
Abstract
Innovations in radiochemistry and pharmacology are opening new vistas for studies of nicotinic acetylcholine receptors (nAChRs) in human brain by positron emission tomography (PET) and by single-photon emission computed tomography (SPECT). In parallel, instrumentation optimized for molecular imaging in rodents facilitates preclinical studies in models of human diseases with perturbed nAChR signalling, notably Alzheimer’s disease and other neurodegenerative conditions, schizophrenia and other neuropsychiatric disorders, substance abuse and traumatic brain injury. The nAChRs are ligand-gated ion channels composed of five subunits forming a central pore for cation flux. The most abundant nAChRs in the central nervous system are heteropentamers (designated α4β2), followed by the α7 homopentamer. We present a systematic review of published findings with the various nAChR ligands using imaging techniques in vivo, emphasizing preclinical models and human studies. Molecular PET imaging of the α4β2 nAChR subtype with the antagonist 2-[18F]fluoro-A-85380 is hampered by the long acquisition times. Newer agents such as (−)-[18F]flubatine, [18F]XTRA or [18F]nifene permit quantitation of α4β2 receptors with PET recordings lasting 90 min or less and without the toxicity risk of earlier epibatidine derivatives. The early PET studies of α7 nAChRs suffered from low pharmacological specificity, further hampered by low natural abundance of the receptor. However, several good α7 nAChR ligands such as [18F]ASEM and [18F]DBT10 have emerged in the past few years. There are still no ligands selective for α6-containing nAChRs, despite their importance for nicotine-induced dopamine release in striatum. Selective α3β4 nAChR radioligands are under development but remain untested in clinical studies of depression and addiction. Several nAChR ligands find use for pharmacological occupancy studies, and competition from endogenous acetylcholine reduces α4β2 binding site availability, a property that enables monitoring by PET of acetylcholine release in living brain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Nicotine is an addictive drug named after the French diplomat Jean Nicot, who introduced cultivation of the tobacco plant (Nicotiana tabacum) in Europe. As early as 1828, nicotine was characterized as the major pharmacologically active substance of this solanaceous herb (Posselt and Reimann 1828). Elucidation of its chemical structure followed 60 years later (Pinner and Wolffenstein 1891; Pinner 1893), and soon thereafter Amé Pictet obtained the first successful synthesis (Pictet 1903). The stimulation of sympathetic ganglia by nicotine, first observed by John Newport Langley in Cambridge (Langley 1901), led to the concept that receptors mediate drug actions (Langley 1905; Bennett 2000) and ultimately to the modern concept of molecular neurotransmission.
Nicotine and related alkaloids are present in other Solanaceae, such as tomato, potato, peppers and eggplant, and also in tea leaves (Schep et al. 2009), such that the mean daily dietary nicotine intake is about 1.4 μg per day (Siegmund et al. 1999). Nicotine absorption in the gut is rapid, and it is highly permeable to the blood-brain barrier (Oldendorf et al. 1979; Allen and Lockman 2003). Indeed, inhaled nicotine enters the brain within seconds (Rose et al. 2010), which may account for the perniciousness of smoking as a vehicle for nicotine self-administration. Within the brain, nicotine binds with high affinity to heteromeric (mainly the α4β2 subtype) and homomeric (mainly the α7 subtype) nicotinic acetylcholine receptors (nAChRs) (Changeux 2010; Bouzat and Sine 2018), which are the focus of this chapter. Signalling by the endogenous agonist acetylcholine via cerebral nAChRs is critically involved in attention, vigilance and cognition, as well as locomotion and reward mechanisms (Changeux 2010; Graef et al. 2011). Activation of nAChRs stimulates dopamine release in the basal ganglia, especially in the ventral striatum (Pradhan et al. 2002; Cumming et al. 2003), which almost certainly underlies the addictive potential of nicotine and tobacco smoking (Hogg et al. 2003). Furthermore, nAChRs play a major role in brain development (Hruska et al. 2009; Ross et al. 2010). Recently allosteric modulators of nAChRs are undergoing intensive consideration for drug development (Chatzidaki and Millar 2015; Wang and Lindstrom 2018).
Nicotinic receptors, in particular the α7 subtype, are expressed by many classes of neurons and indeed by virtually all cell types of the brain, including astrocytes (Sharma and Vijayaraghavan 2001), microglia (De Simone et al. 2005; Suzuki et al. 2006), oligodendrocyte precursor cells (Sharma and Vijayaraghavan 2002) and endothelial cells (Hawkins et al. 2005). Perhaps consistent with this protean cellular distribution, there is evidence for dysfunction of nAChRs in diverse human neurological and psychiatric diseases (Jasinska et al. 2014; Bertrand et al. 2015; Dineley et al. 2015), which motivates the present search for optimal molecular imaging agents.
The nAChRs belong to the Cys-loop superfamily of pentameric ligand-gated ion channels, which also includes the serotonin 5-HT3, γ-aminobutyric acid (GABAA and GABAC) and glycine receptors, as has been reviewed in detail (Paterson and Nordberg 2000; Taly et al. 2009; Plested 2016). Functional nAChRs consist of pentamers of homologous or heterologous subunits forming a central cation channel permeable for Na+, K+ and Ca2+ ions. There are at least 17 genes encoding the following subunits: α1–10, β1–4, δ ε and γ (Karlin 2002), all of which occur in mammals except for the avian α8 subunit. Each monomer possesses an extracellular N-terminal domain, four transmembrane helices and a small cytoplasmic region (Karlin 2002). Also, the 1.76 Å resolution X-ray structure of a nAChR homologue, the prokaryotic acetylcholine-binding protein (AChBP), has been reported (Brejc et al. 2001; Bourne et al. 2005; Hibbs et al. 2009), and a refined model of the membrane-associated nAChR from Torpedo electric organ based on 4 Å resolution electron microscopy data has been presented (Unwin 2005). An X-ray crystal structure is now available for the human α4β2 subtype, bound to nicotine (Giastas et al. 2018).
The consensus nomenclature for nAChRs is based on the predominant subunit composition of the receptor (Alexander et al. 2017). Although most functional receptors are heteromers, the α7–10 subunits form functional homomers in vivo. Most neuronal nAChRs contain α- and β-subunits only (δ, ε and γ are expressed in peripheral tissues). Of the various possible αβ permutations, nAChRs with six of the α-subunits (2–7) and three of the β-subunits (2–4) have been identified in mammalian brain, with heteromeric α4β2 nAChRs predominating (Gotti et al. 2006). The next most abundant cerebral nAChR is the homomeric α7 subtype, which is functionally distinct from the heteromeric nAChRs due to its lower affinity for the agonists acetylcholine and nicotine and higher affinity for α-bungarotoxin, an antagonistic derived from snake venom. Indeed, radiolabelled α-bungarotoxin serves admirably for selective α7 autoradiography in vitro (Clarke et al. 1984). The α7 nAChRs show relatively fast activation and have the highest permeability to Ca2+ of all nAChR subtypes, whereas the α4β2 hetero-oligomer is characterized by a high affinity for ACh and slow desensitization (Changeux 2010). A recent study indicates that heteromeric α7β2 nAChRs are also naturally present in the brain and are functionally distinct from the α7 nAChR (Wu et al. 2016).
In general, activation of nAChRs requires cooperativity between subunits, with the agonist binding sites being located at subunit interfaces (Taylor et al. 1994). Activation of nAChR heteromers requires binding of two ACh molecules at orthosteric binding sites, which are formed within the hydrophilic extracellular domain from three peptide domains on the α-subunit and three domains on the adjacent subunits (β or other) (Kalamida et al. 2007). Homomers have five acetylcholine binding sites, one between each α-α-subunit interface (Millar and Harkness 2008).
As noted above, the nAChRs can shift between functionally distinct conformational states. Four such states have been identified: resting (R), activated (A) with rapid opening within 1 ms and low affinity (μM to mM) for agonists, and two desensitized, which are closed channel states refractory to opening for intervals lasting ms (I, insensitive) or minutes (D, desensitized), although still possessing high affinity (pM to nM) for receptor agonists (Decker et al. 2000; Auerbach 2015). Thus, binding of ligands either at the orthosteric site or any of several allosteric sites alters the functional state of nAChRs by favouring particular conformational states, consequently modifying the equilibrium between the four states of the receptor (Taly et al. 2009). Activation of nAChRs increases cation influx. This has a spectrum of consequences, including (i) immediate effects, such as neurotransmitter release; (ii) short-term effects, such as receptor desensitization and recovery; and (iii) long-lasting adaptive effects, such as neuroprotection or brain plasticity via altered gene expression (Radcliffe and Dani 1998; Leonard 2003; Shen and Yakel 2009). The particular pathways of intracellular signalling evoked by activation of nAChRs are complex and cell-specific (Frazier et al. 1998; Schilström et al. 2000; Berg and Conroy 2002).
Neuronal nAChRs in the brain are localized at post-, pre-, peri- and extrasynaptic sites of cholinergic or other neurones, affording multiple ways in which to modulate brain function. Prolonged exposure of nAChRs to agonists (e.g. nicotine) results in upregulation of functional receptors (i.e. an increase in the number of [3H]nicotine binding sites) rather than the downregulation typically seen with G-protein-coupled metabotropic receptors. This upregulation is thought to occur via altered nAChR turnover, resulting in increased insertion of receptors into the cell membrane, or decreased removal via altered endocytotic trafficking or degradation rates (Peng et al. 1994; Darsow et al. 2005). The predominant nAChRs in the brain, α4β2 and α7, seem particularly sensitive to this form of post-translational regulation. In analogy to dopamine receptors (Cumming 2011), multiple affinity states of nAChRs are differentially sensitive to agonist and antagonist ligands. Exposure to acetylcholine, other agonists and pharmacological chaperones can alter the affinity states of the nAChRs receptor to influence receptor translocation and modulate the cell surface expression of nAChR subtypes (Darsow et al. 2005; Kumari et al. 2008; Govind et al. 2009; Lester et al. 2009; Crespi et al. 2018).
The total concentration of nAChRs in membrane homogenates from rodent brain is between 8 and 15 pmol/g tissue (Wang et al. 2011), and in human brain up to 10 pmol/g tissue (Shimohama et al. 1985; Marutle et al. 1998), which is similar to the abundance of dopamine receptors in striatum (Cumming 2011). The predominant heteromeric α4β2 nAChRs receptors account for some 80% of the total abundance of brain nAChRs (Wang et al. 2011). For the less abundant α7 nAChRs, a Bmax of 5 pmol/g tissue has been described in mice (Whiteaker et al. 1999), while levels of [125I]α-bungarotoxin binding in the temporal cortex were only 1 pmol/g tissue for human and monkey (Gotti et al. 2006). Notably, these studies are based on tissue homogenates. However, the native environment of nAChRs is important for accurate quantitation of nAChRs in the brain (Wang et al. 2011); a very similar phenomenon has been described for other receptors, e.g. dopamine (Cumming 2011) and opioid receptors (Quelch et al. 2014), suggesting that many receptors may be lost during the preparation of membranes or that not all receptors are externalized in the plasma membrane. Therefore, to predict the potential of nAChR radioligands for molecular imaging, binding to nAChRs should in general be tested by autoradiographic analysis of sections from frozen, intact brain.
The relatively few quantitative autoradiographic studies consistently report higher nAChR densities than homogenate-based studies. Autoradiographic studies with ligands for heteromeric α4β2 nAChRs characteristically reveal particularly intense binding in the thalamus (Fig. 18.1). Using [18F]FNEP, the Bmax for heteromeric nAChRs in human thalamus autoradiograms was 20 pmol/g tissue (Gatley et al. 1998), which is comparable to the density of binding sites of 2-[18F]fluoro-A-85380 in porcine thalamus, i.e. 46 pmol/g tissue (Deuther-Conrad et al. 2006). In quantitative receptor autoradiographic studies with the α7 selective ligands [3H]MLA and [125I]α-bungarotoxin, densities were as high as 40 pmol/g tissue in mouse brain (Whiteaker et al. 1999), whereas other studies with [125I]α-bungarotoxin have indicated densities of 4–20 pmol/g tissue in rat and 2–10 pmol/g tissue in neonate pig (Hoffmeister et al. 2011). Across species, autoradiographic studies have revealed a characteristic distribution pattern for α7 nAChRs (Fig. 18.1), which is rather diffuse, but with focally high density in the hippocampus, the colliculi and the hypothalamus (except in newborn piglet), with moderate radiotracer binding in thalamus and low expression in the cerebellum (Breese et al. 1997; Whiteaker et al. 1999; Hoffmeister et al. 2011). The regional distribution of the α7 nAChR ligand [3H]AZ11637326 in rat brain by autoradiography ex vivo (in which the tracer had been administered while the animal was alive) was consistent with autoradiography findings in vitro (Maier et al. 2011).

Representative autoradiograms of rat (upper row) and neonate pig brains (lower row) obtained with an α7 nAChR ligand ([125I]α-bungarotoxin) and the semi-selective α4β2 ligand [3H]epibatidine. For anatomic reference, the right-hand side of the figure shows comparable sagittal planes modified from the rat brain atlas (Paxinos and Watson 1998) and the pig brain atlas (Felix et al. 1999). Structures marked in the references atlases are Co cerebral cortex, Cd caudate nucleus, Cs superior colliculus, Cb cerebellum, Hi hippocampus, Th thalamus, Po pons, Me medulla oblongata
Molecular brain imaging of nAChRs in general refers to the use of radiolabelled receptor ligands, although optical imaging has been used to investigate the cholinergic system (Prakash and Frostig 2005). Furthermore, clinical imaging of α4β2 nAChR in Alzheimer’s disease is reviewed elsewhere (see Sabri et al. in PET and SPECT in Neurology). Therefore, the current review is focussed primarily, albeit not exclusively, on preclinical aspects of PET and SPECT brain imaging of the nAChR in vivo, along with a presentation on instrumentation and radiotracer development. This review is updated and extended from an earlier chapter on the same topic (Brust et al. 2014).
2 Advances in Animal PET and SPECT Technology
As noted above, studies of nAChRs abundance in vitro and ex vivo use selective radioligands in conjunction with quantitative autoradiography. However, molecular imaging with positron emission tomography (PET) or single-photon emission computed tomography (SPECT) enables the detection of neuroreceptors in the living brain. Contrary to autoradiography in vitro, imaging procedures in vivo allow for longitudinal studies in individual animals, thereby reducing intersubject variability and allowing intervention or challenge studies. Animal PET studies using clinical scanners, most having a spatial resolution of approximately 5 mm, are barely adequate for resolution of structures within mouse brain, which measures only 10 mm along its longest axis.
Although dedicated small-animal PET systems such as the Focus series (Siemens), microPET P4 (Concorde Microsystems) or ClearPET (Raytest) enable microPET imaging in rodents and small non-human primates with high resolution and sensitivity, these instruments are no longer commercially available. However, current efforts aim to improve the performance of stand-alone small-animal PET scanners by using detector systems approaching the physical limits of spatial resolution (circa 1 mm) with optimized reconstruction algorithms (Yang et al. 2016). Other approaches are to improve the spatial resolution of commercially available systems by optimizing the arrangement of detectors (Bolwin et al. 2017), or applying new detector material (Abbaszadeh and Levin 2017). Commercial stand-alone PET systems designed for small-animal imaging include the beta-CUBE (Molecubes) or VECTor (MILabs). Contemporary small-animal PET scanners increasingly combine the PET and MRI imaging modalities in one instrument. Sequential PET and MR measurements can be realized with instruments such as the nanoScan PET/MRI systems (Mediso). The PET CLIP-ON system (MR Solutions). The PET/MR 3T system (Bruker) or the PET INSERT system (MR Solutions) give simultaneous multimodal acquisitions.
Despite spatial resolution approaching 1 mm, images from small-animal PET and SPECT instruments suffer from a lack of anatomic information. In common practice, the emission images are registered to digitized brain atlases, based on histology or magnetic resonance imaging (MRI) atlases for rodent brain (Jupp et al. 2007; Rominger et al. 2010). Contemporary multimodal imaging systems combine small-animal PET with SPECT, X-ray computed tomography (CT) and/or MRI. PET-CT presents a great advantage for brain studies in that a high-resolution structural brain image in perfect registration with the PET image is obtained for each individual animal, without resorting to some standard atlas. In addition, the CT scan serves to correct the PET images for attenuation by tissue, thus providing absolute quantitation of radioactivity concentrations in the brain without requiring an additional time-consuming transmission scan. MRI offers better tissue contrast than CT, but the combining of PET and MRI instrumentation initially presented a greater technical challenge.
Initially, the instrumentation for simultaneous small-animal imaging entailed the development of PET inserts, with special efforts made by research teams in academia and industry to improve the PET detector technology with regard to MRI compatibility. An early prototype PET/MRI scanner developed at the University of Cambridge (Lucas et al. 2007) was followed by the commercial PET/MRI system for rodents developed by Mediso; its 1 tesla permanent magnet limits MRI applications but offers great flexibility for animal PET studies. PET inserts using silicone-based photoelectron multipliers are compatible with magnetic fields (Wehner et al. 2015), and a recently developed PET insert can operate within the bore of a 7 T magnet (Thiessen et al. 2016). Likewise, a SPECT camera that can be placed within an MRI magnet has recently been developed for small-animal studies (Meier et al. 2011). Whereas PET attenuation correction is usually obtained by CT scanning, MR-based attenuation correction has been demonstrated for PET and SPECT imaging using clinical scanners (Marshall et al. 2011) and has recently successfully been proven for small-animal studies (Kranz et al. 2014, 2016; Sattler et al. 2014). Despite considerable progress, simultaneous PET/MRI scanners have lower detection sensitivity in comparison with dedicated PET scanners (Cabello and Ziegler 2018; Hallen et al. 2018). However, in recent years the focus has shifted towards the development of fully integrated scanners (Ko et al. 2016; Parl et al. 2019).
Most animal PET studies are confounded by the need to use anaesthesia, which can profoundly alter radiotracer pharmacokinetics. Monkeys are trained to tolerate head fixation during PET recordings lasting as long as 30 min (Sandiego et al. 2013). The “rat conscious animal PET” (ratCAP) with a head-mounted PET detector was developed at Brookhaven for imaging in awake, behaving rats (Schulz et al. 2011). While this technology has been slow to mature, there is a recent report of FDG PET recordings in awake, behaving chickens and rats (Gold et al. 2018).
3 PET and SPECT Radioligands Targeting nAChR
Many autoradiographic studies of nAChRs in human post-mortem brain specimens have employed rather non-selective agonist ligands such as [3H]acetylcholine, [3H]nicotine, [3H/125I]epibatidine or [3H]cytisine, or alternately the antagonist ligand [3H/125I]α-bungarotoxin (Paterson and Nordberg 2000). These studies suggested the existence of at least three native receptor subtypes, i.e. α-bungarotoxin binding at homomeric α7 nAChRs and acetylcholine/nicotine binding mainly at heteromeric α4β2 nAChRs, plus a relatively small population of heteromeric receptors containing the α3 subunit. Given the predominance of α4β2 and the α7 subtypes in the brain as documented above, they have presented the main targets for molecular imaging of nAChRs. The available information on the distribution, density and functional role of other subtypes in the brain is relatively sparse (Gotti et al. 2006; Sharma and Vijayaraghavan 2008; Zoli et al. 2015). Furthermore, the current emphasis on α4β2 and the α7 subtypes as targets for pharmaceutical development may account for the paucity of selective high-affinity drugs for the other subtypes (Gündisch and Eibl 2011) due to a kind of circularity.
3.1 Radioligands for α4β2 nAChRs
Reviews summarizing the history of ligand development for the nAChRs (Sihver et al. 2000a, b; Ding and Fowler 2005; Horti and Villemagne 2006; Horti et al. 2010) have helped galvanize research groups to generate new lead compounds for molecular imaging and therapeutics. Selectivity and high affinity (nM) in vitro are necessary but not sufficient for successful visualization of nAChRs. In general, PET and SPECT tracers, if they are to be successful, must fulfil a multitude of additional criteria. Chief among these are (1) low non-specific binding and absence of brain-penetrating radiometabolites, (2) rapid clearance from non-specific brain regions and plasma to reduce background in the target tissue, (3) high membrane permeability, (4) high permeability and low efflux at the blood-brain barrier (BBB) and (5) attainment of equilibrium binding with a tolerable time interval. Settling upon optimal tracers is often a matter of fierce competition.
The vast majority of novel tracers for α4β2 nAChRs derive from three compounds: nicotine, epibatidine and 3-pyridyl ether. Seven of these tracers have so far been used successfully to image α4β2 receptors in human brain: S-[11C]nicotine (Nordberg 1993); the two halogen-substituted derivatives of A-85380, namely, 2-[18F]fluoro-A-85380 (Kimes et al. 2003) and 6-[18F]fluoro-A-85380 (Ding et al. 2000a; Horti et al. 2000); the two epibatidine derivatives (−)-[18F]flubatine and (+)-[18F]flubatine (Sabri et al. 2018; Tiepolt et al. 2018); [18F]nifene (Betthauser et al. 2017); and [18F]XTRA (Coughlin et al. 2018c). Receptor-ligand interactions frequently entail stereoselective features (Smith and Jakobsen 2007), as has been formally demonstrated for the case of nAChRs in human PET studies with the two stereoisomers of [11C]nicotine (Nordberg et al. 1991, 1992), and also several of the radioligands discussed below. A summary of the compounds that have been investigated for imaging of α4β2 nAChRs with PET and SPECT, their associated references, binding affinities and results of biodistribution ex vivo and PET imaging studies is given in Table 18.1.
3.1.1 Nicotine Derivatives
The tracer S-[11C]nicotine, one of the very first positron-emitting receptor ligands (Maziere et al. 1976), was initially developed to investigate the biodistribution of nicotine in the context of tobacco addiction and for investigation of diabetes insipidus and for insecticide research (Soloway 1976; Gündisch 2000). With the advent of PET, S-[11C]nicotine was tested for imaging nAChRs in human brain (Nordberg 1993). However, co-administration of unlabelled nicotine failed to displace much of the radioligand, indicating that the PET signal did not sensitively reveal specific binding to α4β2 nAChRs (Nybäck et al. 1994); cerebral S-[11C]nicotine uptake proved mainly to be determined by blood flow, rather than local abundance of nAChRs in vivo (Gündisch 2000), this in keeping with its considerable lipophilicity. Indeed, labelled nicotine serves admirably as a cerebral blood flow tracer.
