
Abstract
Aplastic anemia (AA) is a blood disorder characterized by pancytopenia, bone marrow hypoplasia/aplasia, and the absence of underlying malignancy. AA pathophysiology reflects the decrease in the cellularity of bone marrow and the decrease in the pool of hematopoietic stem cells (HSCs) below a threshold that could maintain mature blood cell production, ultimately leading to peripheral pancytopenia. Most patients have no identified underlying cause and are classified as idiopathic, but other etiologies include direct injury to HSCs, inherited genetic disorders, or an immune-mediated process. Treatment strategies include immunosuppressive therapy and/or allogeneic hematopoietic cell transplantation (HCT). Matched related donor (MRD) transplant is considered first-line therapy for young patients (<40 years) and elderly patients without significant comorbidities although improvements in matched unrelated donor (M-URD) transplants have been observed recently. These innovations have resulted in outcomes similar to MRD transplants in younger patients. Additionally, availability of alternative donors, in particular, haploidentical-related (haploID) donors, and the use of reduced-intensity conditioning (RIC) regimens have expanded the availability of allogeneic transplants to older patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Severe aplastic anemia
- Pancytopenia, immune suppressive therapy
- Bone marrow transplantation
- Graft rejection
- Umbilical cord blood transplantation
- Haploidentical transplantation
Introduction
Aplastic anemia (AA) is a blood disorder characterized by pancytopenia, bone marrow hypoplasia/aplasia, and the abscence of underlying malignancy. AA pathophysiology reflects the decrease in the cellularity of bone marrow and the decrease in the pool of hematopoietic stem cells (HSCs) below a threshold that could maintain mature blood cell production, ultimately leading to peripheral pancytopenia. Most patients have no identified underlying cause and are classified as idiopathic, but other etiologies include direct injury to HSCs, inherited genetic disorders, or an immune-mediated process. Treatment strategies include immunosuppressive therapy and/or allogeneic hematopoietic cell transplantation (HCT). Matched related donor (MRD) transplant is considered first-line therapy for young patients (<40 years) and elderly patients without significant comorbidities although improvements in matched unrelated donor (M-URD) transplants have been observed recently. These innovations have resulted in outcomes similar to MRD transplants in younger patients. Additionally, availability of alternative donors, in particular, haploidentical-related (haploID) donors, and the use of reduced-intensity conditioning (RIC) regimens have expanded the availability of allogeneic transplants to older patients.Direct injury to HSCs may occur due to exposure to radiation, chemicals (benzene, solvents, glue vapors), medications (antiseizure medications, gold, arsenic, antithyroid medications, antibiotics such as sulfa and chloramphenicol), and infections (Epstein–Barr virus (EBV), seronegative hepatitis, human immunodeficiency virus (HIV), parvovirus, etc.). Inherited genetic disorders that lead to bone marrow failure include Fanconi anemia, dyskeratosis congenita, megakaryocytic thrombocytopenia, Diamond–Blackfan anemia, Schwachman–Diamond syndrome, thrombocytopenia absent radii, Pearson syndrome, and severe congenital neutropenia. Immune-mediated processes may also result in dysfunctional HSCs [1,2,3,4,5].
Autoimmune damage to HSCs contributes to most cases of acquired aplastic anemia (AA) (typically called idiopathic AA). It has been hypothesized that infections or different triggers could alter the immunologic appearance of HSCs and lead to autoimmune destruction of HSCs. This hypothesis is supported by the observation that many patients with AA respond to immune suppressive therapy [6, 7].
AA is divided into three groups based on marrow cellularity and peripheral blood counts:
-
1.
Severe AA (SAA): Requires both of the following criteria [8]:
-
a.
Bone marrow cellularity <25% (or 25–50% if <30% of residual cells are hematopoietic)
-
b.
At least two of the following:
-
i.
Peripheral blood absolute neutrophil count (ANC) <500/μL (<0.5 × 109/L)
-
ii.
Peripheral blood platelet count <20,000/μL
-
iii.
Peripheral blood reticulocyte count <20,000/μL
-
i.
-
a.
-
2.
Very severe AA (vSAA): Diagnosis includes the same criteria for SAA above but ANC is <200/μL [9]
-
3.
Nonsevere or moderate AA: Hypocellular bone marrow (as described for SAA) but the peripheral blood cytopenias do not fulfill the criteria for SAA or vSAA
Diagnosis of SAA
-
1.
