
Abstract
The thyroid is the location of neuroendocrine C cells that produce calcitonin. The most common thyroid neuroendocrine neoplasm is medullary thyroid carcinoma (MTC) that is composed of C cells. Rare other thyroid neuroendocrine neoplasms include mixed neuroendocrine and non-neuroendocrine neoplasms (MiNENs) that represent a composite MTC and thyroid follicular neoplasm, usually papillary thyroid carcinoma, as well as occasional paragangliomas, intrathyroidal parathyroid tumors, NENs originating from intrathyroidal thymic remnants and metastatic neuroendocrine neoplasms.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Historical Background
The history of the neuroendocrine component of the thyroid dates back to 1894 when Karl Hürthle identified clear cells within the basement membrane of follicles in the thyroid [1]; unfortunately this has been long forgotten, and today many pathologists mistakenly call oncocytes “Hürthle cells.” The cells that Hürthle identified became known as parafollicular or clear cells (C cells) and were largely discounted for more than half a century. Indeed, in the 1953 AFIP Fascicle on Tumors of the Thyroid Gland, there is no mention of these cells or their tumors [2]. However, in 1961 there was a report of an unusual tumor with distinctive morphology [3], and 8 years later Hazard coined the term “medullary” for these solid tumors [4].
The hormone produced by C cells, calcitonin, was purified in 1962 by Copp and Cheney at the University of British Columbia [5]; they thought it was of parathyroid origin and named it for its role in maintaining calcium levels. In 1964 it became clear that calcitonin was secreted by the thyroid and by the parafollicular C cells of Hürthle. In 1966 William proposed that medullary thyroid carcinoma was derived from these calcitonin-producing C cells [6].
Thyroid C cells are prototypic neuroendocrine cells and were thought to be derived from the neural crest [7] but subsequently were shown to be epithelial neuroendocrine cells and, like others in the respiratory and gastroenteropancreatic tract, derive from the endoderm [8, 9]. They produce calcitonin as their main hormone product but also produce calcitonin gene-related peptide (CGRP), somatostatin, gastrin-releasing peptide (GRP), serotonin, and thyrotropin-releasing hormone as well as being a rich source of carcinoembryonic antigen (CEA).
The association of thyroid C cell pathology with pheochromocytoma in a familial disorder was recognized by Williams in 1965 [10]. This description was classified as multiple endocrine neoplasia (MEN) type 2 syndrome. The characterization of this syndrome underwent multiple changes with division into types 2A and 2B or types 2 and 3, but with the recognition that this disease is due to mutations in a single gene, RET, that encodes a tyrosine kinase involved in the migration of neural and neuroendocrine cells [11], the classification has become more complex. Known as MEN2, there are several variants associated with mutations that alter conformation of the molecule in the extracellular and transmembrane domain, all classified as MEN2A, and a more aggressive variant associated with activation of the kinase known as MEN2B [12].
Traditionally diagnosticians have considered medullary thyroid carcinoma (MTC) to be the only NEN of the thyroid gland; however, there is a morphological spectrum of NENs that can be seen in this gland. Thyroid NENs include the following entities: (i) MTC that originates from parafollicular C cells, (ii) mixed neuroendocrine and non-neuroendocrine neoplasms (MiNENs) that often manifest as a composite MTC and papillary thyroid carcinoma, (iii) paraganglioma that originates from dispersed microscopic elements of the laryngeal paraganglia (see Chapter 12), (iv) NENs originating from intrathyroidal parathyroid gland (see Chapter 8), (v) NENs originating from intrathyroidal thymic remnants (i.e., intrathyroidal thymic NENs) (see Chapter 9), and (vi) metastatic neuroendocrine neoplasms. From a patient management perspective, it is important to be aware of these various differential diagnoses and be able to distinguish these neoplasms given their distinct clinicopathologic characteristics.
Epidemiology
Medullary thyroid carcinoma has traditionally been thought to represent about 5% of all thyroid carcinomas [13] and in some series up to 10%, but more recent data suggest that a more accurate number is 1–2% [12]. Despite this low incidence, it is responsible for more than 13% of thyroid cancer-related deaths [12, 14]. Familial syndromes are responsible for a significant proportion of these; in earlier studies, 30–40% were considered to be familial; however more recent studies show a lower incidence of 25–30% suggesting that screening and prophylactic thyroidectomy is causing this proportion to decrease.
Tumor Classification and Morphology
Medullary thyroid carcinoma has characteristic neuroendocrine histologic and cytologic features that should make it an obvious diagnosis in a gland that is otherwise not neuroendocrine. Despite its unique features, it is often misdiagnosed [15, 16], especially on cytology where only about half of cases of this entity are accurately identified [17, 18].
The morphology of medullary carcinoma includes a spectrum of architecture and cytology [15]. Most commonly, these tumors have a typical neuroendocrine pattern of solid nests in a vascular stroma; they are usually infiltrative but can sometimes be well delineated (Fig. 7.1) [19]. Rarely, tumors can have complete or partial encapsulation, but most tumors lack a true capsule as seen in a subset of thyroid follicular epithelial-derived neoplasms. Other tumors can display a nested “zellballen” pattern that can simulate paragangliomas, and such tumors are referred to as paraganglioma-like variants of this disease [20]. They frequently have palisading at the periphery of the solid nests, and occasionally central degeneration results in a pseudopapillary growth pattern that mimics papillary thyroid carcinoma [21, 22], and they can even be cystic [23, 24]. The tumor usually infiltrates around adjacent follicles, and these tumors can sometimes be mistaken for follicular carcinoma; true glandular variants also occur.