3.1.2 Cytisine Derivatives
The nAChR agonist [3H]cytisine is a useful radioligand for characterization of α4β2 nAChRs in vitro. However, the cytisine derivatives [11C]ABT-418 and N-[11C]methylcytisine (Valette et al. 1997) failed in vivo due to their low cerebral uptake and rapid washout. Another derivative, [11C]MPA, showed high-affinity binding to α4β2 nAChRs (~10–100-fold higher than [11C]ABT-418 and S-[11C]nicotine) in membranes from rat forebrain (Sihver et al. 1998). Furthermore [11C]MPA showed rapid uptake into monkey brain, with similar permeability as [11C]ABT-418 and S-[11C]nicotine. Pre-administration of unlabelled S-nicotine (0.02 mg/kg) decreased the peak uptake of [11C]MPA in monkey brain by about 20%, indicating the presence of some specific binding to nAChRs (Sihver et al. 1999b), but no further studies have been published with this ligand. [18F]fluoropyridinylcytisine was developed as another candidate radioligand for α4β2 nAChR imaging. However, its distribution in living rat brain did not match the regional distribution of nAChRs, and blocking studies with nicotine failed to demonstrate specific binding of this tracer (Roger et al. 2003).
3.1.3 Epibatidine Derivatives
Epibatidine, an alkaloid from the skin of the Ecuadoran poison arrow frog Epipedobates anthonyi, has long been known for its high-affinity binding to heteromeric nAChRs (Daly 1998). It has considerable toxicity due to its potent activation of many different neuronal nAChR subtypes (Avalos et al. 2002), notably the α3β4 nAChR (Tomizawa et al. 2001; Avalos et al. 2002). Nonetheless, radiolabelled epibatidine derivatives have found limited use in human PET studies (Bohnen and Frey 2007). Theoretically, subtype-specific analogues of epibatidine might have favourable tracer properties with lesser toxicity (Avalos et al. 2002). Although [18F]NFEP ([18F]FPH, norchlorofluoroepibatidine) and [18F]N-Me-NFEP showed good brain uptake and signal-to-background ratios in mouse and baboon brain (Ding et al. 1996; Dolci et al. 1999), their toxicity, even when prepared at high specific activity, was too high for use in man (Horti et al. 1997; Molina et al. 1997; Villemagne et al. 1997; Ding et al. 1999). However, Horti and co-workers successfully synthesized epibatidine derivatives with lesser toxicity (Horti et al. 1998a).
Fluorine-18-labelled FPhEP (Roger et al. 2006), a functional nAChR antagonist with much reduced toxicity, had faster brain kinetics in baboon than did 2-[18F]fluoro-A-85380 (discussed below), whereas its fluorophenyl analogue [18F]F2PhEP had higher specific binding (Valette et al. 2007). Its binding on PET was quantified as binding potential (BPND), which is proportional to the ratio Bmax/Kd. However, nicotine failed to displace either radioligand, indicating low specific binding. Patt and co-workers developed N-[11C]methylepibatidine and N-[11C]methylhomoepibatidine (Patt et al. 1999, 2001) and compared the uptake and binding of N-[11C]methylepibatidine enantiomers in the brain of living mouse, rat and pig. Whereas the (−)-enantiomer showed slower uptake and gradual accumulation in the brain, the (+)-enantiomer had very rapid uptake and washout, indicating distinct binding mechanisms. Because of its better kinetics and higher selectivity, N-[11C]methyl-(−)-epibatidine was investigated in pigs by PET, showing high brain uptake. However, steady-state binding in the highest binding regions (thalamus) was not attained within 1 h recordings, which is a disadvantage for quantitation of carbon-11-labelled tracers, due to the 20 min physical half-life. Furthermore there was high toxicity, precluding its use in humans (Patt et al. 1999). The analogue N-[11C]methylhomo-epibatidine had a better toxicity profile; as with N-[11C]methylepibatidine, the (−)-enantiomer of N-[11C]methylhomo-epibatidine showed high uptake in pig brain, while the (+)-enantiomer was rapidly washed out. Although findings of N-[11C]methyl-(−)-homoepibatidine binding in pig brain suggested suitability for PET imaging, its toxicity in mice and rats was comparable to that of N-methylepibatidine, again precluding its safe use in humans (Patt et al. 2001).
Further development of epibatidine derivatives has focussed on maintaining or improving kinetic profiles while reducing toxicity. One such compound, 2-(6-chloro-5-phenylpyridin-3-yl)-7-[11C]methyl-7-aza-bicyclo[2.2.1] heptane, showed a thalamus/cerebellum ratio of 4.2 at 90 min after injection, indicating high specific binding in rat brain, which was displaceable by nicotine treatment (1 mg/kg). A preliminary PET study of this tracer in a baboon revealed fast brain uptake and high thalamic binding consistent with α4β2 nAChR distribution. However binding equilibrium was not reached within 2 h, which is the absolute limit for PET recordings with 11C-labelled radioligands (Huang et al. 2004).
Mu et al. synthesized another series of labelled epibatidine and homoepibatidine analogues (Mu et al. 2006). Of these, the 8-[11C]methyl-8-aza-bicyclo[3.2.1]octane derivative had a double bond conjugated with the pyridine nucleus, thus restricting free rotation of the pyridine ring. It showed high affinity (2 nM) in vitro and at least 100-fold selectivity for α4β4 over α7 nAChRs. Furthermore, its toxicity was 50-fold lower than for epibatidine. Although promising for PET studies, there are no further reports on its evaluation. The antagonistic epibatidine derivative (±)-[11C]NMI-EPB (Ding et al. 2006) had 2.5-fold higher uptake in the baboon brain than did the 3-pyridyl ether 2-[18F]fluoro-A-85380, which is discussed in more detail below (Ding et al. 2006). Surprisingly, separation of the (±)-[11C]NMI-EPB enantiomers revealed that (+)-[11C]NMI-EPB had fast kinetics and but low affinity, whereas (−)-[11C]NMI-EPB appeared to be suitable for imaging but had a slow kinetics (Gao et al. 2007a).
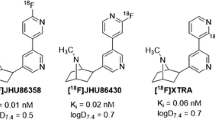
Two further sets of epibatidine analogues (Gao et al. 2007a, 2008b) were subsequently developed by the group from Johns Hopkins University, including (−)-[18F]JHU87522 (now termed [18F]AZAN) (Gao et al. 2008b; Horti et al. 2010), which had promising properties with respect to brain uptake, kinetics, metabolic stability and low toxicity. Results of [18F]AZAN toxicology and human radiation dosimetry have been reported (Horti et al. 2010). PET studies in baboons confirmed that [18F]AZAN rapidly enters brain, attaining steady state within 90 min after injection (Kuwabara et al. 2012). Furthermore, blocking experiments with cytisine showed [18F]AZAN to bind specifically to β2-containing (predominantly α4β2) nAChRs, supporting its suitability for nicotinic drug evaluation. Based on its exceptionally high affinity in vitro and improved lipophilicity over 2-[18F]FA, another epibatidine analogue, (−)-[18F]JHU86428 ([18F]XTRA), was proposed as a potential tracer for the less abundant extrathalamic α4β2 nAChRs (Gao et al. 2008b, Horti et al. 2010). An improved radiosynthesis methodology has been reported (Gao et al. 2010). Following successful imaging in non-human primates (Kuwabara et al. 2017), [18F]XTRA proved fit for quantitation of α4β2 nAChRs in hippocampus of healthy humans and showed an age-dependent decline (Coughlin et al. 2018c).
Toxicity of epibatidine analogues arises from high affinity for the ganglionic α3β4 receptors. This has motivated the search for derivatives with higher α4β2 selectivity. To this end, 18F-labelled stereoisomers of the chloro-fluoro-substituted homoepibatidine analogue, flubatine (previously called NCFHEB), have been synthesized. The flubatine enantiomers both bind with subnanomolar affinity to membranes from rat thalamus or HEK293 cells expressing the human α4β2 nAChR (Deuther-Conrad et al. 2004), with twofold higher affinity for the (+)-enantiomer. Previous work shows that fluoro- and norchloro-analogues of epibatidine have selectivity for β2-containing receptors (Avalos et al. 2002). Indeed, the affinity of both flubatine enantiomers for α4β2 nAChRs was comparable to that of epibatidine, but affinity to ganglionic α3β4 nAChRs was 20–60-fold lower (Deuther-Conrad et al. 2004). The increased subtype selectivity of flubatine seemingly results in lesser pharmacological side effects compared to epibatidine; injection of 25 μg/kg (+)-flubatine or (−)-flubatine to awake mice was without important pharmacological effects (Deuther-Conrad et al. 2008). The doses encountered in a human PET study with [18F]flubatine was 1000-fold lower (Vaupel et al. 2005), which entails a considerable margin of safety. In addition to its selectivity for α4β2, (−)-flubatine and (+)-flubatine also had considerably better selectivity for α4β2 over α7 receptors than did (−)-epibatidine (Deuther-Conrad et al. 2004).
N-methyl and N-ethyl derivatives of flubatine have been synthesized but displayed lower target affinities and were consequently not considered for radiolabelling (Deuther-Conrad et al. 2004). Similar distribution patterns for (+)-[18F]flubatine and (−)-[18F]flubatine were observed in mice, rat and porcine brain (Brust et al. 2008; Deuther-Conrad et al. 2008; Sabri et al. 2008). Allen et al. provided evidence that nicotine analogues are transported into the brain via the blood-brain barrier (BBB) choline transporter (Allen et al. 2003); this mechanism may also be involved in the brain uptake of epibatidine and homoepibatidine derivatives (Deuther-Conrad et al. 2008). Indeed, addition of flubatine to the incubation medium inhibited with an IC50 of 370 ± 90 μM the uptake of [3H]choline in immortalized rat brain endothelial cells, which are known to express the blood-brain barrier (BBB) choline transporter (Deuther-Conrad et al. 2008). This result is comparable to the Ki of 65 μM obtained for hemicholinium-3 in the same experimental system (Friedrich et al. 2001). Furthermore, in vivo experiments in rats have confirmed the postulated interaction of flubatine with the BBB choline transporter; 50 μM flubatine reduced the transport rate of [3H]choline by 21%, whereas equimolar epibatidine resulted in a ~ 40% reduction (Deuther-Conrad et al. 2008). The stronger interaction of epibatidine is consistent with its higher uptake in mouse brain (London et al. 1995) compared to (+)-[18F]flubatine or (−)-[18F]flubatine.
PET studies in young pigs were performed to compare the brain uptake and kinetics of (+)-[18F]flubatine and (−)-[18F]flubatine with that of 2-[18F]fluoro-A-85380, a 3-pyridyl ether discussed in detail below. The brain uptake of both enantiomers proved to be two- to threefold higher than that of 2-[18F]fluoro-A-85380. The binding equilibrium of (−)-[18F]flubatine was reached significantly earlier (~ 60 min p.i.) than that of the (+)-enantiomer (Brust et al. 2008), consistent with its lesser affinity in vitro. The specific binding of (−)-[18F]flubatine in porcine brain was comparable to that of 2-[18F]fluoro-A-85380, but (+)-[18F]flubatine displayed about twofold higher specific binding. Thus, both [18F]flubatine enantiomers may present advantages over 2-[18F]fluoro-A-85380 for application in human PET studies, especially as pertains to the time to equilibrium binding.
The metabolites of (+)-[18F]flubatine in pig urine and plasma have been determined by HPLC-MS (Ludwig et al. 2018). Displaceable binding of (−)-[18F]flubatine in human cerebellum upon smoking (Bhatt et al. 2018) raises a red flag for its quantitation through reference tissue methods, which assume absence of specific binding in the reference region. Nonetheless, (−)-[18F]flubatine PET studies in non-human primate showed complete displacement with nicotine, revealing a non-displaceable distribution volume (VND) of 6 mg/ml, and BPND of 4 in the thalamus, 1 in the frontal cortex and putamen and only 0.1 in the cerebellum (Bois et al. 2015), consistent with an earlier semi-quantitative analysis in non-human primate (Hockley et al. 2013). The extensive displaceable binding in human cerebellum might reflect a species difference.
3.1.4 3-Pyridyl Ethers
First developed as experimental treatments for Alzheimer’s disease, the 3-pyridyl ethers were identified as promising radioligands for nAChRs imaging (Gündisch 2000; Horti and Villemagne 2006; Horti et al. 2010). The 3-pyridyl ethers are equipotent to epibatidine at the mainly α4β2 nAChR [3H]cytisine binding sites in the brain but are 100-fold less potent than epibatidine as agonists at α3β4 nAChRs (Abreo et al. 1996). As noted above, this predicts larger dose safety margins with minimal cardiovascular or other toxic side effects. The prototype compound A-85380 has similar binding affinities at recombinant α2β2, α3β2 and α4β2 nAChRs in vitro (Xiao and Kellar 2004). In addition, the iodinated derivative 5-[125I]iodo-A-85380 binds with high affinity to α6β2β3 nAChRs in monkey and rat striatum (Kulak et al. 2002). Therefore, 3-pyridyl ethers are properly regarded as β2-selective compounds (Jensen et al. 2005; Lai et al. 2005), also considering their lack of affinity for the α7 nAChR (Sullivan et al. 1996).
As noted above, 2-[18F]fluoro-A-85380 and 6-[18F]fluoro-A-85380 (Horti et al. 1998b, 2000) showed early promise for PET imaging, having less toxicity than epibatidine analogues. Of the two, 6-[18F]fluoro-A-85380 (Scheffel et al. 2000) had superior kinetics, characterized by earlier peak and faster clearance from the brain (Ding et al. 2004), and better target-to-background ratios than were obtained with 2-[18F]fluoro-A-85380 in a comparative study in baboon (Ding et al. 2000a). Although 6-[18F]fluoro-A-85380 has nonetheless not yet found wide use, both derivatives proved successful in human brain imaging (see Sabri et al. in PET and SPECT in Neurology). The SPECT analogue 5-[123I]iodo-A-85380 was tested in non-human primates (Chefer et al. 1998), and has also served for nicotine challenge studies in human smokers (Esterlis et al. 2010a), as described below. Although suffering from slow kinetics, 5-[123I]iodo-A-85380 was sensitive to competition from endogenous acetylcholine (Fujita et al. 2003), a property also to be discussed in some detail below. The methyl-substituted derivative 5-[123I]iodo-A-84543 (Henderson et al. 2004) displayed faster kinetics, but homogenous uptake of radioactivity in baboon brain, in contrast to the spatially heterogeneous pattern of specific binding for the analogue [11C]A-84543 seen in mouse brain (Kassiou et al. 1998). PET studies in pigs with [11C]A-186253, a structurally similar tracer, showed little displacement by cytisine, indicating excessive non-specific binding (Itier et al. 2004).
The comparably low brain uptake of 2-[18F]fluoro-A-85380 was similar to that previously found with 5-[11C]methyl-A-85380 (2.2% ID/g brain tissue at 30 min p.i. (Iida et al. 2004)). This may be related to their rather high polarity (Zhang et al. 2004), which is a critical parameter for brain radiotracer uptake (Waterhouse 2003). Nevertheless 2-[18F]fluoro-A-85380 was successfully used to image nAChRs in non-human primates (Chefer et al. 1999, 2003; Valette et al. 1999; Le Foll et al. 2007) and in rat microPET studies (Vaupel et al. 2007). An optimized radiosynthesis of 2-[18F]fluoro-A-85380 and improved analytical techniques (Mitkovski et al. 2005; Schmaljohann et al. 2005; Kimes et al. 2008) have facilitated its use in human PET studies (Ellis et al. 2009b) (Lotfipour et al. 2012b). Despite the disadvantage of its slow binding kinetics, requiring interrupted or continuous PET recordings lasting at least 6 h (Gallezot et al. 2005; Horti and Villemagne 2006), 2-[18F]fluoro-A-85380 remains the most frequently utilized ligand in human PET studies (see Sabri et al. in PET and SPECT in Neurology); despite its limitations, it remains for the present the standard against which other nAChR PET ligands have been compared.
A series of 5-substituted-6-halogeno derivatives of A-85380 have potentially improved lipophilicity and affinity (Zhang et al. 2004). PET studies in rhesus monkey with two such [18F]-labelled derivatives showed higher lipophilicity than for 2-[18F]fluoro-A-85380, resulting in enhanced target-to-background ratios. Imaging studies with another 5-substituted A-85380 derivative, [11C]5-MA (Iida et al. 2004), demonstrated lower total brain uptake and lower target-to-background ratios than for 2- or 6-[18F]fluoro-A-85380. A further analogue, [18F]ZW-104 has had initial testing (Kozikowski et al. 2005; Valette et al. 2009; Saba et al. 2010). In baboon PET studies, [18F]ZW-104 showed regional radioactivity distribution resembling that of 2-[18F]fluoro-A-85380 and some superior properties, including higher accumulation in the brain, earlier peak uptake in the thalamus and faster washout kinetics. However, it also displayed considerable affinity for α3β2 and α2β2 receptors in vitro (Valette et al. 2009) and rather high non-displaceable (by nicotine) uptake in the striatum, a region with comparably low density of nAChRs.
Many further derivatives have been tested in rodent and non-human primate PET studies: [76Br]BAP (Sihver et al. 1999a); [76Br]BrPH (Kassiou et al. 2002); [11C]Me-p-PVC (Brown et al. 2004) and its analogues [11C]JHU85208, [11C]JHU85157 and [11C]JHU85270 (Gao et al. 2007b, 2009); [18F]nifene (Pichika et al. 2006; Easwaramoorthy et al. 2007); and two carbon-11- and fluorine-18-labelled isotopomers of one pyridine-derived ligand. While some of these ligands had better kinetics than 2-[18F]fluoro-A-85380, their low BPND, high non-specific binding or non-selectivity discouraged further development for human imaging (Easwaramoorthy et al. 2007). Despite this, specific binding of [18F]nifene was later attributed entirely to α4β2 nAChRs based on studies in β2-knockout mice (Bieszczad et al. 2012), and subsequent human studies demonstrated the safety and test-retest reliability of [18F]nifene (Betthauser et al. 2017; Lao et al. 2017). This was further supported by a study showing no significant decline in [18F]nifene binding over five decades of healthy human ageing (Mukherjee et al. 2018). Despite the partial selectivity of the tracer for α4β2 nAChRs, there was low binding in human habenula (which contains α3β2 nAChRs) and in the red nucleus (which contains abundant α2β2 nAChRs).
PET imaging of α4β2 nAChRs using the pyridyl ether analogue [18F]nifrolidine has been tested in non-human primate (Chattopadhyay et al. 2005). Although having favourable kinetics, the thalamus-to-cerebellum ratio was lower than that of other α4β2-targeting pyridyl ether analogues (Chattopadhyay et al. 2005), attaining a value of 4 at several hours after administration in non-human primate and showing considerable displaceability by nicotine (Pichika et al. 2013). Similarly, there have been no follow-up reports on the nifrolidine homologue [18F]nifzetidine (Mukherjee et al. 2004), except for a report showing a continuous increase of the thalamus/cerebellum ratio up to 3 h after administration to non-human primates (Pichika et al. 2011). Such slow kinetics is unfavourable for quantitation.
The series of pyridyl ether-based compounds [18F]NIDA52189, [18F]NIDA522131 and [18F]NIDA52289 have been synthesized and evaluated by PET in rhesus monkeys (Horti and Villemagne 2006), especially intended for imaging of the relatively sparse extrathalamic α4β2 nAChRs. Among these, [18F]NIDA52189 (Zhang et al. 2004) and [18F]NIDA522131 (Chefer et al. 2008) were deemed superior to 2-[18F]FA with respect to extrathalamic binding but suffered from slow kinetics in vivo.
3.1.5 Non-Epibatidine-and-Non-A-85380-Related Compounds
Carbon-11-labelled Me-p-PVC (Brown et al. 2002) and p-PVP-MEMA, which were selected from the class of (4-pyridinyl)vinylpyridines developed by Abbott Laboratories, possessed picomolar affinity towards α4β2 receptors. Nevertheless, [11C]Me-p-PVC had low BPND in thalamus of non-human primate (Brown et al. 2004), and [11C]p-PVP-MEMA had a low target-to-background ratio in a preliminary PET study (Dollé et al. 2008).
3.2 Imaging of Heteromeric β4-Containing nAChR Subtype
The putative β4-selective agonist [11C]SIB-1553A was assessed by biodistribution and ex vivo brain autoradiography in rats (Sobrio et al. 2008). Its low specific binding did not encourage further development, and there was no attempt made to separate and individually investigate the enantiomers of this racemic radioligand.
3.3 Radioligands for α7 nAChRs
Efforts to develop a radiopharmaceutical for PET imaging of α7 nAChR have met with growing success in the past few years. Structurally diverse classes of compounds meet the steric and electronic requirements of this binding site, as reviewed recently (Brust et al. 2012; Mo et al. 2014). As noted above, the cerebral expression of α7 nAChR is comparatively low, constituting perhaps one quarter of the density for α4β2 receptors (Spurden et al. 1997; Whiteaker et al. 1999; Hellström-Lindahl and Court 2000). Because of this low natural abundance of α7 sites, high affinity is particularly important for an effective PET tracer. Binding properties of various α7 tracers tested to date are summarized in Table 18.2.
3.3.1 Quinuclidine-Based Ligands
The lead compound of a series of azabicycle carbamate α7 receptor agonists developed by Astra Laboratories has been labelled with carbon-11 and evaluated in rats (Dollé et al. 2001). Despite having relatively good brain uptake, no regionally selective or specific binding could be seen. Another series of potential α7-selective imaging agents based on the quinuclidine moiety has been labelled with carbon-11 and iodine-125 (Pomper et al. 2005). Target selectivities of these compounds were modest, and the most affine compounds had significant binding to the 5-HT3 receptor, a structural homologue of α7 nAChR (Zwart et al. 2004).