The differential diagnosis of AA includes hypoplastic myelodysplastic syndrome (MDS), clonal T-cell disorders, and AA associated with paroxysmal nocturnal hemoglobinuria (PNH). It is important to distinguish AA from hypocellular MDS because the two disease entities are treated differently.
-
2.
Requires exclusion of a variety of inherited or acquired bone marrow failure syndromes with similar phenotypes. A quick and efficient diagnostic plan is important because time from diagnosis to “final” treatment is directly related to outcome regardless of the therapeutic option chosen.
-
3.
Requires careful physical exam to identify any potential dysmorphic features that could be suggestive of inherited bone marrow failure.
-
4.
Comprehensive laboratory work up to identify possible cause should include the following:
-
a.
Complete blood count and manual differential
-
b.
Reticulocyte count
-
c.
CD55/59 screen for PNH
-
d.
Serum aminotransferase
-
e.
Viral serologies for HIV, cytomegalovirus (CMV), Epstein Barr virus (EBV), hepatitis, and herpes simplex virus (HSV)
-
f.
Serum folate and vitamin B12 concentrations
-
g.
Hemoglobin F level
-
h.
Chromosome breakage test to screen for Fanconi anemia
-
i.
Bone marrow aspirate and biopsy with cytogenetics (usually normal in AA but likely to have abnormal karyotype in hypocellular MDS)
-
i.
It is common to find PNH clones of phosphatidylinositol glycan (PIG)-anchored proteins, such as CD55 and CD59, by flow cytometry assay of the bone marrow in approximately 20% of patients with AA [10].
-
Such clones can remain stable, diminish in size, or disappear.
-
Presence of a significant PNH clone with clinical or laboratory evidence of hemolysis or thrombosis is clinically important. Historically, the Ham test was used to support the diagnosis of PNH; however, more recently, diagnosis is made by flow cytometry. Urine should be examined for hemosiderin to exclude intravascular hemolysis, which is an important feature of hemolytic PNH.
-
-
i.
-
j.
Human leukocyte antigen (HLA) testing
-
a.
Treatment
The two major competing treatment strategies for SAA, allogeneic hematopoietic cell transplantation (HCT) and immunosuppressive therapy (IST) with antithymocyte globulin (ATG), date back to 1970 when the first series of successful marrow transplants from HLA-identical sibling donors was reported [11].
-
1.
Generally, it has been accepted that HCT from a matched sibling is considered as the first line of therapy for children and young adult patients (<40 years) [8]. IST is reserved for those patients without a matched sibling or patients ≥40 years of age.
-
a.
The decision is more nuanced because some older patients can tolerate the toxicities of potentially curative HCT. Thus, HCT or IST may be appropriate, depending on disease severity, availability of a donor, and patient comorbidities.
-
b.
This decision is made on a case-by-case basis that considers the degree of cytopenias, life expectancy, and patient preferences [14].
-
a.
-
2.
HCT from an HLA-matched related donor (MRD)
-
a.
One of the early studies that proved that HCT is life saving for SAA is a prospective randomized trial comparing early bone marrow transplantation with conventional treatments.
-
i.
All patients with a matched sibling donor underwent HCT performed within 17–100 (median 33) days of original diagnosis. Patients without an MRD received conventional therapy including transfusion support with or without androgens.
-
ii.
Twenty-four of 36 HCT patients were alive (overall survival [OS] = 66.7%) at a median of 9 months with full marrow reconstitution compared with 12 of 31 patients (OS = 38.7%) who received conventional therapy (p = 0.006) [12].
-
iii.
This study demonstrated that early application of HCT appears to be an effective treatment for SAA.
-
i.
-
b.
Randomized prospective trials comparing HCT with IST in AA are lacking.
-
c.
Meta-analysis by Peinemann et al. [13] reviewed 26 non-randomized controlled trials for patients with AA using either HCT or IST (7955 patients enrolled from 1970 to 2001). Young age and recent year of treatment were identified as factors contributing to improved survival in the HCT group.
-
d.
Conceptual framework of allogeneic HCT is straightforward: replace the aplastic marrow in the patient with a marrow graft from a healthy donor. Three major transplant related problems exist [11]:
-
i.
Graft rejection
-
ii.
Acute graft-versus-host disease (GvHD)
-
iii.
