
Abstract
Based on recent studies, a considerable minority of patients with epilepsy of unknown etiology may have an autoimmune or paraneoplastic cause. Autoimmune seizures or epilepsy usually presents with a subacute onset of refractory seizures along with cognitive, behavioral, or psychiatric dysfunction. Leucine-rich glioma protein1 (LGI-1), N-methyl-D-aspartate receptor (NMDA-R), and glutamic acid decarboxylase 65 (GAD 65) are the most common immunoglobulin G (IgG) specificities associated with autoimmune seizures or epilepsy. Diagnosis of these cases depends on the identification of the clinical syndrome, ancillary studies, and autoantibody testing. Predictive models based on clinical features and initial neurological assessment (antibody prevalence in epilepsy and encephalopathy [APE2] and response to immunotherapy in epilepsy and encephalopathy [RITE2] scores) may be utilized for selection of cases for autoimmune epilepsy evaluation and management. Early initiation of immunotherapy is critical for favorable clinical outcomes. Utilization of immunotherapeutic agents for autoimmune epilepsy management is divided into first-line and second-line treatments. These are incorporated into a proposed treatment algorithm. In the ensuing years, the field of autoimmune epilepsy is likely to expand further with discovery of several novel autoantibodies and improved mechanistic understanding.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Points-
1.
A considerable proportion of patients (10–16%) with epilepsy of unknown etiology may have an autoimmune or paraneoplastic cause.
-
2.
Among patients with epilepsy, coexisting clinical features such as subacute progressive cognitive decline, psychiatric symptoms, viral prodrome, autonomic dysfunction, inflammatory cerebrospinal fluid (CSF), medial temporal lobes hyperintensities on magnetic resonance imaging (MRI), or presence of underlying malignancy are suggestive of an autoimmune etiology.
-
3.
A predictive scoring system (antibody prevalence in epilepsy and encephalopathy [APE2] score) can be utilized in identifying those patients with autoimmune seizures or epilepsy.
-
4.
Early diagnosis and initiation of immunotherapy is critical for favorable clinical outcomes.
-
5.
The field of autoimmune epilepsy is likely to expand further with discovery of several novel autoantibodies and improved mechanistic understanding.
Introduction
Epilepsy is a chronic debilitating disease affecting 0.5–1.0% of the world’s population [1]. Although epilepsy can arise from different structural, metabolic, infectious, or genetic etiologies, the cause of a significant proportion of cases remains unknown [2]. The link between subset of epilepsies and neuroinflammation has been recognized for decades. This includes the suspected inflammatory pathogenesis of epilepsy syndromes such as Rasmussen’s encephalitis [3] and favorable response to immunotherapy of others, for example, Landau-Kleffner syndrome [4]. Furthermore, in the 1960s, initial cases of paraneoplastic limbic encephalitis associated with epilepsy were described [5]. Over the past 2 decades, a plethora of neural autoantibodies targeting cell surface or intracellular antigens associated with encephalopathy and/or epilepsy have been discovered [6]. Many more biomarkers with specific clinical and/or oncological associations are likely to be discovered over the coming years. The rate of discovery may be fueled by development and validation of phage immunoprecipitation sequencing and immunoprecipitation–mass spectrometry techniques [7, 8].
Diagnosis of autoimmune epilepsy, in a majority of the cases, is based on their clinical characteristics, magnetic resonance imaging (MRI) results, cerebrospinal fluid (CSF) analysis, and/or response to immunotherapy trials [9]. The International League Against Epilepsy (ILAE) has recognized autoimmune epilepsy as a distinct entity in the 2017 epilepsy classification [10]. However, diagnostic criteria for autoimmune epilepsy are lacking. A subset of these patients who have coexisting encephalopathy may be characterized using the autoimmune encephalitis diagnostic criteria proposed by Graus et al. in 2016 [11]. Diagnostic criteria for autoimmune encephalitis is further discussed in Chap. 12.
Epidemiology
The true incidence of autoimmune epilepsy remains unknown. Some information can be deduced from a population-based epidemiological study evaluating incidence and prevalence of autoimmune encephalitis. In Olmsted County, Minnesota, the incidence of autoimmune encephalitis was found to be 0.8/100,000 with a prevalence of 13.7/100,000 [12]. This study also showed significant increase in incidence of autoimmune encephalitis in the last decade, with increased recognition of neural-specific antibodies associated with autoimmune encephalitis [13]. However, in this study, proposed autoimmune encephalitis diagnostic criteria were utilized for selection of their cases. Among the selected cases, only a subset had epilepsy as a part of their syndrome. Additionally, autoimmune epilepsy cases without cognitive impairment were excluded.
A hospital-based prospective study reported that 20% of adult patients with epilepsy of unknown etiology were seropositive for neural-specific antibodies associated with autoimmune epilepsy or encephalopathy [14]. In another UK-based retrospective study, estimated frequency of neural-specific antibodies was 15% among patients without a genetic, structural, or metabolic etiology for epilepsy [14,15,16]. Epilepsies of unknown etiology are estimated to constitute one-third of all epilepsies among adults [17]. Therefore, the rate of autoimmune epilepsies based on these studies can be inferred to be around 5–7% of all epilepsies, at least in adults. The frequency of autoantibodies in pediatric epilepsy is more unclear. Wright et al. showed the presence of autoantibodies in about 10% of pediatric patients with new-onset epilepsy [18].