3.3.2 GTS-21
Other potential α7 ligands originate from benzylidene anabasein compounds such as GTS-21 (de Fiebre et al. 1995; Meyer et al. 1998). Indeed, GTS-21 (3-(2,4-dimethoxybenzylidene)-anabaseine) showed early promise as a α7 nAChR agonist medication for improving cognition in patients with schizophrenia (Freedman et al. 2008; Tregellas et al. 2011). It has been labelled with iodine-123 (Zhang et al. 2001) and carbon-11 (Kim et al. 2007b). Consistent with the relatively low affinity and specificity of GTS-21 for α7 nAChRs, the distribution and kinetics of 5- [123I]GTS-21 and 2- [11C]GTS-21 in the brain of living baboon and mice were dominated by non-specific binding (Kim et al. 2007a).
3.3.3 Diazabicyclononane Derivatives
The 1,4-diazabicyclo-[3.2.2]nonane skeleton (Bunnelle et al. 2004) was identified as a motif for α7 nAChR ligands, and the two novel diazabicyclononane-derived PET ligands [76Br]SSR180711 and [11C]CHIBA-1001 were evaluated by PET in conscious non-human primates, with testing in a model of schizophrenia (Hashimoto et al. 2008). Of the two tracers, [11C]CHIBA-1001 demonstrated superior accumulation in the brain, revealing a heterogeneous regional distribution consistent with the localization of α7 in the brain; specific binding was blocked by selective α7 but not α4β2 agonists revealing some potential for measuring occupancy by pharmaceuticals at α7 nAChRs. A first clinical PET study confirmed the suitability of [11C]CHIBA-1001, although the regional binding differences were small in the human brain (Toyohara et al. 2009). Notably, α7-specific binding could not be demonstrated in vitro: there was no displacement of 30 nM [3H]CHIBA binding from rat brain membranes by 1 μM α-bungarotoxin (Tanibuchi et al. 2010). Another rodent study reported low in vitro binding affinity of [3H]CHIBA and poor in vivo selectivity to α7 nAChRs in rodent brain (Ding et al. 2012). Furthermore, differences in the regional distribution of the binding sites of CHIBA-1001 relative to [125I]α-bungarotoxin were evident in monkey and human brain samples (Tanibuchi et al. 2010).
The 1,4-diazabicyclo[3.2.2]nonane derivatives developed by NeuroSearch emerged as promising PET ligands for imaging of cerebral α7 nAChR (Peters et al. 2007). Two carbon-11 radioligands were developed, [11C]NS12857 (Lehel et al. 2009) and [11C]NS14492 (Ettrup et al. 2011), along with the fluorine-18 compounds [18F]NS10743 (Deuther-Conrad et al. 2009) and [18F]NS14490 (Rötering et al. 2014). The cerebral uptake of these three 1,4-diazabicyclo[3.2.2]nonane derivatives exceeded that of [11C]CHIBA-1001. Although the uptake of [11C]NS12857 was not displaced by α7 nAChR-selective compounds, specific binding was clearly evident for [11C]NS14492, [18F]NS14490 and [18F]NS10743, the ligands with higher target affinity (Brust et al. 2012).
NS14492 has been labelled with tritium to allow in vitro autoradiographic studies on α7 nAChR assessing α7 nAChR density in the porcine brain and by extension other species (Magnussen et al. 2015). Figure 18.2 shows the autoradiographic comparison of a saturating concentration of [125I]α-bungarotoxin and [3H]NS14492 in whole brain sections from newborn piglets. Brain region-specific binding pattern of [3H]NS14492 is very similar to that of [125I]α-bungarotoxin, with the non-specific binding being considerably lower. Furthermore, the use of [3H]NS14492 provides a higher resolution, which is especially noticeable in the laminar layers of the neocortex. [3H]NS14492 therefore is an interesting alternative to [125I]α-bungarotoxin when assessing α7 nAChR density in the porcine brain and by extension other species.

Autoradiographic comparison of [125I]α-bungarotoxin and [3H]NS14492 binding to brain sections from newborn piglets. For anatomic reference see Fig. 18.1
The general suitability of the diazabicyclononane derivatives for PET imaging of α7 nAChRs was shown by preclinical PET studies in pigs (Lehel et al. 2009; Deuther-Conrad et al. 2011; Ettrup et al. 2011), although the magnitude of BPND, about 0.5, is rather low for a useful PET tracer. In view of the low natural abundance of the α7 nAChR in the brain, a substantial increase in α7 affinity of PET radiotracers may be required for sensitive quantitation (Brust et al. 2012); target affinities of [11C]CHIBA-1001 (Ki ~ 35 nM) (Hashimoto et al. 2008; Toyohara et al. 2009) and [18F]NS10743 (Ki ~ 10 nM) (Deuther-Conrad et al. 2009) do not predict adequate specific signal in vivo, given the low Bmax (Koeppe 2001). NS14490, a novel diazabicyclononane derivative, with a Ki of 3 nM may be more promising in this regard (Brust and Deuther-Conrad 2012). The distribution of [18F]NS14490 binding in mouse brain autoradiograms correlated with the known pattern of α7 nAChR expression and was displaced with the α7 nAChR ligand methyllycaconitine (Brust and Deuther-Conrad 2012).
With tilorone, an amphiphilic molecule possessing high interferon-inducing potential, a novel α7 nAChR pharmacophore has been identified (Briggs et al. 2008; Schrimpf et al. 2012), leading to a new series of diazabicyclononane-substituted dibenzothiophene derivatives for PET imaging developed independently by two groups (Gao et al. 2013; Scheunemann et al. 2014). The 2-fluoro dibenzothiophene sulfone derivative (DBT10) and 4-fluoro dibenzothiophene sulfone derivative (ASEM) have been identified as potential α7 nAChR imaging agents, as shall be discussed in more detail below. In addition, a small library of further tilorone-based derivatives was synthesized to explore further the impact of the isomeric effect and effects of different cationic centres on the ligand features. However, an isomer, substituted with fluorine and the cationic centre in the same benzo ring, did not achieve criteria for further 18F-labelling. Increased flexibility of the tertiary amine of the cationic centre 9-methyl-3,9-diazabicyclo[3.3.1]nonane and 3-methyl-3,8-diazabicyclo[3.2.1]octane resulted in a remarkable loss of binding affinity (Teodoro et al. 2015). Most recently, a series of novel fluoren-9-one-based diazabicyclononane derivatives has been developed, showing low nM affinity towards the α7 nAChR and >1000-fold selectivity over the α4β2 nAChR (Teodoro et al. 2018; Wang et al. 2018). Two derivatives were radiolabelled leading to the corresponding carbonyl bioisosteres of [18F]DBT10 and [18F]ASEM. The carbonyl derivative of [18F]DBT10 exhibited high initial brain uptake (12% ID/g at 15 min post-injection) and displaceable binding (Wang et al. 2018).
Both [18F]DBT10 and [18F]ASEM have been investigated as PET tracers in non-human primates, showing a favourable kinetic profile for quantitation of the α7 nAChR in living brain (Horti et al. 2014; Hillmer et al. 2016b). Recently, a direct comparison in rhesus monkey of [18F]DBT10 and [18F]ASEM indicated very similar pharmacokinetics (Hillmer et al. 2017). A blocking study with [18F]DBT10 in pigs revealed a 75% decrease of the binding potential BPND after treatment with 3 mg∙kg−1 i.v. of the α7 nAChR partial agonist NS6740 (Fig. 18.3) (Teodoro et al. 2015). The total distribution volume (VT; ml g−1) of [18F]ASEM has been quantified in non-smoking healthy volunteers over a broad range of ages, showing a positive correlation between [18F]ASEM VT and age in various brain regions of interest, with VT increasing from 20 to 30 ml g−1 (Coughlin et al. 2018b). Occupancy by the experimental drug DMXB-A at central α7 nAChRs could be estimated from [18F]ASEM binding changes in the brain of healthy volunteers (Wong et al. 2018).

Specific uptake values (SUV) of [18F]DBT10 in porcine brain under baseline and blocking (3 mg∙kg−1 NS6740 i.v.) conditions
Given this success and in expectation of future human applications, an automated cGMP-compliant radiosynthesis of [18F]DBT10 was established, and toxicity and radiation dosimetry studies were performed. The single-dose toxicity in rats (No-Observed-Effect-Level = 620 μg∙kg−1) and the effective dose estimated from mouse and pig studies (12.7 and 13.7 μSv/MBq, resp.) indicated the safe use of [18F]DBT10 in human PET studies ((Kranz et al. 2014; Teodoro et al. 2015).
3.3.4 [11C]A-582941 and [11C]A-844606
A new series of octahydropyrrolo[3,4-c]pyrrole derivatives was described by Abbott Laboratories, two of which were selected for labelling with carbon-11 (Toyohara et al. 2010). Whereas no regional heterogeneity or displaceable binding was evident in mouse brain ex vivo, pretreatment with an α7-specific agonist decreased the total distribution volumes of both tracers in conscious monkey PET studies, indicative of a BPND close to 0.5, as with the NeuroSearch (NS) compounds cited above.
3.3.5 [125I]I-TSA
A diazabicyclooctane-derived PET ligand with high affinity and selectivity has been radiolabelled and evaluated in mice (Ogawa et al. 2006). Despite a subnanomolar affinity, the high non-specific binding made [125I]I-TSA inadequate for imaging of brain α7 receptors.
3.3.6 R-[11C]MeQAA
The two enantiomers of [11C]MeQAA, an azabicyclooctylester-derived compound, were evaluated in mice and conscious monkey PET studies (Ogawa et al. 2009). Although (R)-[11C]MeQAA showed target-specific accumulation, the in vivo selectivity was insufficient due to binding to the serotonin 5HT3-R. Nonetheless, the tracer was used more recently in a multitracer study of aged monkeys (Nishiyama et al. 2015). The hippocampal binding of (R)-[11C]MeQAA correlated inversely with binding of a marker for mitochondrial complex I and positively with the binding of a marker for beta-amyloid deposition in the aged animals. The authors interpreted their results to indicate a significant (adaptive?) upregulation of α7 nAChRs in metabolically compromised and degenerating brain tissue.
3.4 Radioligands for α3β4 nAChRs
Recently, a series of quinuclidine anti-1,2,3-triazole derivatives was synthetized with the aim of developing an 18F-labelled radioligand for imaging the α3β4 nAChR in the brain. This subtype attracts interest because of its involvement in drug addiction and depression pathways (Rahman et al. 2015). In contrast to α4β2 and α7, the α3β4 subtype expression is mainly in the autonomic ganglia (hence the toxicity of epibatidine) but also in some specific brain regions and neuronal subpopulations. These regions notably include the medial habenula, nucleus interpeduncularis, dorsal medulla, pineal gland and retina (Gotti et al. 2009). Binding studies in vitro revealed that stereochemistry at the C3 position of the quinuclidine scaffold plays an important role in the nAChR subtype selectivity (Sarasamkan et al. 2016). Whereas the (R)-enantiomers are selective to α7 over α4β2 (by factors of 44–225) and to a smaller degree over α3β4 (3–33), their (S)-counterparts prefer α3β4 over α4β2 (62–237) as well as over α7 (5–294). Two potent compounds (S)-T1 and (S)-T2 were identified that bind selectively to α3β4 nAChR over α7 nAChR. The compound (S)-T1 was chosen for radiolabelling and first preclinical evaluation (Sarasamkan et al. 2017). The brain uptake and the brain-to-blood ratio of (S)-[18F]T1 in mice at 30 min post-injection were 2.02 (SUV) and 6.1, respectively. According to an ex vivo analysis, the tracer remained intact (>99%) in the brain. Only one major radiometabolite was detected in plasma and urine samples. In vitro autoradiography on pig brain slices revealed binding of (S)-[18F]T1 to brain regions associated with the expression of α3β4 nAChRs, which could be reduced by the α3β4 nAChR selective drug AT-1001. These findings suggest (S)-[18F]T1 as a promising tool for non-invasive PET imaging of α3β4 nAChRs in the brain.
4 nAChR Imaging of Neurodegenerative Diseases
Reductions in cortical nAChR binding have been found in patients with diverse forms of neurodegeneration, including Alzheimer’s disease, Parkinson’s disease, Lewy body disease, progressive supranuclear palsy and Down’s syndrome (Perry et al. 1986; Picciotto and Zoli 2002) (see also Sabri et al. in PET and SPECT in Neurology). However, there have been few preclinical PET and SPECT studies of nAChRs in animal models of neurodegenerative disease. This is attributable to two considerations: First, models in transgenic mice have only recently become available for some of these diseases, while remaining lacking for others, and second, the spatial resolution of small-animal PET and SPECT instruments has until recently been inadequate for regional analysis of neuroreceptors in rodent brain, as noted in the instrumentation section above. Therefore, the majority of such investigations use autoradiography in vitro. Furthermore, the occurrence of species differences is a hindrance to the interpretation of preclinical imaging studies (Pauly et al. 1989; Quik et al. 2000; Han et al. 2003). Despite these limitations, molecular imaging is emerging as a powerful tool for investigating pathophysiological changes in animal models of neurodegenerative diseases, especially when conducted in conjunction with techniques such as in vivo microdialysis, electrophysiology and histopathology (Higuchi et al. 2012).
4.1 Alzheimer’s Disease
A link between cognitive performance and α4β2 nAChR expression in the forebrain of healthy rats has been demonstrated in a PET study using the ligand [18F]nifene (Bieszczad et al. 2012), as confirmed by autoradiography ex vivo and in vitro. The three imaging methods showed the same rank order of specific binding by brain region. We anticipate that [18F]nifene PET should allow tracking of dynamic changes in nAChRs during learning acquisition and memory consolidation, in rodents and also in large-brained animals. In contrast to the case for humans (Zanardi et al. 2002), normal ageing does not seem to reduce nAChR density in rat brain (Picciotto and Zoli 2002; Schliebs and Arendt 2011).
The hallmark histopathological features of Alzheimer’s disease are extraneuronal amyloid plaques composed of aggregated amyloid-β peptides (Aβ), and intraneuronal neurofibrillary tangles, which are composed largely of hyperphosphorylated forms of tau, a microtubule-associated protein (Thal and Braak 2005). Molecular imaging studies in animal models have primarily targeted fibrillar protein assemblies such as β-amyloid and tau depositions, neuroinflammatory processes and cerebral glucose metabolism (Higuchi et al. 2012). However, these features are closely related to cholinergic hypofunction found in Alzheimer’s disease and relevant animal models (Schliebs and Arendt 2011).
Cognitive impairment in Alzheimer’s disease is at least partially associated with loss of cortical nAChRs, which may arise due to toxicity of soluble β-amyloid (Zanardi et al. 2002; Schliebs 2005; Schliebs and Arendt 2011). Nicotine treatment in a transgenic mouse model (3xTg-AD) can mediate increased tau phosphorylation and decrease β-amyloid load (Rubio et al. 2006). Impaired cholinergic neurotransmission has been described in the brain of Tg2576 mice, which express the Swedish mutation of human β-amyloid precursor protein (Apelt et al. 2002). Enzyme activities for acetylcholine synthesis (choline acetyltransferase) and degradation (acetylcholine esterase; AChE) did not differ between transgenic mice and non-transgenic littermates. However, a reduction of high-affinity choline uptake and M1-muscarinic receptor density was observed. Autoradiography with [3H]cytisine revealed a significant 20% loss of α4-containing nAChRs in cingulate and parietal cortices of these animals at an age of 17 months. However, there was no change in the number of basal forebrain cholinergic neurons in the transgenic mice, compared to age-matched wild-type animals.
Evidence for an involvement of α7 nAChR in Alzheimer’s disease was first presented three decades ago (Davies and Feisullin 1981). More recently, a very high-affinity binding of (soluble) β-amyloid to α7 nAChRs has been described in vitro (Wang et al. 2000), supporting the hypothesis that β-amyloid at very low concentrations may initiate neuronal degeneration via an α7 nAChR-mediated inflammatory process (Bencherif and Lippiello 2010). A 20% reduction in α7 nAChRs labelled with [125I]α-bungarotoxin was evident in the hippocampus, retrosplenial and parietal cortices and thalamus of 3xTg-AD mice at 6 months of age. There was a significant correlation between intraneuronal β-amyloid and reduced α7 nAChR binding in the same mouse model (Oddo et al. 2005). Whereas chronic nicotine administration did not alter α7 nAChR levels in these mice, there was an increase in α4β2 nAChRs labelled with [125I]epibatidine (Oddo et al. 2005). In contrast to the earlier report, no alteration in α7 nAChR binding was noted in a subsequent study of the triple transgenic 3xTg-AD mice, which closely emulate several features of natural Alzheimer’s disease (Hedberg et al. 2010); this unexpected negative finding was attributed to unknown environmental and/or genetic factors.
The abundance of α7 nAChRs was determined using nanogold-conjugated α-bungarotoxin in the APP(SWE) mouse model of Alzheimer’s disease (Jones et al. 2004). Interestingly, the α7 nAChR binding increased in the transgenic animals until 9 months of age but had declined at 12 months, most notably in areas of gliosis associated with β-amyloid plaques (Jones et al. 2006). Also [125I]α-bungarotoxin binding decreased in APP(SWE) mice between 9 months of age and 16 months (Hellström-Lindahl et al. 2004).
With the establishment of reliable cholinergic PET tracers, a number of molecular imaging studies of Alzheimer’s disease have appeared in recent years. The first phase of this research consisted of studies of the α4β2 subtype of nAChR with 2-[18F]fluoro-A-85380 (2-FA). One such quantitative PET study of 14 early AD patients showed no difference in VT compared to healthy controls, nor was there any relationship between tracer uptake and cognitive scores (Ellis et al. 2008). The same research group did not detect any reduction in binding with healthy ageing (Ellis et al. 2009a), and did not find any effect of precognitive treatment with galantamine on 2-FA binding in Alzheimer’s disease patients (Ellis et al. 2009b). Another study with 2-FA using a white matter reference tissue calculation of binding potential (BPND) found a 40% reduction in cerebral cortex of a group of nine patients with moderate Alzheimer’s disease, but no such change in the thalamus (Kendziorra et al. 2011). Similar findings in a group of mild cognitive impairment (MCI) patients suggested that the loss of α4β2 sites was already complete at the prodromal state of the disease. Others reported a 20–40% decline in 2-FA VT/fp (i.e. the distribution volume corrected for the plasma free fraction) in the thalamus, caudate, hippocampus anterior cingulate cortex and insula in a group of 24 Alzheimer’s disease patients compared to a healthy control group (Sultzer et al. 2017). Similarly, a dual tracer study with 2-FA and the beta-amyloid tracer [11C]PiB showed an inverse relationship between the binding ratio of α4β2 tracer and the amyloid BPND in the medial frontal cortex and basal forebrain of Alzheimer’s disease patients (Okada et al. 2013). While the preponderance of such studies indicates a widespread loss of α4β binding sites in Alzheimer’s disease, there is a certain lack of congruence about its spatial extent and magnitude. This may reflect the various endpoints used for quantitation and the very long interval of 4–6 h required for attainment of equilibrium binding with 2-FA. Another PET study with (−)-[18F]flubatine PET in patients with early Alzheimer’s disease showed the reductions in α4β2 nAChR mainly in target regions of the basal forebrain-cortical and septohippocampal cholinergic projections. The same study showed relationship between lower α4β2 nAChR availability and impairment of distinct cognitive domains, notably episodic memory and executive function/working memory (Sabri et al. 2018). Further details, in particular related to the α4β2 radioligands (−)-[18F]flubatine and (+)-[18F]flubatine, are reviewed elsewhere (Sabri et al. 2014).
Very recently, uptake of the α7 nAChR ligand [11C]-(R)-MeQAA was measured in groups of 20 Alzheimer’s disease patients and 10 healthy age-matched controls. In the nucleus basalis magnocellularis and medial prefrontal cortex, the α7 nAChR binding correlated positively with individual beta-amyloid PET results. Furthermore, the α7 nAChR binding correlated positively with memory and executive function (Nakaizumi et al. 2018). This stands in agreement with findings in aged monkeys cited above, where [11C]-(R)-MeQAA binding, ostensibly to α7 NAChR, was associated with increased beta-amyloid binding (Nishiyama et al. 2015). Similar increases in Alzheimer’s disease mice were attributed to microglial activation (Matsumura et al. 2015) at sites of amyloid deposition. At time of writing, the jury is out whether this might represented a compensatory and salutogenic reaction, or another aspect of pathology.
[18F]fluoroethoxybenzovesamicol (FEOBV) is a novel PET radiotracer, which binds selectively to the vesicular acetylcholine transporter in terminals of basal forebrain neurons. SUVR analysis of this tracer showed widespread decreases in tracer binding in a group of Alzheimer’s disease patients (Aghourian et al. 2017). Together with findings reported above, this suggests pre- and postsynaptic elements of basal-forebrain cholinergic pathways are compromised in Alzheimer’s disease.
4.2 Movement Disorders
Parkinson’s disease is, after Alzheimer’s disease, the second most common neurodegenerative disorder. The hallmark neuropathology of Parkinson’s disease is selective degeneration of midbrain dopaminergic neurons of the substantia nigra pars compacta (SNpc) and the presence of intra-cytoplasmic inclusions (Lewy bodies) consisting of aggregated α-synuclein (Spillantini et al. 1997) in surviving dopamine neurons. Neurotoxins such as MPTP and 6-hydroxydopamine have been used in animal models emulating the nigrostriatal degeneration of Parkinson’s disease (Quik 2004). Lesions of the nigrostriatal pathway in rats reveal a population of [3H]nicotine binding sites on dopamine terminals, where they are positioned to influence dopamine release (Clarke and Pert 1985). More recently, the stimulation of striatal dopamine release by nicotine has been linked specifically to α6β2β3 and α6α4β2β3 nAChRs, which predominate in the basal ganglia (Quik et al. 2011). Quantitative analysis with the α6β2 nAChR subtype ligand [125I]α-conotoxin MII in conjunction with plasma membrane dopamine transporter measurements in MPTP-lesioned mice shows an association with presynaptic nigrostriatal terminals (Quik et al. 2003). In that study, much smaller reductions in the binding of [125I]epibatidine (multiple sites) and 5-[125I]iodo-A-85380 (β2-sites) were noted after dopamine lesioning, while no change was detected in α7 nAChRs measured with [125I]α-bungarotoxin binding after nigrostriatal lesions. Displacement of [125I]α-conotoxin MII binding with the analogue E11A was biphasic, allowing resolution of the α6β2β3 and α6α4β2β3 components (Bordia et al. 2007); autoradiographic studies in MPTP-treated mouse and non-human primates, as well as in material from idiopathic Parkinson’s disease patients, revealed the α6α4β2β3 nAChR subtype population to be selectively vulnerable to nigrostriatal damage. Chronic oral nicotine administration was able to protect nicotinic receptors and dopaminergic markers in MPTP-treated monkeys (Bordia et al. 2006; Quik et al. 2006). Treatment of rats with a selective α7 nAChR agonist protected against nigrostriatal degeneration in the 6-OHDA model of parkinsonism, an effect that was linked to attenuated microglial activation (Serriere et al. 2015).