Chronic GvHD
-
Initially a frequent complication among patients with AA who received only cyclophosphamide conditioning; this complication was observed in >35% of patients transplanted in the early 1970s [15].
-
Etiology is related to sensitization to HLA through previous transfusion of blood products. Early studies reported that previously transfused patients had a significantly lower OS compared with patients who had not received transfusions.
-
Decreasing the number of transfused blood products along with irradiation and leukoreduction of platelet and RBC products aided in the reduction of graft rejection [16, 17]
-
Other factors leading to a decrease in graft rejection include the following:
-
Intensifying conditioning regimen with addition of equine ATG at 30 mg/kg/dose × 3 days to cyclophosphamide 50 mg/kg/day × 4 days.
-
Data from the European Society for Blood and Marrow Transplantation (EBMT) showed cyclosporine + methotrexate as GvHD prophylaxis led to a decrease in graft rejection when compared with methotrexate alone [17]. This approach led to engraftment in 95% of patients with an OS of 90% at 2 years post-HCT with good long-term outcomes and limited number of late effects such as avascular necrosis, endocrine dysfunction, and very rare secondary malignancies [18, 19].
-
Immunosuppression aimed at preventing GvHD also had a role in controlling host-versus-graft (HvG) reactions.
-
-
Mixed donor/recipient chimerism occurs in 44–55% of acquired AA patients following MRD HCT [19, 20]. Some patients may exhibit decline in donor chimerism during withdrawal of immune suppression (IS) and are at risk for late graft rejection. Therefore, in contrast to the standard approach following HCT for malignant disorders, guidelines usually reinstitute IS for SAA patients with falling donor chimerism after HCT.
-
Alternatives to high-dose cyclophosphamide (200 mg/kg) with decreased toxicity have been studied.
-
A randomized study comparing fludarabine 120 mg/m2 + cyclophosphamide 100 mg/kg + rabbit ATG 9 mg/kg with cyclophosphamide 200 mg/kg + rabbit ATG showed no significant difference in graft rejection in the two arms (13.4% vs. 16.8%, respectively), or the incidence of acute or chronic GvHD and OS [21]
-
Another conditioning regimen approach is the combination of fludarabine 120 mg/m2 + cyclophosphamide 1200 mg/m2 + alemtuzumab (Campath®) 40–100 mg.
-
The combination of fludarabine + reduced-dose cyclophosphamide + either ATG or alemtuzumab appears to be an alternative conditioning regimen suitable for older patients, but due to the increased risk of graft rejection, the reduced-dose alternative regimens are not recommended for patients, especially children, who can tolerate the high-dose cyclophosphamide 200 mg/m2 + ATG regimen.
-
Table 23.1 summarizes the results of selected studies of MRD HCT for SAA.
-
-
In one report, the substitution of rabbit ATG (total dose 8 mg/kg) in place of equine ATG (90 mg/kg) as part of the conditioning regimen with cyclophosphamide 200 mg/kg prior to HLA-identical sibling bone marrow (BM) transplantation was associated with a decreased incidence of chronic GvHD (0% versus 34%, respectively).
-
However, there was increased risk of invasive fungal disease after transplantation, earlier CMV reactivation, and delayed lymphocyte recovery in rabbit ATG recipients.
-
Despite the decreased incidence of chronic GvHD, there was no difference in post-HCT OS between the two groups.
-
-
Long-term outcomes for MRD HCT in children with SAA have been reported by Seattle group in 148 children (median age 12.8 years) [28].
-
GvHD prophylaxis was methotrexate on days +1, 3, 6 and 11 with cyclosporine.
-
With median follow-up of 25 years, the 5-year survival was 100% in the group that received cyclophosphamide + ATG.
-
The incidence of graft rejection was 7%, acute GvHD grades III–IV were 3%, and chronic GvHD was 10%.
-
This result demonstrates that allogeneic HCT using MRD should be used as the first-line therapy for children and young adults with SAA.
-
-
The same conditioning regimen of cyclophosphamide + ATG in conjunction with a MRD HCT in older patients (> 40 years) is associated with decreased OS.
-
A study from Seattle group evaluated 23 patients (age 40–68) with SAA who underwent MRD HCT between 1988 and 2008.
-
OS was 65% with median follow-up of 9.1 years.
-
It is important to note that 22% of patients died from infection prior to engraftment [29].
