
Summary
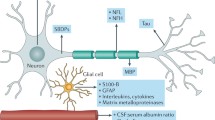
Brain biomarkers (protein S100b and neuron-specific enolase (NSE)), antibodies (aAb) to the NR2 subunit of N-methyl-D-aspartate (NR2(NMDA)) and to the GluR1 subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (GluR1(AMPA)) subtype of glutamate receptors (GluR), NR2 and AMPA peptides, nitrogen oxides (NOx; “nitrites and nitrates”), and 3-nitrotyrosine (NT) were measured in blood from 159 children after mild traumatic brain injury (mTBI), moderate traumatic brain injury (mdTBI), or severe traumatic brain injury (sTBI) within 1–2 days and at intervals during the first 15 days after brain trauma. S100b and NSE levels on the first day were not a strict criterion for injury outcomes. Children with mTBI had the most significant elevations in antibodies to NR2(NMDA) and AMPA peptides, a slight increase in NOx, and, in 25% of cases, appearance of NT in the blood right after TBI. The lowest level of antibodies to NR2(NMDA) GluR detected shortly after the initial TBI was found in children with sTBI, with a negative outcome. The opposite characters of antibodies to NR2(NMDA) on the first day in children with mild and moderate versus severe TBI may be associated with an important mechanism aimed at protecting neurons from Glu excitotoxicity. We hypothesized that a slight increase in NOx after the onset of TBI rapidly activates the innate immune system and contributes to an increase in antibodies to NR2(NMDA). An increase in the AMPA peptide level in mTBI may be early signs of diffuse axonal injury.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Children after traumatic brain injury (TBI)
- S100b
- NSE
- Antibodies (aAb) to glutamate receptors (aAb GluR): aAb NR2(NMDA) and GluR1(AMPA)
- NR2 and AMPA peptides of GluR
- Nitric oxide (NO)
- 3-Nitrotyrosine (NT)
Introduction
Traumatic brain injury (TBI) has been a leading pathology for many years, causing huge social and material damage in society [1]. The search for informative markers of brain damage remains an important challenge for predicting the outcome and treatment of children with TBI. The diagnostic capabilities of magnetic resonance imaging (MRI) and computed tomography (CT) are limited by high capital costs and often do not provide information that can predict the consequences and outcome of TBI, particularly in mild TBI (mTBI) [2, 3]. Many mTBI diagnoses go undetected because of the subtlety of the initial neurological deficit [4]. Problems in the search for adequate brain markers include the need for neuromarkers that reflect the earliest response of the brain and lesions preceding development of secondary damage after TBI. Secondary damage includes excessive release of the excitatory amino acid glutamate (Glu) in the synaptic gap and development of a cascade of excitotoxic reactions, including increased proteolytic enzyme activity, lipid and protein peroxidation, membrane degradation, and mitochondrial de-energization and energy collapse, all of which contribute to neuronal cell death [5, 6]. Many reviews have focused on the pathogenesis of TBI, but the roles of the immune system and oxidative stress remain underestimated [7, 8]. Oxidative stress and damage to glutamate receptors (GluR), with development of an autoimmune response to fragments of GluR, play important roles in the pathological chain of reactions to secondary brain injury [9, 10]. Along with traditional neuromarkers (S100b and neuron-specific enolase (NSE)), brain injury markers such as antibodies (aAb) to GluR, their degradation products (peptides), nitric oxide (NO), and its product 3-nitrotyrosine (NT) may be useful for understanding of TBI pathogenesis and may indicate development of hypoxia and neuroinflammation [11].
