
Abstract
Over the past century, the molecular beam methods pioneered by Otto Stern have advanced our knowledge and understanding of the world enormously. Stern and his colleagues used these new techniques to measure the magnetic dipole moments of fundamental particles with results that challenged the prevailing ideas in fundamental physics at that time. Similarly, recent measurements of fundamental electric dipole moments challenge our present day theories of what lies beyond the Standard Model of particle physics. Measurements of the electron’s electric dipole moment (eEDM) rely on the techniques invented by Stern and later developed by Rabi and Ramsey. We give a brief review of this historical development and the current status of eEDM measurements. These experiments, and many others, are likely to benefit from ultracold molecules produced by laser cooling. We explain how laser cooling can be applied to molecules, review recent progress in this field, and outline some eagerly anticipated applications.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

1 Introduction
It has been nearly a hundred years since Otto Stern and Walther Gerlach used an atomic beam to reveal one of the most striking aspects of the then-burgeoning quantum theory, space quantization [1]. Their work introduced new techniques that would later be used in countless experiments in physics and chemistry. Stern saw clearly the great promise of his new method, stating [2]
The molecular beam method must be made so sensitive that in many instances it will become possible to measure effects and tackle new problems which presently are not accessible with known experimental methods.
He was right. The molecular beam method has been at the heart of atomic and molecular physics ever since and remains the method of choice for a huge number of experiments.
A more recent development—laser cooling—can also be traced back to Stern, via Frisch who used the molecular beam method to measure the photon recoil momentum [3]. By controlling this recoil, modern atomic physics experiments routinely cool atoms and ions to \(\upmu \)K temperatures. Until recently, experiments with molecules lagged behind, usually because of the difficulty of cooling them. Nevertheless, molecules have many useful properties that are increasingly being exploited for a variety of applications including tests of fundamental physics. An important example is the measurement of the electron’s electric dipole moment (eEDM) where the precision of molecular experiments exceeds that achieved using atoms. The desire to improve these measurements provides strong motivation to extend cooling and trapping methods to molecules, and this has been achieved in the last decade. Laser cooling has been used to collimate and decelerate molecular beams, capture molecules in magneto-optical traps, and then cool them to ultracold temperature. These ultracold molecules can be used to address a wide variety of important problems - exploring what new forces lie beyond the Standard Model of particle physics [4, 5], studying collisions and reactions at the quantum level [6, 7], simulating the behaviour of many-body quantum systems [8,9,10], and processing quantum information [11, 12].
In this article, we review some of these past developments and future prospects. We begin in Sect. 2 with a brief review of molecular beam sources. In Sect. 3 we consider how the development of molecular beam methods for measuring magnetic dipole moments eventually enabled measurements of the electric dipole moments of fundamental particles. We briefly review the current status of these experiments in Sect. 4 and explain the importance of laser cooling to the future of this endeavour. In Sects. 5 and 6 we explain how laser cooling works for molecules and present recent achievements in this field. Finally, in Sect. 7, we give a brief overview of some applications of ultracold molecules and how they might be realized.
2 Molecular Beam Sources
The molecular beam method developed by Stern [13, 14] is the foundation for innumerable experiments in atomic and molecular physics. Here, we give a brief review of the three main types of atomic and molecular beam sources in use today: effusive beams, supersonic beams, and cryogenic buffer gas beams. Their velocity distributions and flux are compared in Fig. 1 for the case of YbF molecules, one of the few species for which all three types of beam source have been realized.
Effusive sources typically use heated ovens to generate a sufficient vapour pressure of the atoms or molecules of interest. They operate at low pressure so that there are no collisions in the vicinity of the exit aperture. As first shown experimentally by Stern [13, 14], these sources produce beams with a broad velocity distribution, whose mean and width both scale as \(\sqrt{T/m_\mathrm{s}}\), where T is the temperature of the source and \(m_\mathrm{s}\) is the mass of the species. As a consequence of the high oven temperature, effusive beams are characterized by a wide velocity distribution and low flux in any single quantum state.

Velocity distributions of effusive, supersonic, and buffer-gas beams. The vertical axis indicates the number of molecules per unit solid angle per unit time and per unit interval of velocity. The plots are for YbF molecules in their absolute ground state, chosen because, for this molecule, all three sources have been developed and characterised. Realistic operating conditions have been taken. For the effusive case, molecules are generated from an oven at 1500 K [15]. For the supersonic case, a carrier gas of argon with a reservoir temperature of 300 K is assumed, with an internal beam temperature of 4 K and a repetition rate of 25 Hz [16]. For the cryogenic buffer-gas case, a carrier gas of helium at 4 K creates a beam with moderate hydrodynamic boosting, operating at 10 Hz [17]
In supersonic sources [18, 19], a gas held at high pressure expands through a nozzle into a vacuum chamber. There are a large number of collisions in the vicinity of the nozzle. The slower particles are bumped from behind, while the faster ones bump into those ahead, so that all the particles end up travelling at nearly the same speed. In this way, the random thermal motion is converted into forward kinetic energy, producing a cold, fast beam—the mean velocity is high, but the velocity distribution is narrow, as illustrated in Fig. 1. The collisions also transfer the rotational and vibrational energy of the molecules to forward kinetic energy, resulting in a beam that is cold in all degrees of freedom. The first supersonic beams were continuous, but the method was soon extended by using pulsed valves with short opening times. In this way, intense pulses can be produced without an excessive gas load. A wide variety of methods have been developed to introduce atoms and molecules of interest into the supersonic expansion, including laser ablation, electric discharge, and photodissociation. Translational temperatures of 1 K are typical, and beam speeds are 1800 m/s when the carrier gas is room temperature helium, and 400 m/s for room temperature krypton.
The third type of molecular beam source is the cryogenic buffer gas source [20, 21]. Here, the molecules of interest are formed inside a cryogenically-cooled cell containing a cold buffer gas, often helium at 4 K. The molecules are commonly formed by laser ablation or introduced into the cell through a capillary. The internal and motional degrees of freedom of the molecules thermalize through collisions with the buffer gas, and then the molecules exit through a hole in the cell to form a beam. The density of buffer gas in the cell is determined by the gas flow rate and the size of the exit hole. When the density is low, there are few collisions near the aperture, so the beam tends towards the effusive regime. These beams are slow, especially for heavy species, for then the mass \(m_\mathrm{s}\) is large whereas the temperature T is small, typically two orders of magnitude lower than a standard effusive source. Beam speeds as low as 40 m/s have been achieved this way [22]. However, the molecular flux tends to be low in this regime because most molecules diffuse to the cell walls, where they freeze, instead of passing through the exit aperture. As the buffer gas flow is increased it sweeps more molecules out of the cell, increasing the beam flux. However, collisions near the aperture boost the beam speed. In the limit of high density the speed of the molecules reaches the supersonic speed of the buffer gas which scales as \(\sqrt{T/m_\mathrm{b}}\), where \(m_\mathrm{b}\) is the mass of the buffer gas atoms. Very often, cryogenic buffer gas sources are operated in an intermediate flow regime where the flow is high enough to extract a substantial fraction of the molecules from the cell, but low enough for a moderate beam speed. Speeds in the range 100–200 m/s are typical, as illustrated in Fig. 1. Due to the high flux and low relative beam speeds, these sources are becoming increasingly popular, especially for experiments on laser cooling of molecules and tests of fundamental physics.