-
-
A follow-up study by the EBMT added fludarabine to the cyclophosphamide + ATG regimen with the goal of decreasing the dose of cyclophosphamide in order to reduce organ cytotoxicity and intensify immunosuppression [23].
-
This combination resulted in improved 5-year survival of 77% in the fludarabine cohort compared to 60% in the cohort who did not receive fludarabine.
-
Patients between the ages of 30 and 40 years had a survival probability exceeding 80%.
-
-
-
It is also important to mention that for SAA, it is not appropriate to use peripheral blood stem cells (PBSCs) as donor source, regardless of patients’ age, due to the increased risk of GvHD.
-
A retrospective analysis of 1886 patients with AA who underwent an HCT from a HLA-matched sibling between 1999 and 2009 evaluated either BM (n = 1163) or PBSCs (n = 723) as the source of stem cells [30].
-
Acute and chronic GvHD were more frequent in patients who received PBSCs vs BM.
-
The major cause of death was GvHD with 2% versus 6% in BM vs PBSC recipients, respectively.
-
This contributed to a survival advantage for recipients of BM rather than PBSCs and was statistically significant in patients aged 1–19 years (90% versus 76% p < 0.00001) as well as in patients aged over 20 years (74% versus 64%, p = 0.001). The advantage for recipients of BM over PBSCs was maintained above the age of 50 years (69% versus 39%, p = 0.01).
-
Therefore, unlike in transplantation for pediatric malignancies, whereby PBSC may elicit beneficial graft-versus-leukemia effects, BM is clearly the preferred stem-cell source for acquired SAA patients.
-
-
-
-
-
i.
-
a.
HCT from HLA-Matched Unrelated Donors (M-URD)
-
1.
HCTs using M-URDs have historically been considered second-line therapy for SAA patients who fail IST. Consequences of delayed M-URD include iron overload from chronic RBC transfusions, platelet transfusion refractoriness, potential life-threatening infections, and other comorbidities that can affect OS post-HCT.
-
2.
Outcomes following M-URD HCT have steadily improved since the early 1990s. A retrospective study of 141 patients with SAA who underwent M-URD HCT between 1988 and 1995 showed an OS of 36% [31] compared with a recent pediatric series that showed an OS of 78–95% [23, 32]. This increase in OS is due in large part to improvements in supportive care during the IST phase of therapy, conditioning regimens, and high-resolution HLA typing leading to better unrelated donor matching.
-
3.
An important prospective study sponsored by the National Marrow Donor Program (NMDP) looked into optimization of the conditioning regimen in M-URD HCT [33].
-
a.
The starting conditioning regimen was cyclophosphamide 50 mg/kg/day × 4 doses + equine ATG 30 mg/kg/day × 3 days + total body irradiation (TBI) 600 cGy.
-
b.
In this multicenter study, a total of 87 patients were enrolled between 1994 and 2004 with median age 18.6 years (range 1.3–53.5 years).
-
c.
The optimum TBI dose was 1 × 200 cGy.
-
d.
Graft failure occurred in 5% of patients and OS was 61% at median follow-up of 7 years.
-
e.
However, in the optimized conditioning that included TBI dose of 200 cGy, the 5-year OS in patients < age 20 was 78% compared with 50% for patients > age 20.
-
a.
-
4.
A recently completed phase I/II study by the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) (study 0301, registered at www.clinicaltrials.gov [NCT00326417]) aimed to identify the optimal dose of cyclophosphamide in a M-URD HCT conditioning regimen that incorporated fludarabine, ATG, and low-dose TBI 200 cGy.
-
a.
All patients receive a fixed dose of ATG (either rabbit ATG 3 mg/kg IV or equine ATG 30 mg/kg IV daily on days −4 to −2), fludarabine 30 mg/m2 IV daily on days −5 to −2, and low-dose TBI 200 cGy on day −1.
-
b.
Cyclophosphamide dosing was started at 150 mg/kg and decreased in steps of 50 mg/kg (to 100 mg/kg, 50 mg/kg, and 0 mg/kg).
-
c.
A total of 96 patients (median age 24.5 years, range 0.5–65) underwent M-URD HCT.
-
d.
Median follow-up after HCT was 17 months and 24 months for patients receiving cyclophosphamide 50 mg/kg and 100 mg/kg, respectively.
-
e.
OS at 1 year for patients receiving 50 mg/kg and 100 mg/kg were 97.4% (95% CI 82.8–99.6) and 80.5% (64.8–89.7), respectively [34].