Materials and Methods
In this study, the severity of TBI in 159 children aged >3 years was evaluated on the basis of the Glasgow Coma Scale score (GCS), and the children were divided into the following groups: mTBI (GСS 14–15; 100 children), moderate TBI (mdTBI) (GСS 9–13; 25 children), and severe TBI (sTBI) (GСS <9; 34 children). The outcomes of TBI were evaluated according to the Glasgow Outcome Scale score (GOS): full recovery (GOS 5), moderate disability (GOS 4), high disability (GOS 3), vegetative status (GOS 2), and death (GOS 1). Venous blood samples were investigated on days 1–2, as were the dynamics during the first 2 weeks after TBI. Biomarker levels in blood serum or plasma were determined with an enzyme-linked immunosorbent assay (ELISA) and colorimetric methods: S100b and NSE (CanAg), 3-nitrotyrosine (Hycult Biotech), and the αII-spectrin breakdown product SBDP145 (Cusabio Biotech). Nitrogen oxide (NOx) was measured as “nitrites and nitrates” in plasma (Calbiochem, R&D Systems). The levels of antibodies to NR2(NMDA) and GluR1(AMPA) and NR2 and AMPA peptides of GluR were measured using a method developed by Dambinova et al. [12, 13]. For control values and calculations, we used data on the upper limits of normal marker ranges prescribed by developers and our own data from children without neurological pathology. Statistical evaluation of the data was carried out using Statistica version 6 (StatSoft) and Excel (Microsoft) software. Differences between parameters were compared by means of a Kruskal–Wallis analysis of variance (ANOVA). P values of <0.05 were considered significant and represented as means ± standard errors of the means. Graphics were processed using Prism software (GraphPad).
Results
In the first 2 days, almost all of the children had increases in serum levels of NSE and S100b. In the following days, decreases in these proteins were observed in those with GOS 3, 4, and 5, whereas further increases in their levels was observed in those with a lethal outcome (GOS 1) (Table 1).
We found that immediately after TBI, there was an increase in the level of nitrites and nitrates (NOx); the more severe the damage, the higher the plasma level of these NO metabolites (Table 2). We also noted the appearance of the protein nitrosation product NT in the plasma of children with TBI. At the onset of mTBI, 25% of the children developed a measurable NT level. The highest NT level was found in children with a lethal outcome of combined TBI (the NT level reached 1890 nmol/L in mdTBI and 8101 nmol/L in sTBI).
In the first 2 days, we also detected traces of SBDP145 in the plasma of children with mTBI (0.036 ± 0.012 ng/mL) and mdTBI (0.119 ± 0.023 ng/mL).
Figure 1a, b, c, d presents a more visual demonstration of the levels of antibodies to NR2(NMDA) and peptides, and shows individual data for children with mTBI. The level of antibodies to NR2(NMDA) in 91% of children with mTBI immediately after injury was 2.8 times that in children with mdTBI (GOS 3, 4) and was several times the upper limit of the normal range (Table 2, Fig. 1a). A similar pattern was observed in children with sTBI with different outcomes: the lowest level of antibodies to NR2(NMDA) on the first day after sTBI was found in children with the worst prognosis (GOS 1), and the highest level was found in the group with good recovery (GOS 5) (Fig. 1b). The level of NR2 peptides exceeded the upper limit of the normal range in only 14% of children with mTBI, and this difference versus the normal values was not significant (Fig. 1c). Conversely, the level of AMPA peptides exceeded the upper limit of the normal range in 91% of children with mTBI (Fig. 1d).

NR2(NMDA) antibodies and degradation products of GluR—NR2 and AMPA peptides—in the blood of children with a traumatic brain injury (TBI). (a, c, d) Individual data from 35 children with mild TBI. (b) Days after brain injury. (a) NR2(NMDA) antibody level on the first day after mild TBI. (b) Dynamics of NR2(NMDA) antibodies in children after severe TBI with different Glasgow Outcome Scale scores (GOS). (c, d) NR2 and AMPA peptide levels in blood plasma samples from children with mild TBI. * P < 0.05 for the difference between GOS 1 and GOS 5; a vertical arrow indicates the upper limit of the normal range
The opposite pattern of changes was shown for antibodies to GluR1(AMPA). Children with mdTBI had a higher initial level of antibodies to GluR1(AMPA) and a lower level of AMPA peptides than patients with mTBI. Thus, the more severe the TBI, the lower the blood level of antibodies to NR2(NMDA) and the higher the level of antibodies to (GluR1)AMPA on the first day.