3 Particle Dipole Moments
Stern’s pioneering experiments established the reality of space quantization and determined the magnetic dipole moments of the electron and proton [1, 23,24,25]. It is interesting to consider whether elementary particles might also have electric dipole moments. Just like the magnetic moment, such an electric dipole would have to be oriented along the particle’s spin. Furthermore, this orientation must be fixed, since the particle would otherwise have an additional degree of freedom that would, for example, change the filling of electron orbitals in the periodic table. A spin defines a direction of circulation, as does a magnetic moment, so it seems natural for the two to be associated. Far less natural is the association of an electric dipole – a charge separation – with this direction of circulation. Indeed, such an electric dipole moment (EDM) implies a difference between left- and right-handed coordinate systems, and implies a fundamental arrow of time. To see this, consider the Hamiltonian for a particle with magnetic moment \(\boldsymbol{\mu }\) and EDM \(\boldsymbol{d}\), both fixed relative to the spin, interacting with magnetic and electric fields \(\boldsymbol{B}\) and \(\boldsymbol{E}\):
Reflection in a mirror, equivalent to the parity operation, reverses \(\boldsymbol{E}\) but does not reverse \(\boldsymbol{B}\), \(\boldsymbol{\mu }\) or \(\boldsymbol{d}\). Conversely, reversing the direction of time reverses \(\boldsymbol{B}\), \(\boldsymbol{\mu }\) and \(\boldsymbol{d}\), but not \(\boldsymbol{E}\). We see that while the first term in \(\mathcal {H}\) is even under both the parity and time-reversal operations, the second term is odd and so the existence of an EDM violates both symmetries.
Prior to the 1950s, it was generally considered that nature did not distinguish between left and right, or between forwards and backwards in time, and this seemed to be a powerful argument against the existence of fundamental electric dipoles, implying that \(|\boldsymbol{d}| = 0\). This idea was challenged by Purcell and Ramsey who insisted that it was “a purely experimental matter”, noted that existing evidence was weak, and declared their intention to measure the EDM of the neutron [26]. Regarding the need for experimental evidence to determine whether parity (P), time-reversal (T) and charge conjugation (C) are symmetries of nature, Hermann Weyl was similarly emphatic, writing that [27]
a priori evidence is not sufficient to settle the question; the empirical facts have to be consulted.
Along this line of thought, in 1956 Lee and Yang [28] noted that, for the weak interaction,
parity conservation is so far only an extrapolated hypothesis unsupported by experimental evidence.
Within a few months it was discovered that the weak interaction violates P symmetry [29,30,31]. In 1964 it was found that the weak interaction also violates CP symmetry, the combined symmetry of charge conjugation and parity [32]. CP violation is equivalent to T-violation in most theories, and so the last theoretical objection to the existence of fundamental EDMs was removed. Suddenly, and ever since, the question was not whether particles could have electric dipoles, but why those dipoles are so small.

The evolution of molecular beam methods for measuring dipole moments. (a) Stern and Gerlach’s experiment for measuring magnetic moments and demonstrating space quantization. (b) Rabi’s improved method for measuring magnetic moments. Between the two deflectors there is a uniform magnetic field and an oscillating field which, when resonant, changes the spin state. (c) A further improvement by Ramsey separates the oscillating field into two short regions. (d) Method for determining the electron’s electric dipole moment by measuring the spin precession in an applied electric field. In all its key aspects, the technique is the same as Ramsey’s method
In considering how the electric dipole moment of a particle might be measured, it is instructive to reflect on Stern’s method for measuring magnetic moments, which is illustrated in Fig. 2a. The magnetic moment of an atom is proportional to its internal angular momentum, \(\hbar \boldsymbol{F}\), so we often write \(\boldsymbol{\mu } = -g \mu _\mathrm{B} \boldsymbol{F}\), where \(\mu _\mathrm{B}\) is the Bohr magneton and g is a proportionality factor. Taking \(\boldsymbol{B}\) in the z-direction, the interaction energy is \(W_\mathrm{B} = \langle -\boldsymbol{\mu }\cdot \boldsymbol{B} \rangle = g \mu _\mathrm{B} B \langle F_z \rangle = g \mu _\mathrm{B} B \,m_F \), where \(m_F\) is the projection of the angular momentum onto the z-axis. Space quantization is expressed by only discrete values being allowed for \(m_F\). In the inhomogeneous field of the Stern-Gerlach magnets there is a force on the atoms \(\boldsymbol{F}_\mathrm{B} = -\boldsymbol{\nabla }W_\mathrm{B} = -g \mu _\mathrm{B} \boldsymbol{\nabla }B \,m_F\), leading to the deflection proportional to \(m_F\) observed by Stern and Gerlach. Let us now consider an atom with an unpaired electron that has an eEDM, \(d_e\). If an electric field E is applied in the z-direction, there is an interaction energy \(W_\mathrm{E} = -R d_e E \, m_F/|F|\), analogous to the magnetic interaction. The proportionality factor R depends on the choice of atom and is often called the enhancement factor because, for heavy atoms, |R| can be considerably larger than 1 [33]. For example, for Cs, \(R=120\) [34]. Suppose we pass a beam of Cs atoms through a region of inhomogeneous electric field. Of course, the electric field produces an induced electric dipole in the atom, resulting in a force which is often used to deflect atoms and molecules. This force is the same for states of opposite \(m_F\) and is not the one of interest here. The force due to the permanent EDM is \(\boldsymbol{F}_\mathrm{E} = -\boldsymbol{\nabla }W_\mathrm{E} = R d_e \boldsymbol{\nabla }E \, m_F/|F|\), and deflects states of opposite \(m_F\) in opposite directions. Taking \(d_e = 10^{-29}\) e cm, which is close to the current upper limit [35], a field gradient of 100 kV/cm\(^2\), and a speed of 200 m/s, the deflection after propagating 1 m is about \(10^{-19}\) m. Clearly, this is not a good way to measure \(d_e\). Nevertheless, the subsequent development of molecular beam methods inspired by Stern’s techniques became so sensitive that measurements of \(d_e\) soon became feasible.
In the late 1930s, Rabi introduced a new idea that greatly improved Stern’s method of measuring magnetic moments [36]. Instead of using the magnetic deflector as a measuring device, he used a pair of them as state selectors, as illustrated in Fig. 2b. The deflectors are arranged such that molecules reach a detector provided they remain in the same state so that they have the same magnetic moment throughout. Between the two deflectors, Rabi produced a uniform magnetic field, \(\boldsymbol{B}_0\), so that neighbouring \(m_F\) states are separated by an energy \(\hbar \omega _0 = g\mu B_0\). In this region, an rf field of frequency \(\omega \approx \omega _0\) resonantly drives transitions from one \(m_F\) state to another. Molecules that change \(m_F\) are deflected by the second magnet and miss the detector, resulting in a dip in the detected signal at \(\omega =\omega _0\). This measurement of the resonant frequency, together with a measurement of \(B_0\), determines the magnetic moment. The precision of this measurement is proportional to the interaction time with the rf field, so it’s desirable to make this as long as possible. In practice however, this time is limited by the difficulty of keeping the fields uniform enough.
By the 1950s, Ramsey had solved this problem by separating the rf region into two short sections driven by the same oscillator [37], as illustrated in Fig. 2c. The first deflecting magnet prepares the molecules in a chosen spin state, say spin up. The first rf region rotates the spin so that it is orthogonal to \(\boldsymbol{B}_0\). The spin then precesses in the uniform magnetic field with angular frequency \(\omega _0\) for a time T. When \(\omega = \omega _0\) the rf oscillation is in phase with the spin precession, so the second rf region rotates the spin in the same direction as the first, producing the spin-down state which will miss the detector. If there is a frequency difference \(\omega -\omega _0 = \pm \pi /T\), the extra half rotation means that the spin will be driven back to the spin-up state in the second rf region and will reach the detector. The signal at the detector oscillates as \(\omega \) is scanned, allowing \(\omega _0\) to be determined with an uncertainty inversely proportional to the free precession time T. Ramsey’s method has such high precision that it is suitable for measuring the tiny electric dipole moments of fundamental particles. All that is needed is to add to \(B_0\) a uniform electric field \(E_0\), and then measure the change in the precession frequency when the direction of \(E_0\) is reversed. This change is proportional to the EDM. This was the method used by Smith, Purcell and Ramsey in their first measurement of the neutron EDM [38], and the one used for all subsequent measurements of particle electric dipole moments.