-
f.
Early in the clinical trial, both the 0 mg/kg and 150 mg/kg dose schedules were discontinued due to poor outcomes.
-
g.
This study identified cyclophosphamide 50 mg/kg as the most desirable dose in combination with TBI 200 cGy, fludarabine 120 mg/m2, and ATG for engraftment and early survival for M-URD HCT in patients with acquired SAA.
-
a.
-
5.
Another multicenter, retrospective study from the United Kingdom (UK) evaluated an alemtuzumab (Campath®)-based conditioning regimen [27].
-
a.
Twenty-nine patients received fludarabine 30 mg/m2 for 4 days, cyclophosphamide 300 mg/m2 for 4 days, and alemtuzumab with a median total dose of 60 mg (range 40–100 mg).
-
b.
Median age was 35 years (range 8–62).
-
c.
OS at 2 years was 83% with a cumulative incidence of graft failure of 14.5%.
-
d.
Acute GvHD was observed in only 13.5% patients (all grades I–II) and only 2 patients (4%) developed chronic GvHD.
-
e.
A low incidence of viral infections was seen.
-
f.
Factors influencing OS were HCT comorbidity index (92% with score 0–1 vs 42% with score ≥2, p < 0.001) and age (92% for age < 50 years vs 71% ≥ 50 years, p < 0.001).
-
g.
These data suggest that alemtuzumab-based M-URD HCT regimen for SAA results in durable engraftment with a low incidence of chronic GvHD even in elderly patients.
-
a.
M-URD HCT as First-Line Therapy in Children
-
1.
Despite the initial response to IST in children, there is considerable risk of relapse and long-term side effects of cyclosporine dependence as well as clonal evolution [35, 36].
-
2.
Furthermore, in the case of incomplete response post-IST, children may suffer either from restrictions to sporting and other activities because of subnormal platelet and/or hemoglobin values, or from higher risks for infection due to suboptimal neutrophil count and prolonged cyclosporine treatment.
-
3.
It is clear that event-free survival (EFS) is more meaningful than OS when studying outcomes of SAA in children.
-
4.
These long-term concerns combined with the improvements in outcomes of M-URD HCT have encouraged many investigators to offer upfront transplant to children with SAA.
-
5.
UK investigators reported an excellent estimated 5-year failure-free survival (FFS) of 95% in 44 consecutive children who received a 10/10 allele level HLA-matched unrelated donor; 40 of these children had previously failed IST [37].
-
a.
A follow-up study from the EBMT reported a cohort of 29 consecutive children with SAA who received M-URD HCT as first-line therapy after conditioning with fludarabine–cyclophosphamide–alemtuzumab.
-
i.
OS and EFS were 96% and 92%, respectively [38]. These results demonstrated that upfront M-URD HCT was similar to MRD HCT and superior to IST and M-URD HCT post-IST failure.
-
i.
-
a.
-
6.
There is currently a North American randomized prospective trial of IST vs M-URD HCT in children with SAA (ClinicalTrials.gov number NCT02845596). The conditioning regimen used in this trial is based on BMT CTN results with reduced dose cyclophosphamide 50 mg/kg + fludarabine 120 mg/m2 + ATG 90 mg/m2 + TBI 200 cGy.
-
7.
Bone marrow is the preferred source of HSCs for HCT for SAA.
-
a.
A CIBMTR study compared outcomes of patients with SAA who received 10/10 HLA-M-URDs between 2000 and 2008 after variety of standard conditioning regimens [39].
-
i.
Two hundred twenty-five patients received unmanipulated BM and 71 patients received PBSCs.
-
ii.
Engraftment was similar between the two sources, but the incidence of grades II–IV acute GvHD was 31% in the BM group vs 43% in the PBSC group.
-
iii.
Chronic GvHD was not different after adjusting for age.
-
iv.
Three-year OS was 76% in the BM group compared with 61% in the PBSC group.
-
i.
-
a.
Alternative Donor Transplantation for SAA
-
1.
Fully matched unrelated donors (HLA-A, B, C, DRB1) from worldwide registries can be identified for about 80% of Caucasians; however, identification of a fully matched donor for persons of other races and/or ethnicities is much lower [40].
-
2.