Patterns of growth of medullary thyroid carcinoma. These tumors are usually infiltrative and grow around the follicles of the nontumorous thyroid (top), but occasionally they are well-delineated and expansile lesions that mimic thyroid follicular lesions (bottom)
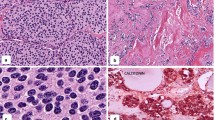
The tumor cells are usually round, polyhedral, or spindle-shaped but they may also be oncocytic or have clear cytoplasm [25, 26] (Fig. 7.2). Absence of distinct cell membranes and discohesive or loosely cohesive appearance with basophilic or amphophilic cytoplasmic granularity are distinctive features. The nuclei are usually bland with a “salt and pepper” appearance, but in some tumors, they develop grooves, resembling papillary thyroid carcinoma or hyalinizing trabecular tumor [27]. Medullary thyroid carcinoma is usually a relatively well-differentiated neuroendocrine tumor, but there is a small cell- or neuroblastoma-like variant that can be mistaken for small blue round cell tumors including but not limited to hematologic malignancy or neuroblastoma [28] and a giant cell variant as well [29]. Pigmented melanin-producing cases occur and rare tumors have an angiosarcoma-like morphology [15, 16, 30, 31].

Architecture and cytology of medullary thyroid carcinoma. The classical variant of this tumor is composed of small nests of discohesive cells in a stroma with amyloid and may have focal calcification (top left). Tumors with less amyloid usually have a more spindle cell morphology (top right). Some tumors are composed of epithelioid cells that appear to be more cohesive but lack the well-defined cell borders of follicular epithelial cells (middle left). When they trap nontumorous follicles, they can be mistaken for follicular cell-derived lesions, but the tumor cells have distinctive morphology including giant cell formation (middle right). Some medullary thyroid carcinomas are composed of oncocytic cells (bottom left) that should be characterized appropriately using immunohistochemistry so that they are not misdiagnosed as “Hürthle cell carcinoma.” The small cell variant of medullary thyroid carcinoma (bottom right) is a more aggressive and less well-differentiated form of this disease
A distinctive feature of this tumor type is the formation of amyloid, beta-pleated sheets of a preprocalcitonin molecule (Fig. 7.3). Amyloid can be identified by its characteristic apple-green birefringence with polarized light that is enhanced by Congo Red staining but can be seen on unstained sections and on tissue stained with H&E. Amyloid is present in just over half of medullary thyroid carcinomas, and it may be only very focal, limited to intracytoplasmic globules. Because of this, it is not a reliable marker of this tumor type. Moreover, amyloid may also be found in benign amyloid goiter and associated with other tumors [32,33,34,35,36].

Amyloid in medullary thyroid carcinoma. The presence of amyloid is identified in approximately half of these tumors. It is usually abundant (top left) but may be scattered and scant (top right); it may be found only within tumor cells that accumulate the material and rupture (top right, arrows). It stains with Congo Red (bottom left), but this stain is not required to elicit the apple-green birefringence with polarized light that is characteristic of amyloid. The amyloid material is composed of preprocalcitonin molecules and stain for calcitonin (bottom right)
Calcification is rare in medullary thyroid carcinomas, and even more rare is the identification of psammoma bodies that have been reported in this tumor type.
Immunohistochemistry is required to confirm the diagnosis (Fig. 7.4). These tumors, as members of the family of neuroendocrine tumors, express synaptophysin and chromogranins as well as the transcription factor regulating neuroendocrine differentiation insulinoma-associated protein 1 (INSM1). They are epithelial NENs and therefore express keratins as seen in other NETs of endodermal origin. Some express TTF1 as detected by the SPT24 antibody; however about one quarter of these neoplasms can be negative for TTF1. The diagnosis must entail identification of the biomarkers that are often considered specific to this entity: calcitonin, CGRP, and carcinoembryonic antigen (CEA) that should be stained using a monoclonal antibody. However, there are several pitfalls that diagnosticians should recognize when using these biomarkers.

Immunohistochemical profile of medullary thyroid carcinoma. These tumors stain strongly for chromogranin (top left) and may have variable nuclear reactivity for TTF1 (top middle). They usually have cytoplasmic positivity for calcitonin (top right), and they stain diffusely for carcinoembryonic antigen (CEA) (bottom left); the importance of CEA cannot be overemphasized, as sometimes aggressive tumors lose expression of calcitonin (bottom middle), while CEA is retained as a valuable tumor marker. Some tumors express hormones ectopically, most commonly ACTH (bottom right) as in this tumor that caused ectopic Cushing syndrome
Calcitonin and CGRP are considered by many to be the specific biomarker of MTC; however a small fraction of MTCs do not express calcitonin and/or CGRP, and more importantly, the expression of these hormones is not specific to this disease; several other neuroendocrine tumors, including parathyroid neoplasms, thymic neuroendocrine neoplasms, head and neck NENs, pancreatic NETs, and paragangliomas, can also express these hormones [20, 37,38,39,40,41]. In addition, tyrosine hydroxylase, which is often used to confirm the paraganglioma diagnosis in a cytokeratin- and transcription factor-negative NEN, can also be expressed in medullary thyroid carcinomas [20, 42]. However, GATA3, which is also expressed in paragangliomas and parathyroid and pituitary NETs, is typically negative in medullary thyroid carcinomas. Since some MTCs can display overlapping features with follicular epithelial neoplasms and these tumors can be positive for TTF1, it is critical to use appropriate tools to distinguish these entities. PAX8 expression in medullary thyroid carcinomas occurs in an antibody-dependent manner that is likely due to cross-reactivity; polyclonal PAX8 antisera and some N-terminus-specific PAX8 monoclonal antibodies can be positive in MTCs, whereas C-terminus-specific monoclonal PAX8 antibodies (clones BC12 and PAX8R1) and N-terminus-specific monoclonal PAX8 (clone MRQ50) are negative in these neoplasms [43].