Clinical Presentation
Clinical presentations of autoimmune epilepsy are variable and evolving as new antibodies are being discovered. However, a majority of these cases have coexisting features of autoimmune encephalitis including subacute progressive cognitive decline, psychiatric symptoms, viral prodrome, autonomic dysfunction, inflammatory CSF, oncological association, or brain MRI changes consistent with autoimmune encephalitis [11]. In this regard, a predictive model based on clinical features and initial neurological assessment (antibody prevalence in epilepsy and encephalopathy [APE2] score) may aid in identification of these patients with autoimmune epilepsy [14, 15, 19]. Furthermore, a scoring system for response to immunotherapy (response to immunotherapy in epilepsy and encephalopathy [RITE2] score) may also be utilized for immunotherapy trials (Tables 13.1 and 13.2). The APE2 score of greater or equal to 4 was 99% sensitive and 93% specific for neural-specific-antibodies, while a RITE2 score of greater than or equal to 7 had 96% sensitivity and 86% specificity for favorable initial immunotherapy response [19].
Neural-Specific Antibodies Associated with Autoimmune Epilepsy
Cell Surface Epitopes
N-methyl-D-aspartate receptor (NMDA-R) encephalitis typically affects young women with a reported median age of 22 years (range: 2 months to 85 years) [20]. Clinical presentation usually begins with a prodrome of a headache or fever, followed by psychiatric manifestations including delusions, hallucinations, mania-like episodes, alternating episodes of extreme agitation, and catatonia. Patients then progress to develop seizures, encephalopathy, oral dyskinesia, choreoathetosis, and autonomic dysfunction [21]. Seizures in NMDA-R encephalitis are usually focal non-motor seizures that might progress to refractory status epilepticus [22]. If untreated, patients will progress to a comatose state [21].
In about half of the patients, a trigger can be identified. The two main triggers are the presence of ovarian teratoma [23] and a history of herpes simplex virus (HSV) encephalitis [24]. Approximately two-thirds of adult women between the ages of 18 to 45 years with NMDA-R encephalitis have been reported to have ovarian teratoma [21]. However, the presence of this tumor is extremely rare in children younger than 12 years or older adults (≥45 years) [6, 21, 25]. Furthermore, prospective evaluation of HSV encephalitis patients showed that 17% of these cases developed NMDA-R encephalitis during follow-up. Three additional patients in this cohort were positive for NMDA-R immunoglobulin G (IgG) without any clinical features of autoimmune encephalitis on follow-up evaluation [24].
Leucine-rich, glioma-inactivated 1 (LGI1) immunoglobulin G is typically associated with seizures and memory deficits usually among older patients (>40 years). However, a few pediatric cases have also been described [26]. One characteristic phenotype described among the adult patients is faciobrachial dystonic seizures (FBDS). These are brief focal dystonic motor seizures and occur multiple times a day. They have a characteristic stereotypic contraction of the face, arm, and leg [27]. Another characteristic seizure semiology is unilateral piloerections episodes. More recently, paroxysmal dizzy spells have also been described in a subset of patients [28]. These “dizzy spells” or “out of body experiences” may precede encephalopathy by 2–12 months.
A minority (~2%) of patients with voltage-gated potassium channel-complex (VGKC) antibodies have coexisting contactin-associated protein-like 2 (CASPR-2) IgG. Peripheral nervous system involvement is more common (neuromyotonia, myokymia, or dysautonomia) among these patients. However, a considerable proportion of patients, especially older patients, may have coexisting epilepsy or encephalitis. Recent studies have highlighted that VGKC IgG in the absence of LGI1 and/or CASPR-2 IgG seropositivity is not a specific biomarker of autoimmunity [29].
α(alpha)-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antibody-associated encephalitis typically presents with classic limbic encephalitis symptoms (anterograde memory deficits, retrograde amnesia, mood changes, and temporal lobe seizures). Recent studies have supported direct antibody-mediated pathogenicity [30, 31]. Median age of onset is around 60 years old (range: 23–81 years), and it occurs more commonly in females (64%) [21]. Two-thirds of the patients have underlying malignancy, mainly small cell lung cancer and thymoma [32]. A considerable proportion of patients have a refractory course and go on to develop diffuse cortical atrophy [30, 31, 33].
γ(gamma)-aminobutyric acid type B (GABA-B) receptor encephalitis usually presents as refractory non-convulsive status epilepticus [21]. Median age of onset is 61 years (range: 16–77 years) and tends to occur more commonly in males [6]. A subset of these cases have underlying malignancy, most commonly small cell lung carcinoma.
Fulminant encephalitis and refractory seizures or status epilepticus have been associated with GABA-A receptor encephalitis. These patients have characteristic multifocal cortical and subcortical T2/FLAIR hyperintensities [22]. Age of symptom onset tends to be younger (median age: 40 years) than cases with GABA-B encephalitis [6].
Patients with dipeptidyl-peptidase-like-protein 6 (DPPX) antibody-associated encephalitis can also have seizures as part of the syndrome. The usual clinical manifestations include gastrointestinal dysfunction, weight loss followed by cognitive dysfunction, hyperekplexia, myoclonus, parasomnias, and occasionally progressive encephalomyelitis with rigidity and myoclonus (PERM) [21, 34].
Metabotropic glutamate receptor 5 (mGluR5) IgG encephalitis patients usually present with subacute onset of encephalopathy, mood changes, movement disorder, and seizures [35]. Status epilepticus has been reported to be a common presenting feature especially among pediatric cases [36].