Despite potential benefits of umbilical cord blood (UCB) as source of HSCs for HCT in SAA (rapid accession of stored UCB units with better tolerance of HLA mismatch), published data for UCB in SAA show limited success due to low dose of HSCs obtained in single UCB associated with higher rates of graft rejection. The use of double cord blood units has decreased the risk of graft rejection but increased the rate of acute GvHD.
-
a.
The Japanese cord blood network reported outcomes of 31 patients with SAA (median age 28 years) who received single UCB. The overall engraftment was 55%, incidence of acute GvHD of 17% and cGvHD of 20%, with an OS of 41% at 2 years [41].
-
b.
A follow-up study in Japan with 12 SAA patients using fludarabine 125 mg/m2 + melphalan 80 mg/m2 + TBI 400 cGy and single CBU with median total nucleated dose (TNC) 2.5 × 107/kg showed an OS of 83.3% at median follow-up of 36 months [42].
-
c.
An EBMT report analyzed the outcomes of 71 patients who received UCB for SAA. This analysis demonstrated an OS of 45% in patients who received a UCB with a TNC > 3.9 × 107/kg compared to 18% for patients who received units with TNC < 3.9 × 107/kg [43].
-
d.
A study by Francophone Society of Bone Marrow Transplantation and Cellular Therapy reported outcomes of 29 consecutive patients with SAA transplanted with UCB with TNC dose ≥4 × 107/kg between 6/11 and 10/15.
-
i.
Conditioning regimen included fludarabine + cyclophosphamide + ATG + TBI.
-
ii.
At a median follow-up of 38.8 months, engraftment was reported in 23 patients (88%) with cumulative incidences of grades II–IV acute and chronic GvHD of 45.8% and 36%, respectively, and OS was 88.5% [43]. Results from this study highlight the importance of UCB with a higher TNC dose/kg.
-
i.
-
a.
-
3.
The use of posttransplant cyclophosphamide (PTCy) in the setting of haploidentical (haploID)-related donors has significantly expanded the access to HCT in malignant disorders; however, the use of haploID HCT in patients with SAA has only recently been attempted. Accordingly, published data are limited.
-
a.
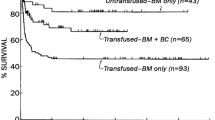
In 2015, a Brazilian group published outcomes of 16 SAA patients (age 5–39 years) who underwent haploID HCT using the modified Hopkins regimen with fludarabine + rabbit ATG 2.5 mg/kg per day on days −4 to −2 + TBI 200–600 cGy with PTCy. HSC sources were BM (N = 13) or PBSCs (N = 3). The rate of neutrophil engraftment was 94% and platelet engraftment was 75%. Three patients developed acute GvHD with grades II–IV GvHD in two. Five patients died, and the 1-year OS was 67.1% [44].
-
b.
A prospective phase II study from the Hopkins group reported 13 patients with a median age 30 years (11–69 years) who underwent haploID HCT [45]
-
i.
A reduced-intensity regimen of rabbit ATG + fludarabine + cyclophosphamide + TBI 200 cGy was used.
-
ii.
All patients received BM with GvHD prophylaxis of PTCy 50 mg/kg/day IV on days +3 and + 4 along with mycophenolate mofetil (Cellcept®, MMF) on days +5 through 35 and tacrolimus from day +5 through 1 year.
-
iii.
G-CSF (Neupogen®) was administered from day +5 until ANC > 1.5 × 109/L × 3 days.
-
iv.
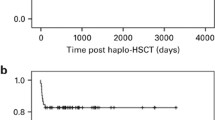
There was no reported graft failure, and mild GvHD occurred in two patients with OS 100%.
-
v.
The very limited transplant-related mortality suggests that this regimen will be feasible in elderly patients with SAA.
-
i.
-
c.
Currently, haploID HCT with PTCy is being studied on a national level in North America by the BMT CTN (CTN 1502 CHAMP study; NCT02918292). This phase II study of haploID HCT uses an RIC regimen with a primary objective of assessing OS at 1-year post-haploID HCT in SAA patients up to the age of 75 years. The study opened in July 2017 and aims to finish enrollment in early 2021.
-
a.
Conclusion
MRD HCT is considered first-line therapy for young patients (<40 years) and elderly patients without significant comorbidities. Improvements in M-URD HCT have been noted using reduced-dose TBI, adding fludarabine to lower-dose cyclophosphamide, and selecting donors who are better HLA matched to patients. These innovations have resulted in outcomes similar to MRD HCT in younger patients. Additionally, availability of alternative donors, in particular, haploID-related donors, using RIC regimens, and PTCy expands the availability of HCT to patients including older patients.
