
Abstract
Thrombotic microangiopathy is a rare, but devastatic complication of hematopoietic cell transplantation characterized by thrombocytopenia, microangiopathic hemolytic anemia, organ failure, and mortality if untreated. Early identification is crucial as withdrawal of offending medications, disease-specific interventions, and potential implementation of complement-directed therapies may prevent organ damage and death. In the following chapter, we outline the common clinical presentation, risk factors, diagnostic steps, and therapeutic interventions for transplant-related thrombotic microangiopathies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The thrombotic microangiopathies (TM) are a group of diseases which share the characteristic clinical features of thrombocytopenia and microangiopathic hemolytic anemia resulting in microvascular occlusion and end organ damage. The “classic” TMs are thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Since the early days of hematopoietic cell transplantation (HCT), many patients have developed a TM-type disease that was often fulminant and fatal [1, 2]. Research has been difficult due to lack of standardized diagnostic criteria, and much controversy remains about optimal therapies.
Clinical Presentation
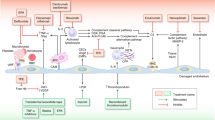
The fundamental problem in all TMs is occlusion of the vasculature by platelet aggregates. This event restricts blood flow which leads to areas of high shear that damage red cells resulting in fragmentation. This is the origin of the “helmet cells” or “schistocytes” component of the diagnostic criteria (microangiopathic hemolytic anemia). This vascular occlusion leads to tissue ischemia and end organ damage. In classic HUS, the predominant pathophysiologic finding involves the kidney leading to renal failure, while in TTP, damage can occur in any organ. The high serum lactate dehydrogenase levels (LDH) seen in TM is due both to red cell destruction and tissue ischemia [3].
In HCT patients, the onset of the TM is often gradual with slowly rising LDH and deteriorating renal function. Often hypertension develops and can be an early clue to the diagnosis. In TM associated with agents such as calcineurin inhibitors (CNI), the onset can be more rapid. As the TM progresses, renal insufficiency and neurological symptoms are the most common findings in many patients running a relentless course until the patient expires [1].
Risk Factors
Many risk factors for TM have been proposed. One difficulty with these factors is that any widespread disease process such as severe infection or graft-versus-host disease (GvHD) can lead to a clinical syndrome similar to TM. This lack of clarity in identification of etiologic events results in the extreme variations in reported posttransplant incidence rates ranging from 0% to 93% of patients [4, 5]!
Risk factors include:
-
1.
Older age
-
2.
Female gender
-
3.
Advanced disease
-
4.
Unrelated donor transplant
-
5.
Radiation-containing conditioning regimens
-
6.
Calcineurin inhibitors
-
7.
Infection
-
8.
GvHD
Classification
Pettit and Clark in 1994 proposed a classification which still provides a useful schema for thinking about transplant-related TM [2].
-
1.
One group is the “multi-organ fulminant” which occurs early (day +20–60), has multi-organ system involvement, and is often fatal.
-
2.
A second type of TTP/HUS is similar to CNI-associated HUS.
-
3.
A third type described as “conditioning” TTP/HUS occurs 6 months or more after total body irradiation and is associated with primary renal involvement.
-
4.
Finally, patients with systemic cytomegaloviral (CMV) infections may present with a TTP/HUS syndrome related to vascular endothelial cell CMV infection.
Etiology
-
1.
In classic TTP, autoimmune destruction leads most patients to have very low levels of ADAMTS-13 (<5%) which is thought to lead to spontaneous platelet aggregation via the failure to cleave the ultrahigh molecular weight multimers of von Willebrand protein.
-
2.
In patients with HCT-related TM:
-
a.
Most reports show reduced but not extremely low levels of ADAMTS-13.
-
b.
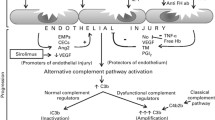
The underlying precipitant is thought to be endothelial damage, either by GvHD, medications, radiation, or infection.
-
i.
This endothelial damage leads to platelet aggregation, microangiopathic hemolytic anemia, and end organ damage.
-
i.
-
c.
Over-activation of complement has been reported, similar to genetic variants of atypical HUS, suggesting inhibition of complement to be a reasonable therapeutic target [6].
-
i.
The fact that many patient will have mutations in genes associated with complement regulation and that clinically patients often quickly respond to anticomplement therapy provides evidence for this concept [7].
-
i.
-
d.
This premise that endothelial injury is the main trigger for HCT-related TM would explain why vascular damage is a shared component of many of the risk factors for TM [8].
-
a.
Diagnosis
Given that the diagnosis of any TM is a clinical one and that HCT patients are prone to have many complications that can mimic a TM, it is easy to appreciate and understand the great center-to-center variation in describing the incidence. Recently two groups have proposed diagnostic consensus criteria that share the common features of evidence of a microangiopathic hemolytic anemia and elevated LDH.
-
1.