Intracellular Epitopes
Glutamic acid decarboxylase (65 kd, GAD65) antibodies (serum titers >20 nmol/L or detection in CSF) are associated with various autoimmune neurological diseases including autoimmune epilepsy, stiff person syndrome, cerebellar ataxia, limbic encephalitis, and PERM [37, 38]. Women are more frequently affected than men, and the median age of symptom onset is 30 years (range: 5–80 years) [22]. In a study of 112 patients with unexplained adult-onset focal epilepsy, 5.4% were found to have high titers of GAD65 antibodies (>1000 U/mL) [39]. Usually patients with GAD65 antibodies (serum titers >20 nmol/L or detection in CSF) are associated with a treatment-refractory course [38, 39]. Refractory nature of the disease is postulated to be secondary to cell-mediated cytotoxicity rather than a direct antibody-mediated pathogenesis.
Patients with antineuronal nuclear antibody type-1(ANNA-1 , a.k.a. anti-Hu) IgG antibodies present with various central and peripheral nervous system manifestations. ANNA-1 IgG seropositivity has a strong association with small cell cancer (81%). Sensory neuronopathy and autonomic dysfunction, especially gastroparesis, are hallmarks of ANNA-1 autoimmunity [13]. However, a considerable proportion (10–17%) of cases present with limbic encephalitis or refractory seizures. Both temporal and extratemporal localization of the seizures have been reported [40].
Seizures and limbic encephalitis are less common among patients with ANNA-2 IgG (a.k.a anti-Ri). These patients usually present with a brainstem or cerebellar syndrome. Initial manifestations include opsoclonus-myoclonus, jaw opening dystonia, and laryngospasms [41].
Ma2 IgG patients usually have a limbic encephalitis or brainstem encephalitis phenotype. In a retrospective study, bilateral tonic-clonic or focal unaware seizures occurred in 12 out of 27 (44%) patients [42]. A majority of Ma2 IgG seropositive have testicular germ cell tumor [43].
Collapsin response-mediator protein-5 (CRMP5) IgG is a paraneoplastic biomarker of small cell lung cancer or thymoma [44]. Patients with CRMP-5 IgG usually manifest with various neurologic signs including chorea, cranial neuropathy, dementia, cerebellar ataxia, myelopathy, and peripheral neuropathy [22, 45, 46]. Focal aware and unaware seizures have also been rarely reported in patients with CRMP-5. Management of underlying malignancy and early initiation of immunotherapy may be associated with favorable outcomes [47].
Rasmussen’s Encephalitis
Rasmussen’s encephalitis is a rare chronic neurological disorder characterized by drug-resistant focal motor epilepsy (epilepsia partialis continua), cognitive decline, hemiplegia, and unilateral hemispheric brain atrophy [5]. Clinical onset is usually during childhood, although it has been reported during adulthood. The disease progresses over three stages, with the first being a “prodromal stage” with a relatively low seizure frequency and rarely a mild hemiparesis. Following that, the patient will enter the “acute stage,” which is characterized by frequent intractable seizures along with progressive neurological decline (hemiparesis, hemianopia, cognitive deterioration, and aphasia if dominant hemisphere). During the final “residual” stage, the patient develops permanent and stable neurological deficits and intractable focal motor seizures [48]. CSF examination may be normal or may show inflammatory changes (lymphocytic pleocytosis and elevated CSF protein). Electroencephalogram (EEG) shows unilateral inter-ictal epileptiform discharges, slowing, and ictal rhythms with occasional spread to the contralateral side due to bilateral synchrony. No specific electrographic signatures have been associated with Rasmussen’s encephalitis. MRI brain scan shows FLAIR/T2 hyperintensity and atrophy involving unilateral cortical and/or subcortical regions with a predilection for perisylvian area. Fluorodeoxyglucose positron emission tomography (FDG-PET) scan may show increased uptake in the affected hemisphere [5, 48]. Multiple case reports have demonstrated favorable response to immunotherapy. In 2013, a randomized trial of tacrolimus and intravenous immunoglobulin showed slowing down of tissue and function loss with either therapies, but without improvement in seizures [49]. At present, hemispherectomy remains the best option of seizure control and arresting the neurological decline [50].
New-Onset Refractory Status Epilepticus
In new-onset refractory status epilepticus (NORSE), a previously healthy individual develops refractory de novo seizures and status epilepticus with no readily identifiable etiology. A retrospective study exploring NORSE in the adult population has shown a significant percentage of these patients to have immune-mediated etiologies—primarily antibody-mediated encephalitis with anti-NMDA receptor being the most common etiology [51]. Various treatments have been tried, including anti-seizure medications, achieving burst suppression with anesthetics, and dietary therapy with modest and variable effects [52]. Use of immunotherapy has been associated with favorable outcomes in a few cases (5–33%) [52].
Steroid Responsive Encephalopathy with Autoimmune Thyroiditis (SREAT) or Hashimoto’s Encephalopathy
Clinical characteristics of Hashimoto’s encephalopathy include encephalopathy, seizures, stroke-like episodes, and myoclonus [13]. These patients typically have thyroid peroxidase (TPO) antibodies but may or may not have a history of thyroiditis. Seizure presentations are variable including new-onset refractory status epilepticus or progressive myoclonic epilepsy [5, 53]. A triad of encephalopathy, evidence of thyroid autoimmunity (clinically or serologically), and a favorable response to steroids have been traditionally utilized for identification of these cases [11].
Electroencephalogram
EEG plays a vital role in diagnosis and management of autoimmune epilepsy and encephalitis. It is essential to look for epileptiform or seizure activity. Long-term monitoring is utilized among patients with subclinical or clinical status epilepticus [54]. Additionally, EEG can also be utilized to evaluate response to immunotherapy and anti-epileptic drugs in some instances.