In a 2-[18F]fluoro-A-85380 PET study, the density of α4β2 nAChRs was slightly reduced in the basal ganglia of non-smoking Parkinson’s disease patients (Kas et al. 2009). Another 2-[18F]fluoro-A-85380 PET study of Parkinson’s disease patients reported a widespread reduction of α4β2 nAChR availability in cortical and subcortical regions, which correlated with the severity of mild cognitive or depressive symptoms (Meyer et al. 2009). There have been no α7 nAChR studies in Parkinson’s disease, despite the preclinical evidence suggesting a role for these receptors in protecting against nigrostriatal degeneration. There is a current lack of agents for molecular imaging of the particular α6-containing nAChRs subtypes present in the basal ganglia, which may be of great relevance to Parkinson’s disease given their involvement in dopamine release.
Huntington’s disease is an autosomal dominant hereditary disorder proceeding to severe cognitive impairment and motor symptoms, notably hyperkinetic involuntary movements (chorea) (Roos 2010). A transgenic rat model of HD, which carries a truncated huntingtin cDNA fragment with 51 CAG repeats under control of the native rat huntingtin promoter, has been developed (von Horsten et al. 2003). Early investigations of nicotinic receptors in post-mortem brain from Huntington’s disease patients did not reveal any significant changes (Perry et al. 1987; Whitehouse and Kellar 1987). However, autoradiographic assessment of 2-year-old transgenic rats revealed significant increase of nAChR in various regions in heterozygous but not homozygous animals (Bauer et al. 2005). There have not yet been any PET studies of nAChR in patients with Huntington’s disease.
5 Epilepsy
Some forms of epilepsy have recently been associated with alterations of α4 nAChR subtype expression (Raggenbass and Bertrand 2002), and there is experimental evidence that the α7 nAChR may play a role in epileptogenesis (Dobelis et al. 2003). The autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) can be caused by mutations in the neuronal nicotinic acetylcholine receptor (nAChR) subunit genes CHRNA4 and CHRNB2 (Steinlein et al. 1995; Phillips et al. 2001). Relative to age-matched non-smoking subjects, there was a 10–20% increase in the binding of 2-[18F]fluoro-A-85380 to α4β2 receptors in the brain of patients with ADNFLE (Picard et al. 2006); knock-in mice bearing a culprit mutant α4 gene have been prepared (Lipovsek et al. 2008), but have not been investigated by receptor autoradiography or PET. A recent clinical PET study of patients with idiopathic generalized epilepsy using the α4β2 nAChR ligand [18F]A-85380 showed focal 25% increases in the binding ratio within the anterior cingulate cortex (ACC), which could distinguish individual patients from healthy controls (Garibotto et al. 2019).
6 nAChR Imaging of Stroke and Neuroinflammation
Stroke is the leading cause of adult disability in the United States and Europe and the second leading cause of death worldwide. Stroke is characterized by a loss of brain functions due to rapid disturbances in cerebral blood supply, either as reduced blood flow by thrombosis or embolism (ischemic stroke) or bleeding (haemorrhagic stroke). Hyperacute mechanisms of stroke-related brain tissue damage, such as excitotoxicity, can be discriminated from delayed factors such as inflammation and apoptosis. All cellular components of the so-called neurovascular unit, which includes neurons, astrocytes and endothelial cells, express nAChRs (Paulson et al. 2010). Insofar as long-term tobacco smoking is a risk factor for ischemic stroke (Hawkins et al. 2002), it may be relevant that nAChRs were altered in a post-mortem study of smokers; α4 expression was increased in neurons and dendritic processes, and α7 expression was decreased in hippocampal neurons and astrocytes (Teaktong et al. 2004). In hypertensive stroke-prone rats, cortical α7 nAChRs are reduced, without concomitant changes in the α4β2 nAChRs (Ferrari et al. 1999). Activation of nAChRs by nicotine promotes endothelial cell proliferation (Villablanca 1998) and leucocyte migration (Yong et al. 1997), which together may increase thrombotic risk. However, nAChR agonism was neuroprotective against excitotoxicity in vitro, an effect mediated by growth factors (Belluardo et al. 1998) and also by inactivation of the toxins (O’Neill et al. 2002). These effects were blocked by the α7 nAChR antagonist α-bungarotoxin (Donnelly-Roberts et al. 1996). Interestingly, nicotine increased oedema/infarct size in a rodent stroke model (Paulson et al. 2010). A particular contribution of α7 nAChRs to stroke-related excitotoxicity might reflect their high Ca2+ permeability, especially under depolarizing conditions. Treatment with the acetylcholinesterase inhibitor methanesulfonyl fluoride attenuated stroke-induced learning and memory deficits in rats (Borlongan et al. 2005).
These rather discordant findings of effects of nAChR agonism in stroke models may reflect the different contributions of excitotoxicity and neuroinflammatory processes. The α7 nAChRs expressed on microglia (Shytle et al. 2004a) seem particularly poised to mediate inflammatory responses. Nicotinic agonism at these sites suppressed inflammation by decreasing TNF-α production, while nicotine antagonists had the opposite effect (Shytle et al. 2004a). In addition to central mechanisms, cholinergic signalling in the “cholinergic anti-inflammatory pathway” involving the vagus nerve may suppress the release of pro-inflammatory cytokines and influence migration of T cells from the periphery to brain areas affected by stroke or multiple sclerosis (Borovikova et al. 2000) by a mechanism sensitive to α–bungarotoxin (Pavlov et al. 2003).
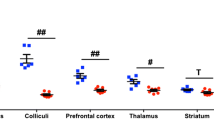
Despite this extensive background, there have been very few molecular imaging studies of nAChRs in relevant neuroinflammation models. We have undertaken PET studies to assess α7 nAChR alterations in the sheep stroke model (Boltze et al. 2008). Here, we measured [18F]DBT10 binding at different time points after permanent medial cerebral artery occlusion (pMCAO) by dynamic imaging using a clinical hybrid PET/MRI system. We found increased tracer uptake in the stroke-border zone 14 days after pMCAO. In these areas, microglia activation and macrophage infiltration were histologically confirmed. Ongoing studies aim to establish better the time course and histological correlates of the α7 nAChR changes in our stroke model. A recent dual tracer study monitored longitudinal changes in 2-[18F]fluoro-A85380 and [11C]PK11195 binding during a month after middle cerebral artery occlusion (MCAO) in rats. In the ischemic territory, both ligands showed progressive binding increase from day 3 to 7 post-injury, followed by a progressive decrease (Martin et al. 2015). Post-mortem analysis linked the changes to increased α4β2 nAChR and TSPO expression on microglia and macrophages.
In PET studies in a rat stroke model with transient MCAO occlusion, the PET signal from the α7 nAChR ligand [11 C]NS14492 increased around the core of the infarct, consistent with activation of microglia and astrocytes following the injury (Colas et al. 2018). In that study, treatment with the α7 agonist PHA 568487 1 week after the stroke lowered TSPO binding (suggesting a rescue from microgliosis) while diminishing the ultimate infarct volume. Thus, the α7 nAChR is a promising target for disease-altering interventions against stroke.
Atherosclerosis is a kind of chronic inflammatory condition that brings a high risk of cardiovascular events. Especially the “vulnerable” atherosclerotic plaques have a high risk of rupture, which is predictable from their avidity on FDG PET, as an index of macrophage infiltration into the vessel wall. As such, α7 nAChR PET presents an unexplored channel for investigating inflammatory changes in atherosclerosis (Boswijk et al. 2017). This follows also for a wide range of chronic inflammatory conditions such as rheumatoid arthritis, Crohn’s disease, etc.
7 nAChR Imaging of Traumatic Brain Injury
Traumatic brain injury is a permanent or temporary impairment of brain functions with an associated diminished or altered state of consciousness caused by external mechanical force transferring kinetic energy to the brain tissue. In developed countries, TBI is the most important cause of death and disability in young adults, in both civilian and military contexts (Olesen and Leonardi 2003) (Dewan et al. 2018). Indeed, TBI causes some 30–50% of all injury-related deaths (de Ramirez et al. 2012; Kamal et al. 2016). Apart from the distinct acute injuries, TBI is a continuous disease process (Masel and DeWitt 2010), with survivors often suffering from persistent or permanent physical and cognitive impairments (Fleminger and Ponsford 2005) occurring as long-term sequelae of the initial injury. This profile imposes a considerable socio-economic burden (Humphreys et al. 2013), which is further exacerbated by the failure of clinical trials aiming to improve outcomes (Loane and Faden 2010; Gruenbaum et al. 2016).
7.1 Animal Models of TBI
Experimental animal models of TBI provide a solid body of evidence for specific cholinergic alterations. A plethora of different TBI models has been developed since the late 1940s. There is a distinction to be made between closed head injuries and open-head models, whereby in the former model the underlying cortical tissue is damaged by impact on the intact dura mater. Most animal studies of cholinergic responses to TBI make use of fluid-percussion injuries (FPI), weight drop and controlled cortical impact (CCI) (Xiong et al. 2013) (O’Connor et al. 2011). This can be performed either through a fluidic wave, as in the FPI models (Thompson et al. 2005), rigid impactors such as weight drop (Marmarou et al. 1994) or CCI models (Lighthall 1988). Unfortunately, there is little data on cholinergic alterations provoked by the more clinically relevant (repeated) closed-head or concussive models. The CCI model, which was first developed in small animals (Lighthall 1988) and later used in pigs (Duhaime et al. 2000; Alessandri et al. 2003; Manley et al. 2006), entails a piston strike, which results in more focal brain injury than is afforded by the FPI model.
Early evidence of the involvement of the cholinergic system in TBI was obtained in dogs and cats, in which increased acetylcholine concentration was noted in cerebrospinal fluid up to 48 h post-injury (Bornstein 1946). In subsequent years, studies of TBI implicate diverse aspects of cholinergic neurotransmissions, including muscarinic and nicotinic receptors, enzymatic pathways and vesicular transporters (Hayes et al. 1992; Arciniegas 2011; Kelso and Oestreich 2012; Shin and Dixon 2015). In an autoradiographic study of rats with TBI, there were widespread and substantial bilateral reductions in cortical and hippocampal α7 nAChRs labelled with [125I]α-bungarotoxin, in contrast to lesser, more focal and ipsilateral effects on heteromeric nAChRs labelled with [3H]epibatidine (Verbois et al. 2000). We have seen a similar decline in α7 binding in the brain of rats and neonate piglets following FP injuries (Donat et al. 2008, 2010a, b; Hoffmeister et al. 2011). In a saturation binding study, the reductions were linked to declines in Bmax, rather than affinity changes (Verbois et al. 2002), an issue that must always be considered in autoradiographic or PET studies performed with a single ligand concentration. Peak effects occurred at 2 days post-injury but persisted for as long as 2 weeks. Brain regions not directly subjected to mechanical damage, such as the thalamus, were found to exhibit reduced density of α7 and α3/4 nAChR, indicating retrograde and anterograde changes.
Most interestingly, brain areas showing reduced nAChR binding after TBI also showed delayed neuroinflammation, as indicated by increased binding of a tracer for the 18 kDA translocator protein, a marker of microglial activation (Donat et al. 2016), suggesting that nAChRs regulate neuroinflammatory responses (Egea et al. 2015). Indeed, in a model of blast-induced TBI, midbrain structures showed lowered gene transcripts of muscarinic and α7 nAChR, accompanied by increased gene transcripts of pro-inflammatory markers (Valiyaveettil et al. 2013). Treatments with agonists and positive allosteric modulators of α7 nAChR attenuated microglia activation after TBI while rescuing blood-brain barrier permeability increases and reducing motor deficits (Gatson et al. 2015; Dash et al. 2016). This is consistent with anti-inflammatory effects α7 nAChR activation on microglia and potentially endothelial cells (Kimura, Dohgu et al. 2018), as indicated before (Cortes et al. 2017; Zhang et al. 2017).
Cognitive impairment was demonstrated in the rat FPI model (Scheff et al. 1997), suggesting that the α7 nAChR reductions may contribute to these cognitive deficits caused by brain trauma. Also in the FPI paradigm, prolonged treatment with nicotine partially attenuated the cognitive deficits seen in the Morris water maze performance and reduced the magnitude and spatial extent of the α7 changes (Verbois et al. 2003a, b). Indeed, nicotine has been effective in a number of other brain lesion models (Visanji et al. 2006; Huang et al. 2009; Zafonte et al. 2009; Quik et al. 2010), even though human patients who are smokers do not show improved outcome after TBI (Ostberg and Tenovuo 2014), because of smoking-associated health impairments such as vascular diseases. Furthermore, investigations in α7 knockout mice failed to show much effect of α7 receptor expression on lesion volume or microglia activation (Kelso et al. 2006), which could be confounded by compensatory mechanisms (Smith et al. 2014). Nonetheless, dietary choline supplements partially rescued the spatial memory deficits and α7 receptor deficits after CCI (Guseva et al. 2008; Guseva et al. 2013).
7.2 Human TBI Studies
Neuroinflammation, a process mediated primarily by glia cells, i.e. microglia and astrocytes, is not just a hallmark pathology of TBI, but also a potential avenue of intervention (Morganti-Kossmann et al. 2019). Activated microglia, which are the resident macrophages of the brain, can assume many different roles post-injury, as characterized by diverse morphologies and molecular expression patterns. The dichotomy of pro-inflammatory M1 microglial phenotype versus anti-inflammatory M2 phenotype (Donat et al. 2017) may be an oversimplification (Ransohoff 2016), but it should not be assumed that all microglia are equal. A similar model of functions and phenotypes has been recently reported for astrocytes (Liddelow et al. 2017).
Glial activation can persist for many years after TBI (Ramlackhansingh et al. 2011). TBI is a risk factor for neurodegenerative diseases including chronic traumatic encephalopathy (McKee et al. 2016) and dementias, including Alzheimer’s disease (AD) (LoBue et al. 2018). The link between TBI and dementia may be mediated either by tau pathology or by altered cholinergic neurotransmission particularly involving hetero- and homomeric nAChR, as proposed for AD in the 1980s (Shimohama et al. 1986), in association with the cholinergic hypothesis of AD (Bohnen et al. 2018; Hampel et al. 2018). Historically, nAChR changes were linked to cognitive symptoms (Wallace and Bertrand 2013), as most α7 and α4/3β2/4 nAChR are expressed on cortical neurons. However, cholinergic enzymes and receptors are also implicated in central and peripheral inflammatory response (Fujii et al. 2017), pointing to the above-mentioned cholinergic anti-inflammatory pathway (Martelli et al. 2014). Acetylcholine receptors may be essential mediators of this pathway and are likely involved in the neuroinflammatory response after TBI (Ren et al. 2017). Indeed, microglia express α7 nAChR (Shytle et al. 2004b), as do astrocytes (Shen and Yakel 2012), where they seem to be directly involved in regulating glia activation. Reduced α7 nAChR expression or antagonist exposure can promote the pro-inflammatory phenotype, while treatment with agonists may shift microglia towards an anti-inflammatory phenotype (Cortes et al. 2017; Zhang et al. 2017).
Several studies have investigated key players of the cholinergic neurotransmission following TBI. However, only very few studies investigated human patients or tissue samples. In a human post-mortem study, performed after traumatic brain injury, reduced choline acetyl transferase activity and synaptophysin immunoreactivity in cerebral cortex were found, indicating damage of the cholinergic innervation, but nAChR binding determined by either [3H]nicotine or [125I]α-bungarotoxin was unchanged (Murdoch et al. 1998), suggesting mainly presynaptic mechanisms. Post-mortem cortical tissue from patients acquired between 1 and 300 h after injury showed a reduction in choline acetyltransferase activity, but no change in muscarinic (M1 and M2 receptors) or nicotinic receptors, determined by either [3H]nicotine or [125I]α-bungarotoxin (Dewar and Graham 1996; Murdoch et al. 1998). Reduced synaptophysin immunoreactivity was seen as an indicator of mainly presynaptic pathomechanisms, even though a follow-up study showed extensive damage in basal forebrain cholinergic neurons (Murdoch et al. 2002). Interestingly, diffusion tensor imaging indicated axonal injury in the same areas (Hong et al. 2012). More importantly, recent studies of CTE showed a gradual emergence of pretangle pathology and oligomeric tau accumulation in cholinergic neurons of the basal forebrain, which might directly relate to cholinergic impairments and ultimately to cognitive dysfunction and immune dysregulation (Mufson et al. 2016). Laser capture microdissection and gene profiling of these neurons showed, among other findings, a reduction in CHRNB2 transcripts, encoding the nAChR β2 subunit (Mufson et al. 2018). Another potential factor in TBI is autoimmune consequences (Raad et al. 2014). One study reported a significant increase in blood autoantibodies to regions of the α7 subunit in children with TBI, which correlated to severity (Sorokina et al. 2012).
In vivo molecular imaging with nAChR subtype-specific tracers is still lacking in clinical TBI. However, PET imaging with [11C]MP4A in chronic TBI patients indicated loss of AChE activity (Östberg et al. 2011; Ostberg et al. 2018). Treatment of post-traumatic cognitive deficits using cholinergic compounds, e.g. acetylcholinesterase inhibitors, has been attempted in several studies of small patient groups. However, most of these exploratory studies did not meet their endpoints justifying larger randomized placebo-controlled studies, and systematic reviews therefore concluded only weak evidence of efficacy (Wheaton et al. 2011; Bengtsson and Godbolt 2016).
8 nAChR Imaging of Addiction and Psychiatric Disorders
8.1 Physiological Effects of Nicotine in the Context of Addiction
Whereas acetylcholine is rapidly inactivated by AChE, nicotine exerts prolonged agonism due to its metabolic stability. This property accounts for the well-known phenomenon of desensitization of nicotinic receptors by nicotine (Govind et al. 2012), in which inactivated receptors concentrate in the plasma membrane due first to agonist-evoked conformational changes, which is followed by decreased degradation of subunits (Govind et al. 2012). In the case of α4β2 nAChRs, upregulation is mediated by activation of protein kinase C, which results in phosphorylation of the α4 subunit (Wecker et al. 2010). This mechanism is supported by studies in rats and 3xTd-AD mice showing upregulation of α4β2 nAChR after chronic nicotine administration (Flores et al. 1992; Oddo et al. 2005). Chronic nicotine treatment persistently increased the binding of the α4β2 ligand 5-[123I]iodo-A-85380 by approximately 50% in the brain of baboons (Kassiou et al. 2001). Similarly, a 2-[18F]fluoro-A-85380 PET study in chronic smokers abstinent for at least 24 h revealed persistent upregulation of α4β2 nAChR throughout the brain (Mukhin et al. 2008); these findings stress the requirement for strict control of exposure to nicotine in the design of clinical and preclinical imaging studies of nAChRs. Thus, exposure of rats to tobacco smoke not only induced nicotine dependence but also increased the α7 nAChR density in the CA2/3 area (+ 25%) and the stratum oriens (+ 18%) of the hippocampus (Small et al. 2010).
Since the nAChRs are ligand-gated cation channels, their activation facilitates depolarization and enhances the release of dopamine and other neurotransmitters. Consistent with the increased energy demands associated with depolarization, acute challenge with nicotine increases the cerebral metabolic rate for glucose (CMRglc) as measured by [14C]-deoxyglucose autoradiography; these effects are most notable in the thalamus and other rat brain regions in which nicotinic receptors are most abundant (London et al. 1988; Marenco et al. 2000). Despite the phenomenon of upregulation/desensitization of nAChRs, stimulation of CMRglc in rat brain was still evident after reinstatement of nicotine following a period of withdrawal (Schröck and Kuschinsky 1991). Effects of nicotine on CMRglc are poorly documented in human brain, but one PET study showed that nicotine-evoked stimulation of 2-[18F]fluorodeoxyglucose uptake in non-smokers was dependent on the hostility trait, as was also seen in smoking subjects treated with a higher dose of nicotine (Fallon et al. 2004). The latter finding seems consistent with desensitization of the cerebrometabolic response to nicotine challenge in the smoking group. Smoking provoked a delayed increased in the cerebral consumption of oxygen in the brain of acutely withdrawn smokers, suggesting a global effect of nicotine or other constituents of tobacco smoke on mitochondrial respiration as distinct from glycolysis (Vafaee et al. 2015).
Baseline levels of 2-[18F]fluoro-A-85380 binding in the brainstem of healthy squirrel monkeys predicted their motivation to subsequently self-administer nicotine, as indicated by number of bar presses (Le Foll et al. 2009), suggesting that nAChRs mediate a trait vulnerability specifically for nicotine abuse. It is unclear whether this association with nicotine addiction generalizes to other addictive drugs. However, activation of nAChRs on dopamine neurons and terminals is central to the reinforcing and addictive properties of nicotine. Stimulation of striatal dopamine release by nicotine has recently been linked to α6β2β3 and α6α4β2β3 nAChRs, which predominate in the basal ganglia (Quik et al. 2011). Activation of these receptors increases the firing rate and augments phasic bursting of midbrain dopamine neurons (De Biasi and Dani 2011). The α7 nAChRs in the ventral tegmental area may mediate nicotine’s stimulatory effect on mesolimbocortical dopaminergic function and consequently its reinforcing and dependence-producing properties (Nomikos et al. 2000).