Discussion
S100b protein and NSE are generally accepted biochemical markers of brain damage. S100b protein is considered a glial protein, predominantly localized in astrocytes, and appears to be involved in signal transduction, energy metabolism, and many other processes, especially through regulation of protein phosphorylation [14, 15]. At nanomole concentrations, S100b stimulates neurite outgrowth and enhances neuron survival; in contrast, micromole levels of extracellular S100b in vitro may have deleterious effects [1, 3]. NSE is a dimer of the cytoplasmic isoenzyme glycolytic enolase, localized in central and peripheral neurons, as well as in neuroendocrine cells [4, 16].
Our results showed that blood levels of the brain injury markers S100b and NSE during the first 2 days after TBI did not have a strong correlation with the severity of brain injury. Our data were supported by the research of Sedaghat and Notopoulos, who found that the S100b level correlated with CT scanning data in only 30% of cases [17]. Kleindienst and Ross reported that in 48% of children with mTBI without cognitive impairment, there was an increase in the serum level of S100b [18], which most likely indicated participation of this protein in adaptive processes developing in response to stress. We found that NSE and S100b levels increased immediately after injury regardless of the severity of TBI, but in cases with a favorable outcome, the levels of both markers decreased to normal within the first 3 days. The maximum S100b protein and NSE levels were observed in children with a lethal outcome of TBI (GOS 1), who had high levels of these proteins throughout the posttraumatic period.
The central excitotoxic roles of GluR and NO in hypoxia are known [10], but there have been very few clinical studies on these aspects of TBI pathogenesis [19,20,21]. Our determination of markers of GluR and their degradation products, together with NO metabolites and nitrotyrosine as a marker of protein nitrosation, was an attempt to assess the development of oxidative stress and the neuroimmunological response to hypoxia. NO has multiple effects and, in different concentrations, plays both protective and damaging roles. Similar dual effects of NO can be observed in the fact that with a small increase, NO can activate the immune system and reduce the excitotoxicity of Glu, but at high levels, NO suppresses the immune response, promotes protein nitrosation, and enhances the damaging effect of Glu [20, 22]. Tisdall et al. [21] showed that in cases of lethal TBI, NOx content in the brain extracellular fluid reached 150 μmol/L in the first 48 h. We also obtained data showing that children with a negative outcome of severe TBI have high levels of NOx and NT soon after the initial injury, which correlates with a decrease in adenosine triphosphate (ATP) content in lymphocytes [23]. At the same time, NO inhalation prevents secondary damage in TBI [24].
Posttraumatic brain injury may be based on immunological mechanisms that sometimes ameliorate the course of TBI and sometimes cause additional damage to brain tissue with development of edema [25, 26]. The appearance in the blood of antibodies to functionally important brain structures, including GluR, indicates their activation. In this case, the increase in the pool of these antibodies should be preceded by the appearance in the blood of the degradation products of these receptors (at the N-terminal site of GluR)—peptides. On the basis of the dual roles of different antibodies [27], it can be assumed that early appearance of antibodies to excitotoxic receptors can reduce their activity and weaken the development of a further cascade of damage. We noted that in cases of mTBI, there was a significant increase in the level of antibodies to NR2(NMDA), while the level of peptides of these receptors in the blood remained within normal limits. These data indicated the existence of mechanisms aimed at early protection of NMDA GluR from development of a cascade of excitotoxic damage. It is possible that with more severe brain damage in sTBI, this mechanism does not work and the level of antibodies to NR2(NMDA) does not increase at the onset of TBI. An adverse outcome of sTBI—especially death—was associated with the lowest level of antibodies to NR2(NMDA) on the first 2 days after the initial injury. The question arises: how can the body react so early to brain damage? Such an early response is most likely associated with activation of innate immunity. The detected significant increase in antibodies to NR2(NMDA) on the first 2 days after mTBI indicated rapid secretion of immunoglobulins by innate-like B-lymphocytes. Activation of such lymphocytes can produce tissue breakdown products—DAMPs (danger-associated molecular patterns)—as well as mediators of activated microglia cells, which, in turn, are stimulated by nitric oxide, the concentration of which increases with mTBI. We demonstrated such a stimulatory effect of subphysiological concentrations of NO in an experiment in which injection of the NO-generated agent NaNO2 to rats led to a rapid increase in the level of antibodies to GluR content in the blood as early as 1 h after administration, and the increase was significant after 24 h [28].
