
Abstract
In this chapter, the clinical aspects of Cushing’s syndrome caused by an ectopic adrenocorticotropic hormone (ACTH)-secreting tumor will be discussed. Following the course of a patient eventually diagnosed with an ACTH-producing atypical neuroendocrine tumor (NET; carcinoid) in the lung, the clinical features, diagnosis, and treatment of ectopic ACTH syndrome will be presented.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara OpeningIn this chapter, the clinical aspects of Cushing’s syndrome caused by an ectopic adrenocorticotropic hormone (ACTH)-secreting tumor will be discussed. Following the course of a patient eventually diagnosed with an ACTH-producing atypical neuroendocrine tumor (NET; carcinoid) in the lung, the clinical features, diagnosis, and treatment of ectopic ACTH syndrome will be presented.
FormalPara Definition of the DiseaseEctopic adrenocorticotropic hormone syndrome is defined as an endogenous Cushing’s syndrome caused by a histologically heterogeneous group of ACTH-secreting neuroendocrine neoplasms (NEN or NET, neuroendocrine tumor). The syndrome belongs to the group of paraneoplastic endocrine syndromes. In exceptionally rare cases, lymphomas or sarcomas have also been shown to be the source of ectopic ACTH production.
The primary tumor may arise from a wide variety of organs, but most commonly in the lung (i.e., small cell lung cancer [SCLC]), pancreas, and thymus.
What feature of this case raises the suspicion of ectopic ACTH syndrome?
The patient had severe life-threatening hypokalemia; however, she had no apparent cause of this electrolyte imbalance. She was not taking diuretics or any other medications. There was a complete absence of gastrointestinal symptoms and signs of kidney disorder. On the other hand, an extraordinarily high potassium dose was needed. These findings pointed to the likelihood of the presence of either primary aldosteronism (see in ► Chap. 28.) or other hormone excess with mineralocorticoid activity. ◘ Table 45.1 summarizes the major causes of hypokalemia.
Is the clinical picture of ectopic ACTH syndrome different from the classical Cushing’s phenotype?
The classical phenotype in Cushing’s syndrome is presented in the chapters on pituitary Cushing’s disease (► Chap. 3) and adrenal Cushing’s syndrome (► Chap. 28). The classical phenotype includes centripetal obesity, striae, buffalo hump, moon-like face, and so on. In ectopic ACTH syndrome associated Cushing’s syndrome, however, these typical phenotypic signs are often lacking, as the syndrome is progressing rapidly and there is not enough time for the development of these symptoms. Severe and rapidly progressing diabetes mellitus, muscle weakness, hypertension, and hypokalemia can be the presenting signs. Hypokalemia is due to the excessive levels of cortisol. The HSD11B2 enzyme (11ß-hydroxysteroid dehydrogenase 2) defends the mineralocorticoid (aldosterone) receptor from the more abundant cortisol by metabolizing it to inactive cortisone in aldosterone-responsive organs. In conditions of excessive cortisol production (e.g., ectopic ACTH syndrome or adrenocortical cancer), the capacity of the enzyme is being exhausted and cortisol binds with high affinity to the mineralocorticoid receptor. If ACTH levels are high, even hyperpigmentation can occur.
What tests should be done to assess the endocrine origin of hypokalemia?
After correcting the electrolyte imbalance, it was key to evaluate the presence of primary aldosteronism (see also in ► Chap. 29.). Even though the patient was lacking the typical signs of Cushing’s syndrome, the possibility of hypercortisolism had also to be investigated (see in ► Chaps. 3 and 27).
What might be the main causes of ACTH-dependent Cushing’s syndrome?
Overall, there are three main causes of ACTH-dependent Cushing’s syndrome. The vast majority of the cases are caused by Cushing’s disease, in which the source of the excess ACTH secretion originates from the pituitary gland. In ectopic ACTH syndrome, ACTH secretion of a non-pituitary tumor is the cause of hypercortisolism. And finally, in exceptionally rare cases (less than 1%), an ectopic corticotropin-releasing hormone (CRH)-secreting tumor may be the cause of Cushing’s syndrome.
Are there any further tests needed to evaluate the cause of Cushing’s syndrome?
Yes, since the treatment options and prognosis of all entities are different, a precise diagnosis is mandatory to plan treatment. Even though—just like in the presented case—glucocorticoid overproduction is usually much more severe, and the presence of hypokalemia is much more common in ectopic ACTH syndrome compared to Cushing’s disease, additional tests are needed to differentiate between these two distinct entities. These tests include noninvasive and invasive venous sampling and imaging studies.