In an early molecular imaging study of nAChRs, binding of the SPECT tracer 5-[123I]-A-85380 at α4β2 sites was increased by one third in the cortex and striatum of smokers with confirmed abstinence for 1 week (Staley et al. 2006). This study recapitulated in human smokers the well-known upregulation and inactivation of nAChRs following repeated nicotine exposure seen in experimental animals. The same group showed normalization of nAChR availability after 6 weeks of abstinence (Cosgrove et al. 2009). The competition paradigm affords the possibility of detecting occupancy by exogenous nicotine or other drugs at nAChR in living individuals. Here, comparison of molecular imaging results at baseline and after drug challenge reveals the percentage of receptors occupied by the drug. In one such 5-[123I]-A-85380 SPECT study, smoking to satiety (about two cigarettes) induced a 67% reduction in their chosen endpoint, VT/fp (Esterlis et al. 2010a). Occupancy after use of a nicotine inhaler (56%) was significantly lower than after smoking cigarettes to satiety (Esterlis et al. 2010b).
These nAChR occupancies may be underestimated due to the slow kinetics of the SPECT tracer relative to the pharmacodynamics of nicotine. Corresponding PET studies with 2-[18F]fluoro-A-85380 showed 79% occupancy after smoking a low-nicotine cigarette and 26% after smoking a denicotinized cigarette (Brody et al. 2009). In another 2-[18F]fluoro-A-85380 PET study, a few puffs on a normal cigarette induced 50% occupancy persisting 3 h later, whereas an entire cigarette provoked 88% occupancy (Brody et al. 2006). Even exposure to second-hand smoke led to 20% occupancy (Brody et al. 2011). A single low dose (0.5 mg) of varenicline provoked complete saturation of 2-[18F]fluoro-A-85380 binding sites in human brain (Lotfipour et al. 2012a). Thus, varencline is apt to evoke full agonism at α7 nAChRs, with partial agonism at other sites, i.e. α4β2, as well as α3β4, and α6β2 subtypes.
Altered dopamine release can be detected in PET studies with [11C]raclopride and other benzamide antagonists of D2/3 receptors, wherein ligand binding is reduced by competition from endogenous dopamine (Laruelle 2000; Cumming et al. 2003). Whereas powerful psychostimulants such as amphetamine can evoke 30% decreases in striatal [11C]raclopride binding, challenge with nicotine reduced this binding by only 5–10% in striatum of anaesthetized pigs, most notably in the ventral striatum (Cumming et al. 2003). There were effects of similar magnitude in the brain of non-human primates (Marenco et al. 2004), although others saw no such effects in PET scans of awake monkeys (Tsukada et al. 2002), suggesting a confounding effect of general anaesthesia. Similarly, intranasal administration of nicotine did not greatly reduce striatal [11C]raclopride binding in healthy habitual smokers, although baseline binding correlated with scores of nicotine dependence, and there was a positive relationship between binding changes and individual reports of pleasant subjective experience (Montgomery et al. 2007), as likewise seen in another [11C]raclopride PET study of smokers (Barrett et al. 2004). In general, preclinical studies with non-contingent pharmacological challenge may not replicate the motivational aspects of self-administered nicotine.
Dopamine synthesis capacity in living striatum can be measured by PET studies with DOPA decarboxylase substrates such as [11C]DOPA or 6-[18F]fluoro-DOPA (FDOPA). Whereas acute nicotine treatment had no effect on [11C]DOPA utilization in striatum of awake monkeys, this utilization was reduced by one third after overnight abstinence in monkey habituated to nicotine and normalized rapidly following reinstatement of nicotine treatment (Domino et al. 2009). On the other hand, in an isolated clinical PET finding, FDOPA utilization was 20–30% higher in striatum of human smokers (Salokangas et al. 2000). Another study showed a 20% reduction in the striatal utilization of FDOPA in a larger group of dependent smokers (Rademacher et al. 2016), which may have greater face validity as an adaptive response to chronic nicotine exposure.
8.2 Alcohol Dependence
There is very high comorbidity between nicotine and alcohol dependence, and considerable overlap between their neural mechanisms of addiction (Larsson and Engel 2004). Indeed, a possible role for nAChR in alcohol addiction has been proposed (Meyerhoff et al. 2006). Altered nACh receptor density in vitro has been observed in rats in response to ethanol using a combination of [3H]nicotine, [3H]MLA and [125I]alpha-bungarotoxin, but the direction of change was inconsistent and region-specific (Yoshida et al. 1982; Booker and Collins 1997; Robles and Sabria 2008). In vivo, binding of the SPECT radioligand [123I]5-IA-85380 in rhesus monkey was unaltered following chronic alcohol self-administration, although decreases in cortical and thalamic binding were observed following abstinence (Cosgrove et al. 2010). More recently, using [18F]nifene in a PET study in non-human primates, reductions in nACh receptor density were observed following chronic alcohol exposure (Hillmer et al. 2014). Further validation work is required in human subjects to resolve these inconsistencies.
8.3 Schizophrenia and Depression
Nicotine addiction is notoriously prevalent among patients with schizophrenia (Lohr and Flynn 1992), a chronic psychiatric disease characterized by behavioural changes, which are difficult to model in experimental animals. The DSM-IV criteria for schizophrenia consist of positive symptoms (such as agitation, paranoia and hallucinations), negative symptoms (including emotional blunting, avolition and social withdrawal) and specific cognitive and psychomotor deficits (Goldman-Rakic and Selemon 1997). An increasing appreciation of the importance of cognitive deterioration has motivated a search for treatments to improve processing speed, attention/vigilance and working memory (Nuechterlein et al. 2004; Heinrichs 2005). This has emerged in the context of a hypothesis implicating gene-mediated dysfunction of α7 nAChRs (Freedman et al. 1997; Stephens et al. 2009; Dome et al. 2010) in the cognitive impairments of schizophrenia (Nomikos et al. 2000). Indeed, in one post-mortem study, the hippocampal [125I]α-bungarotoxin binding was decreased in a schizophrenic patient population consisting mainly of smokers (Freedman et al. 1995). Furthermore there were significant differences in α7 nAChR (CHRNA7) expression at both mRNA and protein levels between smokers and non-smokers with schizophrenia (Mexal et al. 2010).
Nicotinic receptors and treatments have been investigated in a number of murine models emulating some behaviour aspects of schizophrenia. An auditory gating defect in dilute brown non-Agouti (DBA/2) mice is rectified by treatment with an α4β2 agonist (Wildeboer and Stevens 2008), but other nAChR subtypes such as α7 may also mediate sensory gating in these mice (Radek et al. 2006). Mice lacking the plasma membrane dopamine transporter show hyperactivity and cognitive deficits, and are hypersensitive to the locomotor stimulant effect of nicotine, which models schizophrenia. Treatment of these mice with nicotine improved their performance in spatial and cued learning tasks (Weiss et al. 2007a), whereas autoradiography showed a small decline in the β2 subunit, a large decrease in β6 and substantially increased α7 nAChR expression (Weiss et al. 2007b). Deficiency of the microtubule-stabilizing protein STOP results in impaired hippocampal plasticity and behavioural hypersensitivity to psychostimulants, likewise associated with a decrease in β6, and a substantial increase in α7 nAChRs (Bouvrais-Veret et al. 2007), suggesting a common nicotinergic pathway in these two models of schizophrenia.
A preliminary study with the α7-nAChR ligand [18F]ASEM showed a moderate reduction of binding in the brain of a group of patients with schizophrenia (Wong et al. 2018). This finding was recapitulated in another study of recent onset psychosis (Coughlin et al. 2018a). Given the association of α7 nAChR expression with microglia, the resident brain macrophage, it is a matter of interest if there are changes in inflammatory pathways in association with schizophrenia. A recent meta-analysis of many PET TSPO studies in patients with schizophrenia showed an overall finding of reduced microglial activation (Plaven-Sigray et al. 2018). While this was contrary to general expectation of an active inflammatory process during schizophrenia, it may be consistent with the α7-nAChR findings.
Availability of α4β2 nAChR binding sites was globally reduced in a 5-[123I]iodo-A-85380 SPECT study of patients with major depression (Saricicek et al. 2012). Changes in behaviour, and in endocrine, and immune and neurotransmitter systems, modelling symptoms of patients with major depression have been described in rats following bilateral olfactory bulbectomy (Song and Leonard 2005). Such rats present characteristic alterations in cholinergic function (Hozumi et al. 2003). Although degeneration of cholinergic neurons may well underlie the impairments of learning and memory-related behaviour in olfactory bulbectomy rats, densities of nAChRs have not yet been investigated in this model. Interestingly, a recent TSPO PET study showed globally increased microglial expression in cortex of patients with major depression and an association with cognitive deficits (Li et al. 2018). However, there has hitherto been no PET study of α7-nAChR depressed (non-smoking) patients.
9 nAChR Imaging for Measurement of Endogenous Acetylcholine
The competition model for measuring dopamine release by molecular imaging, i.e. the [11C]raclopride binding model described above, has delivered substantial insights into the role of dopamine in the pathophysiologies of schizophrenia and nicotine addiction. However, attempts to generalize the dopamine competition model to other neurotransmitter systems have met with mixed success (Paterson et al. 2010; Finnema et al. 2015). A molecular imaging assay for fluctuations in extracellular acetylcholine would be a useful tool for evaluating new pharmacologic treatments for Alzheimer’s disease and other conditions, and the first demonstration of an acetylcholine sensitive α4β2 PET tracer was recently published (Hillmer et al. 2016a). Competition between endogenous acetylcholine and radioligands targeting muscarinic acetylcholine receptors is reported in the literature (Dewey et al. 1993; Suhara et al. 1994; Ma et al. 2004; Eckelman 2006), but the case is better established for nAChR ligands, the details of which are presented here.
Preclinical studies in rodents suggest that treatment with AChE inhibitors may raise acetylcholine levels sufficiently to elicit changes in binding potential with PET. Treatment with AChE inhibitors, an important class of compounds used for alleviation of Alzheimer’s disease symptoms, affords a convenient way of enhancing acetylcholine levels in living brain in tests of the competition model. In the first such study, the uptake of 2-[18F]fluoro-A-85380 in rat thalamus measured ex vivo was 45% reduced by the AChE inhibitor physostigmine (Dollé et al. 1999); decreases of similar magnitude were seen in animals treated with typical α4β2 agonist ligands (2-fluoro-A-85380, nicotine, epibatidine and cytisine) but not by α7- or 5-HT3-specific antagonists. Previous microdialysis studies had confirmed that the same dose of physostigmine (300 μg/kg) caused large increases in interstitial acetylcholine (Cuadra et al. 1994), thus substantiating the competition model.
In a non-human primate PET study, Ding et al. investigated whether modulation of acetylcholine levels could modulate [18F]NFEP binding (Ding et al. 2000b). As predicted, binding was 25% lower in the thalamus and striatum following physostigmine administration. Furthermore, treatment with the dopamine D2/3 antagonist raclopride reduced the striatal binding of [18F]NFEP by 22%, and the agonist quinpirole increased striatal binding by 26%, but selective dopamine D1 drugs had no such effects. These findings were consistent with results of cerebral microdialysis studies showing that dopamine D2/3 receptors tonically inhibit acetylcholine release in the striatum (Damsma et al. 1991; DeBoer et al. 1996).
In a 5-[123I]iodo-A-85380 SPECT study in non-human primate, a bolus plus constant infusion paradigm was used to obtain equilibrium binding prior to physostigmine treatment (Fujita et al. 2003). Due to instability of the plasma 5-[123I]iodo-A-85380 concentration following physostigmine, the BPND was not calculated, but there was a 14–17% decrease in thalamic tracer uptake. This decline was independent of the physostigmine dose administered, suggesting a ceiling effect in the competition from endogenous acetylcholine. In a subsequent 2-[18F]fluoro-A-85380 study, a more prolonged infusion of physostigmine evoked a 40% reduction in 2-[18F]fluoro-A-85380 VT in putamen and more modest reduction in cerebral cortex (Valette et al. 2005). The effect was dose-dependent, and infusion of galantamine, a weaker AChE inhibitor, did not alter 2-[18F]fluoro-A-85380 binding, which again suggests a fairly narrow relationship between interstitial acetylcholine concentration and increased competition.
In PET studies in rats, cortical and thalamic [18F]nifene binding was found to be significantly reduced (3–10%) following physostigmine and galantamine administration (Hillmer et al. 2013) suggesting this ligand may also be sensitive to changes in endogenous acetylcholine. In vitro binding studies with [18F]nifene and [3H]cytosine showed displacement of both ligands upon addition of an AChE inhibitor to the medium (Easwaramoorthy et al. 2007). This highlights the rapid hydrolysis of endogenous acetylcholine in the presence of native AChE and raises the consideration that inactivation/upregulation of nAChRs may arise from “supernormal” activation during AChE inhibition, or during exposure to nicotine or other long-lived agonists. Breakdown of released acetylcholine is usually complete within a matter of seconds (Bruno et al. 2006). As such, the PET observations described above cannot unambiguously be ascribed to the competition model; some of the decreases in receptor binding after treatment with AChE inhibitors might rather be attributable to transition of the receptors to a low-affinity state or to trafficking mechanisms. This is especially a consideration in studies with ligands with slow kinetics, such as in the above reports.
More recently, (−)-[18F]flubatine ((−)-[18F]NCFHEB) was shown to be sensitive to displacement by endogenous acetylcholine in non-human primates. Treatment with AChE inhibitors donepezil and physostigmine led to dose-dependent, significant reductions (10–34%) in VT in bolus and bolus-infusion protocols, respectively (Gallezot et al. 2014). This finding was soon followed by a similar study in human subjects (Hillmer et al. 2016a), in which a bolus and bolus-infusion protocol with [18F]flubatine revealed small but significant reductions in VT in cortical regions following physostigmine administration.
Overall these data suggest that measuring acetylcholine changes using selective α4β2 nAChR radioligands may be possible and that we should expect future applications of this methodology in clinical populations such as Alzheimer’s disease, substance dependence and schizophrenia.
10 Conclusion
The development of selective ligands for nAChRs has been challenging due to the diversity of subtypes existing in the brain and due to the unfavourable kinetics and toxicity profiles of some lead compounds. Promising novel tracers targeting α4β2 nAChRs include [18F]AZAN, [18F]nifene, [18F]XTRA and both the (−) and (+) enantiomers of [18F]flubatine. For the α7 nAChR, [18F]DBT10 and [18F]ASEM are promising candidates. Despite the importance of α6 nAChRs in the action of nicotine on dopamine release, selective tracers for this target remain elusive.
References
Abbaszadeh S, Levin CS (2017) New-generation small animal positron emission tomography system for molecular imaging. J Med Imaging (Bellingham) 4:011008
Abreo MA, Lin NH, Garvey DS et al (1996) Novel 3-pyridyl ethers with subnanomolar affinity for central neuronal nicotinic acetylcholine receptors. J Med Chem 39:817–825
Aghourian M, Legault-Denis C, Soucy JP et al (2017) Quantification of brain cholinergic denervation in Alzheimer’s disease using PET imaging with [18F]-FEOBV. Mol Psychiatry 22:1531–1538
Alessandri B, Heimann A, Filippi R et al (2003) Moderate controlled cortical contusion in pigs: effects on multi-parametric neuromonitoring and clinical relevance. J Neurotrauma 20:1293–1305
Alexander SP, Peters JA, Kelly E et al (2017) The concise guide to pharmacology 2017/18: Ligand-gated ion channels. Br J Pharmacol 174(Suppl 1):S130–S159
Allen DD, Lockman PR (2003) The blood-brain barrier choline transporter as a brain drug delivery vector. Life Sci 73:1609–1615
Allen DD, Lockman PR, Roder KE et al (2003) Active transport of high-affinity choline and nicotine analogs into the central nervous system by the blood-brain barrier choline transporter. J Pharmacol Exp Ther 304:1268–1274
Apelt J, Kumar A, Schliebs R (2002) Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res 953:17–30
Arciniegas DB (2011) Cholinergic dysfunction and cognitive impairment after traumatic brain injury. Part 2: evidence from basic and clinical investigations. J Head Trauma Rehabil 26:319–323
Arneric SP, Sullivan JP, Briggs CA et al (1994) (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl) isoxazole (ABT 418): a novel cholinergic ligand with cognition-enhancing and anxiolytic activities: I. In vitro characterization. J Pharmacol Exp Ther 270:310–318
Auerbach A (2015) Agonist activation of a nicotinic acetylcholine receptor. Neuropharmacology 96:150–156
Avalos M, Parker MJ, Maddox FN et al (2002) Effects of pyridine ring substitutions on affinity, efficacy, and subtype selectivity of neuronal nicotinic receptor agonist epibatidine. J Pharmacol Exp Ther 302:1246–1252
Barrett SP, Boileau I, Okker J et al (2004) The hedonic response to cigarette smoking is proportional to dopamine release in the human striatum as measured by positron emission tomography and [11C]raclopride. Synapse 54:65–71
Bauer A, Zilles K, Matusch A et al (2005) Regional and subtype selective changes of neurotransmitter receptor density in a rat transgenic for the Huntington’s disease mutation. J Neurochem 94:639–650
Belluardo N, Blum M, Mudo G et al (1998) Acute intermittent nicotine treatment produces regional increases of basic fibroblast growth factor messenger RNA and protein in the tel- and diencephalon of the rat. Neuroscience 83:723–740
Bencherif M, Lippiello PM (2010) Alpha7 neuronal nicotinic receptors: the missing link to understanding Alzheimer’s etiopathology? Med Hypotheses 74:281–285
Bengtsson M, Godbolt AK (2016) Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med 48:1–5
Bennett MR (2000) The concept of transmitter receptors: 100 years on. Neuropharmacology 39:523–546
Berg DK, Conroy WG (2002) Nicotinic α7 receptors: synaptic options and downstream signaling in neurons. J Neurobiol 53:512–523
Bertrand D, Lee CH, Flood D et al (2015) Therapeutic potential of α7 nicotinic acetylcholine receptors. Pharmacol Rev 67:1025–1073
Betthauser TJ, Hillmer AT, Lao PJ et al (2017) Human biodistribution and dosimetry of [18F]nifene, an α4β2* nicotinic acetylcholine receptor PET tracer. Nucl Med Biol 55:7–11
Bhatt S, Hillmer AT, Nabulsi N et al (2018) Evaluation of (−)-[18F]flubatine-specific binding: Implications for reference region approaches. Synapse 72
Bieszczad KM, Kant R, Constantinescu CC et al (2012) Nicotinic acetylcholine receptors in rat forebrain that bind 18F-nifene: relating PET imaging, autoradiography, and behavior. Synapse 66:418–434
Biton B, Bergis OE, Galli F et al (2007) SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (1) binding and functional profile. Neuropsychopharmacology 32:1–16
Bohnen NI, Frey KA (2007) Imaging of cholinergic and monoaminergic neurochemical changes in neurodegenerative disorders. Mol Imaging Biol 9(4):243–257
Bohnen NI, Grothe MJ, Ray NJ et al (2018) Recent advances in cholinergic imaging and cognitive decline-Revisiting the cholinergic hypothesis of dementia. Curr Geriatr Rep 7:1–11
Bois F, Gallezot JD, Zheng MQ et al (2015) Evaluation of [18F]-(−)-norchlorofluorohomoepibatidine ([18F]-(−)-NCFHEB) as a PET radioligand to image the nicotinic acetylcholine receptors in non-human primates. Nucl Med Biol 42:570–577
Boltze J, Forschler A, Nitzsche B et al (2008) Permanent middle cerebral artery occlusion in sheep: a novel large animal model of focal cerebral ischemia. J Cereb Blood Flow Metab 28:1951–1964