We found no increase in the level of antibodies to GluR1(AMPA), while the AMPA peptide level was found to be increased in 91% of children with mTBI. The development of methods for peptide determination and the establishment of the preferred localization of GluR in brain structures indicate a significant presence of NMDA GluR in neurovascular units and AMPA GluR in axonal structures [11]. A high level of AMPA peptides and the appearance of SBDP145 in the blood may be early signs of diffuse axonal injury in children with mTBI. As a rule, children with mTBI have a very short stay in the hospital but, as a result of underestimation of the severity of their condition, they may later have complications in the form of headaches and a decrease in mental abilities. The questions of the regulation of neuroimmune relationships are quite difficult to address, and we are not yet able to explain all of the data. However, we can assume that NO and its products are involved in the immune response and that nitrosative stress accompanies TBI [23]. We suggest that the opposite characters of the NR2(NMDA) antibody level on the first 2 days of mild and moderate versus severe TBI may be associated with an important mechanism aimed at protecting neurons from Glu excitotoxicity.
References
Hergenroeder GW, Redell JB, Moore AN, Dash PK (2008) Biomarkers in the clinical diagnosis and management of traumatic brain injury. Mol Diagn Ther 12:347–358
Beamont A, Gennarelli T (2006) CT prediction of contusion evolution after closed head injury: the role of pericontusional edema. Acta Neurochir Suppl 96:30–32
Mechtler LL, Dhadtri KK, Crutchfield KE (2014) Advanced neuroimaging of mild traumatic brain injury. Neurol Clin 32:31–58
Guzel E, Kemaloglu S, Ceviz A, Kaplan A (2008) Serum neuronspecific enolase as a predictor of short-term outcome and its correlation with Glasgow Coma Scale in traumatic brain injury. Neurosurg Rev 31:439–444
Algattas H, Jason H (2014) Traumatic brain injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int J Mol Sci 15:309–341
Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar A, Shah SB, Duckworth J, Head BP (2017) Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell Mol Neurobiol 37:571–585
Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26:1191–1201
Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK (2019) The immunological response to traumatic brain injury. J Neuroimmunol 332:112–125. https://doi.org/10.1016/j.jneuroim.2019.04.005
Arent AM, de Souza LF, Walz R, Dafre AL (2014) Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. Biomed Res Int 2014:723060. https://doi.org/10.1155/2014/723060
Reutov VP, Samosudova NV, Sorokina EG (2019) A model of glutamate neurotoxicity and mechanisms of the development of the typical pathological process. Biophysics 64:233–250. https://doi.org/10.1134/S0006350919020143
Dambinova SA (2012) Neurodegradomics: the source of biomarkers for mild traumatic brain injury. In: Dambinova SA, Hayes RL, Wang KW (eds) Biomarkers for TBI. RSC Publishing, London, pp 66–86
Dambinova SA, Bettermann K, Glynn T, Tews M, Olson D, Weissman JD, Sowell RL (2012) Diagnostic potential of the NMDA receptor peptide assay for acute ischemic stroke. PLoS One 7(7):e42362. https://doi.org/10.1371/journal.pone.0042362
Dambinova SA, Khounteev GA, Izykenova GA, Zavolokov IG, Ilyukhina AY, Skoromets AA (2003) Blood test detecting autoantibodies to N-methyl-D-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin Chem 49:1752–1762
