
Abstract
It is widely accepted that cancer is driven by genetic mutations that confer uncontrolled cell proliferation and tumor formation. For tumors to take hold and grow, cancer cells evolve mechanisms to favorably shape their microenvironment and avoid being cleared by the immune system. Cancer is not unique to human, but rather affects nearly all multicellular organisms albeit to different degrees. The different degrees of cancer susceptibility across the animal kingdom could be attributed to several factors, which have been the subject of several studies in recent years. The naked mole-rat (NMR, Heterocephalus glaber), an exceptionally long-lived rodent, which, as discussed in detail in the next section, displays significant cancer resistance with only a small number of animals being reported to exhibit spontaneous neoplasms. The reason why studying cancer resistance in NMRs is of particular interest is that not only are they now an established laboratory species, but that NMRs are mammals and thus there is great potential for translating knowledge about their cancer resistance into preventing and/or treating cancer in humans and companion animals.

Photo Credit: Megan Smith
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
14.1 Introduction
Assuming the rate at which cancer-causing mutations arise is the same for all cell types and across species, any increase in body mass (and thus, cell number) or lifespan (increased exposure to toxins and number of cell divisions) should result in increased risk of undergoing malignant transformation. Indeed, within a species, cancer incidence increases with increased body mass. For instance, large-bodied dog species are more prone to development of osteosarcoma compared to small- and medium-sized dog species (Caulin and Maley 2011) and, in humans, greater height is associated with increased risk of developing non-smoking related cancers (Caulin and Maley 2011; Leroi et al. 2003). Similarly, there is a strong correlation between increased cancer rates and age, particularly in humans. Besides this intraspecies correlation between the number of cells/divisions/lifespan and cancer incidence, there is no evidence to suggest that an increase in a species’ body mass or lifespan increases the risk of cancer incidence. Species with large bodies and long lifespans, when compared to species with small-bodies and short-lifespans, do not show increased cancer incidence (Fig. 14.1) (Abegglen et al. 2015; Caulin and Maley 2011). In fact, the lifetime risk of cancer development is similar between humans and laboratory mice (30%) despite vastly different lifespans and body masses of the two species (Rangarajan and Weinberg 2003). Similarly, it is estimated that blue whales, which can have 1000-times more cells than humans, would go extinct if they concomitantly displayed a 1000-fold higher cancer incidence than humans (Caulin and Maley 2011). The lack of such correlation between cancer incidence and a species’ body mass or lifespan is referred to as Peto’s Paradox (Caulin and Maley 2011), named after Sir Richard Peto who first described the phenomenon in 1977. This paradox suggests that cancer resistance increases with increase in species body mass and lifespan . Therefore, large-bodied and long-lived species must have evolved augmented cancer suppression mechanisms. In this book chapter, we discuss cancer susceptibility in a clear violator of Peto’s Paradox, the NMR.

Cancer incidence across 38 mammalian species by lifespan and body mass. Abegglen et al. examined necropsy data for 36 mammalian species and 644 annotated deaths for elephants to compare spontaneous cancer incidence across a panel of mammalian species with varied body masses and lifespans. Each data point derives from at least 10 necropsies and 644 annotated deaths in case of elephant (Abegglen et al. 2015). Data for NMR was derived from reported (n = 210; (Delaney et al. 2013; Taylor et al. 2017)) and unpublished (Grimes et al. 2012) necropsies resulting in 8 cases of spontaneous neoplasia in a total of 2710 necropsies. Body mass and maximum lifespan used for NMR were 40 grams and 37 years respectively. As evident from the linear regression, cancer incidence is not associated body mass and lifespan . Although it is well established that cancer incidence does not correlate with species lifespan and/or body mass, some of the data points in this figure should be taken with caution as the data in Abegglen et al. is from zoo collections and sparse. Nevertheless, naked mole-rats break the rules!) Figure modified from (Abegglen et al. 2015)
Weighing 30–80 grams as adults and living up to 37 years (Lee et al. 2020; Buffenstein and Craft 2021), the NMR is an exceptionally cancer resistant species for its size and longevity that does not follow the rules of Peto’s paradox. Despite being the subject of extensive research for over 30 years, evidence of cancer in this species is rare. This has established the NMR as a research model organism for investigating mechanisms of cancer resistance (Lewis et al. 2016; Seluanov et al. 2018; Shepard and Kissil 2020). The Buffenstein lab has pioneered research on NMR and reported more than 2000 necropsies of captive NMRs with no indication of spontaneous neoplasms in NMR (Buffenstein 2005; Grimes et al. 2012). Recently, that lab has reported the presence of a T cell lymphoma that had spread to lungs, liver, and kidney (see Delaney et al. 2021).This is further supported by findings of Delaney et al. who performed a retrospective pathology study of adult NMRs from a zoo-housed population (Delaney et al. 2013). This study was carried out over a period of 15 years and included gross histologic examination of 138 NMR necropsies. Several preneoplastic lesions were identified in multiple organ systems. This included testicular interstitial hyperplasia in over 30% of nonbreeding males and adrenal hyperplasia in 21% NMRs (♂ = 10, ♀ = 19), in addition to a case of thymic atypical hyperplasia. However, no spontaneous neoplasms were identified, i.e. they appeared to be benign tumors with no signs of metastases.