The importance of monoclonal CEA immunohistochemistry cannot be overemphasized; as discussed earlier other NETs can express calcitonin and/or CGRP and be variably positive for CEA. However, diffuse strong reactivity using a monoclonal antibody to CEA is characteristic of medullary thyroid carcinoma, and while calcitonin can be reduced as tumors dedifferentiate, CEA is typically retained. For this reason, circulating CEA is of clinical value in surveillance, and a reduction in calcitonin levels with persistent or increasing CEA is a feature of tumor progression and dedifferentiation [44, 45]. Other peptides can also be expressed, including somatostatin and some that can give rise to clinical syndromes, for example, derivatives of proopiomelanocortin including ACTH that can cause ectopic Cushing syndrome and serotonin that can be a cause of carcinoid syndrome that can be mimicked by calcitonin. Other unusual hormonal products include glucagon, gastrin, cholecystokinin, vasoactive intestinal peptide (VIP), bombesin, and α-hCG [13, 46,47,48].
Like other neuroendocrine tumors, medullary thyroid carcinomas should have a formal Ki67 labeling index [49], but unlike other neuroendocrine tumors, there is no classification scheme for grading. Prognostic and predictive markers have been identified but are not in routine clinical use [50,51,52]. The poorly differentiated forms such as small cell types tend to be more aggressive [53], and significant tumor necrosis is a feature of more aggressive biology (Fig. 7.5). Angioinvasion, defined as the presence of tumor cells within vascular channels associated with thrombus (Fig. 7.5), is an important predictor of recurrence and distant metastasis [50]. Expression of somatostatin receptors (SSTRs) may be of value in determining therapy including administration of somatostatin analogues to inhibit hormone secretion and restrain growth and somatostatin-based peptide receptor radiotherapy (PRRT) in the treatment of unresectable disease [54,55,56,57]. Somatostatin-labeled imaging is also useful to identify metastatic deposits [58, 59].

Prognostic features in medullary thyroid carcinoma. The presence of extensive tumor necrosis (top) and angioinvasion, defined by tumor cells within vascular channels associated with thrombus (bottom), are adverse features seen within the primary tumor
The differential diagnosis includes intrathyroidal paraganglioma that can be identified by nuclear reactivity for GATA3, cytoplasmic staining for tyrosine hydroxylase, and lack of keratin and monoclonal CEA reactivity. Since tyrosine hydroxylase reactivity can sometimes be focal or absent depending on functional status of a paraganglioma, the use of a panel approach combining GATA3, TTF1, keratins, and monoclonal CEA should be used in the diagnostic workup.
The rare intrathyroidal thymic NEN can pose diagnostic challenges [41]. These tumors are often thought not to express diffuse monoclonal CEA, but can be positive for CGRP and calcitonin.
Medullary thyroid carcinomas with clear cell change and/or oncocytic change can simulate an intrathyroidal parathyroid neoplasm. Parathyroid tumors express GATA3, GCM2, keratins, and parathyroid hormone. Rarely, calcitonin and CGRP can be expressed in parathyroid neoplasms [40] (references); however, the parathyroid-specific transcription factors and lack of monoclonal CEA expression distinguish parathyroid origin.
Mixed follicular-C cell tumors constitute the only well-recognized mixed neuroendocrine and non-neuroendocrine tumor (MiNEN) of the thyroid gland. These unusual neoplasms may be composite or collision tumors [60,61,62,63,64] or the exceptional monomorphous proliferations with dual differentiation [65,66,67]. It is important to recognize that most medullary thyroid carcinomas have trapped nontumorous thyroid follicles (Fig. 7.6) and the follicular epithelium may show reactive atypia, but this does not qualify as a composite tumor. There must be clear evidence of two malignant components, and when in doubt, rely only on the presence of metastasis of both components to a regional node (Fig. 7.6) [63, 64, 68]. The application of biomarkers of malignancy of follicular epithelial neoplasms (e.g., HBME-1, galectin-3 NRASQ61R- or BRAFV600E-mutation-specific antibodies) [69] can assist in proving malignancy of the follicular component, but this must be coupled with the clear knowledge that the medullary thyroid carcinoma may express some of these markers.

