
Abstract
Myoepithelial tumors represent a family of lesions with variable terminology, based on anatomic location: pleomorphic adenoma in the salivary gland (where myoepithelioma was first described), benign mixed tumor in the skin, and myoepithelial tumor or parachordoma in the soft tissues.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Definition
Myoepithelial tumors represent a family of lesions with variable terminology, based on anatomic location: pleomorphic adenoma in the salivary gland (where myoepithelioma was first described), benign mixed tumor in the skin, and myoepithelial tumor or parachordoma in the soft tissues.
Myoepithelioma was included in the World Health Organization classification of soft tissue tumors in 2002 and classified as intermediate malignancy tumor of uncertain line of differentiation in 2013; in fact, in contrast to the salivary gland, primary myoepithelial tumors in the soft tissue, bone, and skin have no known normal cellular counterpart.
It is also known as myoepithelioma of soft tissue, ectomesenchymal chondromyxoid tumor, myoepithelial tumor, parachordoma, and myoepithelial carcinoma (the malignant variant).
Some authors believe that parachordoma (which should not be confused with extra-axial chordoma or chordoma periphericium) is a separate entity, although most investigators support the hypothesis that myoepithelioma and parachordoma represent morphological variants of a single tumor type.
Epidemiology and Predisposition
Myoepithelioma of soft tissues is a rare tumor occurring in a wide age range but mainly in young to middle-aged adults (median: 40 years), without gender predilection. Of note, myoepithelial carcinoma predominates in the pediatric population. The body sites most frequently involved are the limbs and limb girdles, trunk, and head and neck; rarely other sites (including bone and viscera) are affected.
Clinically, the lesion typically presents as a palpable painless mass (1–20 cm in diameter; mean: 5 cm) involving the subcutaneous tissue and less frequently deep soft tissues; sometimes the tumor develops primarily in the skin.
Pathology
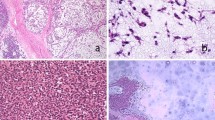
Macroscopically, most tumors are well circumscribed and nodular, but rarely they have infiltrative margins.
Microscopically, myoepithelioma shows a wide morphological spectrum, in analogy to its salivary counterpart. The tumor shows reticular or trabecular architecture with myxoid stroma and focal areas with nested or solid architecture and hyalinized or chondroid stroma that may predominate in some cases. Myoepithelioma is composed exclusively (or predominantly) of myoepithelial cells, as opposed to mixed tumors which also show ductal differentiation. Plasmacytoid “hyaline cell,” spindle, epithelioid, clear cell, and mixed tumor cell variants of myoepithelioma have been described.
Differential diagnosis may be needed with the following: extraskeletal myxoid chondrosarcoma(NR4A3 rearrangement); malignant mixed tumor (malignant cartilage or bone are present); neurothekeoma (nested architecture, sclerotic dermal collagen; negative for S100, EMA, and cytokeratins); epithelioid benign fibrous histiocytoma (plump and often binucleate epithelioid cells; negative for cytokeratins and S100); epithelioid sarcoma (it develops in distal extremities of young adults; S100 and GFAP negative); and Spitz nevus (large epithelioid melanocytes, junctional component, HMB45 and S100 positive; EMA and cytokeratin negative).
Biomarkers
More than 90% of cases stain positive for cytokeratins, S100, and calponin. Desmin, CD34, and brachyury are consistently negative.
Chromosomal translocations leading to the formation of EWSR1-basedFootnote 1 gene fusions have been reported in about 45% of cases: EWSR1-POU5F1 fusion gene (from t(6;22)(p21;q12)), EWSR1-ZNF444 fusion gene (from t(19;22)(q13;q12)), EWSR1-PBX1 fusion gene (from t(1;22)(q23;q12)), EWSR1-PBX3 fusion gene (from t(9;22)(q33;q12)), and EWSR1-KLF17 fusion gene (from t(1;22)(p34;q12)).
In mixed tumors, PLAG1 is frequently expressed, which correlates with PLAG1 gene rearrangement.
Prognosis
Myoepithelial tumors of soft tissue are classified as benign (myoepithelioma and mixed tumor/chondroid syringoma) and malignant (myoepithelial carcinoma).
Most myoepitheliomas are benign tumors. The only reliable criterion for malignancy is cytologic atypia (moderate to severe nuclear atypia), which is in contrast to salivary tumors for which malignancy is defined by the presence of capsular invasion and infiltrative growth. Benign tumors are referred to as “myoepitheliomas,” which overall lack cytologic atypia (if present, it is at most mild). Necrosis and increased mitotic activity alone are not predictive of malignant behavior.
Histologically benign tumors have been associated with a 20% local recurrence rate and rarely metastasize. In contrast, myoepithelial carcinoma recur and metastasize in about 50% of cases (mainly to lungs, lymph nodes, bone, and soft tissues).
Therapy
The primary treatment modality of soft tissue myoepithelioma is complete excision. Some data support the use of perioperative radiation therapy in the management of myoepithelial carcinoma. Metastatic disease is poorly to moderately sensitive to systemic chemotherapy (e.g., doxorubicin).
Notes
- 1.
EWSR1 (Ewing Sarcoma Breakpoint Region 1) encodes a multifunctional protein that is involved in various cellular processes, including gene expression, cell signaling, and RNA processing and transport. EWSR1 is a “promiscuous” gene because it can fuse with different partner genes in phenotypically similar neoplasms or with the same genes in morphologically and behaviorally different tumors. In fact, EWSR1-based chimeric genes can be found not only in Ewing and Ewing-like sarcomas but also in other tumors such as angiomatoid fibrous histiocytoma, clear cell sarcoma, low-grade fibromyxoid sarcoma, sclerosing epithelioid fibrosarcoma, hemangioma of bone, desmoplastic small round cell tumor, extraskeletal myxoid chondrosarcoma, myoepithelial tumor of soft tissue, and myxoid liposarcoma.
It must be underscored that fluorescence in situ hybridization (FISH) analysis has a significant risk of false-negative results, making next-generation sequencing (NGS)-based diagnostic tools more sensitive for detecting EWSR1 rearrangements.
Suggested Readings
Chamberlain (2019) Adult soft tissue myoepithelial carcinoma: treatment outcomes and efficacy of chemotherapy. Med Oncol 37(2):13
Fletcher (2020) WHO classification of tumours of soft tissue and bone (5th edition)
Jo (2020) Soft tissue special issue: myoepithelial neoplasms of soft tissue: an updated review with emphasis on diagnostic considerations in the head and neck. Head Neck Pathol 14(1):121–131
Koyama (2020) Metachronous pancreatic and thyroid metastases from primary soft-tissue myoepithelioma in the clavicular region: a case report of a long-term survivor. Am J Case Rep 21:e920702
Kravtsov (2017) Myoepithelioma of soft tissue: a cytological-pathological correlation with literature review. Ann Diagn Pathol 27:14–17
Rastrelli (2019) Myoepithelioma of the soft tissue: a systematic review of clinical reports. Eur J Surg Oncol 45(9):1520–1526
Verma (2017) Myoepithelial tumor of soft tissue and bone: a current perspective. Histol Histopathol 32(9):861–877
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2021 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mocellin, S. (2021). Myoepithelioma. In: Soft Tissue Tumors . Springer, Cham. https://doi.org/10.1007/978-3-030-58710-9_175
Download citation
DOI: https://doi.org/10.1007/978-3-030-58710-9_175
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-58709-3
Online ISBN: 978-3-030-58710-9
eBook Packages: MedicineMedicine (R0)