Figure 2d illustrates a typical electron EDM measurement that uses Ramsey’s molecular beam method. A molecular beam passes through a state selector, which could be a magnetic or electric deflector but in modern experiments is often a laser beam that optically pumps molecules to the desired state. Next, a first region of oscillating field aligns the spin along x. This can be done using an rf field, or a laser field that drives a Raman transition or optical pumping process. The spin now precesses around z due to the combination of \(\mu \) interacting with \(B_0\) and \(d_e\) interacting with \(E_0\). Finally the spin direction is measured, for example by mapping its direction to a pair of states that are easily distinguished spectroscopically. The change in the spin precession angle that correlates with the reversal of \(E_0\) determines \(d_e\).
4 Current Status and Future Directions of eEDM Experiments
In the Standard Model of particle physics, the eEDM is predicted to be \(d_e \approx 10^{-38}\) e cm [39]. Theories that extend the Standard Model often introduce new CP-violating interactions, which are needed to explain the observed asymmetry between matter and antimatter in the universe [40], and these new interactions lead to much larger eEDM values. Thus, eEDM measurements can be excellent probes of these theories. Early measurements used heavy atoms and yielded results consistent with zero, eventually assigning an upper limit of \(|d_e| < 1.6 \times 10^{-27}\) e cm [41, 42]. Although more difficult to produce and control, heavy polar molecules can be far more sensitive to the eEDM than atoms [43, 44]. The sensitivity is proportional to the degree of polarization and therefore to the electric-field-induced mixing of opposite parity states. In atoms, these are different electronic states whose spacing is typically \(\sim 1\) eV, but in molecules they are neighbouring rotational states whose energy spacing is about four orders of magnitude smaller, or the opposite parity states of an \(\varOmega \)-doublet where the spacing is even smaller still. Because the levels are closely spaced, only a modest electric field is needed to fully polarize the molecule. In this case, it is common to define an effective electric field \(E_\mathrm{eff} = R E\) which saturates to a maximum value \(E_\mathrm{eff}^\mathrm{max}\). The effective field is enormous for some species, and its maximum value is often easy to reach. For example, \(E_\mathrm{eff}^\mathrm{max} \approx 26\) GV/cm for YbF and \(\approx 78\) GV/cm for ThO.
The enormous effective fields make eEDM experiments with molecules very attractive, and measurements have been made using beams of YbF [45, 46], beams of ThO [35, 47], a cell of PbO vapour [48], and trapped HfF\(^{+}\) molecular ions [49]. The results of these measurements are all consistent with zero, and the best upper limit is currently \(|d_e| < 1.1 \times 10^{-29}\) e cm at the 90% confidence level [35]. Remarkably, this experiment and ones like it test theories that extend the Standard Model at an energy scale similar to, and even exceeding, the maximum collision energy of the Large Hadron Collider.
Given the great significance of eEDM experiments, it is natural to consider how to make the next leap in sensitivity. The uncertainty in measuring \(d_e\) scales as \(1/(T\sqrt{N})\) where T is the spin precession time and N is the number of molecules used in the measurement. In a molecular beam experiment of length L, where the spin precession region occupies most of the space and the beam has diverged sufficiently that it fills the detector, T is proportional to L but N falls as \(1/L^2\) because of the divergence of the beam. Consequently, there is no benefit in increasing L. This can be circumvented by cooling the molecules to much lower temperatures. A beam that is cooled in the transverse directions is collimated and can travel for long distances without spreading, allowing T to increase without reducing N. Going further, molecules that are cold enough can be launched into a fountain [50] or stored in a trap [51], giving access to even longer spin precession times. These ideas require molecules cooled to \(\mu \)K temperatures. Such low temperatures can be reached either by associating ultracold atoms into molecules [52], or by direct laser cooling [53]. EDM experiments using laser-cooled YbF, BaF, YbOH and TlF are all currently being developed [54,55,56,57]. They will have unprecedented sensitivity and tremendously exciting potential for new discoveries in fundamental physics.
5 Laser Cooling of Molecules: Principles
5.1 Laser Cooling Scheme
Figure 3 illustrates the energy level structure of a typical diatomic molecule, showing the electronic, vibrational, rotational, and hyperfine structure and the notation used to label the levels. For molecules to be slowed, cooled and trapped by radiation pressure, they must scatter many photons from the light, typically \(10^4\) or more. This calls for a cooling scheme where an upper level decays to only a few lower levels, so that only a few transitions need to be addressed. The inset to Fig. 3 shows an example of such a scheme. The upper level is the lowest level of positive parity in the first electronically excited state, labelled here as \(\mathrm{A}, v=0, R=0, +\). Electric dipole transitions to the X state must change the parity and obey the selection rule \(\varDelta R=0,\pm 1\), which means that only the \(R=1\) rotational state is accessible.Footnote 1 However, the molecule can decay to any vibrational state, since there is no selection rule dictating how v can change in an electronic transition. The branching ratio to each vibrational state is mainly given by the square of the overlap integral between the vibrational wavefunctions in the lower and upper electronic states, which is known as the Franck Condon factor. For molecules where the optically active electron is not involved in the bonding, the sets of vibrational wavefunctions for the two electronic states are very similar. In this case, the branching ratio is close to 1 for the \(\varDelta v=0\) transition and diminishes rapidly with increasing \(\varDelta v\). These molecules are the ones best suited to laser cooling because only a few vibrational bands need to be addressed, requiring just a few lasers, as indicated in the figure. Hyperfine components of these transitions can usually be addressed by adding radio-frequency sidebands to each laser using acousto-optic or electro-optic modulators.

Energy level structure of a typical diatomic molecule, with indicative transition frequencies. The ground electronic state is labelled X, and the excited states are A, B, etc. Transitions between electronic states are usually in, or near, the visible part of the electromagnetic spectrum. Each electronic state has a set of vibrational states, labelled by v. Vibrational transitions are usually in the mid-infrared. Each vibrational state has a set of rotational states, labelled by the rotational angular momentum, R, and the parity p. Rotational transitions are in the microwave regime. Each rotational state usually has a set of hyperfine states with total angular momentum F determined by the electronic angular momentum and the nuclear spin. In the example shown, both are 1/2. The inset shows the laser cooling scheme discussed in the text. Arrows show transitions driven by lasers, and the weights of the dotted lines indicate the relative branching ratios of the decay channels
5.2 Doppler and Sub-Doppler Cooling
Despite the complexity of the molecular structure outlined above, and the need to drive many transitions using several laser frequencies, the basic principles of laser cooling can be understood by focussing on just two or three levels. Figure 4a illustrates the principle of Doppler cooling applied in one dimension to a hypothetical two-level molecule. A molecule moving to the right with speed v interacts with a pair of identical, counter-propagating laser beams with wavevector k. The frequency of the light, \(\omega \), is slightly smaller than the molecular transition frequency, \(\omega _0\). The laser beam from the right is Doppler shifted closer to resonance, so the molecule scatters more photons from this beam and slows down as a result of this imbalanced radiation pressure. The force on the molecule due to each of the beams is
where \(\delta _0 = \omega - \omega _0\) is the detuning, \(\varGamma \) is the natural linewidth of the transition, I is the laser intensity and \(I_\mathrm{s}\) is a characteristic intensity known as the saturation intensity. The graph in Fig. 4a shows these two forces as a function of v in the case where \(I=I_\mathrm{s}\) and \(\delta _0 = -2 \varGamma \). The solid line is their sum and shows that there is a force driving the molecule towards zero velocity. In addition to this cooling force, there is heating due to the randomly-directed momentum kicks associated with the photon scattering events. When the heating and cooling rates are balanced, the molecule reaches its equilibrium temperature. The minimum temperature for Doppler cooling is known as the Doppler limit and is \(T_\mathrm{D,min} = \hbar \varGamma /(2 k_\mathrm{B}).\)

Principles of a Doppler cooling, where a negative detuning is used and b sub-Doppler cooling, where a positive detuning is used
Figure 4b illustrates a method of sub-Doppler cooling. Here, we distinguish two Zeeman sub-levels of the ground state. Due to the choice of states and the angular momentum selection rules, laser light of a given polarization cannot drive transitions from one of the ground states. We call this the dark state because it does not couple to the light. The other state is called the bright state. The bright state has an ac Stark shift which is positive when the detuning is positive. The dark state has no ac Stark shift because it does not couple to the light. If the two counter-propagating laser beams have different polarizations, neither parallel nor orthogonal, both the intensity and polarization of the light field will vary with position. This causes the ac Stark shift of the bright state to be modulated in position, setting up potential energy hills and valleys which the molecules move through. A molecule in the bright state will be excited by the light and optically pumped into the dark state. This is most likely to happen at positions where the intensity of the light field is high, which are also the positions where the ac Stark shift is largest, i.e. near the tops of the hills. A moving molecule can make a non-adiabatic transition back to the bright state because the polarization changes as it moves. This is most likely to happen where the energy gap between bright and dark states is smallest, i.e. near the bottom of the hills. As a result, molecules moving through the light field lose energy because they climb hills more often than they descend into valleys. The graph in Fig. 4b shows the typical force produced by this mechanism. It operates over a smaller range of velocities than Doppler cooling, and produces a smaller maximum force. Crucially however, the gradient near zero velocity, which is the damping constant, is substantially higher than for Doppler cooling. Furthermore, because the molecule spends much of its time in the dark state, there is less photon scattering, and thus a lower heating rate. Thus, for small velocities, the cooling rate is higher while the heating rate is lower, leading to much lower temperatures.