Trapped thyroid or composite tumor? The presence of thyroid follicles within a medullary thyroid carcinoma (top left) does not indicate the presence of a composite tumor, since these lesions grow by surrounding the adjacent nontumorous gland. The follicular cells may even exhibit nuclear atypia that resembles papillary thyroid carcinoma but this is usually reactive. Careful examination will confirm the presence of cells that are negative for calcitonin (top middle) and monoclonal CEA and positive for thyroglobulin (top right) in such cases, confirming that these are two separate populations of cells. However, when a lesion metastasizes to a lymph node with both C cell and follicular cell components (bottom), that confirms that the tumor was indeed a composite lesion
The familial nature of medullary thyroid carcinoma can be detected by careful pathologic examination to identify C cell hyperplasia to neoplasia that may be associated with progression to multifocal primary microtumors (medullary microcarcinomas ) (Fig. 7.7) [70, 71]. Normal C cells are present as scattered single cells at the junction of the upper third and lower two thirds of the lateral thyroid lobes. C cell hyperplasia, an increase in the population of C cells, has two clinicopathologic variants: (i) reactive or secondary and (ii) precursor or primary forms. The criteria used to define C cell hyperplasia are variable and include an increased number of C cells with (i) more than 7 cells per cluster leading to 50 C cells per low-power field, (ii) complete follicles surrounded by C cells, or (iii) C cells outside the normal location, including in the lower pole of the thyroid lobes and isthmus [72]. The two cytomorphologic variants are linear and nodular hyperplasia; the former is usually associated with reactive (secondary) to other lesions, whereas nodular C cell hyperplasia is a feature of germline RET mutation and can progress to microtumors (medullary microcarcinomas) and clinical medullary thyroid carcinoma. The potential for metastasis is thought to be achieved once C cells invade the basement membrane of a follicle. The distinction of medullary microcarcinoma from nodular C cell hyperplasia can be challenging. The identification of stromal desmoplasia and single cell infiltration can help in this distinction. Collagen type IV immunohistochemistry can also facilitate the assessment of basement membrane breakdown in microinvasive tumors [69, 72].

C cell hyperplasia and micromedullary thyroid carcinoma. In patients with germline RET mutations, these precursor lesions develop multifocally throughout the thyroid. C cell hyperplasia is difficult to see on routine H&E staining, but immunohistochemistry for calcitonin (shown, top) or monoclonal CEA will identify the increased number of C cells forming clusters and completely surrounding follicles. With progression, they form small tumors that are visible on H&E (bottom left) and stain for calcitonin (bottom right) and monoclonal CEA (not shown)
It is important to note that C cell hyperplasia cannot be assessed in the nontumorous tissue surrounding a medullary thyroid carcinoma, since this may represent invasive tumor; therefore, this analysis should be carried out on the lobe opposite a tumor. Other causes of C cell hyperplasia including chronic hypercalcemia, thyroiditis, and reaction to nodular follicular lesions [73,74,75,76] as well as PTEN hamartoma tumor syndrome (PHTS) usually are characterized by linear C cell hyperplasia [77] that does not appear to progress to malignancy. Interestingly, in animals, antidiabetic incretins (glucagon-like peptide-1 analogues such as exenatide, liraglutide, and taspoglutide) have been implicated as causing C cell hyperplasia and MTC [78], but the data in humans have not supported this finding.
Since RET and RAS mutations are mutually exclusive in MTC, immunolocalization of NRASQ61R using the mutation-specific SP174 antibody [79] can assist in screening for sporadic MTCs (see pathogenesis below).
Molecular Pathogenesis
A significant proportion of MTCs are hereditary [12] as integral components of MEN2 syndrome. In MEN2A, they are associated with pheochromocytomas and parathyroid proliferations. In MEN2B, the thyroid, adrenal, and parathyroid proliferations are also associated with mucosal ganglioneuromas and a Marfanoid habitus. Some patients with MEN2A also have cutaneous lichen amyloidosis (CLA) and/or Hirschsprung’s disease [12]. The syndrome formerly known as “familial medullary thyroid carcinoma” (FMTC) is now classified as a variant of MEN2A syndrome that rarely is associated with parathyroid disease or pheochromocytoma, but screening for these other entities is still warranted as it may occur [12]. These syndromes are all caused by germline mutations in the RET proto-oncogene. Familial transmission of MEN2A is associated with activating mutations in the ligand-binding regions of the extracellular domain or in the transmembrane or cytoplasmic domains. The most common mutations are in exon 10, codons 609, 611, 618, and 620; exon 11, codons 630 and 634; and exons 8, 13, 14, 15, and 16. In contrast, MEN2B is not usually familial, but rather is due to sporadic (de novo) germline mutation, most frequently in codon 918 of exon 16 and occasionally in codon 883 in exon 15 [12].
The identification of germline MET mutations in two siblings with wild-type RET harboring inherited medullary thyroid carcinomas has expanded germline correlates of this disease and can open potential use of MET-inhibitor therapies in affected patients [80].
The management of patients with this disorder includes assessment of relatives, and members of known kindreds should undergo genetic screening early in life. As this represents a unique situation of inheritance of an activated oncogene (unlike most familial cancer syndromes that involve a mutant tumor suppressor requiring a second hit), affected individuals have an almost 100% chance of developing medullary thyroid carcinoma. For this reason, screening is critically important and affected individuals should undergo prophylactic thyroidectomy. The age at which this procedure is undertaken should be determined by the specific mutation and family history; however there is also occasional “genetic anticipation” which can cause earlier onset of tumors in following generations [12, 81, 82].