Bolwin K, Vernekohl D, Luhder J et al (2017) Development of a clear sub-millimeter small animal PET scanner by reducing the influence of the non-collinearity effect. J Instrum 12
Booker TK, Collins AC (1997) Long-term ethanol treatment elicits changes in nicotinic receptor binding in only a few brain regions. Alcohol 14:131–140
Bordia T, Grady SR, McIntosh JM et al (2007) Nigrostriatal damage preferentially decreases a subpopulation of alpha6beta2* nAChRs in mouse, monkey, and Parkinson’s disease striatum. Mol Pharmacol 72:52–61
Bordia T, Parameswaran N, Fan H et al (2006) Partial recovery of striatal nicotinic receptors in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned monkeys with chronic oral nicotine. J Pharmacol Exp Ther 319:285–292
Borlongan CV, Sumaya IC, Moss DE (2005) Methanesulfonyl fluoride, an acetylcholinesterase inhibitor, attenuates simple learning and memory deficits in ischemic rats. Brain Res 1038:50–58
Bornstein MB (1946) Presence and action of acetylcholine in experimental brain trauma. J Neurophysiol 9:349–366
Borovikova LV, Ivanova S, Zhang M et al (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458–462
Boswijk E, Bauwens M, Mottaghy FM et al (2017) Potential of α7 nicotinic acetylcholine receptor PET imaging in atherosclerosis. Methods
Bourne Y, Talley TT, Hansen SB et al (2005) Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J 24:1512–1522
Bouvrais-Veret C, Weiss S, Andrieux A et al (2007) Sustained increase of alpha7 nicotinic receptors and choline-induced improvement of learning deficit in STOP knock-out mice. Neuropharmacology 52:1691–1700
Bouzat C, Sine SM (2018) Nicotinic acetylcholine receptors at the single-channel level. Br J Pharmacol 175:1789–1804
Breese CR, Adams C, Logel J et al (1997) Comparison of the regional expression of nicotinic acetylcholine receptor alpha7 mRNA and [125I]-α-bungarotoxin binding in human postmortem brain. J Comp Neurol 387:385–398
Brejc K, van Dijk WJ, Klaassen RV et al (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411:269–276
Briggs CA, Schrimpf MR, Anderson DJ et al (2008) α7 nicotinic acetylcholine receptor agonist properties of tilorone and related tricyclic analogues. Br J Pharmacol 153:1054–1061
Brody AL, Mandelkern MA, Costello MR et al (2009) Brain nicotinic acetylcholine receptor occupancy: effect of smoking a denicotinized cigarette. Int J Neuropsychopharmacol 12:305–316
Brody AL, Mandelkern MA, London ED et al (2011) Effect of secondhand smoke on occupancy of nicotinic acetylcholine receptors in brain. Arch Gen Psychiatry 68:953–960
Brody AL, Mandelkern MA, London ED et al (2006) Cigarette smoking saturates brain α4β2 nicotinic acetylcholine receptors. Arch Gen Psychiatry 63:907–915
Brown L, Chefer S, Pavlova O et al (2004) Evaluation of 5-(2-(4-pyridinyl)vinyl)-6-chloro-3-(1-methyl-2-(S)-pyrrolidinylmethoxy)py ridine and its analogues as PET radioligands for imaging nicotinic acetylcholine receptors. J Neurochem 91:600–612
Brown LL, Kulkarni S, Pavlova OA et al (2002) Synthesis and evaluation of a novel series of 2-chloro-5-((1-methyl-2-(S)-pyrrolidinyl)methoxy)-3-(2-(4-pyridinyl)vinyl) pyridine analogues as potential positron emission tomography imaging agents for nicotinic acetylcholine receptors. J Med Chem 45:2841–2849
Bruno JP, Gash C, Martin B et al (2006) Second-by-second measurement of acetylcholine release in prefrontal cortex. Eur J Neurosci 24:2749–2757
Brust P, Deuther-Conrad W (2012) Molecular imaging of α7 nicotinic acetylcholine receptors in vivo: current status and perspectives. In: Bright P (ed) Neuroimaging - clinical applications. InTech, Rijeka, Croatia
Brust P, Deuther-Conrad W, Donat CK et al (2014) Preclinical aspects of nicotinic acetylcholine receptor imaging. In: Dierckx RAJO (ed) PET and SPECT of neurobiological systems. Springer, Berlin, Heidelberg
Brust P, Patt JT, Deuther-Conrad W et al (2008) In vivo measurement of nicotinic acetylcholine receptors with [18F]norchloro-fluoro-homoepibatidine. Synapse 62:205–218
Brust P, Peters D, Deuther-Conrad W (2012) Development of radioligands for the imaging of α7 nicotinic acetylcholine receptors with positron emission tomography. Curr Drug Targets 13:594–601
Bunnelle WH, Dart MJ, Schrimpf MR (2004) Design of ligands for the nicotinic acetylcholine receptors: the quest for selectivity. Curr Top Med Chem 4:299–334
Cabello J, Ziegler SI (2018) Advances in PET/MR instrumentation and image reconstruction. Br J Radiol 91:20160363
Changeux JP (2010) Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci 11:389–401
Chattopadhyay S, Xue B, Collins D et al (2005) Synthesis and evaluation of nicotine α4β2 receptor radioligand, 5-(3’-18F-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine, in rodents and PET in nonhuman primate. J Nucl Med 46:130–140
Chatzidaki A, Millar NS (2015) Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol 97:408–417
Chefer SI, Horti AG, Koren AO et al (1999) 2-[18F]F-A-85380: a PET radioligand for α4β2 nicotinic acetylcholine receptors. Neuroreport 10:2715–2721
Chefer SI, Horti AG, Lee KS et al (1998) In vivo imaging of brain nicotinic acetylcholine receptors with 5-[123I]iodo-A-85380 using single photon emission computed tomography. Life Sci 63:PL355–PL360
Chefer SI, London ED, Koren AO et al (2003) Graphical analysis of 2-[18F]FA binding to nicotinic acetylcholine receptors in rhesus monkey brain. Synapse 48:25–34
Chefer SI, Pavlova OA, Zhang Y et al (2008) NIDA522131, a new radioligand for imaging extrathalamic nicotinic acetylcholine receptors: in vitro and in vivo evaluation. J Neurochem 104:306–315
Clarke PB, Pert A (1985) Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res 348:355–358
Clarke PB, Pert CB, Pert A (1984) Autoradiographic distribution of nicotine receptors in rat brain. Brain Res 323:390–395
Colas L, Domercq M, Ramos-Cabrer P et al (2018) In vivo imaging of α7 nicotinic receptors as a novel method to monitor neuroinflammation after cerebral ischemia. Glia
Cortes M, Cao M, Liu HL et al (2017) alpha7 nicotinic acetylcholine receptor signaling modulates the inflammatory phenotype of fetal brain microglia: first evidence of interference by iron homeostasis. Sci Rep 7:10645
Cosgrove KP, Batis J, Bois F et al (2009) beta2-Nicotinic acetylcholine receptor availability during acute and prolonged abstinence from tobacco smoking. Arch Gen Psychiatry 66:666–676
Cosgrove KP, Kloczynski T, Bois F et al (2010) Decreased Beta2*-nicotinic acetylcholine receptor availability after chronic ethanol exposure in nonhuman primates. Synapse 64:729–732
Coughlin J, Du Y, Crawford JL et al (2018a) The availability of the α7 nicotinic acetylcholine receptor in recent-onset psychosis: a study using 18F-ASEM PET. J Nucl Med
Coughlin JM, Du Y, Rosenthal HB et al (2018b) The distribution of the alpha7 nicotinic acetylcholine receptor in healthy aging: an in vivo positron emission tomography study with [18F]ASEM. NeuroImage 165:118–124
Coughlin JM, Slania S, Du Y et al (2018c) 18F-XTRA PET for enhanced imaging of the extrathalamic alpha4beta2 nicotinic acetylcholine receptor. J Nucl Med 59:1603–1608
Crespi A, Colombo SF, Gotti C (2018) Proteins and chemical chaperones involved in neuronal nicotinic receptor expression and function: an update. Br J Pharmacol 175:1869–1879
Cuadra G, Summers K, Giacobini E (1994) Cholinesterase inhibitor effects on neurotransmitters in rat cortex in vivo. J Pharmacol Exp Ther 270:277–284
Cumming P (2011) Absolute abundances and affinity states of dopamine receptors in mammalian brain: a review. Synapse 65:892–909
Cumming P, Rosa-Neto P, Watanabe H et al (2003) Effects of acute nicotine on hemodynamics and binding of [11C]raclopride to dopamine D2,3 receptors in pig brain. NeuroImage 19:1127–1136
Daly JW (1998) Thirty years of discovering arthropod alkaloids in amphibian skin. J Nat Prod 61:162–172
Damsma G, Robertson GS, Tham CS et al (1991) Dopaminergic regulation of striatal acetylcholine release: importance of D1 and N-methyl-D-aspartate receptors. J Pharmacol Exp Ther 259:1064–1072
Darsow T, Booker TK, Pina-Crespo JC et al (2005) Exocytic trafficking is required for nicotine-induced up-regulation of α4β2 nicotinic acetylcholine receptors. J Biol Chem 280:18311–18320
Dash PK, Zhao J, Kobori N et al (2016) Activation of alpha 7 cholinergic nicotinic receptors reduce blood-brain barrier permeability following experimental traumatic brain injury. J Neurosci 36:2809–2818
Davies P, Feisullin S (1981) Postmortem stability of α-bungarotoxin binding sites in mouse and human brain. Brain Res 216:449–454
De Biasi M, Dani JA (2011) Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci 34:105–130
de Fiebre CM, Meyer EM, Henry JC et al (1995) Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic α7/125I-alpha-bungarotoxin receptor subtypes. Mol Pharmacol 47:164–171
de Ramirez SS, Hyder AA, Herbert HK et al (2012) Unintentional injuries: magnitude, prevention, and control. Annu Rev Public Health 33:175–191
De Simone R, Ajmone-Cat MA, Carnevale D et al (2005) Activation of α7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation 2:1–10
DeBoer P, Heeringa MJ, Abercrombie ED (1996) Spontaneous release of acetylcholine in striatum is preferentially regulated by inhibitory dopamine D2 receptors. Eur J Pharmacol 317:257–262
Decker MW, Sullivan JP, Arneric SP et al (2000) Neuronal nicotinic acetylcholine receptors: novel targets for CNS therapeutics. In: Bloom FE, Kupfer DJ (eds) Psychopharmacology: fourth generation of progress. American College of Neuropsychopharmacology, New York. http://www.acnp.org/g4/gn401000009/default.htm
Deuther-Conrad W, Fischer S, Hiller A et al (2011) Assessment of α7 nicotinic acetylcholine receptor availability in juvenile pig brain with [18F]NS10743. Eur J Nucl Med Mol Imaging 38:1541–1549
Deuther-Conrad W, Fischer S, Hiller A et al (2009) Molecular imaging of α7 nicotinic acetylcholine receptors: design and evaluation of the potent radioligand [18F]NS10743. Eur J Nucl Med Mol Imaging 36:791–800
Deuther-Conrad W, Patt JT, Feuerbach D et al (2004) Norchloro-fluoro-homoepibatidine: specificity to neuronal nicotinic acetylcholine receptor subtypes in vitro. Farmacoterapia 59:785–792
Deuther-Conrad W, Patt JT, Lockman PR et al (2008) Norchloro-fluoro-homoepibatidine (NCFHEB) - a promising radioligand for neuroimaging nicotinic acetylcholine receptors with PET. Eur Neuropsychopharmacol 18:222–229
Deuther-Conrad W, Wevers A, Becker G et al (2006) Autoradiography of 2-[18F]F-A-85380 on nicotinic acetylcholine receptors in the porcine brain in vitro. Synapse 59:201–210
Dewan MC, Rattani A, Gupta S et al (2018) Estimating the global incidence of traumatic brain injury. J Neurosurg:1–18
Dewar D, Graham DI (1996) Depletion of choline acetyltransferase activity but preservation of M1 and M2 muscarinic receptor binding sites in temporal cortex following head injury: a preliminary human postmortem study. J Neurotrauma 13:181–187
Dewey SL, Smith GS, Logan J et al (1993) Modulation of central cholinergic activity by GABA and serotonin: PET studies with 11C-benztropine in primates. Neuropsychopharmacology 8:371–376
Dineley KT, Pandya AA, Yakel JL (2015) Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36:96–108
Ding M, Ghanekar S, Elmore CS et al (2012) [3H]Chiba-1001(methyl-SSR180711) has low in vitro binding affinity and poor in vivo selectivity to nicotinic alpha-7 receptor in rodent brain. Synapse 66:315–322
Ding Y, Liu N, Wang T et al (2000a) Synthesis and evaluation of 6-[18F]fluoro-3-(2(S)-azetidinylmethoxy)pyridine as a PET tracer for nicotinic acetylcholine receptors. Nucl Med Biol 27:381–389
Ding YS, Fowler J (2005) New-generation radiotracers for nAChR and NET. Nucl Med Biol 32:707–718
Ding YS, Fowler JS, Logan J et al (2004) 6-[18F]Fluoro-A-85380, a new PET tracer for the nicotinic acetylcholine receptor: studies in the human brain and in vivo demonstration of specific binding in white matter. Synapse 53:184–189
Ding YS, Gatley SJ, Fowler JS et al (1996) Mapping nicotinic acetylcholine receptors with PET. Synapse 24:403–407
Ding YS, Kil KE, Lin KS et al (2006) A novel nicotinic acetylcholine receptor antagonist radioligand for PET studies. Bioorg Med Chem Lett 16:1049–1053
Ding YS, Logan J, Bermel R et al (2000b) Dopamine receptor-mediated regulation of striatal cholinergic activity: positron emission tomography studies with norchloro[18F]fluoroepibatidine. J Neurochem 74:1514–1521
Ding YS, Molina PE, Fowler JS et al (1999) Comparative studies of epibatidine derivatives [18F]NFEP and [18F]N-methyl-NFEP: kinetics, nicotine effect, and toxicity. Nucl Med Biol 26:139–148
Dobelis P, Hutton S, Lu Y et al (2003) GABAergic systems modulate nicotinic receptor-mediated seizures in mice. J Pharmacol Exp Ther 306:1159–1166
Dolci L, Dolle F, Valette H et al (1999) Synthesis of a fluorine-18 labeled derivative of epibatidine for in vivo nicotinic acetylcholine receptor PET imaging. Bioorg Med Chem 7:467–479
Dollé F, Dolci L, Valette H et al (1999) Synthesis and nicotinic acetylcholine receptor in vivo binding properties of 2-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine: a new positron emission tomography ligand for nicotinic receptors. J Med Chem 42:2251–2259
Dollé F, Langle S, Roger G et al (2008) Synthesis and in-vivo evaluation of [(11)C]p-PVP-MEMA as a PET radioligand for imaging nicotinic receptors. Aust J Chem 61:438–445
Dollé F, Valette H, Hinnen F et al (2001) Synthesis and preliminary evaluation of a carbon-11-labelled agonist of the α7 nicotinic acetylcholine receptor. J Label Compd Radiopharm 44:785–795
Dolle RE, Le Bourdonnec B, Goodman AJ et al (2008) Comprehensive survey of chemical libraries for drug discovery and chemical biology: 2007. J Comb Chem 10:753–802
Dome P, Lazary J, Kalapos MP et al (2010) Smoking, nicotine and neuropsychiatric disorders. Neurosci Biobehav Rev 34:295–342
Domino EF, Tsukada H, Harada N (2009) Positron emission tomographic measure of brain dopamine dependence to nicotine as a model of drugs of abuse. Psychopharmacology 204:149–153
Donat CK, Gaber K, Meixensberger J et al (2016) Changes in binding of [123I]CLINDE, a high-affinity translocator protein 18 kDa (TSPO) selective radioligand in a rat model of traumatic brain injury. NeuroMolecular Med 18:158–169
Donat CK, Schuhmann MU, Voigt C et al (2008) Time-dependent alterations of cholinergic markers after experimental traumatic brain injury. Brain Res 1246:167–177
Donat CK, Scott G, Gentleman SM et al (2017) Microglial activation in traumatic brain injury. Front Aging Neurosci 9:208
Donat CK, Walter B, Deuther-Conrad W et al (2010a) Alterations of cholinergic receptors and the vesicular acetylcholine transporter after lateral fluid percussion injury in newborn piglets. Neuropathol Appl Neurobiol 36:225–236
Donat CK, Walter B, Kayser T et al (2010b) Effects of lateral fluid percussion injury on cholinergic markers in the newborn piglet brain. Int J Dev Neurosci 28:31–38
Donnelly-Roberts DL, Xue IC, Arneric SP et al (1996) In vitro neuroprotective properties of the novel cholinergic channel activator (ChCA), ABT-418. Brain Res 719:36–44
Duhaime AC, Margulies SS, Durham SR et al (2000) Maturation-dependent response of the piglet brain to scaled cortical impact. J Neurosurg 93:455–462
Easwaramoorthy B, Pichika R, Collins D et al (2007) Effect of acetylcholinesterase inhibitors on the binding of nicotinic α4β2 receptor PET radiotracer, 18F-nifene: a measure of acetylcholine competition. Synapse 61:29–36
Eckelman WC (2006) Imaging of muscarinic receptors in the central nervous system. Curr Pharm Des 12:3901–3913
Egea J, Buendia I, Parada E et al (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol
Ellis JR, Nathan PJ, Villemagne VL et al (2009a) The relationship between nicotinic receptors and cognitive functioning in healthy aging: an in vivo positron emission tomography (PET) study with 2-[(18)F]fluoro-A-85380. Synapse 63:752–763
Ellis JR, Nathan PJ, Villemagne VL et al (2009b) Galantamine-induced improvements in cognitive function are not related to alterations in α4β2 nicotinic receptors in early Alzheimer’s disease as measured in vivo by 2-[ 18F]Fluoro-A-85380 PET. Psychopharmacology 202:79–91
Ellis JR, Villemagne VL, Nathan PJ et al (2008) Relationship between nicotinic receptors and cognitive function in early Alzheimer’s disease: A 2-[F-18]fluoro-A-85380 PET study. Neurobiol Learn Mem 90:404–412
Esterlis I, Cosgrove KP, Batis JC et al (2010a) Quantification of smoking-induced occupancy of beta2-nicotinic acetylcholine receptors: estimation of nondisplaceable binding. J Nucl Med 51:1226–1233
Esterlis I, Mitsis EM, Batis JC et al (2010b) Brain beta2*-nicotinic acetylcholine receptor occupancy after use of a nicotine inhaler. Int J Neuropsychopharmacol:1–10
Ettrup A, Mikkelsen JD, Lehel S et al (2011) [11C]NS14492 as a novel PET ligand for imaging cerebral α7 nicotinic receptors: in vivo evaluation and drug occupancy measurements. J Nucl Med 52:1449–1456
Fallon JH, Keator DB, Mbogori J et al (2004) Hostility differentiates the brain metabolic effects of nicotine. Brain Res Cogn Brain Res 18:142–148
Fan H, Scheffel UA, Rauseo P et al (2001) [125/123I] 5-Iodo-3-pyridyl ethers. syntheses and binding to neuronal nicotinic acetylcholine receptors. Nucl Med Biol 28:911–921
Felix B, Leger ME, Albe-Fessard D et al (1999) Stereotaxic atlas of the pig brain. Brain Res Bull 49:1–137
Ferrari R, Frasoldati A, Leo G et al (1999) Changes in nicotinic acetylcholine receptor subunit mRNAs and nicotinic binding in spontaneously hypertensive stroke prone rats. Neurosci Lett 277:169–172
Finnema SJ, Scheinin M, Shahid M et al (2015) Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology 232:4129–4157
Fleminger S, Ponsford J (2005) Long term outcome after traumatic brain injury. BMJ (Clin Res ed) 331:1419–1420
Flores CM, Rogers SW, Pabreza LA et al (1992) A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol 41:31–37
Frazier CJ, Rollins YD, Breese CR et al (1998) Acetylcholine activates an α-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci 18:1187–1195
Freedman R, Coon H, Myles-Worsley M et al (1997) Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci U S A 94:587–592
Freedman R, Hall M, Adler LE et al (1995) Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 38:22–33
Freedman R, Olincy A, Buchanan RW et al (2008) Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry 165:1040–1047
Friedrich A, George RL, Bridges CC et al (2001) Transport of choline and its relationship to the expression of the organic cation transporters in a rat brain microvessel endothelial cell line (RBE4). Biochim Biophys Acta 1512:299–307
Fujii T, Mashimo M, Moriwaki Y et al (2017) Expression and function of the cholinergic system in immune cells. Front Immunol 8:1085
Fujita M, Al-Tikriti MS, Tamagnan G et al (2003) Influence of acetylcholine levels on the binding of a SPECT nicotinic acetylcholine receptor ligand [123I]5-I-A-85380. Synapse 48:116–122
Fujita M, Tamagnan G, Zoghbi SS et al (2000) Measurement of α4β2 nicotinic acetylcholine receptors with [123I]5-I-A-85380 SPECT. J Nucl Med 41:1552–1560
Gallezot JD, Bottlaender M, Gregoire MC et al (2005) In vivo imaging of human cerebral nicotinic acetylcholine receptors with 2-[18F]-fluoro-A-85380 and PET. J Nucl Med 46:240–247
Gallezot JD, Esterlis I, Bois F et al (2014) Evaluation of the sensitivity of the novel α4β2* nicotinic acetylcholine receptor PET radioligand 18F-(−)-NCFHEB to increases in synaptic acetylcholine levels in rhesus monkeys. Synapse 68:556–564
Gao Y, Horti AG, Kuwabara H et al (2007a) Derivatives of (−)-7-methyl-2-(5-(pyridinyl)pyridin-3-yl)-7-azabicyclo[2.2.1]heptane are potential ligands for positron emission tomography imaging of extrathalamic nicotinic acetylcholine receptors. J Med Chem 50:3814–3824
Gao Y, Horti AG, Kuwabara H et al (2008a) New synthesis and evaluation of enantiomers of 7-methyl-2-exo-(3′-iodo-5′-pyridinyl)-7-azabicyclo[2.2.1]heptane as stereoselective ligands for PET imaging of nicotinic acetylcholine receptors. Bioorg Med Chem Lett 18:6168–6170
Gao Y, Kellar KJ, Yasuda RP et al (2013) Derivatives of dibenzothiophene for positron emission tomography imaging of alpha7-nicotinic acetylcholine receptors. J Med Chem
Gao Y, Kuwabara H, Spivak CE et al (2008b) Discovery of (−)-7-methyl-2-exo-[3′-(6-[18F]fluoropyridin-2-yl)-5′-pyridinyl]-7-azabicyclo[2.2 .1]heptane, a radiolabeled antagonist for cerebral nicotinic acetylcholine receptor (α4β2-nAChR) with optimal positron emission tomography imaging properties. J Med Chem 51:4751–4764
Gao Y, Ravert HT, Holt D et al (2007b) 6-Chloro-3-(((1-[11C]methyl)-2-(S)-pyrrolidinyl)methoxy)-5-(2-fluoropyri din-4-yl)pyridine ([11C]JHU85270), a potent ligand for nicotinic acetylcholine receptor imaging by positron emission tomography. Appl Radiat Isot 65:947–951