Although the retrospective study by Delaney et al. did not identify spontaneous neoplasms in NMRs, the authors did find two histologic lesions that could be considered neoplasia (Delaney et al. 2013, 2021). In one case, changes consistent with diagnosis of follicular hyperplasia or follicular lymphoma were seen in the spleen of a subordinate female NMR. However, definitive diagnosis was not possible due to lack of specific antibodies. In a separate female, a case of presumptive tubular papillary hyperplasia with atypia was identified (Delaney et al. 2013). More recently three separate studies have reported the presence of a small number of spontaneous tumors in captive NMRs primarily housed in zoological institutions (see Chap. 15). The first report identified a subcutaneous adenocarcinoma, likely of salivary or mammary origin, in an otherwise healthy 22-year old worker male and a case of gastric neuroendocrine carcinoma in a 20-year old worker male (Delaney et al. 2015). In the second report, Taylor et al. described four cases of spontaneous neoplasia in addition to a case of presumptive neoplasia in adult NMRs (Taylor et al. 2017). They diagnosed a hepatocellular carcinoma with lung metastasis and peritoneal carcinomatosis in a 17–24-year-old female. They further describe the presence of a large mass on a kidney of a 6–13-year-old male. The mass, which made up 20% of the animal’s body mass, was diagnosed as Wilms’ tumor (nephroblastoma). The authors also found multicentric lymphosarcoma in the lymph nodes, spleen , lungs, kidneys, intestinal tract and salivary gland of an 8–11-year old male. Lastly, this study also identified a case of presumptive esophageal adenocarcinoma in a 16–19-year old male in addition to diagnosing cutaneous hemangioma in a 14-year old male via biopsy (Taylor et al. 2017). Most recently Cole et al. have reported a case sacral chordoma in a 15+ year old female from a small zoo-housed colony (Cole et al. 2019). To our knowledge, there has been no genetic evaluation of any of the tumors that have been observed in NMRs and yet such analysis could shed light on molecular events governing their cancer resistance.
Given the scarcity of evidence of spontaneous neoplasms in the NMR, the high rate (2%, 1/57 of the living population and 8%, 3/37 of nonautolyzed necropsied animals) of tumors identified in the study by Taylor et al. is surprising (Taylor et al. 2017). Considering that the entire study population was derived from a single breeding pair, it may indicate that this NMR population is especially susceptible to cancer development. However, since the tumors reported in the other publications (Cole et al. 2019; Delaney et al. 2015) came from different zoo populations, this seems unlikely. Nonetheless, the rate of cancer incidence reported in the study by Taylor et al. is still low compared to cancer incidence in other rodents such as mice and rats (Andervont and Dunn 1962; Burek and Hollander 1977; Chrisp et al. 1996; Lipman et al. 2004). Regardless of the actual incidence rate, in total these reports indicate that, although rare, spontaneous tumors do occur in the NMR. However, considering its small body mass and long lifespan , the cancer resistance that NMRs display is still remarkable.
In addition to the rarity of spontaneous neoplasia in the NMR, experimental efforts to induce tumorigenesis in this species through chemical mutagenesis has not been successful thus far. The Buffenstein lab performed the two-stage skin carcinogenesis protocol using DMBA/TPA (7,12-dimethylbenz[a]anthracene)/(12-O-tetradecanoylphorbol-13-acetate; also known as phorbol 12-myristate 13-acetate [PMA]) to experimentally induce tumors in NMRs. Typically in this protocol, a sub-carcinogenic dose of the tumor initiator DMBA is topically applied to animals 2 weeks before the tumor promoter TPA is repeatedly applied until tumors develop (Abel et al. 2009). This protocol successfully produced papillomas in all the mice (C57BL/6 strain) within 2–4 months of treatment but failed to do so in the NMRs even after 6 months (unpublished data from K.N. Lewis in the Buffenstein lab). The strain of mice used is considered to be one of the more resistant to this treatment, yet all developed papillomas or black dysplastic nevi. The papillomas are clonal outgrowths of the skin which can progress to squamous cell carcinomas (Abel et al. 2009). This further confirms the relative resistance of the NMR to tumor development. However, it is worth highlighting that these chemical mutagenesis studies employed the standard doses (25 ug/kg) DMBA and (4 ug/kg) TPA used to induce carcinogenesis in mice and NMRs were only monitored for an additional 2 months after all the mice had developed tumors. Given that sensitivity to chemical mutagens varies even for different mouse strains (Abel et al. 2009), the sensitivity of NMRs to DMBA/TPA first needs to be evaluated before performing the mutagenesis protocol for a longer period of time to establish its resistance to chemical carcinogenesis further. Similarly, it is important to assess if NMRs have the cytochromes needed to metabolize this carcinogen to its more toxic moiety. Additionally, chemical carcinogenesis with a different agent such as 3-methylcholantrene (3-MCA), a potent carcinogen used for induction of fibrosarcomas, should be used to evaluate any tissue-specificity of the NMR’s cancer resistance. Of relevance here is the observation of Manov et al. who performed carcinogenesis with 3-MCA, as well as the two-stage skin carcinogenesis with DMBA/TPA, on the blind mole-rat (BMR, Spalax), another highly cancer resistant rodent species unrelated to NMR (Manov et al. 2013). The DMBA/TPA treatment produced skin lesions in BMRs, however, these wounds completely healed within weeks leaving behind thickened epidermis and did not progress to papillomas or carcinomas (Manov et al. 2013). This is despite the fact that the BMRs in the study received much higher doses of DMBA/TPA and the duration of the application of these mutagens was longer for BMRs than the control mice (C57BL/6 strain). The 3-MCA treatment, on the other hand, resulted in development of fibrosarcoma in one of the BMRs out of 12 animals tested (Manov et al. 2013).