Typical form of the total force as a function of speed due to the combination of Doppler and sub-Doppler processes illustrated in Fig. 4. The two lines are for equal and opposite detunings: dashed red for negative, and solid blue for positive
The Doppler cooling mechanism shown in Fig. 4a requires a negative detuning, while the sub-Doppler cooling mechanism requires a positive detuning.Footnote 2 The two mechanisms often appear together, resulting in the typical velocity-dependent force illustrated in Fig. 5. A negative detuning is useful for capturing molecules with a wide range of initial velocities and cooling them to lower velocity. However, the lowest temperatures are not reached because the total force has the wrong sign at low velocity. Once molecules are slow enough, the frequency of the light can be switched to a positive detuning so that the sub-Doppler mechanism cools them further. In this way, molecules have been cooled to temperatures far below the Doppler limit.
6 Laser Cooling of Molecules: Practice
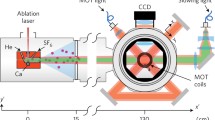
Figure 6 illustrates an apparatus for laser cooling and trapping of molecules. The experiments begin with a molecular beam, a testament to the experimental power of the method developed by Stern and subsequent researchers. The cryogenic buffer gas sources described in Sect. 2 are ideal for this application because they deliver the critical combination of a high flux of molecules with a low initial speed. The illustration shows how this beam is laser cooled in the transverse directions, decelerated to low speed using radiation pressure, and then captured and cooled in a magneto-optical trap. We discuss each of these steps in turn.

An illustration of an apparatus for laser cooling and trapping of molecules. A beam of molecules from a cryogenic buffer gas source is cooled in the transverse directions, decelerated by the radiation pressure of a counter-propagating laser beam, and captured in a magneto-optical trap

Adapted from [54]
Transverse laser cooling of YbF molecules. a Schematic of experiment. b Density distribution in one transverse direction following laser cooling in that direction. The lines are fits to a sum of four Gaussians.
6.1 Transverse Laser Cooling of a Molecular Beam
The density in a molecular beam drops with distance from the source because the beam spreads out as it propagates. Laser cooling can reduce the transverse temperature enormously, resulting in an intense, highly collimated molecular beam. The pioneering work on laser cooling of molecules was done at Yale [58]. They worked with a beam of SrF molecules and showed how to cool the beam in one transverse direction using both Doppler cooling and sub-Doppler cooling. Several other diatomic and polyatomic molecular species have since been cooled using similar methods [54, 59,60,61,62,63,64]. Figure 7a illustrates an experiment [54] to cool YbF molecules in one transverse direction, x. A beam of molecules from a cryogenic buffer gas source passes through a 20 cm long sheet of laser cooling light that forms a standing wave in the x direction. All molecules are then optically pumped to the lowest vibrational level by the clean-up light, and then detected by laser-induced fluorescence on a camera. Figure 7b shows the resulting density distribution of these molecules along x. When the detuning of the laser light is positive, a narrow peak appears at the centre of the distribution, corresponding to molecules that have been cooled to low temperature by sub-Doppler cooling. In this experiment, the transverse temperature of the cooled molecules was found to be below 100 \(\mu \)K. When the detuning is negative, a dip appears at the centre with broad wings on either side where the molecules have accumulated. The reason for this profile can be appreciated from the form of the force curve in Fig. 5. For negative detuning, the sub-Doppler mechanism forces molecules near zero velocity to a higher velocity, while Doppler cooling forces high velocity molecules towards lower velocities. As a result, molecules accumulate around the non-zero velocity where the force curve crosses zero.
6.2 Slowing a Molecular Beam with Radiation Pressure
Transverse laser cooling produces a highly collimated molecular beam, but the molecules still have a high forward speed. They can be decelerated using the radiation pressure of a laser beam propagating in the opposite direction. Here, it is essential to account for the changing Doppler shift as the molecules slow down. This can be done by chirping the frequency of the laser so that it follows the changing Doppler shift, or by broadening the frequency spectrum of the laser to cover the full range of Doppler shifts. Laser slowing of molecules was first demonstrated by the group at Yale using SrF [65], and similar methods have been applied to other molecules [66,67,68,69].

Adapted from [69]
Radiation pressure slowing of a beam of CaF molecules. The laser light propagates in the opposite direction to the molecular beam and is frequency chirped. Black lines show the velocity distributions without slowing, and coloured lines are the distributions when the slowing is applied with various frequency chirps. The dashed lines show the resonant velocity at the beginning and end of the chirp.
Figure 8 illustrates frequency-chirped laser slowing of a cryogenic buffer-gas beam of CaF molecules [69]. The black curves show the velocity distributions with no slowing applied, and the coloured curves show the distributions after slowing using various frequency chirps. The initial frequency of the laser is tuned to be resonant with molecules moving at about 180 m/s. When there is no chirp, the molecules are decelerated to about 100 m/s. They bunch up around this speed because the faster molecules are initially closer to resonance so are decelerated more than the slower ones. The distribution is shifted to lower velocities as the chirp increases, but the number of detected molecules drops at low velocities. This is because there is no transverse cooling in these experiments, so the beam diverges rapidly as it slows down, reducing the number of molecules that pass through the detector.
6.3 Trapping the Molecules
With the molecules slowed to low velocity, it becomes possible to trap them. Magneto-optical traps (MOTs) have been used to cool and trap atoms for decades [71]. In a MOT, counter-propagating laser beams result in a velocity-dependent force which cools the atoms, as described in Sect. 5.2. The detuning of the light is usually negative so that Doppler cooling, with its large capture velocity, is the dominant process. This alone does not trap the atoms because the force does not depend on position. To produce a position-dependent force, a magnetic field gradient is added, typically by using a pair of coils as illustrated in Fig. 6. The current flows in opposite directions in the two coils so that the magnitude of the field is zero at the centre and increases linearly in all directions away from this point. For a stationary atom at the field-zero, there is no net force because the atom is equally likely to scatter photons from any of the laser beams. When the atom is displaced, the Zeeman effect shifts the frequencies of transitions with \(\varDelta m_{F}=\pm 1\) in opposite directions, one closer to resonance and the other further away. These two transitions are driven by circularly polarized light of opposite handedness. By choosing the handedness of the beams in the correct way, the transition closest to resonance is always driven by the beam that pushes the atom back towards the field-zero. This traps the atoms.