EEG findings in autoimmune encephalitis are variable and may be nonspecific. Extreme delta brush (EDB) was first described in NMDA encephalitis patients by Schmitt et al. [55]. This EEG pattern consists of rhythmic delta activity at 1–3 Hz with superimposed burst of rhythmic beta activity at 20–30 Hz riding on each delta wave [55]. EDB has been reported in about 30% of acutely ill or hospitalized NMDA encephalitis patients and is associated with a more prolonged illness. Although it was thought to be fairly specific initially, recent studies have described the presence of EDB with other metabolic and structural causes of encephalopathy [56].
FBDS, a pathognomonic feature of LGI1 autoimmune epilepsy, usually has no ictal EEG correlate. Occasionally preceding electro-decrement or sharply contoured rhythmic delta activity has been reported over the contralateral frontotemporal region [57]. In a small series of LGI1 patients, frequent subclinical temporal lobe seizures associated with hyperventilation were reported. This ictal activity had similar morphology to subclinical rhythmic electrographic discharges of adults (SREDA) [58]. A more recent study of EEGs with 16 LGI1 encephalitis patients reported multiple frequent seizure semiologies or subclinical seizures associated with temporal and frontal discharges [57]. Additionally, multifocal interictal epileptiform discharges and interictal slow-wave activity were observed in 25% and 69%, respectively.
Imaging
Brain MRI is usually included in initial evaluation of autoimmune epilepsy and encephalitis. Features considered suggestive of autoimmune encephalitis include T2/FLAIR hyperintensity restricted to one or both medial temporal lobes (Fig. 13.1) or multifocal T2/FLAIR hyperintensities in the gray matter, white matter, or both compatible with demyelination or inflammation [11]. However, MRI may be normal, especially early in the course of the disease [59, 60]. Brain MRI also provides valuable information regarding differential diagnosis of new-onset epilepsy and/or subacute onset cognitive dysfunction such as tumors, brain abscess, neuro-sarcoidosis, and other inflammatory and infectious diseases. Beside brain MRI, abnormalities in functional MRI [61], diffusion tensor imaging (DTI) [61], FDG-PET/CT [62], and single photon emission tomography (SPECT) [63] have been described in patients with autoimmune epilepsy and can provide valuable diagnostic and at times prognostic values.

Patient 1 with LGI1 IgG limbic encephalitis. MRI brain (FLAIR sequence) showing bilateral medial temporal hyperintensities on axial (a) and sagittal sections (b). Patient 2 with ANNA1 IgG limbic encephalitis. MRI brain (FLAIR sequence) showing bilateral medial temporal hyperintensities on axial (c) and sagittal sections (d). Patient 3 with Ma2 IgG limbic encephalitis. MRI brain (FLAIR sequence) showing bilateral medial temporal (right greater than left) hyperintensities on axial (e) and sagittal sections (f). Abbreviations: ANNA-1 antineuronal nuclear antibody-1, MRI magnetic resonance imaging, FLAIR fluid attenuated inversion recovery, LGI1 leucine-rich, glioma inactivated 1
In the initial phase of NMDA-R encephalitis, the brain MRI abnormalities are typically discrete and nonspecific [20, 64]. However, resting state functional MRI shows disrupted hippocampal functional connectivity. Moreover, DTI has detected a more widespread white matter damage that correlated with disease severity [61]. Decreased occipital lobe metabolism on FDG-PET/CT has been described as a unique finding in these patients [65]. Resolution of lateral and medial occipital hypometabolism may correlate with better outcomes.
Brain MRI findings in patients with LGI1 encephalitis varies depending on the stage of the disease and the progression [65]. In the early phase of the disease, brain MRI is typically normal, although basal ganglia abnormalities including increased FLAIR signal, restricted diffusion, and contrast enhancement have been reported [66]. Patients with FBDS may develop T1 hyperintensity in region of basal ganglia [67]. As the disease progresses, unilateral or bilateral T2/FLAIR hyperintensities of the medial temporal lobes and basal ganglia are observed. On follow-up imaging, hippocampal atrophy is frequently seen [65]. FDG-PET/CT reveals basal ganglia hypermetabolism, a specific finding, and frequently the earliest finding on imaging studies [68].
Among patients with GAD65 antibody-associated autoimmune epilepsy, brain MRI demonstrates disproportionate parenchymal atrophy for age and abnormal cortical/subcortical T2 hyperintensities. Hippocampal abnormalities are seen only in a minority (26%) of patients [69].
Patients with mGluR5 antibodies have abnormal brain MRI in 45% of the cases, with involvement of both limbic and extralimbic (thalamus, pons, cerebral, and cerebellar cortices) regions. Additionally, FDG-PET in some of these cases demonstrates hypometabolism of the temporoparietal cortex or cerebellum [36]. Whereas GABA-A receptor encephalitis has a unique pattern of widespread and extensive cortical and subcortical FLAIR hyperintensity [70].
Medial temporal lobe involvement has been reported in multiple antibody specificities including AMPA-R [71], GABA-B receptor IgG [72], ANNA-1 IgG [40], Ma2-IgG [42], adenylate kinase 5 [73], etc. A majority of these cases do not have associated gadolinium enhancement except for Ma2 IgG-associated limbic encephalitis [42].
Cancer Screening
CT of the chest, abdomen, and pelvis with contrast is recommended as initial evaluation for cancer association. Scrotal ultrasounds should be performed in all males, especially those presenting with brainstem, diencephalic or limbic encephalitis. In women, mammograms should be performed for evaluation of breast cancer. Pelvic sonography and pelvic MRI are recommended for ovarian teratoma or adenocarcinoma screening. If initial radiological evaluations did not reveal any malignancies and clinical suspicion for paraneoplastic neurological syndrome is high or the patient has neural specific antibody with strong oncological association (Table 13.3), PET-CT should be pursued [74, 75]. If the patient’s evaluation reveals a neoplasm other than that predicted by the antibody present, further cancer evaluation should be performed as more than one cancer can coexist [74]. Paraneoplastic syndromes and associated malignancies are further discussed in Chap. 16.