Whereas there is clearly a paucity of spontaneous neoplasia and resistance to chemical carcinogenesis in NMRs, there has been conflicting evidence in the literature about the ability of NMR cells in vitro to undergo tumorigenesis in response to SV40 large T antigen (encoded by SV40LT ) and oncogenic HRAS (HRASG12V), a combination of oncogenes sufficient to transform mouse and rat cells. As discussed in greater detail further on, whereas Seluanov et al. have reported that NMR cells are not transformed by SV40LT and HRASG12V (Seluanov et al. 2009), we have observed robust tumorigenesis in >70 NMR cell lines (Hadi et al. 2020) and in a reply to our work, the group of Seluanov and Gorbunova observe a similar result (Zhao et al. 2020). Furthermore, it has been reported that NMR cells respond to introduction of SV40LT and HRASG12V by undergoing ‘crisis’ as evident from the presence of cells with giant nuclei, anaphase bridges, abnormal chromosome content (Liang et al. 2010). Similar ‘crisis’ onset was observed in NMR cells upon loss of attachment substrate. This ‘crisis’ phenotype could be rescued by ectopic expression of human telomerase reverse transcriptase (hTERT) and NMR cells expressing SV40LT and HRASG12V together with hTERT formed tumors when injected into immunodeficient mice (Liang et al. 2010). However, the use of TERT seems paradoxical since NMR cells have been reported to constitutively express TERT (Kim et al. 2011) albeit in most tissues at very low levels.
The body of research on tumor incidence in the NMRs thus far suggests that, if not entirely cancer free, it is undeniably a highly cancer resistant species. This has resulted in efforts to uncover the mechanisms underlying the pronounced cancer resistance of the NMR. In the next section, we describe the mechanisms that likely contribute to the NMR’s cancer resistance.
14.2 Enhanced Cytoprotection
Cells isolated from NMR are more resistant to forms of cellular stresses such as heavy metal poisoning, acidosis and DNA damaging agents compared to mouse cells (Husson and Smith 2018; Lewis et al. 2012; Salmon et al. 2008). Salmon et al. have shown that the median lethal dose (LD50) of multiple cell stressors including methyl methanesulfonate, cadmium, and heat is significantly higher for NMR cells than mouse cells (Salmon et al. 2008). In contrast, NMR cells are more sensitive to ultraviolet light (UVC) and hydrogen peroxide (H2O2) and undergo apoptosis at much lower doses (Salmon et al. 2008). Similarly, much higher doses of chromium and the DNA damaging agents adriamycin and camptothecin are required to kill NMR cells than mouse cells (Lewis et al. 2012). Additionally, nonlethal doses of genotoxins at which most NMR cells survive cause NMR cells to halt cellular growth (Lewis et al. 2012). As measured by a BrdU (bromodeoxyuridine) incorporation assay, the surviving mouse cells continue to proliferate. In contrast, surviving NMR cells show little BrdU incorporation. This suggests that unlike mouse cells, NMR cells undergo prolonged cell cycle arrest and repair any DNA damage before resuming growth (Lewis et al. 2012). Moreover, even under basal conditions NMR cells have high expression of p53, ~ 50-times higher than mouse cells. The p53 levels show a further 15-fold increase response to genotoxic stress, whereas the p53 expression in mouse cells rise only fivefold the basal levels (Lewis et al. 2012). More recently, it has been demonstrated that even under basal conditions the NMR p53 protein is more stable than mouse p53 and that DNA damage induced by gamma irradiation failed to increase the stability of NMR p53 beyond the stability observed at basal conditions (Deuker et al. 2020). Additionally, a significant amount of NMR p53 is located in the nucleus (considered to be active p53) even under basal conditions and its nuclear localization is not increased further by DNA damage (Deuker et al. 2020). This enhanced expression of NMR p53, together with its increased stability and nuclear localization under normal conditions likely provides NMR cells with enhanced genome protection and cell cycle regulation, which contribute to the remarkable cancer resistance observed in this species.
Cells from NMR also express high levels of the cytoprotective nuclear factor erythroid 2-related factor 2 (NRF2) protein compared to mouse (Lewis et al. 2012). NRF2 is a ubiquitously expressed transcription factor that is negatively regulated by KEAP1 and degraded through the ubiquitin proteasome pathway. Under stressful conditions, degradation of NRF2 is relieved and it migrates to the nucleus where it drives transcription of genes that possess antioxidant response element (ARE). This includes genes involved in detoxification, proteome maintenance and cell cycle regulation. In addition to higher basal expression of NRF2 (Lewis et al. 2012), NMR cells show higher NRF2:ARE binding than mouse cells and even under basal conditions have elevated expression of NRF2 cytoprotective target genes (Lewis et al. 2015; Narayan et al. 2021). The role of NRF2 in tumorigenesis is context dependent with evidence supporting both tumor suppressive and oncogenic roles (Menegon et al. 2016). For example in KEAP-1 mutant lung cancer NRF2 constitutive activity promotes tumorigenesis (Menegon et al. 2016). While Nrf2−/− mice are more susceptible to cancers DMBA mediated skin carcinomas (Kensler et al. 2007; Pearson et al. 2007), the increased expression of NRF2 together with its enhanced NRF2:ARE binding and elevated expression of NRF2 target genes are likely to play a role in NMRs cancer resistance.