Non-invasive tests:
The CRH stimulation test
The physiologic response to IV-administered CRH is a 35–900% increase in serum ACTH and a 20–600% increase in serum cortisol within 120 minutes. As opposite to normal individuals and Cushing’s disease patients, patients with ectopic ACTH syndrome do not respond to exogenous CRH stimulation. This is due to suppression of pituitary ACTH stimulation seen in these patients. The test is done in a supine position after a few hours of fasting. And 1 μg of synthetic corticotropin-releasing hormone is administered intravenously. Blood sampling for serum cortisol and ACTH is done from a peripheral vein in every 15 minutes for 60 minutes and at 90 and 120 minutes.
The high-dose dexamethasone suppression test
Since ectopic ACTH-producing tumors are completely resistant to the cortisol-induced feedback mechanism as opposite to the only relative resistance seen in ACTH-producing pituitary adenomas, the high-dose dexamethasone may also be a useful tool in differentiating between the causes of hypercortisolism. In its overnight form, the test is carried out by administering 8 mg of dexamethasone orally at 23:00 and peripheral venous blood sampling the following morning cortisol levels. Non-suppressed morning cortisol (>140 nmol/L) values indicate ectopic ACTH-secreting malignant tumor.
Despite being easy and safe to perform these noninvasive tests, alone they are neither sensitive nor specific enough to categorize every patient precisely. Thus, both tests should be performed, and the results are required to be unanimous and supported with imaging studies. Otherwise, invasive testing is indicated.
Imaging:
A native and gadolinium contrast-enhanced MRI of the pituitary gland is the best choice for visualizing an ACTH-producing adenoma in the sellar region.
In the present case, a pituitary magnetic resonance imaging (MRI) (see ◘ Fig. 45.1) has been performed to assess the cause of Cushing’s syndrome. The sella MRI showed no visible adenoma in the pituitary, which strengthened the likelihood of ectopic ACTH syndrome and was necessary to do before performing invasive sampling.

Coronal a and sagittal b cross-section of a normal sella MRI showing no signs of a micro- or macroadenoma
Invasive testing:
Bilateral inferior petrosal sinus sampling (BIPSS) is the gold standard test to demonstrate pituitary ACTH hypersecretion and to rule out or prove ectopic ACTH secretion. The test is performed after cannulation of both inferior petrosal sinus (IPS) via the femoral or jugular veins. During the test, bilateral central and unilateral peripheral serum ACTH is measured before and after administration of CRH. In case of a central-to-peripheral plasma ACTH gradient of <2.0 before CRH administration or <3.0 after CRH is diagnostic of an ectopic source of ACTH. (Vice versa, ACTH gradients over 2.0 and 3.0 before and after CRH, respectively, are characteristic for a pituitary ACTH source.)
How to find the ACTH-secreting tumor?
One of the most difficult challenges in managing patients with ectopic ACTH syndrome is to localize the primary tumor.
Unfortunately, there is no clear guidance yet on which radiological method is best suited for locating the neoplasm. Most tumors associated with ectopic ACTH syndrome reside in the thorax. Contrast-enhanced computed tomography (CT) of the thorax seems to be the best modality based on common etiology. Abdominal CT or MRI can be a sensitive method to assess distant metastasizes. Also, nuclear imaging, such as 18F-fluorodeoxyglucose (FDG)-PET-CT and 111In-octreotide scintigraphy or 68Ga-DOTATATE-PET-CT, may be helpful in tumor detection in poorly or well-differentiated neuroendocrine neoplasms, respectively.
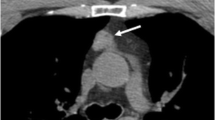
In the present case, a contrast-enhanced thoraco-abdomino-pelvical CT scan has been performed. The images have revealed a small mass of about 30 × 12 mm in size surrounding the bronchi in the eighth segment of the right lung (see ◘ Fig. 45.2). Additionally, an about 20 mm large solitary bone metastasis has been found in the fourth rib on the right side. The abdominal CT has also revealed adrenal glands showing signs of adrenal hyperplasia (◘ Fig. 45.2). This was most likely caused by the constant adrenal stimulation caused by the ectopic ACTH.

Ectopic ACTH-secreting bronchial tumor in S8 of the right lung a and hyperplastic adrenal gland b
Should histological sampling be performed in lung neuroendocrine tumors?