Gao Y, Ravert HT, Kuwabara H et al (2009) Synthesis and biological evaluation of novel carbon-11 labeled pyridyl ethers: candidate ligands for in vivo imaging of α4β2 nicotinic acetylcholine receptors (α4β2-nAChRs) in the brain with positron emission tomography. Bioorg Med Chem 17:4367–4377
Gao Y, Wang H, Mease RC et al (2010) Improved syntheses of precursors for PET radioligands [18F]XTRA and [18F]AZAN. Tetrahedron Lett 51:5333–5335
Garibotto V, Wissmeyer M, Giavri Z et al (2019) Nicotinic receptor abnormalities as a biomarker in idiopathic generalized epilepsy. Eur J Nucl Med Mol Imaging 46:385–395
Gatley SJ, Ding YS, Brady D et al (1998) In vitro and ex vivo autoradiographic studies of nicotinic acetylcholine receptors using 18F fluoronochloroepibatidine in rodent and human brain. Nucl Med Biol 25:449–454
Gatson JW, Simpkins JW, Uteshev VV (2015) High therapeutic potential of positive allosteric modulation of alpha7 nAChRs in a rat model of traumatic brain injury: proof-of-concept. Brain Res Bull 112:35–41
Giastas P, Zouridakis M, Tzartos SJ (2018) Understanding structure-function relationships of the human neuronal acetylcholine receptor: insights from the first crystal structures of neuronal subunits. Br J Pharmacol 175:1880–1891
Gold MEL, Norell MA, Budassi M et al (2018) Rapid 18F-FDG uptake in brain of awake, behaving rat and anesthetized chicken has implications for behavioral PET studies in species with high metabolisms. Front Behav Neurosci 12:115
Goldman-Rakic PS, Selemon LD (1997) Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull 23:437–458
Gotti C, Clementi F, Fornari A et al (2009) Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol 78:703–711
Gotti C, Zoli M, Clementi F (2006) Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 27:482–491
Govind AP, Vezina P, Green WN (2009) Nicotine-induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem Pharmacol 78:756–765
Govind AP, Walsh H, Green WN (2012) Nicotine-induced upregulation of native neuronal nicotinic receptors is caused by multiple mechanisms. J Neurosci 32:2227–2238
Graef S, Schönknecht P, Sabri O et al (2011) Cholinergic receptor subtypes and their role in cognition, emotion, and vigilance control: an overview of preclinical and clinical findings. Psychopharmacology 215:205–229
Gruenbaum SE, Zlotnik A, Gruenbaum BF et al (2016) Pharmacologic neuroprotection for functional outcomes after traumatic brain injury: a systematic review of the clinical literature. CNS Drugs 30:791–806
Gündisch D (2000) Nicotinic acetylcholine receptors and imaging. Curr Pharm Des 6:1143–1157
Gündisch D, Eibl C (2011) Nicotinic acetylcholine receptor ligands, a patent review (2006-2011). Expert Opin Ther Pat 21:1867–1896
Guseva MV, Hopkins DM, Scheff SW et al (2008) Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma 25:975–983
Guseva MV, Kamenskii AA, Gusev VB (2013) Optimization of choline administration regimen for correction of cognitive functions in rats after brain injury. Bull Exp Biol Med. 2013 155(2):197–9
Hallen P, Schug D, Weissler B et al (2018) PET performance evaluation of the small-animal Hyperion II(D) PET/MRI insert based on the NEMA NU-4 standard. Biomed Phys Eng Express 4:065027
Hampel H, Mesulam MM, Cuello AC et al (2018) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141:1917–1933
Han ZY, Zoli M, Cardona A et al (2003) Localization of [3H]nicotine, [3H]cytisine, [3H]epibatidine, and [125I]alpha-bungarotoxin binding sites in the brain of Macaca mulatta. J Comp Neurol 461:49–60
Hashimoto K, Nishiyama S, Ohba H et al (2008) [11C]CHIBA-1001 as a novel PET ligand for α7 nicotinic receptors in the brain: a PET study in conscious monkeys. PLoS One 3:e3231
Hawkins BT, Brown RC, Davis TP (2002) Smoking and ischemic stroke: a role for nicotine? Trends Pharmacol Forensic Sci 23:78–82
Hawkins BT, Egleton RD, Davis TP (2005) Modulation of cerebral microvascular permeability by endothelial nicotinic acetylcholine receptors. Am J Physiol Heart Circ Physiol 289:H212–H219
Hayes RL, Jenkins LW, Lyeth BG (1992) Neurotransmitter-mediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J Neurotrauma 9(Suppl 1):S173–S187
Hedberg MM, Clos MV, Ratia M et al (2010) Effect of huprine X on β-amyloid, synaptophysin and α7 neuronal nicotinic acetylcholine receptors in the brain of 3xTg-AD and APPswe transgenic mice. Neurodegener Dis 7:379–388
Heinrichs RW (2005) The primacy of cognition in schizophrenia. Am Psychol 60:229–242
Hellström-Lindahl E, Court J, Keverne J et al (2004) Nicotine reduces Abeta in the brain and cerebral vessels of APPsw mice. Eur J Neurosci 19:2703–2710
Hellström-Lindahl E, Court JA (2000) Nicotinic acetylcholine receptors during prenatal development and brain pathology in human aging. Behav Brain Res 113:159–168
Henderson DJ, Eberl S, Thomson S et al (2004) 3-Pyridyl ethers as SPECT radioligands for imaging nicotinic acetylcholine receptors. Appl Radiat Isot 60:669–676
Hibbs RE, Sulzenbacher G, Shi J et al (2009) Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal α7 nicotinic acetylcholine receptor. EMBO J 28:3040–3051
Higuchi M, Maeda J, Ji B et al (2012) PET applications in animal models of neurodegenerative and neuroinflammatory disorders. Curr Top Behav Neurosci 11:45–64
Hillmer AT, Esterlis I, Gallezot JD et al (2016a) Imaging of cerebral α4β2* nicotinic acetylcholine receptors with (−)-[18F]Flubatine PET: Implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. NeuroImage 141:71–80
Hillmer AT, Li S, Zheng MQ et al (2017) PET imaging of α7 nicotinic acetylcholine receptors: a comparative study of [18F]ASEM and [18F]DBT-10 in nonhuman primates, and further evaluation of [18F]ASEM in humans. Eur J Nucl Med Mol Imaging
Hillmer AT, Tudorascu DL, Wooten DW et al (2014) Changes in the α4β2* nicotinic acetylcholine system during chronic controlled alcohol exposure in nonhuman primates. Drug Alcohol Depend 138:216–219
Hillmer AT, Wooten DW, Farhoud M et al (2013) PET imaging of acetylcholinesterase inhibitor induced effects on α4β2 nicotinic acetylcholine receptor binding. Synapse 67:882–886
Hillmer AT, Zheng MQ, Li S et al (2016b) PET imaging evaluation of [18F]DBT-10, a novel radioligand specific to α7 nicotinic acetylcholine receptors, in nonhuman primates. Eur J Nucl Med Mol Imaging 43:537–547
Hockley BG, Stewart MN, Sherman P et al (2013) (−)-[18F]Flubatine: evaluation in rhesus monkeys and a report of the first fully automated radiosynthesis validated for clinical use. PETSerotoninpdf 56:595–599
Hoffmeister PG, Donat CK, Schuhmann MU et al (2011) Traumatic brain injury elicits similar alterations in α7 nicotinic receptor density in two different experimental models. NeuroMolecular Med 13:44–53
Hogg RC, Raggenbass M, Bertrand D (2003) Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol 147:1–46
Hong JH, Jang SH, Kim OL et al (2012) Neuronal loss in the medial cholinergic pathway from the nucleus basalis of Meynert in patients with traumatic axonal injury: a preliminary diffusion tensor imaging study. J Head Trauma Rehabil 27:172–176
Horti A, Scheffel U, Stathis M et al (1997) Fluorine-18-FPH for PET imaging of nicotinic acetylcholine receptors. J Nucl Med 38:1260–1265
Horti AG, Chefer SI, Mukhin AG et al (2000) 6-[18F]fluoro-A-85380, a novel radioligand for in vivo imaging of central nicotinic acetylcholine receptors. Life Sci 67:463–469
Horti AG, Gao Y, Kuwabara H et al (2010) Development of radioligands with optimized imaging properties for quantification of nicotinic acetylcholine receptors by positron emission tomography. Life Sci 86:575–584
Horti AG, Gao Y, Kuwabara H et al (2014) 18F-ASEM, a radiolabeled antagonist for imaging the α7-nicotinic acetylcholine receptor with PET. J Nucl Med 55:672–677
Horti AG, Scheffel U, Kimes AS et al (1998a) Synthesis and evaluation of N-[11C]methylated analogues of epibatidine as tracers for positron emission tomographic studies of nicotinic acetylcholine receptors. J Med Chem 41:4199–4206
Horti AG, Scheffel U, Koren AO et al (1998b) 2-[18F]Fluoro-A-85380, an in vivo tracer for the nicotinic acetylcholine receptors. Nucl Med Biol 25:599–603
Horti AG, Villemagne VL (2006) The quest for Eldorado: development of radioligands for in vivo imaging of nicotinic acetylcholine receptors in human brain. Curr Pharm Des 12:3877–3900
Horti AG, Wong DF (2009) Clinical perspective and recent development of pet radioligands for imaging cerebral nicotinic acetylcholine receptors. PET Rev 4:89–100
Hozumi S, Nakagawasai O, Tan-No K et al (2003) Characteristics of changes in cholinergic function and impairment of learning and memory-related behavior induced by olfactory bulbectomy. Behav Brain Res 138:9–15
Hruska M, Keefe J, Wert D et al (2009) Prostate stem cell antigen is an endogenous lynx1-like prototoxin that antagonizes α7-containing nicotinic receptors and prevents programmed cell death of parasympathetic neurons. J Neurosci 29:14847–14854
Huang LZ, Parameswaran N, Bordia T et al (2009) Nicotine is neuroprotective when administered before but not after nigrostriatal damage in rats and monkeys. J Neurochem 109:826–837
Huang Y, Zhu Z, Narendran R et al (2004) Pharmacological evaluation of [C-11]2-[3-(6-chloro-5-phenyl)pyridinyl]-7-methyl-7-aza-bicyclo[2.2.1]heptane, a new PET radioligand for the nicotinic acetylcholine receptors. NeuroImage 22:S2–T113
Huang Y, Zhu Z, Xiao Y et al (2005) Epibatidine analogues as selective ligands for the α(x)β2-containing subtypes of nicotinic acetylcholine receptors. Bioorg Med Chem Lett 15:4385–4388
Humphreys I, Wood RL, Phillips CJ et al (2013) The costs of traumatic brain injury: a literature review. Clinicoecon Outcomes Res 5:281–287
Iida Y, Ogawa M, Ueda M et al (2004) Evaluation of 5-11C-methyl-A-85380 as an imaging agent for PET investigations of brain nicotinic acetylcholine receptors. J Nucl Med 45:878–884
Imming P, Klaperski P, Stubbs MT et al (2001) Syntheses and evaluation of halogenated cytisine derivatives and of bioisosteric thiocytisine as potent and selective nAChR ligands. Eur J Med Chem 36:375–388
Itier V, Schonbachler R, Tribollet E et al (2004) A-186253, a specific antagonist of the alpha4beta2 nAChRs: its properties and potential to study brain nicotinic acetylcholine receptors. Neuropharmacology 47:538–557
Jasinska AJ, Zorick T, Brody AL et al (2014) Dual role of nicotine in addiction and cognition: a review of neuroimaging studies in humans. Neuropharmacology 84:111–122
Jensen AA, Frolund B, Liljefors T et al (2005) Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 48:4705–4745
Jones IW, Barik J, O’Neill MJ et al (2004) Alpha bungarotoxin-1.4 nm gold: a novel conjugate for visualising the precise subcellular distribution of α7* nicotinic acetylcholine receptors. J Neurosci Methods 134:65–74
Jones IW, Westmacott A, Chan E et al (2006) α7 nicotinic acetylcholine receptor expression in Alzheimer’s disease: receptor densities in brain regions of the APP(SWE) mouse model and in human peripheral blood lymphocytes. J Mol Neurosci 30:83–84
Jupp B, Williams J, Binns D et al (2007) Imaging small animal models of epileptogenesis. Neurol Asia 12(Suppl. 1):51–54
Kalamida D, Poulas K, Avramopoulou V et al (2007) Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J 274:3799–3845
Kamal VK, Agrawal D, Pandey RM (2016) Epidemiology, clinical characteristics and outcomes of traumatic brain injury: Evidences from integrated level 1 trauma center in India. J Neurosci Rural Pract 7:515–525
Karlin A (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci 3:102–114
Kas A, Bottlaender M, Gallezot JD et al (2009) Decrease of nicotinic receptors in the nigrostriatal system in Parkinson’s disease. J Cereb Blood Flow Metab 29:1601–1608
Kassiou M, Bottlaender M, Loc’h C et al (2002) Pharmacological evaluation of a Br-76 analog of epibatidine: a potent ligand for studying brain nicotinic acetylcholine receptors. Synapse 45:95–104
Kassiou M, Eberl S, Meikle SR et al (2001) In vivo imaging of nicotinic receptor upregulation following chronic (−)-nicotine treatment in baboon using SPECT. Nucl Med Biol 28:165–175
Kassiou M, Scheffel UA, Ravert HT et al (1998) Pharmacological evaluation of [11C]A-84543: an enantioselective ligand for in vivo studies of neuronal nicotinic acetylcholine receptors. Life Sci 63:PL13–PL18
Kelso ML, Oestreich JH (2012) Traumatic brain injury: central and peripheral role of alpha7 nicotinic acetylcholine receptors. Curr Drug Targets 13:631–636
Kelso ML, Wehner JM, Collins AC et al (2006) The pathophysiology of traumatic brain injury in alpha7 nicotinic cholinergic receptor knockout mice. Brain Res 1083:204–210
Kendziorra K, Wolf H, Meyer PM et al (2011) Decreased cerebral α4β2* nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer’s disease assessed with positron emission tomography. Eur J Nucl Med Mol Imaging 38:515–525
Kim SW, Ding YS, Alexoff D et al (2007a) Synthesis and positron emission tomography studies of C-11-labeled isotopomers and metabolites of GTS-21, a partial α7 nicotinic cholinergic agonist drug. Nucl Med Biol 34:541–551
Kim SW, Ding YS, Alexoff D et al (2007b) Synthesis and positron emission tomography studies of C-11-labeled isotopomers and metabolites of GTS-21, a partial alpha7 nicotinic cholinergic agonist drug. Nucl Med Biol 34:541–551
Kimes AS, Chefer SI, Matochik JA et al (2008) Quantification of nicotinic acetylcholine receptors in the human brain with PET: bolus plus infusion administration of 2-[18F]F-A85380. NeuroImage 39:717–727
Kimes AS, Horti AG, London ED et al (2003) 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. FASEB J 17:1331–1333
Ko GB, Yoon HS, Kim KY et al (2016) Simultaneous multiparametric PET/MRI with silicon photomultiplier PET and ultra-high-field MRI for small-animal imaging. J Nucl Med 57:1309–1315
Koeppe RA (2001) A panel discussion on the future of pharmacology and experimental tomography. In: Gjedde A, Hansen SB, Knudsen GM, Paulson OB (eds) Physiological imaging of the brain with PET. Academic Press, New York
Kozikowski A, Musachio J, Kellar K et al (2005) Ligands for nicotinic acetylcholine receptors, and methods of making and using them. Georgetetown University
Kranz M, Sattler B, Deuther-Conrad W et al (2014) Preclinical dose assessment and biodistribution of [18F]DBT10, a new α7 nicotinic acetylcholine receptor (α7-nAChR) imaging ligand. J Nucl Med 55(Suppl. 1):1143
Kranz M, Sattler B, Wüst N et al (2016) Evaluation of the enantiomer specific biokinetics and radiation doses of [18F]fluspidine-a new tracer in clinical translation for imaging of σ1 receptors. Molecules 21:1164
Kulak JM, Sum J, Musachio JL et al (2002) 5-Iodo-A-85380 binds to α-conotoxin MII-sensitive nicotinic acetylcholine receptors (nAChRs) as well as α4β2 subtypes. J Neurochem 81:403–406
Kumari S, Borroni V, Chaudhry A et al (2008) Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J Cell Biol 181:1179–1193
Kuwabara H, Gao Y, Stabin M et al (2017) Imaging α4β2 nicotinic acetylcholine receptors (nAChRs) in baboons with [18F]XTRA, a radioligand with improved specific binding in extra-thalamic regions. Mol Imaging Biol 19:280–288
Kuwabara H, Wong DF, Gao Y et al (2012) PET Imaging of nicotinic acetylcholine receptors in baboons with 18F-AZAN, a radioligand with improved brain kinetics. J Nucl Med 53:121–129
Lai A, Parameswaran N, Khwaja M et al (2005) Long-term nicotine treatment decreases striatal α6* nicotinic acetylcholine receptor sites and function in mice. Mol Pharmacol 67:1639–1647
Langley JN (1901) On the stimulation and paralysis of nerve-cells and of nerve-endings: Part I. J Physiol 27:224–236
Langley JN (1905) On the reaction of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curari. J Physiol 33:374–413
Lao PJ, Betthauser TJ, Tudorascu DL et al (2017) [18F]Nifene test-retest reproducibility in first-in-human imaging of α4β2* nicotinic acetylcholine receptors. Synapse 71. https://doi.org/10.1002/syn.21981
Larsson A, Engel JA (2004) Neurochemical and behavioral studies on ethanol and nicotine interactions. Neurosci Biobehav Rev 27:713–720
Laruelle M (2000) Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 20:423–451
Le Foll B, Chefer SI, Kimes AS et al (2007) Validation of an extracerebral reference region approach for the quantification of brain nicotinic acetylcholine receptors in squirrel monkeys with PET and 2-18F-fluoro-A-85380. J Nucl Med 48:1492–1500
Le Foll B, Chefer SI, Kimes AS et al (2009) Baseline expression of α4β2* nicotinic acetylcholine receptors predicts motivation to self-administer nicotine. Biol Psychiatry 65:714–716
Lehel S, Madsen J, Ettrup A et al (2009) [11C]NS-12857: a novel PET ligand for α7-nicotinergic receptors. J Labelled Compd Rad 52:S379–S379
Leonard S (2003) Consequences of low levels of nicotinic acetylcholine receptors in schizophrenia for drug development. Drug Dev Res 60:127–136
Lester HA, Xiao C, Srinivasan R et al (2009) Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J 11:167–177
Li H, Sagar AP, Keri S (2018) Microglial markers in the frontal cortex are related to cognitive dysfunctions in major depressive disorder. J Affect Disord 241:305–310
Liang F, Navarro HA, Abraham P et al (1997) Synthesis and nicotinic acetylcholine receptor binding properties of exo-2-(2′-fluoro-5′-pyridinyl)-7-azabicyclo- 2.2.1 heptane: a new positron emission tomography ligand for nicotinic receptors. J Med Chem 40:2293–2295
Liddelow SA, Guttenplan KA, Clarke LE et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487
Lighthall JW (1988) Controlled cortical impact: a new experimental brain injury model. J Neurotrauma 5:1–15
Lipovsek M, Plazas P, Savino J et al (2008) Properties of mutated murine α4β2 nicotinic receptors linked to partial epilepsy. Neurosci Lett 434:165–169
Loane DJ, Faden AI (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 31:596–604
LoBue C, Cullum CM, Didehbani N et al (2018) Neurodegenerative dementias after traumatic brain injury. J Neuropsychiatry Clin Neurosci 30:7–13
Lohr JB, Flynn K (1992) Smoking and schizophrenia. Schizophr Res 8:93–102
London ED, Connolly RJ, Szikszay M et al (1988) Effects of nicotine on local cerebral glucose utilization in the rat. J Neurosci 8:3920–3928
London ED, Scheffel U, Kimes AS et al (1995) In vivo labeling of nicotinic acetylcholine receptors in brain with [3H]epibatidine. Eur J Pharmacol 278:R1–R2
Lotfipour S, Mandelkern M, Alvarez-Estrada M et al (2012a) A single administration of low-dose varenicline saturates α4β2* nicotinic acetylcholine receptors in the human brain. Neuropsychopharmacology 37:1738–1748
Lotfipour S, Mandelkern M, Alvarez-Estrada M et al (2012b) A single administration of low-dose varenicline saturates alpha4beta2(*) nicotinic acetylcholine receptors in the human brain. Neuropsychopharmacology 37:1738–1748
Lucas A, Hawkes R, Ansorge R et al (2007) Development of a combined microPET(R)MR system. Workshop on Medical Instrumentation Signal and Imaging, 11-12 April 2007 Aveiro. Portugal 827
Ludwig FA, Fischer S, Smits R et al (2018) Exploring the metabolism of (+)-[18F]flubatine in vitro and in vivo: LC-MS/MS aided identification of radiometabolites in a clinical PET study. Molecules 23(2):464
Ma B, Sherman PS, Moskwa JE et al (2004) Sensitivity of [11C]N-methylpyrrolidinyl benzilate ([11C]NMPYB) to endogenous acetylcholine: PET imaging vs tissue sampling methods. Nucl Med Biol 31:393–397
Magnussen JH, Ettrup A, Donat CK et al (2015) Radiosynthesis and in vitro validation of 3H-NS14492 as a novel high affinity alpha7 nicotinic receptor radioligand. Eur J Pharmacol 762:35–41
Maier DL, Hill G, Ding M et al (2011) Pre-clinical validation of a novel alpha-7 nicotinic receptor radiotracer, [3H]AZ11637326: target localization, biodistribution and ligand occupancy in the rat brain. Neuropharmacology 61:161–171
Malpass JR, Hemmings DA, Wallis AL et al (2001) Synthesis and nicotinic acetylcholine-binding properties of epibatidine homologues: homoepibatidine and dihomoepibatidine. Nicotin1 2001:1044–1050
Manley GT, Rosenthal G, Lam M et al (2006) Controlled cortical impact in swine: pathophysiology and biomechanics. J Neurotrauma 23:128–139
Marenco S, Carson RE, Berman KF et al (2004) Nicotine-induced dopamine release in primates measured with [11C]raclopride PET. Neuropsychopharmacology 29:259–268
Marenco T, Bernstein S, Cumming P et al (2000) Effects of nicotine and chlorisondamine on cerebral glucose utilization in immobilized and freely-moving rats. Br J Pharmacol 129:147–155
Marmarou A, Foda MA, van den Brink W et al (1994) A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg 80:291–300
Marshall HR, Stodilka RZ, Theberge J et al (2011) A comparison of MR-based attenuation correction in PET versus SPECT. Phys Med Biol 56:4613–4629