Figure taken from [70]
Damped oscillations of CaF molecules in a magneto-optical trap. a Fluorescence images of the trapped molecules at various times after giving the molecules a radial push. b Red points: position of the centre of the cloud versus time. Blue line: fit to a damped harmonic oscillator model.
The first three-dimensional magneto-optical trap of molecules was demonstrated in 2014 by the Yale group [72]. They captured a few hundred SrF molecules for about 50 ms at a density of \(6\times 10^{2}\) cm\(^{-3}\), and cooled them to 2.3 mK. They went on to investigate several other trapping configurations [73,74,75] and were soon able to trap about \(10^{4}\) molecules for 500 ms at a density of \(2.5 \times 10^{5}\) cm\(^{-3}\) and temperatures down to \(250~\mu \)K. MOTs of CaF [70, 76, 77] and YO [78] molecules have also been produced, with steadily increasing number densities. Figure 9a is a sequence of pictures showing CaF molecules trapped in a MOT. Each picture is made by imaging the fluorescence of the trapped molecules onto a camera. The molecules were given a sudden push in the horizontal direction at time T=0 and the subsequent images show them oscillating in the trap. Figure 9b shows the displacement of the cloud versus T together with a fit to the motion of a damped harmonic oscillator. The results show that the MOT exhibits both a restoring force and a damping force, as expected.
Once molecules have been trapped in a MOT, they can be cooled to much lower temperature using the sub-Doppler cooling method described in Sect. 5.2. Typically, the magnetic field is turned off and the detuning of the lasers is switched from negative to positive. The molecules typically cool below the Doppler limit [76, 79, 80] in about 1 ms, and temperatures as low as 4 \(\mu \)K have been reached in this way [81,82,83]. At this point the cooling can be turned off and the molecules stored in conservative traps where their quantum states can be controlled and preserved for long periods. Ensembles of laser-cooled molecules have recently been confined in pure magnetic traps [80, 84] and in optical dipole traps [79], and single molecules have been held in tightly-focussed tweezer traps [85]. Coherent control of the hyperfine and rotational states of molecules has been studied [10, 84] and rotational superpositions with long coherence times have been demonstrated for trapped molecules [86].
7 Applications of Laser-Cooled Molecules
The ultracold molecules produced by direct laser cooling are well suited to a wide variety of exciting applications. Many of these applications require molecules trapped for long periods, long-lived coherences, and control over all degrees of freedom at the quantum level, often including the motional degree of freedom. Figure 10 illustrates four experimental approaches that satisfy some, or all, of these requirements. The molecules may be launched into a molecular fountain so that they are in free fall throughout a measurement [50, 87], or they could be trapped near the surface of a chip that integrates microscopic traps with superconducting microwave resonators [12]. Small, reconfigurable arrays of molecules can be produced using optical tweezer traps [85], while larger arrays can be made using optical lattices in one, two or three dimensions [88]. Here, we give an overview of future research directions using these platforms.

Techniques for controlling ultracold molecules. a Molecules are launched into a fountain. b Molecules are stored on the surface of a chip using either electric or magnetic potentials created by planar electrodes or current-carrying wires. c Molecules are trapped in a 1D, 2D, or 3D lattice created by interfering counter-propagating lasers. d Molecules are trapped using an array of optical tweezers
7.1 Testing Fundamental Physics
Ultracold molecules provide several avenues to constrain new theories or discover new physics [5]. As discussed in Sects. 3 and 4, the use of molecules to measure fundamental electric dipole moments is an amazingly powerful probe of symmetry-violating physics beyond the Standard Model. Other kinds of symmetry tests can also be done using molecules. For example, they can be used to explore parity violation in nuclei with unprecedented sensitivity [89]. Of particular interest is the measurement of nuclear anapole moments which arise from weak interactions within nuclei. A recent experiment with a beam of BaF has demonstrated the exceptional sensitivity achievable [90]. It is also of fundamental interest to measure the parity-violating energy difference between left- and right-handed chiral molecules, which is predicted but has not yet been observed [91]. The recent extension of laser cooling to polyatomic symmetric top molecules [63] shows that quite complex molecules can be cooled to sub-millikelvin temperature, making a parity-violation measurement using a laser cooled chiral species feasible in the future.
Precise measurements of molecular transition frequencies can also be used to test the idea that the fundamental constants may actually vary in time or space, or according to the local density of matter. Such variations are predicted by theories that aim to unify gravity with the other forces, and by some theories of dark energy [92, 93]. The frequencies of molecular transitions depend primarily on two fundamental constants, the fine-structure constant \(\alpha \) and the proton-to-electron mass ratio \(\mu =m_p/m_e\). The rotational and vibrational frequencies of molecules scale as \(\mu ^{-1}\) and \(\mu ^{-1/2}\) respectively, a direct dependence that an electronic transition in an atom does not have. Moreover, certain transitions have enhanced sensitivity to \(\alpha \) or \(\mu \) [94], sometimes because the transition energy results from a near cancellation between two large contributions of different origin. Astrophysical observations of atomic and molecular spectra can be used to study variations on a cosmological timescale [5]. Here, laboratory measurements are important for establishing the present-day frequencies to high precision, as has been done using cold molecular beams of OH [95] and CH [96]. Atomic and molecular clocks can be used to set limits on present-day variations on a timescale of a few years. So far, the most stringent limits come from atomic clock measurements [97, 98], but molecular clocks are likely to contribute valuable information in the near future. For example, ultracold KRb molecules were recently used to set limits on the temporal variation of \(\mu \) [99], a lattice clock of Sr\(_2\) molecules is being developed [100], a molecular fountain of ultracold ammonia molecules has been demonstrated and could be used to search for variations in \(\mu \) [87], and clocks based on the vibrational transitions of laser-cooled molecules look promising [101].
7.2 Collisions and Ultracold Chemistry
Molecules prepared at ultracold temperature in a single quantum state are ideal for studying how those molecules interact and what happens in a collision or chemical reaction [102]. With such a high degree of control, it becomes possible to explore how the rotational or hyperfine state influences the outcome of a collision, and to study collisions in a single partial wave regime. Electric or magnetic fields can be used to tune through collision resonances, and electric fields can be used to control the relative orientation of the colliding molecules. A fascinating recent advance in this direction is the study of collisions between individual laser-cooled molecules trapped in optical tweezers [103]. Two molecules, each prepared in a single quantum state, were brought together in a highly controlled way by merging the two separate tweezers, and the collisional loss rate measured for several choices of state. This experiment marks the first contribution of laser-cooled molecules to understanding ultracold chemistry. Some work in the ultracold regime has already been done using molecules assembled from ultracold atoms [52]. Examples include the control of chemical reactions though the choice of quantum state [104] or molecule orientation [105], and the controlled formation of a single molecule from a pair of atoms [106]. Direct laser cooling diversifies the set of ultracold molecules and molecular properties available for these studies, which is an exciting prospect for future research.