Neuroendocrine tumors of the lung include a diverse spectrum of neoplasms showing differences in differentiation and mitotic activity. Different pulmonary NETs require different therapy and have different overall prognosis. Thus, it is of great importance to perform histologic analysis of the tumor.
What are the differences among bronchial NETs?
Neuroendocrine tumors of the lung may be well-differentiated, also known as pulmonary carcinoids (PCs). PCs may be low-grade, also called typical carcinoids (TCs), or intermediate-grade, also known as atypical carcinoids (ACs). (The term carcinoid is still in use with bronchial neuroendocrine neoplasms.) While TCs grow slowly and rarely give distant metastasizes and have a better prognosis, ACs have a higher likelihood of recurrence, grow faster, are prone to give distant metastases, and have a worse prognosis. PCs are rare and account for about 1–2% of all lung malignancies in adults. TCs are eight to ten times more common compared to ACs, making ACs the most uncommon of lung NETs. On the other end of the spectrum are poorly differentiated neuroendocrine carcinomas such as small cell lung cancer and large cell neuroendocrine cancer (LCNEC), both of which generally grow very rapidly and, because of their potential to give distant metastases, have a poor prognosis.
Do all lung NET-s produce ACTH?
No. In fact, most of them do not. The most common paraneoplastic endocrine syndrome in PCs is the carcinoid syndrome (► Chap. 45) found in about 2–5% of well-differentiated tumor cases. The second most frequent functioning syndrome is Cushing’s syndrome. It is worth noting here that the carcinoid syndrome associated with pulmonary carcinoids can be different from the classical carcinoid syndrome, the so-called lung NET-variant or atypical carcinoid syndrome. In this variant form, flushes can last for hours or even days, and various symptoms such as anxiety, disorientation, hypersalivation, hypotension, and tachycardia can be observed. Histamine may play a major role in this variant. Carcinoid crisis (► Chap. 44) is very rare in patients with PCs.
In rare and extremely rare cases, acromegaly and ectopic insulin secretion has also seen in PCs. It is also worth mentioning that the syndrome of inappropriate antidiuretic hormone secretion (SIADH) is seen in 5% in SCLC cases but rarely in PC (for further reading, see ► Chap. 10).
Are there any curative treatment options in this patient’s case?
The only curative treatment option that might free the patient of the malignant tumor as well as put an end to the ectopic ACTH production is surgical. Imaging studies suggested that the primary tumor in S8 of the right lung could be removed by lobectomy and lymph node dissection along with the solitary bone metastasis.
What needs to be done prior to surgery?
In this patient’s case, Cushing’s syndrome was severe, highlighted by the more than ten times upper normal limit 24-hour urinary cortisol excretions. Thus, prior to surgery, the symptoms of hypercortisolism should be alleviated to avoid pre and postoperative complications such as impaired wound healing, infections, thrombosis, and arrhythmias due to severe hypokalemia. Thus, steroid biosynthesis inhibitors should be introduced. Preoperative medical therapeutic options included ketoconazole, metyrapone, intravenous etomidate, or the glucocorticoid receptor antagonist mifepristone. Ketoconazole, given at a daily dose of 600–800 mg, is the most commonly used and effective. Metyrapone is also often used in this setting, and therapy starts at 1 g/day divided into four doses and increased to a maximum dose of 6 g/day. Anticoagulant therapy should also be started with low molecular weight heparin.
In the present case, the patient was started on metyrapone and titrated up to a dose of 4.5 g/day. Despite steroid biosynthesis inhibition, eucortisolemia could not be reached (see ◘ Fig. 45.4), and the patient required oral potassium supplementation and potassium-sparing diuretics to fully correct serum potassium levels.
How can the success of surgery be evaluated?
Histological examination of the removed specimens, tumor markers, such as chromogranin A (CgA), or neuron-specific enolase (NSE) and re-evaluation of the patient for ectopic ACTH syndrome are the key components of evaluating surgical success. A total resection, normalized serum tumor markers (if these were elevated before surgery), and complete remission of hormonal symptoms suggest surgical success.
Unfortunately, in the present case, despite both tumors seeming to be macroscopically removed during the surgical process, histology indicated that the cortical layer of the removed rib was infiltrated by tumor cells and local soft tissue invasion was present confirming R1 resection. These findings were also confirmed by persistent ectopic ACTH syndrome during the postoperative period (see ◘ Fig. 45.4). It is worth mentioning that even though the procedure did not turn out to be curative, the debulking of most of the tumor made eucortisolemia achievable with metyrapone.