14.3 Contact Inhibition
Adherent mammalian cells, when cultured in vitro form a monolayer. When cells in the monolayer come into contact with other cells, they stop dividing. This state of arrested cell division is referred to as contact inhibition and is mediated by the cyclin dependent kinase inhibitor 1B (CDKN1B, p27KIP1) (Seluanov et al. 2009). Contact inhibition is a potent tumor suppression mechanism. It has been observed that NMR cells are hypersensitive to contact inhibition and arrest growth at very low densities in vitro (Seluanov et al. 2009). This phenomenon, termed ‘Early Contact Inhibition’ (ECI) is mediated by cell-cell contact, rather than secreted factors, and requires functional p53 and pRb pathways since inhibition of either of these pathways alone is not sufficient to abrogate the ECI phenotype (Seluanov et al. 2009). Contact inhibition in human and mouse cells is mediated by p27KIP1 and contact-inhibited cells accumulate higher levels of this protein. However, NMR cells that undergo ECI do not show elevated levels of p27KIP1. On the contrary, ECI NMR cells have higher levels of another CDK (Cyclin Dependent Kinase) inhibitor, p16INK4a (Seluanov et al. 2009). Similar to contact-inhibited mouse and human cells, NMR cells that have lost the ECI phenotype arrest growth upon contact with other cells i.e. they display contact inhibition and accumulate high levels of p27KIP1. This suggests that NMR cells have two contact inhibition mechanisms: ECI mediated by p16INK4a and normal contact inhibition mediated by p27KIP1 (Seluanov et al. 2009; see Narayan et al. 2021).
14.4 Presence of High Molecular Weight Hyaluronan
It has been reported that NMR cells, particularly those derived from skin, produce high molecular weight hyaluronan (HMW-HA), a glycosaminoglycan polysaccharide, which mediates its cancer resistance (Tian et al. 2013). According to Tian et al. the hyaluronan (HA) produced by NMR cells is higher in molecular weight than that produced by mouse, human and guinea pig. Specifically, the authors have reported that the molecular weight of the NMR HA is 6–12 MDa, whereas the HA produced by mouse, human and guinea pig ranges from 0.5 to 2 MDa (Tian et al. 2013). Cells grown in the presence of bacterial hyaluronidase (HAase), the enzyme that catalyzes HA degradation, could reach full confluency in culture and it is thought that NMR cell ECI is induced by HMW-HA through inducing expression of p16INK4a , the CDK inhibitor responsible for ECI (Tian et al. 2013). Furthermore, the study shows that the gene encoding hyaluronan synthase 2 (Has2), the enzyme responsible for synthesis HMW-HA, has a special sequence in the NMR: two asparagine residues that are conserved among other mammals analyzed in the study are replaced with serine residues in the NMR. Since ectopic expression of the NMR Has2 cDNA in human embryonic kidney (HEK293) cell line resulted in production of HMW-HA, the authors claim that these substitutions (N178S, N301S) likely provide the NMR’s Has2 with high processivity. Additionally, compared to mouse, human and guinea pig, NMR cells have low HAase activity. Thus, the increased HA production together with its reduced clearance is thought to result in accumulation of HMW-HA in the extracellular environment surrounding NMR cells, which triggers ECI by binding to CD44, a known HA receptor (Tian et al. 2013). The study has further reported that binding of the HMW-HA to CD44 results in accumulation of growth inhibitory unphosphorylated form of NF2 (Neurofibromin 2) and removal of HMA-HA results build-up of phosphorylated NF2 which is growth promoting. Moreover, the presence of HMW-HA is not sufficient to induce ECI in NMR cells, instead ECI induction requires functional HA signaling leading to induction of p16INK4a expression. This indicates that HMW-HA could mediate NMR’s cancer resistance by inducing ECI through the HA-CD44-NF2 pathway. According to this report removal of HMW-HA renders the NMR cells susceptible to malignant transformation by SV40LT and HRASG12V.
Effects of expression of NMR Has2 in cancer cell lines have also been reported (Zhao et al. 2019). Zhao et al. expressed the NMR Has2 gene in 4 T1 and BT549 breast cancer cell lines of murine and human origin respectively. They found that expression of NMR Has2 produced HMW-HA which subsequently resulted in slower cell proliferation in vitro and caused a five-fold reduction in mean tumor weight in vivo (Zhao et al. 2019). The slower cell proliferation following expression of the NMR Has2, however, is not surprising and perhaps not limited to NMR HMW-HA. A similar phenomenon has been reported in vascular smooth muscle cells (VSMCs) wherein HMW-HA keeps the VSMCs in quiescent state by providing anti-mitogenic signals and degraded low molecular weight HA (LMW-HA) eliminates these signals (Kothapalli et al. 2008). One point to consider with the report from Zhao et al. is that the authors associate the reduced cell proliferation and increased apoptosis seen in NMR Has2 expressing 4 T1 cells, which are widely accepted to have mutated p53, with increased expression p53 and its downstream target genes. Recently Takasugi et al. have reported that NMR HMW-HA promotes cytoprotection by affecting expression of CD44-dependent genes including those linked to p53 pathway (Takasugi et al. 2020).