7.3 Quantum Simulation
It is important to understand the behaviour of systems consisting of many quantum particles all interacting with one another. These many-body quantum systems exhibit remarkable phenomena that are poorly understood at present, such as high temperature superconductivity, magnetism, the fractional Hall effect, and the structure of nuclei. We often use computer simulations to help understand complicated systems, but it is impossible for a (classical) computer to simulate more than a few tens of interacting quantum particles. Instead, we may try to engineer a well-controlled quantum system in such a way that it simulates a many-body problem that we wish to understand [107]. One system that has been developed with considerable success is an optical lattice of ultracold atoms. These lattices have been used to study important problems in condensed matter physics, such as the quantum phase transition between a superfluid and a Mott insulator [108], and models of antiferromagnetism [109]. However, the variety of systems that can be simulated is limited because atoms only have short-range interactions, meaning that they only interact appreciably when they are on the same site of the lattice. Complex many-body phenomena usually arise from long-range interactions among many particles, which atoms in a lattice struggle to emulate. A lattice of ultracold polar molecules solves this problem because the molecules interact through the long-range dipole-dipole interaction. This interaction has two main effects. First, the energy of the system depends on the configuration of the dipoles, and second, the interaction can mediate the transport of excitations from one site to another. Both effects have a long-range, anisotropic character, and can be controlled using a dc electric field or a microwave field resonant with a rotational transition. This makes for a tremendously rich environment for exploring the behaviour of interacting quantum systems [9]. Taking a first step in this direction, the effects of spin-exchange mediated by dipole-dipole interactions have been studied in a lattice of polar molecules [110]. Recent progress that will advance this field includes the formation of a Fermi degenerate gas of molecules [111], collisional cooling methods for molecules [112], the ever-increasing variety of polar molecules being brought into the ultracold regime, and the improvements in controlling their hyperfine and rotational states [10, 86, 113, 114]. It seems likely that, in the near future, lattices of ultracold polar molecules will significantly advance our understanding of strongly-interacting many-body quantum systems.
7.4 Quantum Information Processing
There are many proposals for using ultracold molecules for quantum information processing [11, 12, 115,116,117]. The hyperfine and rotational states of molecules have extremely long lifetimes and so can serve as stable qubits or qudits. By using microwave fields to drive rotational transitions, single qubit operations can be done rapidly and robustly using very mature microwave technology. The dipole-dipole interaction can be used to entangle pairs of molecules and perform two-qubit operations. Each molecule in an array can be addressed separately either by using a field gradient to shift the frequency of the qubit transition differently for each molecule, or by using an addressing laser to produce an ac Stark shift at a chosen site. One interesting approach for quantum information processing is an array of optical tweezer traps with a single molecule in each trap, as illustrated in Fig. 10(d) and recently demonstrated [85]. The molecules can be tightly confined and cooled to the motional ground state of the trap [118], and the array of molecules can be reconfigured as needed [119] in order to implement quantum gates between selected pairs. Another interesting approach is to trap the molecules near a surface using microscopic electric or magnetic traps, as illustrated in Fig. 10(b) and proposed in [12]. The same chip can support superconducting microwave resonators with small mode volumes and at frequencies that match the rotational frequency of the chosen molecule. By reaching the regime of strong coupling between trapped molecules and the resonator it becomes possible to transfer quantum information between a molecule and a microwave photon in the resonator, and to use the resonator to couple distant molecules to one another. This architecture is thus a hybrid quantum processor that combines the advantages of molecules for storing and processing quantum information with the advantages of photons for exchanging that information.
8 Summary
Over the past century, the humble molecular beam method has pushed forward the frontiers of knowledge in physics and chemistry. Today, molecules laser cooled to ultracold temperatures are an exciting and powerful platform for investigating the boundaries of modern scientific knowledge, including what might lie beyond the Standard Model of particle physics, how chemistry works at a fundamental level, and how quantum phenomena lead to emergent collective behaviors. This rich history and bright future has been strongly shaped by the visionary work of Otto Stern and his colleagues.
Notes
- 1.
Often, R is not a good quantum number because it is coupled strongly to other angular momenta in the molecule, such as the orbital angular momenta of the electrons. In this case, R may be replaced by the relevant coupled angular momentum and the same principles apply.
- 2.
Note that there are other methods of sub-Doppler cooling, commonly used to cool atoms, that work for negative detunings. For molecules, it appears that sub-Doppler cooling always requires a positive detuning.
References
O. Stern, W. Gerlach, Z. Phys. 9, 349 (1922)
O. Stern, Z. Phys 39, 751 (1926)
R. Frisch, Z. Phys. 86, 42 (1933)
D. DeMille, J.M. Doyle, A.O. Sushkov, Science 357, 990 (2017)
M.S. Safronova, D. Budker, D. DeMille, D.F.J. Kimball, A. Derevianko, C.W. Clark, Rev. Mod. Phys. 90, 025008 (2018)
D.S. Jin, J. Ye, Chem. Rev. 112, 4801 (2012)