Donato R (2001) S100: a multigenic family of calcium-modulated proteins of EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 33:637–668
Rothermundt M, Peters M, Prehn JHM, Arolt V (2003) S100b in brain damage and neurodegeneration. Microsc Res Tech 6:614–632
Berger R, Adelson P, Dulani T, Cassidy L, Kochanek P (2005) Serum neuron-specific enolase, S100b, and myelin basic protein concentrations after inflicted and noninflicted traumatic brain injury in children. J Neurosurg 103:61–68
Sedaghat F, Notopoulos A (2008) S100 protein family and its application in clinical practice. Hippokratia 12:198–204
Kleindienst A, Ross BM (2006) A critical analysis of the role of the neurotrophic protein S100b in acute brain injury. J Neurotrauma 23:1185–1200
Carpenter KLH, Timofeev I, Al-Rawi PG, Menon DK, Pickard JD, Hutchinson PJ (2008) Nitric oxide in acute brain injury: a pilot study of NOx concentrations in human brain microdialysates and their relationship with energy metabolism. Acta Neurochir Suppl 102:207–213
Sorokina EG, Semenova ZB, Bazarnaya NA, Meshcheryakov SV, Reutov VP, Goryunova AV, Pinelis VG, Granstrem OK, Roshal LM (2009) Autoantibodies to glutamate receptors and products of nitric oxide metabolism in serum in children in the acute phase of craniocerebral trauma. Neurosci Behav Physiol 39:329–334
Tisdall MM, Konrad R, Kitchen ND, Smith M, Petzold A (2013) The prognostic value of brain extracellular fluid nitric oxide. Metabolites after traumatic brain injury. Neurocrit Care 19:65–68
Bal-Price A, Brown GC (2001) Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 21:6480–6491
Zakirov RS, Sorokina EG, Karaseva OV, Semenova ZB, Petrichuk SV, Roshal LM, Pinelis VG (2015) Peripheral blood lymphocytes mitochondrial function in children with traumatic brain injury [in Russian]. Vestn Ross Akad Med Nauk 6:710–717. https://doi.org/10.15690/vramn568
Terpolilli NA, Kim S-W, Thal SC, Kuebler WM, Plesnila N (2013) Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab 33:311–318
Gannushkina IV (1974) Immunological aspects of trauma and vascular brain lesions [in Russian]. Meditsina, Moscow
Smith RM, Giannoudis PV (1998) Trauma and immune response. J R Soc Med 91:417–420
Archelos JJ, Hartung HP (2000) Pathogenic role of autoantibodies in neurological diseases. Trends Nerosci 23:317–327
Sorokina EG, Reutov VP, Granstrem OK, Fadyukova OE, Obrezchikova MN, Krushinsky AL, Kuzenkov VS, Koshelev VB, Dambinova SA, Pinelis VG (2003) A possible role of nitric oxide in damage of glutamate receptors in epilepsy. Vesti Nats Acad Navuk Belarusi Ser Med-Biol Nauk 1:18–22
Acknowledgments
The authors thank Professor G. A. Ignatieva for valuable tips in discussing the results of this work.
This work was supported by the Russian Ministry of Health (grant number AAA-A19-119012590190-6 injury).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors have no conflict of interest.
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sorokina, E.G. et al. (2021). Brain Biomarkers in Children After Mild and Severe Traumatic Brain Injury. In: Depreitere, B., Meyfroidt, G., Güiza, F. (eds) Intracranial Pressure and Neuromonitoring XVII. Acta Neurochirurgica Supplement, vol 131. Springer, Cham. https://doi.org/10.1007/978-3-030-59436-7_22
Download citation
DOI: https://doi.org/10.1007/978-3-030-59436-7_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-59435-0
Online ISBN: 978-3-030-59436-7
eBook Packages: MedicineMedicine (R0)