The report that HMW-HA in the NMR is produced by a unique version of the Has2 gene (Tian et al. 2013) has resulted in further investigation of the evolution of the Has2 gene and properties of the HMW-HA in the NMR (Faulkes et al. 2015; Kulaberoglu et al. 2019). Whereas Kulaberoglu and colleagues identified that NMR HA displays highly folded structures and readily forms gels (properties that might assist in presenting a barrier to cell invasion), Faulkes et al. focused on the evolution of the Has2 gene sequence from 70 different mammalian species. This study identified that the serine residue at position 178, one of the residues in the NMR Has2 thought to be responsible for synthesis of the HMW-HA, occurs only in one of the other species analyzed, namely the cane rat (Thryonomys swinderianus), another member of the same parvorder Phiomopha (within the Hystricognathi) and close relative of the sub-Saharan mole-rats where the NMR is grouped. The second serine residue considered important in the NMR Has2 gene at position 301 was found to be common to all members of the Bathyergidae family analyzed (Faulkes et al. 2015). Interestingly, the Has2 sequence in the BMR, another highly cancer resistant species and one that produces HMW-HA at higher Has2 levels than the NMR, is highly similar to the mouse Has2 sequence (Faulkes et al. 2015). Additionally, it has also been reported that the high levels of HMW-HA produced by BMR cells neither affects their growth in vitro or their anticancer properties (Manov et al. 2013). This casts doubts on the importance of the NMR Has2 sequence in the production of the HMW-HA and its tumor suppressive function (Tian et al. 2013). It is also worth highlighting that the proposed mechanisms of cancer resistance including ECI and presence of HMW-HA in the tumors do not explain the lack of nonsolid tumors (e.g. hematological cancers) in this species.
More recently Hadi et al. have called into question the apparent resistant of the NMR cells to SV40LT and HRASG12V induced malignant transformation and have raised further concerns about the importance of the HMW-HA in the cancer resistance of NMRs (Hadi et al. 2020). Unlike previous reports (Liang et al. 2010; Seluanov et al. 2009; Tian et al. 2013), the authors show that like mouse and rat cells, NMR cells are not resistant to, and can be robustly transformed by, SV40LT and HRASG12V (Hadi et al. 2020). They analyzed over 70 different cell lines from 5 different tissues of 11 NMRs and have shown that irrespective of the promoter used for driving transcription of these oncogenes, the mode of delivery of the oncogenes, or basal medium used for culturing these cells, NMR cells expressing SV40LT and HRASG12V form colonies in soft agar and tumors in immunodeficient mice. Further support to Hadi et al.’s observation that, like other rodent cells, the NMR cells can be transformed by classical oncogenes comes from Deuker et al. who have reported that loss of p53 together with ectopic expression of KRASG12V results in malignant transformation of NEFs (NMR embryonic fibroblasts) (Deuker et al. 2020). Hadi et al. further suggest that the key mechanisms behind the NMR’s cancer resistance are likely to be non-cell-autonomous and may include the presence of a unique cellular microenvironment and/or immune system (Hadi et al. 2020). The NMR’s extreme susceptibility to pathogens such as coronavirus (Ross-Gillespie et al. 2007) and herpes simplex virus Type 1 (HSV1) (Artwohl et al. 2009) could indicate that this species does not have a protective immune system. However, recently, Hilton et al. have performed a comprehensive analysis of the NMR’s immune system with a single cell transcriptomic approach and described unique features of the immune system in this species (Hilton et al. 2019; Lin and Buffenstein 2021). Peculiar features of note of the NMR’s immune system include a lack of immune cell population corresponding to natural killer cells (NK) and unlike mouse a heavy reliance on myeloid-based innate immunity. Further studies to address whether other immune lineages are able to compensate for the lack of NK cells in the context of cancer immune-surveillance are warranted.
14.5 Presence of Additional Tumor Suppressor Genes
One likely explanation for the pronounced resistance of the NMR tumor incidence is the presence of additional tumor suppressor genes. For example, Tian et al. have reported that the INK4 locus , which encodes three distinct tumor suppressors in humans (p14ARF, p15INK4b, p16INK4a) and mice (p19ARF, p15INK4b, p16INK4a ) encodes a fourth tumor suppressor in NMR (Tian et al. 2015). This additional tumor suppressor, called pALTINK4a/b that is not found in human or mouse cells, consists of exon 1 of p15INK4b and exon 2 and 3 and 3’ UTR (untranslated region) of p16INK4a (Tian et al. 2015). Reportedly, the pALTINK4a/b isoform is more efficient than p15INK4b and p16INK4a at inducing cell cycle arrest and its transcription is triggered by a variety of stimuli including oncogene overexpression, loss of anchorage, UV or gamma irradiation, and high cell density (Tian et al. 2015). Moreover, NMR cells grown in the presence of HAase show reduced pALTINK4a/b expression, which indicates a role for this transcript in the HA-induced ECI. It is further suggested that the presence of this additional CDK inhibitor may enable NMR cells to better adjust their cell cycle checkpoints and find a balance between tumor suppression and growth (Tian et al. 2015).