J. Toscano, H.J. Lewandowski, B.R. Heazlewood, Phys. Chem. Chem. Phys. 22, 9180 (2020)
A. Micheli, G. Pupillo, H.P. Büchler, P. Zoller, Phys. Rev. A 76, 043604 (2007)
A.V. Gorshkov, S.R. Manmana, G. Chen, J. Ye, E. Demler, M.D. Lukin, A.M. Rey, Phys. Rev. Lett. 107, 115301 (2011)
J.A. Blackmore, L. Caldwell, P.D. Gregory, E.M. Bridge, R. Sawant, J. Aldegunde, J. Mur-Petit, D. Jaksch, J.M. Hutson, B.E. Sauer, M.R. Tarbutt, S.L. Cornish, Quantum. Sci. Technol. 4, 014010 (2018)
D. DeMille, Phys. Rev. Lett. 88, 067901 (2002)
A. André, D. DeMille, J.M. Doyle, M.D. Lukin, S.E. Maxwell, P. Rabl, R.J. Schoelkopf, P. Zoller, Nat. Phys. 2, 636 (2006)
O. Stern, Z. Phys. 2, 49 (1920)
O. Stern, Z. Phys. 3, 417 (1921)
B.E. Sauer, J. Wang, E.A. Hinds, Phys. Rev. Lett. 74, 1554 (1995)
M.R. Tarbutt, J.J. Hudson, B.E. Sauer, E.A. Hinds, V.A. Ryzhov, V.L. Ryabov, V.F. Ezhov, J. Phys. B 35, 5013 (2002)
N.E. Bulleid, S.M. Skoff, R.J. Hendricks, B.E. Sauer, E.A. Hinds, M.R. Tarbutt, Phys. Chem. Chem. Phys. 15(29), 12299 (2013)
G. Scoles (ed.), Atomic and Molecular Beam Methods (Oxford University Press, Oxford, 1988)
R. Campargue (ed.), Atomic and Molecular Beams: The State of the Art 2000 (Springer, Berlin, 2001)
S.E. Maxwell, N. Brahms, R. deCarvalho, D.R. Glenn, J.S. Helton, S.V. Nguyen, D. Patterson, J. Petricka, D. DeMille, J.M. Doyle, Phys. Rev. Lett. 95, 173201 (2005)
N.R. Hutzler, H.I. Lu, J.M. Doyle, Chem. Rev. 112, 4803 (2012)
H.I. Lu, J. Rasmussen, M.J. Wright, D. Patterson, J.M. Doyle, Phys. Chem. Chem. Phys. 13, 18986 (2011)
O.R. Frisch, O. Stern, Z. Phys. 85, 4 (1933)
I. Estermann, O. Stern, Z. Phys. 86, 132 (1933)
I. Estermann, O. Stern, Z. Phys. 86, 135 (1933)
E.M. Purcell, N.F. Ramsey, Phys. Rev. 78, 807 (1950)
H. Weyl, Symmetry (Princeton University Press, Princeton, New Jersey, 1952)
T.D. Lee, C.N. Yang, Phys. Rev. 104, 254 (1956)
C.S. Wu, E. Ambler, R.W. Hayward, D.D. Hoppes, R.P. Hudson, Phys. Rev. 105, 1413 (1957)
R.L. Garwin, L.M. Lederman, M. Weinrich, Phys. Rev. 105, 1415 (1957)
J.I. Friedman, V.L. Telgedi, Phys. Rev 105, 1681 (1957)
J.H. Christenson, J.W. Cronin, V.L. Fitch, R. Turlay, Phys. Rev. Lett. 13, 138 (1964)
P.G.H. Sandars, Phys. Lett. 14, 196 (1965)
H.S. Nataraj, B.K. Sahoo, B.P. Das, D. Mukherjee, Phys. Rev. Lett. 101, 033002 (2008)
V. Andreev, D.G. Ang, D. DeMille, J.M. Doyle, G. Gabrielse, J. Haefner, N.R. Hutzler, Z. Lasner, C. Meisenhelder, B.R. O’Leary, C.D. Panda, A.D. West, E.P. West, X. Wu, Nature 562, 355 (2018)
I.I. Rabi, S. Millman, P. Kusch, J.R. Zacharias, Phys. Rev. 55, 526 (1939)
N.F. Ramsey, Phys. Rev. 78, 695 (1950)
J.H. Smith, E.M. Purcell, N.F. Ramsey, Phys. Rev. 108, 120 (1957)
M. Pospelov, A. Ritz, Phys. Rev. D 89, 056006 (2014)
A.D. Sakharov, Phys.-Uspekhi 34(5), 392 (1991)
S.A. Murthy, J.D. Krause, Z.L. Li, L.R. Hunter, Phys. Rev. Lett. 63, 965 (1989)
B.C. Regan, E.D. Commins, C.J. Schmidt, D. DeMille, Phys. Rev. Lett. 88, 071805 (2002)
P.G.H. Sandars, in Atomic Physics 4, ed. by G. zu Pulitz (Plenum, New York, 1975), p. 71
E.A. Hinds, Physica Scripta T70, 34 (1997)
J.J. Hudson, B.E. Sauer, M.R. Tarbutt, E.A. Hinds, Phys. Rev. Lett. 89, 023003 (2002)
J.J. Hudson, D.M. Kara, I.J. Smallman, B.E. Sauer, M.R. Tarbutt, E.A. Hinds, Nature 473, 493 (2011)
J. Baron, W.C. Campbell, D. DeMille, J.M. Doyle, G. Gabrielse, Y.V. Gurevich, P.W. Hess, N.R. Hutzler, E. Kirilov, I. Kozyryev, B.R. O’Leary, C.D. Panda, M.F. Parsons, E.S. Petrik, B. Spaun, A.C. Vutha, A.D. West, Science 343, 269 (2014)
S. Eckel, P. Hamilton, E. Kirilov, H.W. Smith, D. DeMille, Phys. Rev. A 87, 052130 (2013)
W.B. Cairncross, D.N. Gresh, M. Grau, K.C. Cossel, T.S. Roussy, Y. Ni, Y. Zhou, J. Ye, E.A. Cornell, Phys. Rev. Lett. 119, 153001 (2017)
M.R. Tarbutt, B.E. Sauer, J.J. Hudson, E.A. Hinds, New J. Phys. 15, 053034 (2013)
M.R. Tarbutt, J.J. Hudson, B.E. Sauer, E.A. Hinds, Faraday Discuss. 142, 37 (2009)
K.K. Ni, S. Ospelkaus, M.H.G. de Miranda, A. Pe’er, B. Neyenhuis, J.J. Zirbel, S. Kotochigova, P.S. Julienne, D.S. Jin, J. Ye, Science 322, 231 (2008)
M.R. Tarbutt, Contemp. Phys. 59, 356 (2018)
J. Lim, J.R. Almond, M.A. Trigatzis, J.A. Devlin, N.J. Fitch, B.E. Sauer, M.R. Tarbutt, E.A. Hinds, Phys. Rev. Lett. 120, 123201 (2018)
The NL-eEDM collaboration, P. Aggarwal, H.L. Bethlem, A. Borschevsky, M. Denis, K. Esajas, P.A.B. Haase, Y. Hao, S. Hoekstra, K. Jungmann, T.B. Meijknecht, M.C. Mooij, R.G.E. Timmermans, W. Ubachs, L. Willmann, A. Zapara, Eur. Phys. J. D 72, 197 (2018)
I. Kozyryev, N.R. Hutzler, Phys. Rev. Lett. 119, 133002 (2017)
E.B. Norrgard, E.R. Edwards, D.J. McCarron, M.H. Steinecker, D. DeMille, S.S. Alam, S.K. Peck, N.S. Wadia, L.R. Hunter, Phys. Rev. A 95, 062506 (2017)
E.S. Shuman, J.F. Barry, D. DeMille, Nature 467, 820 (2010)
M.T. Hummon, M. Yeo, B.K. Stuhl, A.L. Collopy, Y. Xia, J. Ye, Phys. Rev. Lett. 110, 143001 (2013)
I. Kozyryev, L. Baum, K. Matsuda, B.L. Augenbraun, L. Anderegg, A.P. Sedlack, J.M. Doyle, Phys. Rev. Lett. 118, 173201 (2017)
B.L. Augenbraun, Z.D. Lasner, A. Frenett, H. Sawaoka, C. Miller, T.C. Steimle, J.M. Doyle, New J. Phys. 22, 022003 (2020)
L. Baum, N.B. Vilas, C. Hallas, B.L. Augenbraun, S. Raval, D. Mitra, J.M. Doyle, Phys. Rev. Lett 124, 133201 (2020)
D. Mitra, N.B. Vilas, C. Hallas, L. Anderegg, B.L. Augenbraun, L. Baum, C. Miller, S. Raval, J.M. Doyle, Science 369, 1366 (2020)
R.L. McNally, I. Kozyryev, S. Vazquez-Carson, K. Wenz, T. Wang, T. Zelevinsky, New J. Phys. 22, 083047 (2020)
J.F. Barry, E.S. Shuman, E.B. Norrgard, D. DeMille, Phys. Rev. Lett. 108, 103002 (2012)
V. Zhelyazkova, A. Cournol, T.E. Wall, A. Matsushima, J.J. Hudson, E.A. Hinds, M.R. Tarbutt, B.E. Sauer, Phys. Rev. A 89, 053416 (2014)
M. Yeo, M.T. Hummon, A.L. Collopy, B. Yan, B. Hemmerling, E. Chae, J.M. Doyle, J. Ye, Phys. Rev. Lett. 114(22), 223003 (2015)
B. Hemmerling, E. Chae, A. Ravi, L. Anderegg, G.K. Drayna, N.R. Hutzler, A.L. Collopy, J. Ye, W. Ketterle, J.M. Doyle, J. Phys. B 49, 174001 (2016)
S. Truppe, H.J. Williams, N.J. Fitch, M. Hambach, T.E. Wall, E.A. Hinds, B.E. Sauer, M.R. Tarbutt, New J. Phys. 19, 022001 (2017)