In case of the residual atypical carcinoid and persistent ectopic ACTH-dependent syndrome, what treatment options are available for hormone control?
Cushing’s syndrome can be treated with previously mentioned steroid biosynthesis inhibitors as well as somatostatin analogues (SSA). If hormonal control cannot be achieved via medical therapy, other more invasive anti-tumor therapy such as metastasis resection, radiofrequency ablation, arterial chemoembolization, or in uncontrollable cases bilateral adrenalectomy should be considered.
What treatment options are there to control tumor growth or induce regression?
In asymptomatic patients with slowly growing low tumor burden, options include only observation, as well. In patients in need of tumor stabilization, SSA treatment may be an option due to the expression of somatostatin receptors by the tumor. In progressive, well-differentiated PCs expressing somatostatin receptors, systemic peptide receptor radionuclide therapy (PRRT) with 90Yttrium-DOTA octreotide or 177Lutetium DOTA octreotide may also be effective. Side effects of PRRT include hematological toxicities and renal toxicity (► Chap. 45). Advanced well-differentiated intermediate-grade PCs not expressing somatostatin receptors should be treated with everolimus, a mammalian target of rapamycin (mTOR) inhibitor, or anti-angiogenetic agents such as pazopanib. And finally, in advanced intermediate-grade lung ACs, systemic chemotherapy with 5-fluorouracil (5-FU) and temozolomide-based chemotherapy can be advised. Etoposide and cisplatin combination due to its high toxicity is preserved for high-grade lung neuroendocrine cancer such as SCLC.
Is routine genetic testing necessary in patients with PC?
No. PCs are rarely (<5%) associated with multiple endocrine neoplasia type 1 (MEN1). Clinical assessment such as family history and screening routine lab tests such as serum calcium, parathyroid hormone, and prolactin is sufficient to follow up and select patient who should be tested for MEN1.
The reader is advised to read (or repeat) the chapters on Adrenal Cushing’s syndrome (► Chap. 27) and pituitary Cushing’s disease (► Chap. 3). Moreover, the previous chapter on a small intestinal neuroendocrine tumor with carcinoid syndrome is interesting to read, and this includes more pieces of information on somatostatin analogues and peptide receptor radionuclide treatment (► Chap. 44). The syndrome of inappropriate secretion of antidiuretic hormone is discussed in ► Chap. 10.
FormalPara Take Home Messages-
Ectopic ACTH syndrome is caused by malignant ACTH-producing non-pituitary tumors, most commonly in the lungs, thymus, and pancreas.
-
The clinical picture is markedly different compared to other causes of Cushing’s syndrome, as patients usually lack the typical symptoms of Cushing’s disease.
-
Ectopic ACTH syndrome is associated with elevated ACTH levels and usually markedly elevated cortisol levels.
-
Ectopic ACTH syndrome is associated with severe, commonly life-threatening hypercortisolism complicated with severe hypokalemia, serious infections, thromboembolism, and impaired wound healing.
-
Imaging needed to locate the tumor includes CT, MRI, FDG-PET-CT, and Octreoscan.
-
The only curative option is surgery.
-
Palliative treatment options include steroid biosynthesis inhibitors and depending on the grade anti-cancer therapy (somatostatin analogues, interferon, mTOR inhibitors, PRRT, or systemic chemotherapy).
Case Presentation Continued
In the present case, even though tumor burden was undetectable, the patient had persistent hypercortisolism caused by an intermediate-grade AC. Thus, on the top of steroid biosynthesis inhibitor metyrapone, SSA has been started to inhibit tumor cell growth. As presented in ◘ Fig. 45.4, eucortisolemia could be reached using this combination therapy. On follow-up imaging, we have seen no signs of relapse either.
Case Presentation
A 60-year-old postmenopausal female patient presented herself at our department in an emergency setting. Her main complaints started a few days ago and included severe general weakness and symmetrical swelling of both lower extremities. She had no relevant previous medical history and did not take any medications regularly.