Genome and transcriptome sequencing of the NMR has unveiled some interesting features of the NMR genome, which might play a role in mediating NMR’s cancer resistance (Fang et al. 2014; Kim et al. 2011). In particular, several designated cancer-related genes including Ccne1 (cyclin E1), Apex1 (a DNA repair enzyme), Rfc1 (replication factor C) and Top2a (DNA topoisomerase) in the NMR carry unique amino acids substitutions which might alter their activities, such that they contribute to the NMR’s cancer resistance (Kim et al. 2011), but these remain to be tested experimentally. Additionally, several DNA damage response and repair genes including mutS homolog 3 (Msh3), ubiquitin specific peptidase 1 and 5 (Usp1 and Usp5) have elevated expression in NMRs compared to non-fossorial, surface-dwelling rodents (Fang et al. 2014). Furthermore, the Fastk gene is inactivated by multiple indel (insertion, deletion) events in the NMR (Fang et al. 2014). This gene encodes Fas-activated serine/threonine kinase that serves as a regulator of Fas-mediated apoptosis. Given that FASTK is overexpressed in cancers and is associated with cell survival, inactivation of this gene is likely to contribute the cancer resistance phenotype of NMRs (Fang et al. 2014). Furthermore, NMRs have been reported to have low nucleotide diversity, which is comparable to that observed in humans, but is much lower than that of mouse and rat. This low nucleotide diversity, among other things, maybe the result of low mutation rates or an efficient DNA repair system in the NMR (Kim et al. 2011).
Like NMR somatic cells, induced pluripotent stem cells (iPSCs) generated from NMR cells also exhibit pronounced tumor resistance, and strikingly, unlike iPSCs of other mammals, NMR iPSCs do not form teratomas in vivo (Miyawaki et al. 2016; Miura et al. 2021). This resistance of NMR iPSCs is likely due to species-specific activation of Arf tumor suppressor gene and inactivation of Eras (ES cell-expressed Ras ) oncogene . Indeed, knocking down NMR Arf and expression of mouse Eras cause NMR iPSCs to form teratomas in vivo. Furthermore, suppression of Arf during reprogramming which is known to increase reprogramming efficiency in mouse cells induces senescence in NMR cells, a phenomenon termed as ARF suppression-induced senescence (ASIS ). ASIS is reported to be NMR-specific and is suggested to play an important role in protecting somatic NMR cells and NMR iPSCs against tumor formation (Miyawaki et al. 2016).
14.6 Summary
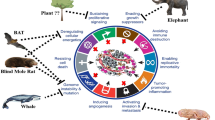
In summary, the NMR is remarkably cancer resistant and ever more is being learned about potential molecular mechanisms that underpin this resistance. Whilst it no longer appears to be the case that NMR cells are themselves unusually resistant to malignant transformation, they clearly have mechanisms enabling greater control of cell cycle progression and enhanced cytoprotection. Genome-wide CRISPR screens in this species can be very informative. Hadi et al. have used a previously described computational pipeline (Tzelepis et al. 2016) and developed a CRISPR guide RNA (gRNA) library for the NMR genome, a set of ~55,000 putative genes (unpublished data). This library contains more than 240,000 gRNAs with most genes being targeted by five unique gRNAs. This library can be used to perform genome-wide CRISPR screens to gain further insights into cell-autonomous cancer suppression mechanisms and uncover yet unknown tumor suppressor genes (if any) in the NMR genome. Given that NMR cells are transformed by expression of SV40LT and HRAS, an hTERT (human telomerase reverse transcriptase) immortalized cell line can be used for such screens. Moreover, given that the NMR FASTK gene is inactivated through indel mutations (Fang et al. 2014) and carries a premature stop codon leading to inactivation of Eras (ES cell expressed Ras) (Miyawaki et al. 2016), it suggests that inactivation and/or activity modulation of proto-oncogenes may in part be responsible for the NMR’s cancer resistance. Since single nucleotide changes can result in amino acid substitution which can in turn alter protein activity, the role of the reported NMR specific amino acid changes (Kim et al. 2011; Yang et al. 2015) and other yet unexplored substitutions in the NMR’s cancer resistance is worth investigating. This can be achieved by combining a chemical mutagenesis screen using ENU (N-ethyl-N-nitrosourea) (Acevedo-Arozena et al. 2008) on primary NMR cells with next-generation sequencing (NGS). Lastly, future examination of how the microenvironment, immune system and certain cancer-related genes are involved in cancer resistance at the whole organism level will be of particular interest (Fig. 14.2).