Sporadic medullary carcinomas may harbor mutations of RET, usually in codon 918 encoding the cytoplasmic tyrosine kinase domain, providing a target for therapy. The majority of sporadic tumors that lack RET mutations harbor RAS mutations, many of which can be identified using an immunohistochemical assay for mutant NRASQ61R [79]. Rare tumors have been reported with a RET fusion [83,84,85], ALK fusion [86], sequence variants of NTRK1 [87], BRAF mutations or fusions [88, 89], telomerase activation [90], and microRNA abnormalities [91].
Prognosis
The prognosis of patients with medullary thyroid carcinoma varies with a number of parameters including age at diagnosis and tumor stage including extrathyroidal extension, lymph node status, and distant metastases [92].
Surgical resection is the only hope for cure of this disease, and patients with the diagnosis of MTC should undergo total thyroidectomy with central-compartment lymph node dissection if there is no evidence of disseminated disease biochemically and on imaging. There is a significant role for lateral neck dissection if there is any evidence of involved cervical lymph nodes or if the patient’s calcitonin level is >200 ng/L [12]. Those with evidence of local residual disease after surgery or high-risk findings on pathology are thought to benefit from postoperative external beam radiation therapy (EBRT) to the neck.
Distant metastasis requires a tailored approach to therapy. Surgical resection has been used for solitary metastasis to the lung, brain, or liver. Radiofrequency ablation is recommended for hepatic metastases. Metastatic disease to brain or vertebral lesions that result in spinal cord compression may require glucocorticoid therapy in addition to surgical decompression and/or EBRT. Systemic therapy with somatostatin analogues is used to restrain tumor growth and alleviate symptoms of hormone excess. The tyrosine kinase inhibitors vandetanib or cabozantinib can provide a significant increase in progression-free survival as shown in prospective randomized double-blind clinical trials [12] but may have adverse effects. The use of a specific RET inhibitor LOXO-292 is currently under investigation (libretto trial, NCT03157128 at https://ClinicalTrials.gov/) and BLU-667 (arrow trial, NCT03037385). The role of somatostatin-based peptide receptor radiotherapy (PRRT) offers promise for the treatment of unresectable disease [54,55,56,57].
Symptomatic relief of diarrhea induced by calcitonin can be obtained with anti-motility agents such as loperamide or codeine, and the pain from bone metastases can be treated with denosumab or bisphosphonates. Patients with ectopic hormone excess such as Cushing syndrome due to tumor production of ACTH and/or CRH should be treated with medical therapies to inhibit glucocorticoids (e.g., ketoconazole, mifepristone, aminoglutethimide, metyrapone, or mitotane) or may be helped by bilateral adrenalectomy.
References
Hurthle K. Beitrage zur Kenntiss der Secretionsvorgangs in der Schilddruse. Arch Gesamte Physiol. 1894;56:1–44.
Warren S, Meissner WA. Tumors of the thyroid gland. Atlas of tumor pathology, series 1, fascicle 14. Washington, D.C.: Armed Forces Institute of Pathology; 1953.
Horn RC. Carcinoma of the thyroid. Description of a distinctive morphologic variant and report of 7 cases. Cancer. 1951;4:697–707.
Hazard JB, Hawk WA, Crile G Jr. Medullary (solid) carcinoma of the thyroid; a clinicopathologic entity. J Clin Endocrinol Metab. 1959;19(1):152–61.
Copp DH, Cheney B. Calcitonin-a hormone from the parathyroid which lowers the calcium-level of the blood. Nature. 1962;193:381–2.
Williams ED. Histogenesis of medullary carcinoma of the thyroid. J Clin Pathol. 1966;19:114–8.
Le DN, Fontaine J, Le LC. New studies on the neural crest origin of the avian ultimobranchial glandular cells--interspecific combinations and cytochemical characterization of C cells based on the uptake of biogenic amine precursors. Histochemistry. 1974;38(4):297–305.
Johansson E, Andersson L, Ornros J, et al. Revising the embryonic origin of thyroid C cells in mice and humans. Development. 2015;142(20):3519–28.
Kameda Y. Cellular and molecular events on the development of mammalian thyroid C cells. Dev Dyn. 2016;245(3):323–41.
Williams ED. A review of 17 cases of carcinoma of the thyroid and phaeochromocytoma. J Clin Pathol. 1965;18:288–92.
Mulligan LM, Kwok JBJ, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–60.
Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567–610.
Rosai J, Carcangiu ML, DeLellis RA. Tumors of the thyroid gland. Atlas of tumor pathology, third series, fascicle 5. Armed Forces Institute of Pathology: Washington, D.C; 1992.
Jimenez C, Hu MI, Gagel RF. Management of medullary thyroid carcinoma. Endocrinol Metab Clin N Am. 2008;37(2):481–96.
DeLellis RA, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of Tumours of endocrine organs. Lyons: IARC Press; 2004.
Baloch ZW, LiVolsi VA. C-cells and their associated lesions and conditions: a pathologists perspective. Turk Patoloji Derg. 2015;31(Suppl 1):60–79.
Trimboli P, Treglia G, Guidobaldi L, et al. Detection rate of FNA cytology in medullary thyroid carcinoma: a meta-analysis. Clin Endocrinol. 2015;82(2):280–5.
Essig GF Jr, Porter K, Schneider D, et al. Fine needle aspiration and medullary thyroid carcinoma: the risk of inadequate preoperative evaluation and initial surgery when relying upon FNAB cytology alone. Endocr Pract. 2013;19(6):920–7.