Blood and Marrow Transplant Clinical Trial Network (BMT-CTN) Criteria [5]
-
a.
RBC fragmentation and ≥ 2 schistocytes per high-powered field
-
b.
Concurrent increase in LDH from institutional baseline
-
c.
Concurrent renal and/or neurological dysfunction with no other explanation
-
d.
Negative Coombs test
-
a.
-
2.
International Working Group Criteria [4]
-
a.
Increased percentage (>4%) of schistocytes in the blood
-
b.
New, prolonged, or progressive thrombocytopenia (<50,000/uL or >50% decrease from previous counts)
-
c.
Sudden and persistent increase in LDH
-
d.
Decreased hemoglobin or increased transfusion requirements
-
e.
Decrease in serum haptoglobin
-
a.
More recently, it has been observed that many patients will have rising LDH and signs of renal damage (proteinuria, severe hypotension) before the onset of overt TM. There is also increasing use of sC5-9 to detect complement activation [9].
Treatment
-
1.
CNI-associated TM: This disorder often occurs within days after the introduction of these medications or with a significant increase in blood levels of these agents. The renal and neurological manifestation can be rapid and severe including malignant hypertension, seizures, and cortical blindness. Therapy is discontinuation of the medications and to manage the closely associated hypertension. In patients with mild TM and high serum levels, one can lower the dose to see if the symptoms abate [7].
-
2.
Conditioning-associated TM: This subtype is rare and may be a manifestation of radiation damage to the vasculature. Usually the course is progressive with no specific therapy available [7, 10].
-
3.
Systemic CMV-associated TM: CMV is trophic to the endothelium and aggressive therapy of CMV is the cornerstone of therapy [7, 9].
-
4.
Multi-organ fulminant TM: Therapy remains unsatisfactory. The first step is to maximize treatment of any process that may be aggravating the TM (GvHD, infection, etc.). Unlike classic TTP, the role of plasma exchange remains controversial. Most series report very poor response rates with poor outcomes and high rates of complications [11]. Increasingly utilized for these patients, prompt initiation of the complement inhibitor eculizumab (Soliris®) has been reported to improve outcomes. A reasonable approach would be for a patient with signs of TM (rising LDH, signs of renal dysfunction [hypertension, proteinuria]) is to initiate eculizumab at a HUS dosing (900 mg IV once weekly × 4 doses then 1200 mg IV every other week). If the patient responds, there is uncertainly how long therapy should be continued, but many will continue for 6 months minimum prior to reevaluation [8, 9, 12]. Narsoplimab is another antibody therapy, targeting the mannan-binding lectin-associated serine protease-2 (MASP-2) in the lectin pathway for complement activation that is in advanced clinical trials and could emerge as another therapeutic option for advanced TM.
References
Choi CM, Schmaier AH, Snell MR, Lazarus HM. Thrombotic microangiopathy in haematopoietic stem cell transplantation: diagnosis and treatment. Drugs. 2009;69(2):183–98.
Pettitt AR, Clark RE. Thrombotic microangiopathy following bone marrow transplantation. Bone Marrow Transplant. 1994;14:495–504.
Shatzel JJ, Taylor JA. Syndromes of thrombotic microangiopathy. Med Clin North Am. 2017;101:395–415.
Ruutu T, Barosi G, Benjamin RJ, et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica. 2007;92:95–100.
Ho VT, Cutler C, Carter S, et al. Blood and marrow transplant clinical trials network toxicity committee consensus summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11:571–5.
Jodele S, Licht C, Goebel J, et al. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood. 2013;122:2003–7.
Jodele S. Complement in pathophysiology and treatment of transplant-associated thrombotic microangiopathies. Semin Hematol. 2018;55:159–66.
Jodele S, Dandoy CE, Myers KC, et al. New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54:181–90.
Wanchoo R, Bayer RL, Bassil C, Jhaveri KD. Emerging concepts in hematopoietic stem cell transplantation-associated renal thrombotic microangiopathy and prospects for new treatments. Am J Kidney Dis. 2018;72:857–65.
Weitz IC. Thrombotic microangiopathy in cancer. Thromb Res. 2018;164(Suppl1):S103–5.
Sartain S, Shubert S, Wu MF, et al. Therapeutic plasma exchange does not improve renal function in hematopoietic stem cell transplantation-associated thrombotic microangiopathy: an institutional experience. Biol Blood Marrow Transplant. 2019;25(1):157–62.
Olson SR, Lu E, Sulpizio E, et al. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48:96–107.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Shatzel, J.J., DeLoughery, T.G. (2021). Thrombotic Microangiopathies. In: Maziarz, R.T., Slater, S.S. (eds) Blood and Marrow Transplant Handbook. Springer, Cham. https://doi.org/10.1007/978-3-030-53626-8_38
Download citation
DOI: https://doi.org/10.1007/978-3-030-53626-8_38
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53625-1
Online ISBN: 978-3-030-53626-8
eBook Packages: MedicineMedicine (R0)