References
Camitta BM. Pathogenesis and treatment of aplastic anemia. Rinsho Ketsueki. 1984;25(4):459–69.
Ehrlich P. Ueber einer Fall von Ana¨mie mit Bemerkungen u¨ber regenerative Vera¨nderungen des Knochenmarks. Charite Annalen. 1888;13:300–9.
Young NS, Kaufman DW. The epidemiology of acquired aplastic anemia. Haematologica. 2008;93(4):489–92.
Montane E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93(4):518–23.
Marsh JC, Ball SE, Darbyshire P, et al. Guidelines for the diagnosis and management of acquired aplastic anaemia. Br J Haematol. 2003;123(5):782–801.
Young NS. Current concepts in the pathophysiology and treatment of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2013;2013:76–81.
Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. New Engl J Med. 1997;336(19):1365–72.
Davies JK, Guinan EC. An update on the management of severe idiopathic aplastic anaemia in children. Br J Haematol. 2007;136(4):549–64.
Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70(2):177–82.
Dunn DE, Tanawattanacharoen P, Boccuni P, et al. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med. 1999;131(6):401–8.
Thomas ED, Storb R, Fefer A, et al. Aplastic anaemia treated by marrow transplantation. Lancet. 1972;1(7745):284–9.
Camitta BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48(1):63–70.
Peinemann F, Grouven U, Kroger N, et al. First-line matched related donor hematopoietic stem cell transplantation compared to immunosuppressive therapy in acquired severe aplastic anemia. PLoS One. 2011;6(4):e18572.
Young NS, Barrett AJ. The treatment of severe acquired aplastic anemia. Blood. 1995;85(12):3367–77.
Storb R, Prentice RL, Thomas ED. Marrow transplantation for treatment of aplastic anemia. An analysis of factors associated with graft rejection. New Engl J Med. 1977;296(2):61–6.
Schuening F, Bean MA, Deeg HJ, Storb R. Prevention of graft failure in patients with aplastic anemia. Bone Marrow Transplant. 1993;12(Suppl 3):S48–9.
McCann SR, Bacigalupo A, Gluckman E, et al. Graft rejection and second bone marrow transplants for acquired aplastic anaemia: a report from the Aplastic Anaemia Working Party of the European Bone Marrow Transplant Group. Bone Marrow Transplant. 1994;13(3):233–7.
Bacigalupo A, Socie G, Hamladji RM, et al. Current outcome of HLA identical sibling versus unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015;100(5):696–702.
Konopacki J, Porcher R, Robin M, et al. Long-term follow up after allogeneic stem cell transplantation in patients with severe aplastic anemia after cyclophosphamide plus antithymocyte globulin conditioning. Haematologica. 2012;97(5):710–6.
Lawler M, McCann SR, Marsh JC, et al. Serial chimerism analyses indicate that mixed haemopoietic chimerism influences the probability of graft rejection and disease recurrence following allogeneic stem cell transplantation (SCT) for severe aplastic anaemia (SAA): indication for routine assessment of chimerism post SCT for SAA. Br J Haematol. 2009;144(6):933–45.
Kim H, Lee JH, Joo YD, et al. A randomized comparison of cyclophosphamide vs. reduced dose cyclophosphamide plus fludarabine for allogeneic hematopoietic cell transplantation in patients with aplastic anemia and hypoplastic myelodysplastic syndrome. Ann Hematol. 2012;91(9):1459–69.
Kahl C, Leisenring W, Deeg HJ, et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long-term follow-up. Br J Haematol. 2005;130(5):747–51.
Maury S, Bacigalupo A, Anderlini P, et al. Improved outcome of patients older than 30 years receiving HLA-identical sibling hematopoietic stem cell transplantation for severe acquired aplastic anemia using fludarabine-based conditioning: a comparison with conventional conditioning regimen. Haematologica. 2009;94(9):1312–5.
Champlin RE, Perez WS, Passweg JR, et al. Bone marrow transplantation for severe aplastic anemia: a randomized controlled study of conditioning regimens. Blood. 2007;109(10):4582–5.
Dufour C, Pillon M, Socie G, et al. Outcome of aplastic anaemia in children. A study by the severe aplastic anaemia and paediatric disease working parties of the European group blood and bone marrow transplant. Br J Haematol. 2015;169(4):565–73.