Driman D, Murray D, Kovacs K, Stefaneanu L, Higgins HP. Encapsulated medullary carcinoma of the thyroid. A morphologic study including immunohistochemistry, electron microscopy flow cytometry, and in situ hybridization. Am J Surg Pathol. 1991;15:1089–95.
Mete O, Essa A, Bramdev A, Govender N, Chetty R. MEN2 syndrome-related medullary thyroid carcinoma with focal tyrosine hydroxylase expression: does it represent a hybrid cellular phenotype or functional state of tumor cells? Endocr Pathol. 2017;28(4):362–6.
Harach HR, Williams ED. Glandular (tubular and follicular) variants of medullary carcinoma of the thyroid. Histopathology. 1983;7:83–97.
Kawanishi N, Norimatsu Y, Ohsaki H, et al. Diagnosis of pseudopapillary variant of medullary thyroid carcinoma by fine-needle aspiration cytology. Diagn Cytopathol. 2014;42(9):823–6.
Ozkara SK, Gurbuz Y, Muezzinoglu B, Yumbal Z. Encapsulated cystic papillary variant of medullary carcinoma of thyroid gland. Endocr Pathol. 2002;13(2):167–71.
Hyrcza MD, Winer D, Mete O. Images in endocrine pathology: papillary variant of medullary thyroid carcinoma with cystic change. Endocr Pathol. 2015;26(1):87–9.
Asa SL. My approach to oncocytic tumours of the thyroid. J Clin Pathol. 2004;57(3):225–32.
Mete O, Asa SL. Oncocytes, oxyphils, Hurthle, and Askanazy cells: morphological and molecular features of oncocytic thyroid nodules. Endocr Pathol. 2010;21(1):16–24.
Santosh KV, Raychaudhuri S, Subramanya H, Naveen Kumar BJ. Cytology of hyalinising trabecular adenoma-like variant of medullary thyroid carcinoma. J Cancer Res Ther. 2011;7(2):189–91.
Bhat V, Shariff S, Reddy RA. Extramedullary plasmacytoma of thyroid – a mimicker of medullary carcinoma at fine needle aspiration cytology: a case report. J Cytol. 2014;31(1):53–6.
Rekhi B, Kane SV, D’Cruz A. Cytomorphology of anaplastic giant cell type of medullary thyroid carcinoma–a diagnostic dilemma in an elderly female: a case report. Diagn Cytopathol. 2008;36(2):136–8.
Ikeda T, Satoh M, Azuma K, Sawada N, Mori M. Medullary thyroid carcinoma with a paraganglioma-like pattern and melanin production: a case report with ultrastructural and immunohistochemical studies. Arch Pathol Lab Med. 1998;122(6):555–8.
Laforga JB, Aranda FI. Pseudoangiosarcomatous features in medullary thyroid carcinoma spindle-cell variant. Report of a case studied by FNA and immunohistochemistry. Diagn Cytopathol. 2007;35(7):424–8.
Valenta LJ, Michel-Bechet M, Mattson JC, Singer FR. Microfollicular thyroid carcinoma with amyloid rich stroma, resembling the medullary carcinoma of the thyroid (MCT). Cancer. 1977;39:1573–86.
Polliack A, Freund U. Mixed papillary and follicular carcinoma of the thyroid gland with stromal amyloid. Am J Clin Pathol. 1970;53:592–5.
Di CV, Garzi A, Petruzziello F, et al. Nodular goiter with amyloid deposition in an elderly patient: fine-needle cytology diagnosis and review of the literature. BMC Surg. 2013;13 Suppl 2:S43.
Nobuoka Y, Hirokawa M, Kuma S, et al. Cytologic findings and differential diagnoses of primary thyroid MALT lymphoma with striking plasma cell differentiation and amyloid deposition. Diagn Cytopathol. 2014;42(1):73–7.
Wang AR, Baloch ZW, Montone KT. Amyloid goiter in a patient with progressive thyromegaly. Endocr Pathol. 2016;27(1):76–8.
Asa SL, Kovacs K, Killinger DW, Marcon N, Platts M. Pancreatic islet cell carcinoma producing gastrin, ACTH, α-endorphin, somatostatin and calcitonin. Am J Gastroenterol. 1980;74:30–5.
Nozieres C, Chardon L, Goichot B, et al. Neuroendocrine tumors producing calcitonin: characteristics, prognosis and potential interest of calcitonin monitoring during follow-up. Eur J Endocrinol. 2016;174(3):335–41.
Uccella S, Blank A, Maragliano R, Sessa F, Perren A, La RS. Calcitonin-producing neuroendocrine neoplasms of the pancreas: clinicopathological study of 25 cases and review of the literature. Endocr Pathol. 2017;28(4):351–61.
Hodgson A, Pakbaz S, Tayyari F, Young JEM, Mete O. Diagnostic pitfall: parathyroid carcinoma expands the spectrum of calcitonin and calcitonin gene-related peptide expressing neuroendocrine neoplasms. Endocr Pathol. 2019;30(2):168–72.
Szybowska M, Mete O, Weber E, Silver J, Kim RH. Neuroendocrine neoplasms associated with germline pathogenic variants in the homologous recombination pathway. Endocr Pathol. 2019;30(3):237–45.
Zabel M, Grzeszkowiak J. Characterisation of thyroid medullary carcinoma TT cell line. Histol Histopathol. 1997;12(1):283–9.