Overview of potential mechanisms of cancer resistance in naked mole-rats
References
Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, Kiso WK, Schmitt DL, Waddell PJ, Bhaskara S et al (2015) Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. J Am Med Assoc 314:1850–1860
Abel EL, Angel JM, Kiguchi K, DiGiovanni J (2009) Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 4:1350–1362
Acevedo-Arozena A, Wells S, Potter P, Kelly M, Cox RD, Brown SDM (2008) ENU mutagenesis, a way forward to understand gene function. Annu Rev Genomics Hum Genet 9:49–69
Albuquerque TAF, Drummond do Val L, Doherty A, de Magalhães JP (2018) From humans to hydra: patterns of cancer across the tree of life. Biol Rev 93(3):1715–1734
Andervont HB, Dunn TB (1962) Occurrence of tumors in wild house mice. J Natl Cancer Inst 28:1153–1163
Artwohl J, Ball-Kell S, Valyi-Nagy T, Wilson SP, Lu Y, Park TJ (2009) Extreme susceptibility of African naked mole rats (Heterocephalus glaber) to experimental infection with herpes simplex virus type 1. Comp Med 59(1):83–90
Buffenstein R (2005) The naked mole-rat: a new long-living model for human aging research. J Gerontol Ser A Biol Sci Med Sci 60:1369–1377
Buffenstein R, Craft W (2021) The idiosyncratic physiological traits of the naked mole-rat; a resilient animal model of aging, longevity, and healthspan. In: Buffenstein R, Park TJ, Holmes MM (eds) The Extraordinary Biology of the Naked Mole-Rat. Springer, New York, pp 221–254
Burek JD, Hollander CF (1977) Incidence patterns of spontaneous tumors in BN/Bi rats. J Natl Cancer Inst 58:99–105
Caulin AF, Maley CC (2011) Peto’s paradox: evolution’s prescription for cancer prevention. Trends Ecol Evol 26(4):175–182
Chrisp CE, Turke P, Luciano A, Swalwell S, Peterson J, Miller RA (1996) Lifespan and lesions in genetically heterogeneous (four-way cross) mice: a new model for aging research. Vet Pathol 33:735–743
Cole JE, Steeil JC, Sarro SJ, Kerns KL, Cartoceti A (2019) Chordoma of the sacrum of an adult naked mole-rat. J Vet Diagn Investig 1:104063871989498
Delaney MA, Nagy L, Kinsel MJ, Treuting PM (2013) Spontaneous histologic lesions of the adult naked mole rat (Heterocephalus glaber): a retrospective survey of lesions in a zoo population. Vet Pathol 50:607–621
Delaney MA, Ward JM, Walsh TF, Chinnadurai SK, Kerns K, Kinsel MJ, Treuting PM (2015) Initial case reports of cancer in naked mole-rats (Heterocephalus glaber). Vet Pathol 53:691–696
Delaney MA, Imai DM, Buffenstein R (2021) Spontaneous disease and pathology of naked mole-rats. In: Buffenstein R, Park TJ, Holmes MM (eds) The Extraordinary Biology of the Naked Mole-Rat. Springer, New York, pp 353–380
Deuker MM, Lewis KN, Ingaramo M, Kimmel J, Buffenstein R, Settleman J (2020) Unprovoked stabilization and nuclear accumulation of the naked mole-rat p53 protein. Sci Rep 10(1):6966
Fang X, Seim I, Huang Z, Gerashchenko MV, Xiong Z, Turanov AA, Zhu Y, Lobanov AV, Fan D, Yim SH et al (2014) Adaptations to a subterranean environment and longevity revealed by the analysis of mole rat genomes. Cell Rep 8:1354–1364
Faulkes CG, Davies KTJ, Rossiter SJ, Bennett NC (2015) Molecular evolution of the hyaluronan synthase 2 gene in mammals: implications for adaptations to the subterranean niche and cancer resistance. Biol Lett 11(5):20150185
Grimes KM, Lindsey ML, Gelfond JAL, Buffenstein R (2012) Getting to the heart of the matter: age-related changes in diastolic heart function in the longest-lived rodent, the naked mole rat. J Gerontol Ser A Biol Sci Med Sci 67 A:384–394
Hadi F, Kulaberoglu Y, Lazarus KA, Bach K, Ugur R, Beattie P, Smith ESJ, Khaled WT (2020) Transformation of naked mole-rat cells. Nature 583(7814):E1–E7
Hilton HG, Rubinstein ND, Janki P, Ireland AT, Bernstein N, Fong NL, Wright KM, Smith M, Finkle D, Martin-McNulty B, Roy M, Imai DM, Jojic V, Buffenstein R (2019) Single-cell transcriptomics of the naked mole-rat reveals unexpected features of mammalian immunity. PLoS Biol 17(11):e3000528
Husson Z, Smith ESJ (2018) Naked mole-rat cortical neurons are resistant to acid-induced cell death. Mol Brain 11(1):26
Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47:89–116
Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM, Sun X, Turanov AA, Yang P et al (2011) Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479(7372):223–227
Kothapalli D, Flowers J, Xu T, Puré E, Assoian RK (2008) Differential activation of ERK and Rac mediates the proliferative and anti-proliferative effects of hyaluronan and CD44. J Biol Chem 283:31823–31829
Kulaberoglu Y, Bhushan B, Hadi F, Chakrabarti S, Khaled WT, Rankin KS, Smith ESJ, Frankel D (2019) The material properties of naked mole-rat hyaluronan. Sci Rep 9(1):6632
Lee BP, Smith M, Buffenstein R, Harries LW (2020) Negligible senescence in naked mole rats may be a consequence of well-maintained splicing regulation. GeroScience 42:633–651
Leroi AM, Koufopanou V, Burt A (2003) Cancer selection. Nat Rev Cancer 3(3):226–231
Lewis KN, Mele J, Hornsby PJ, Buffenstein R (2012) Stress resistance in the naked mole-rat: the bare essentials – a mini-review. Gerontology 58(5):453–462
Lewis KN, Wason E, Edrey YH, Kristan DM, Nevo E, Buffenstein R (2015) Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc Natl Acad Sci 27:259–278
