In: Blau, N., Dionisi Vici, C., Ferreira, C.R., Vianey-Saban, C., van Karnebeek, C.D.M. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-67727-5_37
Download citation
DOI: https://doi.org/10.1007/978-3-030-67727-5_37
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67726-8
Online ISBN: 978-3-030-67727-5
eBook Packages: MedicineMedicine (R0)