Treatment
Management of autoimmune epilepsy is focused on immunotherapies. Multiple studies have demonstrated favorable effects of early immunotherapy on seizure frequency and cognition [9, 15, 76, 77]. However, randomized control trials evaluating efficacy immunotherapy in autoimmune epilepsy are limited. Therefore, immunotherapy recommendations are largely based on case series and clinical experience [20, 78]. Recently, a placebo-controlled trial of IVIg for LGI1 and CASPR2 IgG-associated autoimmune epilepsy was published [79]. In this study of 17 patients, 75% of the patients receiving IVIg achieved more than 50% seizure reduction by 5 weeks compared to only 22% of patients in the placebo arm [79]. In some instances, a positive response to a treatment trial of immunotherapy can aid in the diagnosis of seronegative autoimmune epilepsy [9]. In this regard, the RITE2 score may be a useful scoring system for managing clinician prior to immunotherapy initiation [15].
Immunotherapeutic agents are classically divided into first-line and second-line therapies (Table 13.4). First-line therapies include high-dose intravenous methylprednisolone (IVMP), intravenous immunoglobulin, or plasmapheresis. Second-line agents such as rituximab, cyclophosphamide, mycophenolate, azathioprine, or bortezomib are used in refractory cases or as a maintenance therapy to prevent relapses.
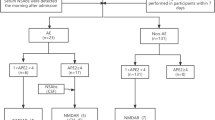
Treatment of autoimmune epilepsy should be based on the severity of the clinical course (Fig. 13.2). In patients with rapid progression and refractory course, more aggressive immunotherapy is needed including both first- and second-line therapies. Conversely, some patients with autoimmune epilepsy have a more benign course, and their epilepsy can be controlled with antiepileptic drugs and a short course of immunotherapy. In all cases, cancer surveillance, as discussed previously should be pursued as treatment of the underlying cancer, is pivotal for the successful treatment of autoimmune epilepsy.

Management algorithm for autoimmune epilepsy. Abbreviations: Ab antibody, CSF cerebrospinal fluid, EEG electroencephalogram, HIV human immunodeficiency virus, IVIg intravenous immunoglobulin, IVMP intravenous methylprednisone, PLEX plasmapheresis, PO per oral
Proposed immunotherapy trials include 6 or 12 weeks of high-dose IV methylprednisolone (IVMP). Intravenous methylprednisolone 1000 mg per day for 3 days followed by once weekly for 5 weeks (6 IVMP week trial), followed by once every 2 weeks for 6 weeks (12 IVMP week trial). If the patient has contraindications for IVMP (active infections, poorly controlled diabetes, chronic hepatitis, or tuberculosis), a 6- or 12-week course of intravenous immunoglobulin1 may be considered. These include 0.4 g/kg IVIg daily for 3 days followed by 0.4 g/kg every week for 6 weeks (6 IVIg week trial) and then every 2 weeks for 6 weeks (12 IVIg week trial). A treatment response can be ascertained using a seizure diary to assess seizure frequency and/or change in semiology and neurological examination including screening mental status examination after completion of immunotherapy trial. The quality of life in epilepsy (QOLIE-31) can be utilized as well. EEG, brain MRI with gadolinium, PET brain, and formal cognitive tests are additional parameters that can be monitored. Seizures in autoimmune epilepsy may show early improvement within 4–6 weeks of initiating immunotherapy. Conversely, cognitive impairment and amnesia, if present, recover more slowly. For patients who have incomplete or lack of response to IVMP or IV, or those who have contraindication to IVMP and/or IVIg, a course of plasmapheresis (5–7 cycles) should be considered followed by a second-line immunotherapy (rituximab or cyclophosphamide). A gradual taper of prednisone if feasible is advised following the initial treatment, as abrupt discontinuation might lead to relapses. Chronic maintenance immunotherapy should be initiated to decrease the likelihood of a relapse. Azathioprine, mycophenolate mofetil, rituximab, and cyclophosphamide are agents typically utilized. The exact duration required for maintenance immunotherapy is not known, although a trial of immunotherapy withdrawal may be considered after 2 years of treatment if the patient has not had any relapses.
Antiepileptic Drugs (AEDS)
Even though seizures in autoimmune epilepsy are characteristically resistant to antiepileptic drugs (AEDs) alone, they continue to play an important role in symptomatic management. In all autoimmune epilepsy patients, AEDs should be used along with immunotherapy treatment. There are no randomized trial data to support one AED over another. Levetiracetam is commonly employed for management of seizures given the favorable side effect profile and minimal drug-to-drug interaction. In some instances, it can be difficult to ascertain if the psychiatric manifestations are due to levetiracetam adverse effects [80] or due to disease pathology. Anti-inflammatory effect for levetiracetam has been hypothesized to play a beneficial role [81]. However, a recent retrospective study evaluating AED in autoimmune epilepsy found none of the patients on levetiracetam achieved seizure freedom [82]. Whereas seizure freedom rates were considerably higher with the use of sodium channel blocking AEDs (carbamazepine, phenytoin, oxcarbazepine, and lacosamide). [82]. The reason for better efficacy of sodium channel-blocking AEDs remains unclear. Interestingly, both carbamazepine and oxcarbazepine have been shown to reduce levels of interleukin-1 (IL)-1 and IL-2 in healthy subjects [81]. Medications such as carbamazepine and phenytoin have enzyme induction properties, which can alter the pharmacokinetics of immunosuppressive therapies. Therefore, newer sodium channel blocking AEDs with more favorable pharmacokinetic profiles (such as oxcarbazepine and lacosamide) could be preferred in management of autoimmune epilepsy.