Besides apparent symmetric edemas on both legs, no abnormalities have been found on a physical examination. Her blood pressure was normal. There were no signs of kidney or liver failure. Her blood glucose values showed a pre-diabetic state in her carbohydrate metabolism. The cause of her symptoms was soon unveiled by a routine lab evaluation showing severe hypokalemia (serum potassium 1.6 mmol/L, normal: 3.5–5.0). Interestingly, she had no signs of arrhythmias or any other ECG abnormalities. Urgent supplementation of intravenous and oral potassium chloride has been commenced. In an effort to reach physiologic serum potassium levels, it has been established that extraordinarily high dose (20 g/day) of replacement therapy was required to reach normokalemia.
Case Presentation Continued
◘ Table 45.2 shows the hormonal results of the patient.
Primary aldosteronism was ruled out (normal aldosterone) on one hand. On the other hand, the patient has severe ACTH-dependent Cushing’s disease (elevated late-night cortisol values and 24-hour urinary free cortisol [UFC], no suppression on low-dose dexamethasone test, and ACTH is elevated), which is most possibly causing her symptoms. (Suppressed renin is due to the mineralocorticoid activity of excessive glucocorticoid production.)
Case Presentation Continued
In the present case, in view of a negative pituitary MRI and due to the high likelihood of ectopic ACTH syndrome based on the clinical setting, BIPSS has been performed. The results (presented in ◘ Table 45.3) show a low central to peripheral ratio on both basal and CRH-stimulated ACTH levels confirming an ectopic ACTH syndrome.
Case Presentation Continued
In the present case, due to its peripheral localization, the tumor was inaccessible via bronchoscopy. Thus, CT-guided biopsy has been performed. The histological evaluation of the samples revealed a tumor with cytologically bland immature cells, containing regular round nuclei with finely dispersed chromatin and inconspicuous small nucleoli arranged in distinct organoid growth patterns with a delicate vascular stroma. Within the tumor, dot-like necrosis was visible. Immunohistochemical (IH) staining showed positivity for thyroid transcription factor (TTF-1), cytokeratin (CK8-18), CD56, chromogranin A (CgA), synaptophysin, and ACTH. The mitotic activity of the tumor cells was approximately 10–15%. Based on these findings, the diagnosis of an atypical bronchial carcinoid was made (see ◘ Fig. 45.3).

Histological pictures of an atypical bronchial carcinoid, 20x magnification. a Hematoxylin-eosin staining. b Synaptophysin immunohistochemistry. c Chromogranin A immunohistochemistry. d ACTH immunohistochemistry. e Ki-67 immunohistochemistry. (Courtesy of Katalin Borka MD PhD [2nd Department of Pathology, Semmelweis University], and János Szőke MD PhD [National Institute of Oncology])

Overall change of UFC levels in the presented case during the different treatment periods. Note: The rise of UFC in the postoperative period is due to the discontinuation of metyrapone therapy
Suggested Reading
Caplin ME, Baudin E, Ferolla P, Filosso P, Garcia-Yuste M, Lim E. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604–20. https://doi.org/10.1093/annonc/mdv041.
Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, Yao JC. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017;3(10):1335–42. https://doi.org/10.1001/jamaoncol.2017.0589.
de Herder WW, Lamberts SW. Tumor localization--the ectopic ACTH syndrome. J Clin Endocrinol Metab. 1999;84(4):1184–5. https://doi.org/10.1210/jcem.84.4.5690.
Igaz P, Tombol Z, Szabo PM, Liko I, Racz K. Steroid biosynthesis inhibitors in the therapy of hypercortisolism: theory and practice. Curr Med Chem. 2008;15(26):2734–47. https://doi.org/10.2174/092986708786242921.
Loriaux DL. Diagnosis and differential diagnosis of Cushing’s syndrome. N Engl J Med. 2017;376(15):1451–9. https://doi.org/10.1056/NEJMra1505550.
Nieman LK. Diagnosis of Cushing’s syndrome in the modern era. Endocrinol Metab Clin North Am. 2018;47(2):259–73. https://doi.org/10.1016/j.ecl.2018.02.001.
Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Endocrine S. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(8):2807–31. https://doi.org/10.1210/jc.2015-1818.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nagy, G. (2021). Ectopic ACTH Syndrome Caused by a Bronchial Neuroendocrine Tumor. In: Igaz, P. (eds) Practical Clinical Endocrinology. Springer, Cham. https://doi.org/10.1007/978-3-030-62011-0_45
Download citation
DOI: https://doi.org/10.1007/978-3-030-62011-0_45
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-62010-3
Online ISBN: 978-3-030-62011-0
eBook Packages: MedicineMedicine (R0)