Lewis KN, Soifer I, Melamud E, Roy M, McIsaac RS, Hibbs M, Buffenstein R (2016) Unraveling the message: insights into comparative genomics of the naked mole-rat. Mamm Genome 27:259–278
Liang S, Mele J, Wu Y, Buffenstein R, Hornsby PJ (2010) Resistance to experimental tumorigenesis in cells of a long-lived mammal, the naked mole-rat (Heterocephalus glaber). Aging Cell 9(4):626–635
Lin TD, Buffenstein R (2021) The unusual immune system of the naked mole-rat. In: Buffenstein R, Park TJ, Holmes MM (eds) The Extraordinary Biology of the Naked Mole-Rat. Springer, New York, pp 315–327
Lipman R, Galecki A, Burke DT, Miller RA (2004) Genetic loci that influence cause of death in a heterogeneous mouse stock. J Gerontol Biol Sci 59:B977–B983
Manov I, Hirsh M, Iancu TC, Malik A, Sotnichenko N, Band M, Avivi A, Shams I (2013) Pronounced cancer resistance in a subterranean rodent, the blind mole-rat, Spalax: in vivo and in vitro evidence. BMC Biol 11:91
Menegon S, Columbano A, Giordano S (2016) The dual roles of NRF2 in cancer. Trends Mol Med 22:578–593
Miura K, Oiwa Y, Kawamura Y (2021) Induced pluripotent stem cells from cancer-resistant naked mole-rats. In: Buffenstein R, Park TJ, Holmes MM (eds) The Extraordinary Biology of the Naked Mole-Rat. Springer, New York, pp 329–339
Miyawaki S, Kawamura Y, Oiwa Y, Shimizu A, Hachiya T, Bono H, Koya I, Okada Y, Kimura T, Tsuchiya Y et al (2016) Tumour resistance in induced pluripotent stem cells derived from naked mole-rats. Nat Commun 7:11471
Narayan V, McMahon M, O’Brien J, McAllister F, Buffenstein R (2021) Insights into the molecular basis of genome stability and pristine proteostasis in naked mole-rats. In: Buffenstein R, Park TJ, Holmes MM (eds) The Extraordinary Biology of the Naked Mole-Rat. Springer, New York, pp 287–314
Pearson KJ, Lewis KN, Price NL, Chang JW, Perez E, Victoria Cascajo M, Tamashiro KL, Poosala S, Csiszar A, Ungvari Z et al (2007) Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc Natl Acad Sci 105:2325–2330
Rangarajan A, Weinberg RA (2003) Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer 3(12):952–959
Ross-Gillespie A, O’Riain MJ, Keller LF (2007) Viral epizootic reveals inbreeding depression in a habitually inbreeding mammal. Evolution (N Y) 61(9):2268–2273
Salmon AB, Akha AAS, Buffenstein R, Miller RA (2008) Fibroblasts from naked mole-rats are resistant to multiple forms of cell injury, but sensitive to peroxide, ultraviolet light, and endoplasmic reticulum stress. J Gerontol Biol Sci 63A(3):232–241
Seluanov A, Hine C, Azpurua J, Feigenson M, Bozzella M, Mao Z, Catania KC, Gorbunova V (2009) Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc Natl Acad Sci 106(46):19352–19357
Seluanov A, Gladyshev VN, Vijg J, Gorbunova V (2018) Mechanisms of cancer resistance in long-lived mammals. Nat Rev Cancer 18:433–441
Shepard A, Kissil JL (2020) The use of non-traditional models in the study of cancer resistance – the case of the naked mole rat. Oncogene 39:5083–5097
Takasugi M, Firsanov D, Tombline G, Ning H, Ablaeva J, Seluanov A, Gorbunova V (2020) Naked mole-rat very-high-molecular-mass hyaluronan exhibits superior cytoprotective properties. Nat Commun 11(1):2376
Taylor KR, Milone NA, Rodriguez CE (2017) Four cases of spontaneous neoplasia in the naked mole-rat (Heterocephalus glaber), a putative cancer-resistant species. J Gerontol Ser A Biol Sci Med Sci 72:38–43
Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, Mao Z, Nevo E, Gorbunova V, Seluanov A (2013) High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499(7458):346–349
Tian X, Azpurua J, Ke Z, Augereau A, Zhang ZD, Vijg J, Gladyshev VN, Gorbunova V, Seluanov A (2015) INK4 locus of the tumor-resistant rodent, the naked mole rat, expresses a functional p15/p16 hybrid isoform. Proc Natl Acad Sci 112(4):1053–1058
Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M et al (2016) A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep 17:1193–1205
Yang Z, Zhang Y, Chen L (2015) Single amino acid changes in naked mole rat may reveal new anti-cancer mechanisms in mammals. Gene 572(1):101–107
Zhao Y, Qiao S, Hou X, Tian H, Deng S, Ye K, Nie Y, Chen X, Yan H, Tian W (2019) Bioengineered tumor microenvironments with naked mole rats high-molecular-weight hyaluronan induces apoptosis in breast cancer cells. Oncogene 38:4297–4309
Zhao J, Tian X, Zhu Y, Zhang Z, Rydkina E, Yuan Y, Zhang H, Roy B, Cornwell A, Nevo E et al (2020) Reply to: transformation of naked mole-rat cells. Nature 583:E8–E13
Acknowledgments
FH was funded by a Gates Cambridge Trust PhD scholarship. E.S.J.S is funded by CRUK Grant (C56829/A22053) and W.T.K is funded by CRUK Grant (C47525/A17348).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hadi, F., Smith, E.S.J., Khaled, W.T. (2021). Naked Mole-Rats: Resistant to Developing Cancer or Good at Avoiding It?. In: Buffenstein, R., Park, T.J., Holmes, M.M. (eds) The Extraordinary Biology of the Naked Mole-Rat. Advances in Experimental Medicine and Biology, vol 1319. Springer, Cham. https://doi.org/10.1007/978-3-030-65943-1_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-65943-1_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65942-4
Online ISBN: 978-3-030-65943-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)