Follow-Up
Patients with autoimmune epilepsy should be followed up regularly, preferably by an epileptologist in conjunction with a neuroimmunologist. On long-term follow-up, some patients continue to have drug-resistant epilepsy, despite initial treatment with immunotherapy and continued treatment with AEDs. Whether the disease is now at a “burn out” stage or an acute inflammatory process continues is hard to discern. Ancillary data including repeating brain MRI with gadolinium contrast, CSF analysis, and PET brain can be helpful in guiding treatment decisions. Serum and/or CSF antibody titers have poor relationship to clinical course. Epilepsy surgery has been tried in select cases of autoimmune epilepsy [83]. However, outcomes seem to be worse than that expected in other etiologies of drug-resistant epilepsy.
Conclusion
Future Directions
Many challenges continue to face us as we move forward in the field of autoimmune epilepsy. Several novel biomarkers are likely to be discovered over the next few years. The use of phage immunocepitation sequencing and protein microarrays might help us to identify novel antibodies that were not discovered using traditional immunoprecipitation mass spectrometry techniques [84,85,86].
Further insights into the mechanisms of antibody-mediated epilepsy syndromes will allow us to better utilize the various AEDs and immunotherapies. Additionally, investigations into the role of cytokines and chemokines in diagnosis and prognostication of autoimmune epilepsy will also aid in seizure management and relapse prevention [87, 88]. Finally, randomized controlled trials are needed to determine optimal therapeutic agents, doses, and treatment duration for autoimmune epilepsy management.
References
Ong MS, Kohane IS, Cai T, Gorman MP, Mandl KD. Population-level evidence for an autoimmune etiology of epilepsy. JAMA Neurol. 2014;71(5):569–74.
Brodie MJ, Zuberi SM, Scheffer IE, Fisher RS. The 2017 ILAE classification of seizure types and the epilepsies: what do people with epilepsy and their caregivers need to know? Epileptic Disord. 2018;20(2):77–87.
Granata T, Andermann F. Rasmussen encephalitis. Handb Clin Neurol. 2013;111:511–9.
Sinclair DB, Snyder TJ. Corticosteroids for the treatment of Landau-kleffner syndrome and continuous spike-wave discharge during sleep. Pediatr Neurol. 2005;32(5):300–6.
Britton J. Autoimmune epilepsy. Handb Clin Neurol. 2016;133:219–45.
Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med. 2018;378(9):840–51.
Larman HB, Zhao Z, Laserson U, Li MZ, Ciccia A, Gakidis MA, et al. Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol. 2011;29(6):535–41.
Scharf M, Miske R, Kade S, Hahn S, Denno Y, Begemann N, et al. A spectrum of neural autoantigens, newly identified by histo-immunoprecipitation, mass spectrometry, and recombinant cell-based indirect immunofluorescence. Front Immunol. 2018;9:1447.
Toledano M, Britton JW, McKeon A, Shin C, Lennon VA, Quek AM, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology. 2014;82(18):1578–86.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21.
Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391–404.
Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. 2018;83(1):166–77.
Dubey D, Toledano M, McKeon A. Clinical presentation of autoimmune and viral encephalitides. Curr Opin Crit Care. 2018;24(2):80–90.
Dubey D, Alqallaf A, Hays R, Freeman M, Chen K, Ding K, et al. Neurological autoantibody prevalence in epilepsy of unknown etiology. JAMA Neurol. 2017;74(4):397–402.
Dubey D, Singh J, Britton JW, Pittock SJ, Flanagan EP, Lennon VA, et al. Predictive models in the diagnosis and treatment of autoimmune epilepsy. Epilepsia. 2017;58(7):1181–9.
Brenner T, Sills GJ, Hart Y, Howell S, Waters P, Brodie MJ, et al. Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia. 2013;54(6):1028–35.
Abramovici S, Bagic A. Epidemiology of epilepsy. Handb Clin Neurol. 2016;138:159–71.
Wright S, Geerts AT, Jol-van der Zijde CM, Jacobson L, Leang B, Waters P, van Tol MJ, Stroink H, Neuteboom RF, Brouwer OF, Vincent A. Neuronal antibodies in pediatric epilepsy: clinical features and long-term outcomes of a historical cohort not treated with immunotherapy. Epilepsia. 2016;57(5):823–31.
Dubey D, Kothapalli N, McKeon A, Flanagan EP, Lennon VA, Klein CJ, et al. Predictors of neural-specific autoantibodies and immunotherapy response in patients with cognitive dysfunction. J Neuroimmunol. 2018;323:62–72.
Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157–65.
Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. 2017;97(2):839–87.
Quek AML, O’Toole O. Autoimmune epilepsy: the evolving science of neural autoimmunity and its impact on epilepsy management. Semin Neurol. 2018;38(3):290–302.
Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25–36.
Armangue T, Spatola M, Vlagea A, Mattozzi S, Carceles-Cordon M, Martinez-Heras E, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 2018;17:760.
Titulaer MJ, McCracken L, Gabilondo I, Iizuka T, Kawachi I, Bataller L, et al. Late-onset anti-NMDA receptor encephalitis. Neurology. 2013;81(12):1058–63.