Gucer H, Caliskan S, Kefeli M, Mete O. Do you know the details of your PAX8 antibody? Monoclonal PAX8 (MRQ-50) is not expressed in a series of 45 medullary thyroid carcinomas. Endocr Pathol. 2020;31(1):33–8.
Nelkin BD, de Bustros AC, Mabry M, Baylin SB. The molecular biology of medullary thyroid carcinoma. A model for cancer development and progression. JAMA. 1989;261:3130–5.
Mendelsohn G, Wells SA, Baylin SB. Relationship of tissue carcinoembryonic antigen and calcitonin to tumor virulence in medullary thyroid carcinoma. An immunohistochemical study in early, localized and virulent disseminated stages of disease. Cancer. 1984;54:657–62.
Williams ED, Morales AM, Horn RC. Thyroid carcinoma and Cushing's syndrome. A report of two cases with a review of the common features of the non-endocrine tumours associated with Cushing's syndrome. J Clin Pathol. 1968;21:129–35.
Birkenhäger JC, Upton GV, Seldenrath HJ, Kreiger DT, Tashjian AH Jr. Medullary thyroid carcinoma: ectopic production of peptides with ACTH-like, corticotrophin releasing factor-like and prolactin production-stimulating activities. Acta Endocrinol (Copen). 1976;83:280–92.
Goltzman D, Huang S-N, Browne C, Solomon S. Adrenocorticotropin and calcitonin in medullary thyroid carcinoma: frequency of occurrence and localization in the same cell type by immunohistochemistry. J Clin Endocrinol Metab. 1979;49:364–9.
Rindi G, Klimstra DS, Abedi-Ardekani B, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol. 2018;31(12):1770–86.
Erovic BM, Kim D, Cassol C, et al. Prognostic and predictive markers in medullary thyroid carcinoma. Endocr Pathol. 2012;23(4):232–42.
Werner TA, Tamkan-Olcek Y, Dizdar L, et al. Survivin and XIAP: two valuable biomarkers in medullary thyroid carcinoma. Br J Cancer. 2016;114(4):427–34.
Ferreira LB, Eloy C, Pestana A, et al. Osteopontin expression is correlated with differentiation and good prognosis in medullary thyroid carcinoma. Eur J Endocrinol. 2016;174(4):551–61.
Shi W, Zhao QY, Liu ZM, Wang ST, Liu CP. Small cell carcinoma: a rare subtype of thyroid cancer with unanticipated prognosis. Curr Med Sci. 2019;39(2):265–9.
Lamberts SWJ, Krenning EP, Reubi JC. The role of somatostatin and its analogs in the diagnosis and treatment of tumors. Endocr Rev. 1991;12:450.
Krenning EP, Kwekkeboom DJ, Valkema R, Pauwels S, Kvols LK, de Jong M. Peptide receptor radionuclide therapy. Ann N Y Acad Sci. 2004;1014:234–45.
Makis W, McCann K, McEwan AJ. Medullary thyroid carcinoma (MTC) treated with 177Lu-DOTATATE PRRT: a report of two cases. Clin Nucl Med. 2015;40(5):408–12.
Beukhof CM, Brabander T, van Nederveen FH, et al. Peptide receptor radionuclide therapy in patients with medullary thyroid carcinoma: predictors and pitfalls. BMC Cancer. 2019;19(1):325.
Lamberts SWJ, Bakker WH, Reubi JC, Krenning EP. Somatostatin-receptor imaging in the localization of endocrine tumors. N Engl J Med. 1990;323:1246–9.
Tran K, Khan S, Taghizadehasl M, et al. Gallium-68 Dotatate PET/CT is superior to other imaging modalities in the detection of medullary carcinoma of the thyroid in the presence of high serum calcitonin. Hell J Nucl Med. 2015;18(1):19–24.
Mizukami Y, Michigishi T, Nonomura A, et al. Mixed medullary-follicular carcinoma of the thyroid occurring in familial form. Histopathology. 1993;22:284–7.
Apel RL, Alpert LC, Rizzo A, LiVolsi VA, Asa SL. A metastasizing composite carcinoma of the thyroid with distinct medullary and papillary components. Arch Pathol Lab Med. 1994;118:1143–7.
Fink A, Tomlinson G, Freeman JL, Rosen IB, Asa SL. Occult micropapillary carcinoma associated with benign follicular thyroid disease and unrelated thyroid neoplasms. Mod Pathol. 1996;9(8):816–20.
Pastolero GC, Coire CI, Asa SL. Concurrent medullary and papillary carcinomas of thyroid with lymph node metastases. Am J Surg Pathol. 1996;20:245–50.
Mete O, Asa SL. Composite medullary and papillary thyroid carcinoma in a patient with MEN 2B. Case report and review of C-cell lesions of the thyroid. Pathol Case Rev. 2009;14(6):208–13.
LiVolsi VA. Mixed thyroid carcinoma: a real entity? Lab Investig. 1987;57:237–9.
Holm R, Sobrinho-Simoes M, Nesland JM, Johannessen J-V. Concurrent production of calcitonin and thyroglobulin by the same neoplastic cells. Ultrastruct Pathol. 1986;10:241–8.