Dufour C, Pillon M, Passweg J, et al. Outcome of aplastic anemia in adolescence: a survey of the Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation. Haematologica. 2014;99(10):1574–81.
Marsh JC, Gupta V, Lim Z, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft-versus-host disease after allogeneic stem cell transplantation for acquired aplastic anemia. Blood. 2011;118(8):2351–7.
Burroughs LM, Woolfrey AE, Storer BE, et al. Success of allogeneic marrow transplantation for children with severe aplastic anaemia. Br J Haematol. 2012;158(1):120–8.
Sangiolo D, Storb R, Deeg HJ, et al. Outcome of allogeneic hematopoietic cell transplantation from HLA-identical siblings for severe aplastic anemia in patients over 40 years of age. Biol Blood Marrow Transplant. 2010;16(10):1411–8.
Bacigalupo A, Socie G, Schrezenmeier H, et al. Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anemia: survival advantage for bone marrow in all age groups. Haematologica. 2012;97(8):1142–8.
Deeg HJ, Seidel K, Casper J, et al. Marrow transplantation from unrelated donors for patients with severe aplastic anemia who have failed immunosuppressive therapy. Biol Blood Marrow Transplant. 1999;5(4):243–52.
Yagasaki H, Takahashi Y, Hama A, et al. Comparison of matched-sibling donor BMT and unrelated donor BMT in children and adolescent with acquired severe aplastic anemia. Bone Marrow Transplant. 2010;45(10):1508–13.
Deeg HJ, O'Donnell M, Tolar J, et al. Optimization of conditioning for marrow transplantation from unrelated donors for patients with aplastic anemia after failure of immunosuppressive therapy. Blood. 2006;108(5):1485–91.
Anderlini P, Wu J, Gersten I, et al. Cyclophosphamide conditioning in patients with severe aplastic anaemia given unrelated marrow transplantation: a phase 1-2 dose de-escalation study. Lancet Haematol. 2015;2(9):e367–75.
Rogers ZR, Nakano TA, Olson TS, et al. Immunosuppressive therapy for pediatric aplastic anemia: a North American Pediatric Aplastic Anemia Consortium study. Haematologica. 2019;104(10):1974–83.
Hartung HD, Olson TS, Bessler M. Acquired aplastic anemia in children. Pediatr Clin N Am. 2013;60(6):1311–36.
Samarasinghe S, Steward C, Hiwarkar P, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol. 2012;157(3):339–46.
Dufour C, Veys P, Carraro E, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of EBMT. Br J Haematol. 2015;171(4):585–94.
Eapen M, Le Rademacher J, Antin JH, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. 2011;118(9):2618–21.
Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014;371(4):339–48.
Yoshimi A, Kojima S, Taniguchi S, et al. Unrelated cord blood transplantation for severe aplastic anemia. Biol Blood Marrow Transplant. 2008;14(9):1057–63.
Yamamoto H, Kato D, Uchida N, et al. Successful sustained engraftment after reduced-intensity umbilical cord blood transplantation for adult patients with severe aplastic anemia. Blood. 2011;117(11):3240–2.
Peffault de Latour R, Purtill D, Ruggeri A, et al. Influence of nucleated cell dose on overall survival of unrelated cord blood transplantation for patients with severe acquired aplastic anemia: a study by Eurocord and the aplastic anemia working party of the European group for blood and marrow transplantation. Biol Blood Marrow Transplant. 2011;17(1):78–85.
Esteves I, Bonfim C, Pasquini R, et al. Haploidentical BMT and post-transplant Cy for severe aplastic anemia: a multicenter retrospective study. Bone Marrow Transplant. 2015;50(5):685–9.
DeZern AE, Zahurak M, Symons H, et al. Alternative donor transplantation with high-dose post-transplantation cyclophosphamide for refractory severe aplastic anemia. Biol Blood Marrow Transplant. 2017;23(3):498–504.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hanna, R. (2021). Hematopoietic Cell Transplant for Severe Aplastic Anemia. In: Maziarz, R.T., Slater, S.S. (eds) Blood and Marrow Transplant Handbook. Springer, Cham. https://doi.org/10.1007/978-3-030-53626-8_23
Download citation
DOI: https://doi.org/10.1007/978-3-030-53626-8_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53625-1
Online ISBN: 978-3-030-53626-8
eBook Packages: MedicineMedicine (R0)