Sebastian Lopez-Chiriboga A, Klein C, Zekeridou A, McKeon A, Dubey D, Flanagan EP, et al. LGI1 and CASPR2 neurological autoimmunity in children. Ann Neurol. 2018;84(3):473–80.
Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69(5):892–900.
Gadoth A, Pittock SJ, Dubey D, McKeon A, Britton JW, Schmeling JE, et al. Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG-positive patients. Ann Neurol. 2017;82(1):79–92.
van Sonderen A, Schreurs MW, de Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology. 2016;86(18):1692–9.
Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65(4):424–34.
Haselmann H, Mannara F, Werner C, Planaguma J, Miguez-Cabello F, Schmidl L, et al. Human autoantibodies against the AMPA receptor subunit GluA2 induce receptor reorganization and memory dysfunction. Neuron. 2018;100:91.
Hoftberger R, van Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. 2015;84(24):2403–12.
Joubert B, Kerschen P, Zekeridou A, Desestret V, Rogemond V, Chaffois MO, et al. Clinical spectrum of encephalitis associated with antibodies against the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor: case series and review of the literature. JAMA Neurol. 2015;72(10):1163–9.
Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M, Roberts K, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73(1):120–8.
Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine JC, et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77(18):1698–701.
Spatola M, Sabater L, Planaguma J, Martinez-Hernandez E, Armangue T, Pruss H, et al. Encephalitis with mGluR5 antibodies: symptoms and antibody effects. Neurology. 2018;90(22):e1964–e72.
Pittock SJ, Yoshikawa H, Ahlskog JE, Tisch SH, Benarroch EE, Kryzer TJ, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc. 2006;81(9):1207–14.
Peltola J, Kulmala P, Isojarvi J, Saiz A, Latvala K, Palmio J, et al. Autoantibodies to glutamic acid decarboxylase in patients with therapy-resistant epilepsy. Neurology. 2000;55(1):46–50.
Lilleker JB, Biswas V, Mohanraj R. Glutamic acid decarboxylase (GAD) antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure. 2014;23(8):598–602.
Rudzinski LA, Pittock SJ, McKeon A, Lennon VA, Britton JW. Extratemporal EEG and MRI findings in ANNA-1 (anti-Hu) encephalitis. Epilepsy Res. 2011;95(3):255–62.
Pittock SJ, Lucchinetti CF, Lennon VA. Anti-neuronal nuclear autoantibody type 2: paraneoplastic accompaniments. Ann Neurol. 2003;53(5):580–7.
Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127(Pt 8):1831–44.
Voltz R, Gultekin SH, Rosenfeld MR, Gerstner E, Eichen J, Posner JB, et al. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med. 1999;340(23):1788–95.
Yu Z, Kryzer TJ, Griesmann GE, Kim K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49(2):146–54.
Dubey D, Lennon VA, Gadoth A, Pittock SJ, Flanagan EP, Schmeling JE, et al. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurology. 2018;90(2):e103–e10.
Vernino S, Tuite P, Adler CH, Meschia JF, Boeve BF, Boasberg P, et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol. 2002;51(5):625–30.
Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69(5):582–93.
Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, Kurthen M, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. 2005;128(Pt 3):454–71.
Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, von Lehe M, et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia. 2013;54(3):543–50.
Vining EP, Freeman JM, Pillas DJ, Uematsu S, Carson BS, Brandt J, et al. Why would you remove half a brain? The outcome of 58 children after hemispherectomy-the Johns Hopkins experience: 1968 to 1996. Pediatrics. 1997;100(2 Pt 1):163–71.
Gaspard N, Foreman BP, Alvarez V, Cabrera Kang C, Probasco JC, Jongeling AC, et al. New-onset refractory status epilepticus: etiology, clinical features, and outcome. Neurology. 2015;85(18):1604–13.
Gaspard N, Hirsch LJ, Sculier C, Loddenkemper T, van Baalen A, Lancrenon J, et al. New-onset refractory status epilepticus2 and febrile infection-related epilepsy syndrome (FIRES): state of the art and perspectives. Epilepsia. 2018;59(4):745–52.
Arya R, Anand V, Chansoria M. Hashimoto encephalopathy presenting as progressive myoclonus epilepsy syndrome. Eur J Paediatr Neurol. 2013;17(1):102–4.
Kaplan PW, Sutter R. Electroencephalography of autoimmune limbic encephalopathy. J Clin Neurophysiol. 2013;30(5):490–504.
Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79(11):1094–100.
Baykan B, Gungor Tuncer O, Vanli-Yavuz EN, Baysal Kirac L, Gundogdu G, Bebek N, et al. Delta brush pattern is not unique to NMDAR encephalitis: evaluation of two independent long-term EEG cohorts. Clin EEG Neurosci. 2018;49(4):278–84.
Aurangzeb S, Symmonds M, Knight RK, Kennett R, Wehner T, Irani SR. LGI1-antibody encephalitis is characterised by frequent, multifocal clinical and subclinical seizures. Seizure. 2017;50:14–7.
Steriade C, Mirsattari SM, Murray BJ, Wennberg R. Subclinical temporal EEG seizure pattern in LGI1-antibody-mediated encephalitis. Epilepsia. 2016;57(8):e155–60.
Escudero D, Guasp M, Arino H, Gaig C, Martinez-Hernandez E, Dalmau J, et al. Antibody-associated CNS syndromes without signs of inflammation in the elderly. Neurology. 2017;89(14):1471–5.
Dubey D, Sawhney A, Greenberg B, Lowden A, Warnack W, Khemani P, et al. The spectrum of autoimmune encephalopathies. J Neuroimmunol. 2015;287:93–7.