H.J. Williams, S. Truppe, M. Hambach, L. Caldwell, N.J. Fitch, E.A. Hinds, B.E. Sauer, M.R. Tarbutt, New J. Phys. 19, 113035 (2017)
E. Raab, M. Prentiss, A. Cable, S. Chu, D. Pritchard, Phys. Rev. Lett. 59, 2631 (1987)
J.F. Barry, D.J. McCarron, E.B. Norrgard, M.H. Steinecker, D. DeMille, Nature 512, 286 (2014)
D.J. McCarron, E.B. Norrgard, M.H. Steinecker, D. DeMille, New J. Phys. 17, 035014 (2015)
E.B. Norrgard, D.J. McCarron, M.H. Steinecker, M.R. Tarbutt, D. DeMille, Phys. Rev. Lett. 116, 063004 (2016)
M.H. Steinecker, D.J. McCarron, Y. Zhu, D. DeMille, Chem. Phys. Chem. 17, 3664 (2016)
S. Truppe, H.J. Williams, M. Hambach, L. Caldwell, N.J. Fitch, E.A. Hinds, B.E. Sauer, M.R. Tarbutt, Nat. Phys. 13, 1173 (2017)
L. Anderegg, B.L. Augenbraun, E. Chae, B. Hemmerling, N.R. Hutzler, A. Ravi, A. Collopy, J. Ye, W. Ketterle, J.M. Doyle, Phys. Rev. Lett. 119, 103201 (2017)
A.L. Collopy, S. Ding, Y. Wu, I.A. Finneran, L. Anderegg, B.L. Augenbraun, J.M. Doyle, J. Ye, Phys. Rev. Lett. 121, 213201 (2018)
L. Anderegg, B.L. Augenbraun, Y. Bao, S. Burchesky, L.W. Cheuk, W. Ketterle, J.M. Doyle, Nat. Phys. 14, 890 (2018)
D.J. McCarron, M.H. Steinecker, Y. Zhu, D. DeMille, Phys. Rev. Lett. 121, 013202 (2018)
L.W. Cheuk, L. Anderegg, B.L. Augenbraun, Y. Bao, S. Burchesky, W. Ketterle, J.M. Doyle, Phys. Rev. Lett. 121, 083201 (2018)
L. Caldwell, J.A. Devlin, H.J. Williams, N.J. Fitch, E.A. Hinds, B.E. Sauer, M.R. Tarbutt, Phys. Rev. Lett. 123, 033202 (2019)
S. Ding, Y. Wu, I.A. Finneran, J.J. Burau, J. Ye, Phys. Rev. X 10, 021049 (2020)
H.J. Williams, L. Caldwell, N.J. Fitch, S. Truppe, J. Rodewald, E.A. Hinds, B.E. Sauer, M.R. Tarbutt, Phys. Rev. Lett. 120, 163201 (2018)
L. Anderegg, L.W. Cheuk, Y. Bao, S. Burchesky, W. Ketterle, K.K. Ni, J.M. Doyle, Science 365, 1156 (2019)
L. Caldwell, H.J. Williams, N.J. Fitch, J. Aldegunde, J.M. Hutson, B.E. Sauer, M.R. Tarbutt, Phys. Rev. Lett. 124, 063001 (2020)
C. Cheng, A.P.P. van der Poel, P. Jansen, M. Quintero-Pérez, T.E. Wall, W. Ubachs, H.L. Bethlem, Phys. Rev. Lett. 117, 253201 (2016)
S.A. Moses, J.P. Covey, M.T. Miecnikowski, B. Yan, B. Gadway, J. Ye, D.S. Jin, Science 350, 659 (2015)
S.B. Cahn, J. Ammon, E. Kirilov, Y.V. Gurevich, D. Murphree, R. Paolino, D.A. Rahmlow, M.G. Kozlov, D. DeMille, Phys. Rev. Lett. 112, 163002 (2014)
E. Altuntaş, J. Ammon, S.B. Cahn, D. DeMille, Phys. Rev. Lett. 120, 142501 (2018)
S. Tokunaga, C. Stoeffler, F. Auguste, A. Shelkovnikov, C. Daussy, A. Amy-Klein, C. Chardonnet, B. Darquié, Mol. Phys. 111, 2363 (2013)
J.P. Uzan, Living Rev. Relativity 14, 1 (2011)
K.A. Olive, M. Pospelov, Phys. Rev. D 77, 043524 (2010)
C. Chin, V.V. Flambaum, M.G. Kozlov, New J. Phys. 11, 055048 (2009)
E.R. Hudson, H.J. Lewandowski, B.C. Sawyer, J. Ye, Phys. Rev. Lett. 96, 143004 (2006)
S. Truppe, R.J. Hendricks, S.K. Tokunaga, H.J. Lewandowski, M.G. Kozlov, C. Henkel, E.A. Hinds, M.R. Tarbutt, Nat. Commun. 4, 2600 (2013)
N. Huntemann, B. Lipphardt, C. Tamm, V. Gerginov, S. Weyers, E. Peik, Phys. Rev. Lett. 113, 210802 (2014)
R.M. Godun, P.B.R. Nisbet-Jones, J.M. Jones, S.A. King, L.A.M. Johnson, H.S. Margolis, K. Szymaniec, S.N. Lea, K. Bongs, P. Gill, Phys. Rev. Lett. 113, 210801 (2014)
J. Kobayashi, A. Ogino, S. Inouye, Nat. Comms. 10, 3771 (2019)
S.S. Kondov, C.H. Lee, K.H. Leung, C. Liedl, I. Majewska, R. Moszynski, T. Zelevinsky, Nat. Phys. 15, 1118 (2019)
M. Kajita, J. Phys. Soc. Jpn. 87, 104301 (2018)
J.L. Bohn, A.M. Rey, J. Ye, Science 357, 1002 (2017)
L.W. Cheuk, L. Anderegg, Y. Bao, S. Burchesky, S. Yu, W. Ketterle, K.K. Ni, J.M. Doyle, Phys. Rev. Lett. 125, 043401 (2020)
S. Ospelkaus, K.K. Ni, D. Wang, M.H.G. de Miranda, B. Neyenhuis, G. Quéméner, P.S. Julienne, J.L. Bohn, D.S. Jin, J. Ye, Science 327, 853 (2010)
M.H.G. de Miranda, A. Chotia, B. Neyenhuis, D. Wang, G. Quéméner, S. Ospelkaus, J.L. Bohn, J. Ye, D.S. Jin, Nat. Phys. 7, 502 (2011)
L.R. Liu, J.D. Hood, Y. Yu, J.T. Zhang, N.R. Hutzler, T. Rosenband, K.K. Ni, Science 360, 900 (2018)
R.P. Feynman, Int. J. Theo. Phys. 21, 467 (1982)
M. Greiner, O. Mandel, T. Esslinger, T.W. Hänsch, I. Bloch, Nature 415, 39 (2002)
A. Mazurenko, C.S. Chiu, G. Ji, M.F. Parsons, M. Kanász-Nagy, R. Schmidt, F. Grusdt, E. Demler, D. Greif, M. Greiner, Nature 545, 462 (2017)
B. Yan, S.A. Moses, B. Gadway, J.P. Covey, K.R.A. Hazzard, A.M. Rey, D.S. Jin, J. Ye, Nature 501, 521 (2013)
L.D. Marco, G. Valtolina, K. Matsuda, W.G. Tobias, J.P. Covey, J. Ye, Science 363, 853 (2019)
H. Son, J.J. Park, W. Ketterle, A.O. Jamison, Nature 580, 197 (2020)
J.W. Park, Z.Z. Yan, H. Loh, S.A. Will, M.W. Zwierlein, Science 357, 372 (2017)
F. Seeßelberg, X.Y. Luo, M. Li, R. Bause, S. Kotochigova, I. Bloch, C. Gohle, Phys. Rev. Lett. 121, 253401 (2018)
S.F. Yelin, K. Kirby, R. Côté, Phys. Rev. A 74, 050301 (2006)
K.K. Ni, T. Rosenband, D.D. Grimes, Chem. Sci. 9, 6830 (2018)
R. Sawant, J.A. Blackmore, P.D. Gregory, J. Mur-Petit, D. Jaksch, J. Aldegunde, J.M. Hutson, M.R. Tarbutt, S.L. Cornish, New J. Phys. 22, 013027 (2020)
L. Caldwell, M.R. Tarbutt, Phys. Rev. Res. 2, 013251 (2020)
D. Barredo, S. de Léséleuc, V. Lienhard, T. Lahaye, A. Browaeys, Science 354, 1021 (2016)
Acknowledgements
We are grateful to Bretislav Friedrich and Horst Schmidt-Böcking for inviting us to contribute to this Festschrift, and to Luke Caldwell and Ed Hinds for their helpful feedback. The authors are supported by EPSRC (EP/P01058X/1), STFC (ST/S000011/1), the Royal Society, the John Templeton Foundation (grant 61104), the Gordon and Betty Moore Foundation (grant 8864), and the Alfred P. Sloan Foundation (grant G-2019-12505).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Fitch, N.J., Tarbutt, M.R. (2021). From Hot Beams to Trapped Ultracold Molecules: Motivations, Methods and Future Directions. In: Friedrich, B., Schmidt-Böcking, H. (eds) Molecular Beams in Physics and Chemistry. Springer, Cham. https://doi.org/10.1007/978-3-030-63963-1_22
Download citation
DOI: https://doi.org/10.1007/978-3-030-63963-1_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63962-4
Online ISBN: 978-3-030-63963-1
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)