Holm R, Sobrinho-Simoes M, Nesland JM, Sambade C, Johannessen J-V. Medullary thyroid carcinoma with thyroglobulin immunoreactivity. A special entity? Lab Investig. 1987;57:258–68.
González-Cámpora R, Lopez-Garrido J, Martin-Lacave I, Miralles-Sánchez EJ, Villar JL. Concurrence of a symptomatic encapsulated follicular carcinoma, an occult papillary carcinoma and a medullary carcinoma in the same patient. Histopathology. 1992;21:380–2.
Baloch Z, Mete O, Asa SL. Immunohistochemical biomarkers in thyroid pathology. Endocr Pathol. 2018;29(2):91–112.
Wolfe HJ, Melvin KEW, Cervi-Skinner SJ. C-cell hyperplasia preceding medullary thyroid carcinoma. N Engl J Med. 1973;289:437–41.
DeLellis RA, Wolfe HJ. The pathobiology of the human calcitonin (C)-cell: a review. Pathol Annu. 1981;16:25–52.
Asa SL, de Jesus AC, Kerr D, et al. Thyroid. In: Mete O, Asa SL, editors. Endocrine pathology. Cambridge: Cambridge University Press; 2016. p. 398–572.
Albores-Saavedra J, Monforte H, Nadji M, Morales AR. C-cell hyperplasia in thyroid tissue adjacent to follicular cell tumors. Hum Pathol. 1988;19:795–9.
Biddinger PW, Brennan MF, Rosen PP. Symptomatic C-cell hyperplasia associated with chronic lymphocytic thyroiditis. Am J Surg Pathol. 1991;15:599–604.
Scopsi L, Di Palma S, Ferrari C, Holst JJ, Rehfeld JF, Rilke F. C-cell hyperplasia accompanying thyroid diseases other than medullary carcinoma: an immunocytochemical study by means of antibodies to calcitonin and somatostatin. Mod Pathol. 1991;4:297–304.
Libbey NP, Nowakowski KJ, Tucci JR. C-cell hyperplasia of the thyroid in a patient with goitrous hypothyroidism and Hashimoto's thyroiditis. Am J Surg Pathol. 1989;13:71–7.
Nose V. Familial non-medullary thyroid carcinoma: an update. Endocr Pathol. 2008;19(4):226–40.
Waser B, Beetschen K, Pellegata NS, Reubi JC. Incretin receptors in non-neoplastic and neoplastic thyroid C cells in rodents and humans: relevance for incretin-based diabetes therapy. Neuroendocrinology. 2011;94(4):291–301.
Reagh J, Bullock M, Andrici J, et al. NRASQ61R mutation-specific immunohistochemistry also identifies the HRASQ61R mutation in medullary thyroid cancer and may have a role in triaging genetic testing for MEN2. Am J Surg Pathol. 2017;41(1):75–81.
Sponziello M, Benvenuti S, Gentile A, et al. Whole exome sequencing identifies a germline MET mutation in two siblings with hereditary wild-type RET medullary thyroid cancer. Hum Mutat. 2018;39(3):371–7.
Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658–71.
Smallridge RC, Ain KB, Asa SL, et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22(11):1104–39.
Grubbs EG, Ng PK, Bui J, et al. RET fusion as a novel driver of medullary thyroid carcinoma. J Clin Endocrinol Metab. 2015;100(3):788–93.
Zedenius J, Larsson C, Bergholm U, et al. Mutations of codon 918 in the RET proto-oncogene correlate to poor prognosis in sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab. 1995;80:3088–90.
Elisei R, Cosci B, Romei C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93(3):682–7.
Ji JH, Oh YL, Hong M, et al. Identification of driving ALK fusion genes and genomic landscape of medullary thyroid cancer. PLoS Genet. 2015;11(8):e1005467.
Gimm O, Greco A, Hoang-Vu C, Dralle H, Pierotti MA, Eng C. Mutation analysis reveals novel sequence variants in NTRK1 in sporadic human medullary thyroid carcinoma. J Clin Endocrinol Metab. 1999;84(8):2784–7.
Robbins RJ, Thomas JS, Osuna PM, Shakil J. A BRAF V600E mutation in RET-negative medullary thyroid cancer. Case Rep Endocrinol. 2020;2020:7641940.
Kasaian K, Wiseman SM, Walker BA, et al. Putative BRAF activating fusion in a medullary thyroid cancer. Cold Spring Harb Mol Case Stud. 2016;2(2):a000729.
Wang N, Xu D, Sofiadis A, et al. Telomerase-dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. J Clin Endocrinol Metab. 2014;99(8):E1571–9.
Santarpia L, Calin GA, Adam L, et al. A miRNA signature associated with human metastatic medullary thyroid carcinoma. Endocr Relat Cancer. 2013;20(6):809–23.
Meng K, Luo H, Chen H, Guo H, Xia W. Prognostic value of numbers of metastatic lymph node in medullary thyroid carcinoma: a population-based study using the SEER 18 database. Medicine (Baltimore). 2019;98(1):e13884.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Asa, S.L., Mete, O. (2021). Thyroid Neuroendocrine Neoplasms. In: Asa, S.L., La Rosa, S., Mete, O. (eds) The Spectrum of Neuroendocrine Neoplasia. Springer, Cham. https://doi.org/10.1007/978-3-030-54391-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-54391-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-54390-7
Online ISBN: 978-3-030-54391-4
eBook Packages: MedicineMedicine (R0)