Finke C, Kopp UA, Scheel M, Pech LM, Soemmer C, Schlichting J, et al. Functional and structural brain changes in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2013;74(2):284–96.
Solnes LB, Jones KM, Rowe SP, Pattanayak P, Nalluri A, Venkatesan A, et al. Diagnostic value of (18)F-FDG PET/CT versus MRI in the setting of antibody-specific autoimmune encephalitis. J Nucl Med. 2017;58(8):1307–13.
Ohta K, Seki M, Dalmau J, Shinohara Y. Perfusion IMP-SPECT shows reversible abnormalities in GABA(B) receptor antibody associated encephalitis with normal MRI. Brain Behav. 2011;1(2):70–2.
Heine J, Pruss H, Bartsch T, Ploner CJ, Paul F, Finke C. Imaging of autoimmune encephalitis–relevance for clinical practice and hippocampal function. Neuroscience. 2015;309:68–83.
Probasco JC, Solnes L, Nalluri A, Cohen J, Jones KM, Zan E, et al. Decreased occipital lobe metabolism by FDG-PET/CT: an anti-NMDA receptor encephalitis biomarker. Neurol Neuroimmunol Neuroinflamm. 2018;5(1):e413.
Irani SR, Vincent A. Voltage-gated potassium channel-complex autoimmunity and associated clinical syndromes. Handb Clin Neurol. 2016;133:185–97.
Flanagan EP, Kotsenas AL, Britton JW, McKeon A, Watson RE, Klein CJ, et al. Basal ganglia T1 hyperintensity in LGI1-autoantibody faciobrachial dystonic seizures. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):e161.
Shin YW, Lee ST, Shin JW, Moon J, Lim JA, Byun JI, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol. 2013;265(1–2):75–81.
Fredriksen JR, Carr CM, Koeller KK, Verdoorn JT, Gadoth A, Pittock SJ, et al. MRI findings in glutamic acid decarboxylase associated autoimmune epilepsy. Neuroradiology. 2018;60(3):239–45.
Spatola M, Petit-Pedrol M, Simabukuro MM, Armangue T, Castro FJ, Barcelo Artigues MI, et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology. 2017;88(11):1012–20.
Dogan Onugoren M, Deuretzbacher D, Haensch CA, Hagedorn HJ, Halve S, Isenmann S, et al. Limbic encephalitis due to GABAB and AMPA receptor antibodies: a case series. J Neurol Neurosurg Psychiatry. 2015;86(9):965–72.
Hoftberger R, Titulaer MJ, Sabater L, Dome B, Rozsas A, Hegedus B, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81(17):1500–6.
Do LD, Chanson E, Desestret V, Joubert B, Ducray F, Brugiere S, et al. Characteristics in limbic encephalitis with anti-adenylate kinase 5 autoantibodies. Neurology. 2017;88(6):514–24.
Pittock SJ, Palace J. Paraneoplastic and idiopathic autoimmune neurologic disorders: approach to diagnosis and treatment. Handb Clin Neurol. 2016;133:165–83.
Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135–40.
Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA, Pettingill P, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. 2013;136(Pt 10):3151–62.
Byun JI, Lee ST, Jung KH, Sunwoo JS, Moon J, Lim JA, et al. Effect of immunotherapy on seizure outcome in patients with autoimmune encephalitis: a prospective observational registry study. PLoS One. 2016;11(1):e0146455.
Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain. 2018;141(2):348–56.
Dubey D, Britton J, McKeon A, Gadoth A, Zekeridou A, Lopez Chiriboga SA, et al. Randomized placebo-controlled trial of intravenous immunoglobulin in autoimmune LGI1/CASPR2 epilepsy. Ann Neurol. 2020;87:313–23.
Abou-Khalil BW. Antiepileptic drugs. Continuum (Minneap Minn). 2016;22(1 Epilepsy):132–56.
Beghi E, Shorvon S. Antiepileptic drugs and the immune system. Epilepsia. 2011;52(Suppl 3):40–4.
Feyissa AM, Lopez Chiriboga AS, Britton JW. Antiepileptic drug therapy in patients with autoimmune epilepsy. Neurol Neuroimmunol Neuroinflamm. 2017;4(4):e353.
Carreno M, Bien CG, Asadi-Pooya AA, Sperling M, Marusic P, Elisak M, et al. Epilepsy surgery in drug resistant temporal lobe epilepsy associated with neuronal antibodies. Epilepsy Res. 2017;129:101–5.
Schubert RD, Wilson MR. A tale of two approaches: how metagenomics and proteomics are shaping the future of encephalitis diagnostics. Curr Opin Neurol. 2015;28(3):283–7.
Mohan D, Wansley DL, Sie BM, Noon MS, Baer AN, Laserson U, et al. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat Protoc. 2018;13(9):1958–78.
Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. 2013;73(3):408–18.
Vezzani A, Fujinami RS, White HS, Preux PM, Blumcke I, Sander JW, et al. Infections, inflammation and epilepsy. Acta Neuropathol. 2016;131(2):211–34.
Vezzani A. Epilepsy and inflammation in the brain: overview and pathophysiology. Epilepsy Curr. 2014;14(1 Suppl):3–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Husari, K., Dubey, D. (2021). Autoimmune Epilepsy. In: Piquet, A.L., Alvarez, E. (eds) Neuroimmunology. Springer, Cham. https://doi.org/10.1007/978-3-030-61883-4_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-61883-4_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-61882-7
Online ISBN: 978-3-030-61883-4
eBook Packages: MedicineMedicine (R0)