Martelli D, McKinley MJ, McAllen RM (2014) The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci 182:65–69
Martin A, Szczupak B, Gomez-Vallejo V et al (2015) In vivo PET imaging of the alpha4beta2 nicotinic acetylcholine receptor as a marker for brain inflammation after cerebral ischemia. J Neurosci 35:5998–6009
Marutle A, Warpman U, Bogdanovic N et al (1998) Regional distribution of subtypes of nicotinic receptors in human brain and effect of aging studied by (+/−)- 3H epibatidine. Brain Res 801:143–149
Masel BE, DeWitt DS (2010) Traumatic brain injury: a disease process, not an event. J Neurotrauma 27:1529–1540
Matsumura A, Suzuki S, Iwahara N et al (2015) Temporal changes of CD68 and α7 nicotinic acetylcholine receptor expression in microglia in Alzheimer’s disease-like mouse models. J Alzheimers Dis 44:409–423
Maziere M, Comar D, Marazano C et al (1976) Nicotine-11C: synthesis and distribution kinetics in animals. Eur J Nucl Med 1:255–258
McKee AC, Alosco ML, Huber BR (2016) Repetitive head impacts and chronic traumatic encephalopathy. Neurosurg Clin N Am 27:529–535
Meier D, Wagenaar DJ, Chen S et al (2011) A SPECT camera for combined MRI and SPECT for small animals. Nucl Instrum Methods Phys Res 652:731–734
Mexal S, Berger R, Logel J et al (2010) Differential regulation of α7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J Mol Neurosci 40:185–195
Meyer EM, Kuryatov A, Gerzanich V et al (1998) Analysis of 3-(4-hydroxy, 2-methoxybenzylidene)anabaseine selectivity and activity at human and rat α7 nicotinic receptors. J Pharmacol Exp Ther 287:918–925
Meyer PM, Strecker K, Kendziorra K et al (2009) Reduced α4β2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Arch Gen Psychiatry 66:866–877
Meyerhoff DJ, Tizabi Y, Staley JK et al (2006) Smoking comorbidity in alcoholism: neurobiological and neurocognitive consequences. Alcohol Clin Exp Res 30:253–264
Millar NS, Harkness PC (2008) Assembly and trafficking of nicotinic acetylcholine receptors (Review). Mol Membr Biol 25:279–292
Mitkovski S, Villemagne VL, Novakovic KE et al (2005) Simplified quantification of nicotinic receptors with 2[18F]F-A-85380 PET. Nucl Med Biol 32:585–591
Mo YX, Yin YF, Li YM (2014) Neural nAChRs PET imaging probes. Nucl Med Commun 35:135–143
Molina PE, Ding YS, Carroll FI et al (1997) Fluoro-norchloroepibatidine: preclinical assessment of acute toxicity. Nucl Med Biol 24:743–747
Montgomery AJ, Lingford-Hughes AR, Egerton A et al (2007) The effect of nicotine on striatal dopamine release in man: A [11C]raclopride PET study. Synapse 61:637–645
Morganti-Kossmann MC, Semple BD, Hellewell SC et al (2019) The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol 137:731–755
Mu L, Drandarov K, Bisson WH et al (2006) Synthesis and binding studies of epibatidine analogues as ligands for the nicotinic acetylcholine receptors. Eur J Med Chem 41:640–650
Mufson EJ, He B, Ginsberg SD et al (2018) Gene profiling of nucleus basalis tau containing neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. J Neurotrauma 35:1260–1271
Mufson EJ, Perez SE, Nadeem M et al (2016) Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Brain Inj 30:1399–1413
Mukherjee J, Lao PJ, Betthauser TJ et al (2018) Human brain imaging of nicotinic acetylcholine α4β2* receptors using [18F]Nifene: Selectivity, functional activity, toxicity, aging effects, gender effects, and extrathalamic pathways. J Comp Neurol 526:80–95
Mukherjee J, Pichika R, Leslie FM et al (2004) Design and development of novel new PET imaging agents for α4β2 nicotinic recpetors: 18F-nifrolidine and 18F-nifzetidine. NeuroImage 22:S2–T124
Mukhin AG, Kimes AS, Chefer SI et al (2008) Greater nicotinic acetylcholine receptor density in smokers than in nonsmokers: a PET study with 2-18F-FA-85380. J Nucl Med 49:1628–1635
Murdoch I, Nicoll JAR, Graham DI et al (2002) Nucleus basalis of Meynert pathology in the human brain after fatal head injury. J Neurotrauma 19:279–284
Murdoch I, Perry EK, Court JA et al (1998) Cortical cholinergic dysfunction after human head injury. J Neurotrauma 15:295–305
Nakaizumi K, Ouchi Y, Terada T et al (2018) In vivo depiction of α7 nicotinic receptor loss for cognitive decline in Alzheimer’s disease. J Alzheimers Dis 61:1355–1365
Nishiyama S, Ohba H, Kanazawa M et al (2015) Comparing α7 nicotinic acetylcholine receptor binding, amyloid-β deposition, and mitochondria complex-I function in living brain: a PET study in aged monkeys. Synapse 69:475–483
Nomikos GG, Schilström B, Hildebrand BE et al (2000) Role of α7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behav Brain Res 113:97–103
Nordberg A (1993) Clinical studies in Alzheimer patients with positron emission tomography. Behav Brain Res 57:215–224
Nordberg A, Hartvig P, Lilja A et al (1991) Nicotine receptors in the brain of patients with Alzheimer’s disease. Studies with 11C-nicotine and positron emission tomography. Acta Radiol Suppl 376:165–166
Nordberg A, Lilja A, Lundqvist H et al (1992) Tacrine restores cholinergic nicotinic receptors and glucose metabolism in Alzheimer patients as visualized by positron emission tomography. Neurobiol Aging 13:747–758
Nuechterlein KH, Barch DM, Gold JM et al (2004) Identification of separable cognitive factors in schizophrenia. Schizophr Res 72:29–39
Nybäck H, Halldin C, Ahlin A et al (1994) PET studies of the uptake of (S)- and (R)-[11C]nicotine in the human brain: difficulties in visualizing specific receptor binding in vivo. Psychopharmacology 115:31–36
O’Connor WT, Smyth A, Gilchrist MD (2011) Animal models of traumatic brain injury: a critical evaluation. Pharmacol Ther 130:106–113
O’Neill MJ, Murray TK, Lakics V et al (2002) The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Targets CNS Neurol Disord 1:399–411
Oddo S, Caccamo A, Green KN et al (2005) Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc Natl Acad Sci U S A 102:3046–3051
Ogawa M, Nishiyama S, Tsukada H et al (2010) Synthesis and evaluation of new imaging agent for central nicotinic acetylcholine receptor α7 subtype. Nucl Med Biol 37:347–355
Ogawa M, Tatsumi R, Fujio M et al (2006) Synthesis and evaluation of [125I]I-TSA as a brain nicotinic acetylcholine receptor α7 subtype imaging agent. Nucl Med Biol 33:311–316
Ogawa M, Tsukada H, Hatano K et al (2009) Central in vivo nicotinic acetylcholine receptor imaging agents for positron emission tomography (PET) and single photon emission computed tomography (SPECT). Biol Pharm Bull 32:337–340
Okada H, Ouchi Y, Ogawa M et al (2013) Alterations in α4β2 nicotinic receptors in cognitive decline in Alzheimer’s aetiopathology. Brain 136:3004–3017
Oldendorf W, Braun L, Cornford E (1979) pH dependence of blood-brain barrier permeability to lactate and nicotine. Stroke 10:577–581
Olesen J, Leonardi M (2003) The burden of brain diseases in Europe. Eur J Neurol 10:471–477
Ostberg A, Tenovuo O (2014) Smoking and outcome of traumatic brain injury. Brain Inj 28:155–160
Ostberg A, Virta J, Rinne JO et al (2018) Brain cholinergic function and response to rivastigmine in patients with chronic sequels of traumatic brain injury: A PET study. J Head Trauma Rehabil 33:25–32
Östberg A, Virta J, Rinne JO et al (2011) Cholinergic dysfunction after traumatic brain injury: preliminary findings from a PET study. Neurology 76:1046–1050
Parl C, Kolb A, Stricker-Shaver D et al (2019) Dual layer doI detector modules for a dedicated mouse brain PET/MRI. Phys Med Biol 64:055004
Paterson D, Nordberg A (2000) Neuronal nicotinic receptors in the human brain. Prog Neurobiol 61:75–111
Paterson LM, Tyacke RJ, Nutt DJ et al (2010) Measuring endogenous 5-HT release by emission tomography: promises and pitfalls. J Cereb Blood Flow Metab 30:1682–1706
Patt JT, Spang JE, Buck A et al (2001) Synthesis and in vivo studies of the stereoisomers of N-[11C]methyl-homoepibatidine. Nucl Med Biol 28:645–655
Patt JT, Spang JE, Westera G et al (1999) Synthesis and in Vivo studies of [C-11]N-methylepibatidine: comparison of the stereoisomers. Nucl Med Biol 26:165–173
Paulson JR, Yang T, Selvaraj PK et al (2010) Nicotine exacerbates brain edema during in vitro and in vivo focal ischemic conditions. J Pharmacol Exp Ther 332:371–379
Pauly JR, Stitzel JA, Marks MJ et al (1989) An autoradiographic analysis of cholinergic receptors in mouse brain. Brain Res Bull 22:453–459
Pavlov VA, Wang H, Czura CJ et al (2003) The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med 9:125–134
Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. Academic Press, New York
Peng X, Gerzanich V, Anand R et al (1994) Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol 46:523–530
Perry EK, Perry RH, Smith CJ et al (1987) Nicotinic receptor abnormalities in Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry 50:806–809
Perry EK, Perry RH, Smith CJ et al (1986) Cholinergic receptors in cognitive disorders. Can J Neurol Sci 13:521–527
Peters D, Olsen GM, Nielsen EO et al (2007) Novel 1,4-diaza-bicyclo[3.2.2]nonyl oxadiazolyl derviatives and their medical use, WO/2007/138037.
Phillips HA, Favre I, Kirkpatrick M et al (2001) CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet 68:225–231
Picard F, Bruel D, Servent D et al (2006) Alteration of the in vivo nicotinic receptor density in ADNFLE patients: a PET study. Brain 129:2047–2060
Picciotto MR, Zoli M (2002) Nicotinic receptors in aging and dementia. J Neurobiol 53:641–655
Pichika R, Easwaramoorthy B, Christian BT et al (2011) Nicotinic alpha4beta2 receptor imaging agents. Part III. Synthesis and biological evaluation of 3-(2-(S)-azetidinylmethoxy)-5-(3′-(18)F-fluoropropyl)pyridine ((18)F-nifzetidine). Nucl Med Biol 38:1183–1192
Pichika R, Easwaramoorthy B, Collins D et al (2006) Nicotinic α4β2 receptor imaging agents Part II. Synthesis and biological evaluation of 2-[18F]fluoro-3-[2-((S)-3-pyrrolinyl)methoxy]pyridine (18F-nifene) in rodents and imaging by PET in nonhuman primate. Nucl Med Biol 33:295–304
Pichika R, Kuruvilla SA, Patel N et al (2013) Nicotinic α4β2 receptor imaging agents. Part IV. Synthesis and Biological Evaluation of 3-(2-(S)-3,4-dehydropyrrolinyl methoxy)-5-(3′-18F-Fluoropropyl)pyridine (18F-Nifrolene) using PET. Nucl Med Biol 40:117–125
Pictet A (1903) Synthese de la nicotine. Nicotin7 137:860–862
Pin F, Vercouillie J, Ouach A et al (2014) Design of α7 nicotinic acetylcholine receptor ligands in quinuclidine, tropane and quinazoline series. Chemistry, molecular modeling, radiochemistry, in vitro and in rats evaluations of a [18F] quinuclidine derivative. Eur J Med Chem 82:214–224
Pinner A (1893) Ueber Nicotin. Die Constitution des Alkaloids V Mittheilung Berichte der deutschen chemischen Gesellschaft 26:292–305
Pinner A, Wolffenstein R (1891) Ueber Nicotin. Ber Dtsch Chem Ges 24:61–67
Plaven-Sigray P, Matheson GJ, Collste K et al (2018) Positron emission tomography studies of the glial cell marker translocator protein in patients with psychosis: a meta-analysis using individual participant data. Biol Psychiatry 84:433–442
Plested AJ (2016) Structural mechanisms of activation and desensitization in neurotransmitter-gated ion channels. Nat Struct Mol Biol 23:494–502
Pomper MG, Phillips E, Fan H et al (2005) Synthesis and biodistribution of radiolabeled α7 nicotinic acetylcholine receptor ligands. J Nucl Med 46:326–334
Posselt W, Reimann L (1828) Chemische Untersuchungen des Tabaks und Darstellung des eigenthümlichen wirksamen Princips dieser Pflanze. Nicotin6 24:138–161
Pradhan AA, Cumming P, Clarke PB (2002) [125I]Epibatidine-labelled nicotinic receptors in the extended striatum and cerebral cortex: lack of association with serotonergic afferents. Brain Res 954:227–236
Prakash N, Frostig RD (2005) What has intrinsic signal optical imaging taught us about NGF-induced rapid plasticity in adult cortex and its relationship to the cholinergic system? Mol Imaging Biol 7:14–21
Quelch DR, Katsouri L, Nutt DJ et al (2014) Imaging endogenous opioid peptide release with [11C]carfentanil and [3H]diprenorphine: influence of agonist-induced internalization. J Cereb Blood Flow Metab 34:1604–1612
Quik M (2004) Smoking, nicotine and Parkinson’s disease. Trends Neurosci 27:561–568
Quik M, Campos C, Parameswaran N et al (2010) Chronic nicotine treatment increases nAChRs and microglial expression in monkey substantia nigra after nigrostriatal damage. J Mol Neurosci 40:105–113
Quik M, Parameswaran N, McCallum SE et al (2006) Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J Neurochem 98:1866–1875
Quik M, Perez XA, Grady SR (2011) Role of alpha6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem Pharmacol 82:873–882
Quik M, Polonskaya Y, Gillespie A et al (2000) Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol 425:58–69
Quik M, Sum JD, Whiteaker P et al (2003) Differential declines in striatal nicotinic receptor subtype function after nigrostriatal damage in mice. Mol Pharmacol 63:1169–1179
Raad M, Nohra E, Chams N et al (2014) Autoantibodies in traumatic brain injury and central nervous system trauma. Neuroscience 281:16–23
Radcliffe KA, Dani JA (1998) Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci 18:7075–7083
Radek RJ, Miner HM, Bratcher NA et al (2006) Alpha4beta2 nicotinic receptor stimulation contributes to the effects of nicotine in the DBA/2 mouse model of sensory gating. Psychopharmacology 187:47–55
Rademacher L, Prinz S, Winz O et al (2016) Effects of smoking cessation on presynaptic dopamine function of addicted male smokers. Biol Psychiatry 80:198–206
Raggenbass M, Bertrand D (2002) Nicotinic receptors in circuit excitability and epilepsy. J Neurobiol 53:580–589
Rahman S, Engleman EA, Bell RL (2015) Nicotinic receptor modulation to treat alcohol and drug dependence. Front Neurosci 8:426
Ramlackhansingh AF, Brooks DJ, Greenwood RJ et al (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70:374–383
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19:987–991
Ren C, Tong YL, Li JC et al (2017) The protective effect of alpha 7 nicotinic acetylcholine receptor activation on critical illness and its mechanism. Int J Biol Sci 13:46–56
Robles N, Sabria J (2008) Effects of moderate chronic ethanol consumption on hippocampal nicotinic receptors and associative learning. Neurobiol Learn Mem 89:497–503
Roger G, Lagnel B, Rouden J et al (2003) Synthesis of a [2-pyridinyl-18F]-labelled fluoro derivative of (−)-cytisine as a candidate radioligand for brain nicotinic α4β2 receptor imaging with PET. Bioorg Med Chem 11:5333–5343
Roger G, Saba W, Valette H et al (2006) Synthesis and radiosynthesis of [18F]FPhEP, a novel α4β2-selective, epibatidine-based antagonist for PET imaging of nicotinic acetylcholine receptors. Bioorg Med Chem 14:3848–3858
Rominger A, Wagner E, Mille E et al (2010) Endogenous competition against binding of [18F]DMFP and [18F]fallypride to dopamine D2/3 receptors in brain of living mouse. Synapse 64:313–322
Roos RA (2010) Huntington’s disease: a clinical review. Orphanet J Rare Dis 5:40
Rose JE, Mukhin AG, Lokitz SJ et al (2010) Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci U S A 107:5190–5195
Ross RG, Stevens KE, Proctor WR et al (2010) Research review: cholinergic mechanisms, early brain development, and risk for schizophrenia. J Child Psychol Psychiatry 51:535–549
Rötering S, Deuther-Conrad W, Cumming P et al (2014) Imaging of α7 nicotinic acetylcholine receptors in brain and cerebral vasculature of juvenile pigs with [18F]NS14490. EJNMMI Res 4:43
Rubio A, Perez M, Avila J (2006) Acetylcholine receptors and tau phosphorylation. Curr Mol Med 6:423–428
Saba W, Valette H, Granon S et al (2010) [18F]ZW-104, a new radioligand for imaging α2-α3-α4/β2 central nicotinic acetylcholine receptors: Evaluation in mutant mice. Synapse 64:570–572
Sabri O, Kendziorra K, Wolf H et al (2008) Acetylcholine receptors in dementia and mild cognitive impairment. Eur J Nucl Med Mol Imaging 35(Suppl 1):S30–S45
Sabri O, Meyer PM, Gertz H-J et al (2014) PET imaging of the α4β2* nicotinic acetylcholine receptors in Alzheimer’s disease. In: Dierckx RAJO (ed) PET and SPECT in neurology. Springer, Berlin Heidelberg
Sabri O, Meyer PM, Graf S et al (2018) Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer’s dementia. Brain 141:1840–1854
Salokangas RK, Vilkman H, Ilonen T et al (2000) High levels of dopamine activity in the basal ganglia of cigarette smokers. Am J Psychiatry 157:632–634
Sandiego CM, Jin X, Mulnix T et al (2013) Awake nonhuman primate brain PET imaging with minimal head restraint: evaluation of GABAA-benzodiazepine binding with 11C-flumazenil in awake and anesthetized animals. J Nucl Med 54:1962–1968
Sarasamkan J, Fischer S, Deuther-Conrad W et al (2017) Radiosynthesis of (S)-[18F]T1: The first PET radioligand for molecular imaging of α3β4 nicotinic acetylcholine receptors. Appl Radiat Isot 124:106–113
Sarasamkan J, Scheunemann M, Apaijai N et al (2016) Varying chirality across nicotinic acetylcholine receptor subtypes: selective binding of quinuclidine triazole compounds. ACS Med Chem Lett 7:890–895
Saricicek A, Esterlis I, Maloney KH et al (2012) Persistent beta2*-nicotinic acetylcholinergic receptor dysfunction in major depressive disorder. Am J Psychiatry 169:851–859
Sattler B, Kranz M, Starke A et al (2014) Internal dose assessment of (−)-18F-flubatine, comparing animal model datasets of mice and piglets with first-in-human results. J Nucl Med 55:1885–1892
Scheff SW, Baldwin SA, Brown RW et al (1997) Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. J Neurotrauma 14:615–627
Scheffel U, Horti AG, Koren AO et al (2000) 6-[18F]Fluoro-A-85380: an in vivo tracer for the nicotinic acetylcholine receptor. Nucl Med Biol 27:51–56
Schep LJ, Slaughter RJ, Beasley DM (2009) Nicotinic plant poisoning. Clin Toxicol (Phila) 47:771–781
Scheunemann M, Teodoro R, Wenzel B et al (2014) Synthesis and F-18 labeling of a 2-fluoro dibenzothiophene sulfone derivative, as a potential alpha-7 nicotinic acetylcholine receptor (α7 nAChR) imaging agent. Nuklearmedizin 53:A26
Schilström B, Fagerquist MV, Zhang X et al (2000) Putative role of presynaptic α7* nicotinic receptors in nicotine stimulated increases of extracellular levels of glutamate and aspartate in the ventral tegmental area. Synapse 38:375–383
Schliebs R (2005) Basal forebrain cholinergic dysfunction in Alzheimer’s disease—interrelationship with beta-amyloid, inflammation and neurotrophin signaling. Neurochem Res 30:895–908
Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221:555–563
Schmaljohann J, Gundisch D, Minnerop M et al (2005) A simple and fast method for the preparation of n.c.a. 2-[18F]F-A85380 for human use. Appl Radiat Isot 63:433–435
Schrimpf MR, Sippy KB, Briggs CA et al (2012) SAR of α7 nicotinic receptor agonists derived from tilorone: exploration of a novel nicotinic pharmacophore. Bioorg Med Chem Lett 22:1633–1638
Schröck H, Kuschinsky W (1991) Effects of nicotine withdrawal on the local cerebral glucose utilization in conscious rats. Brain Res 545:234–238
Schulz D, Southekal S, Junnarkar SS et al (2011) Simultaneous assessment of rodent behavior and neurochemistry using a miniature positron emission tomograph. Nat Methods 8:347–352
Serriere S, Domene A, Vercouillie J et al (2015) Assessment of the protection of dopaminergic neurons by an alpha7 nicotinic receptor agonist, PHA 543613 using [(18)F]LBT-999 in a Parkinson’s disease rat model. Front Med (Lausanne) 2:61
Sharma G, Vijayaraghavan S (2001) Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci U S A 98:4148–4153
Sharma G, Vijayaraghavan S (2002) Nicotinic receptor signaling in nonexcitable cells. J Neurobiol 53:524–534
Sharma G, Vijayaraghavan S (2008) Nicotinic receptors: role in addiction and other disorders of the brain. Subst Abus 2008:81
Shen JX, Yakel JL (2009) Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol Sin 30:673–680
Shen JX, Yakel JL (2012) Functional alpha7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci 48:14–21
Shimohama S, Taniguchi T, Fujiwara M et al (1985) Biochemical characterization of the nicotinic cholinergic receptors in human brain: binding of (−)-[3H]nicotine. J Neurochem 45:604–610
Shimohama S, Taniguchi T, Fujiwara M et al (1986) Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J Neurochem 46:288–293
Shin SS, Dixon CE (2015) Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J Neurotrauma 32:1429–1440
Shytle RD, Mori T, Townsend K et al (2004a) Cholinergic modulation of microglial activation by α7 nicotinic receptors. J Neurochem 89:337–343
Shytle RD, Mori T, Townsend K et al (2004b) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 89:337–343
Siegmund B, Leitner E, Pfannhauser W (1999) Determination of the nicotine content of various edible nightshades (Solanaceae) and their products and estimation of the associated dietary nicotine intake. J Agric Food Chem 47:3113–3120
Sihver W, Fasth KJ, Horti AG et al (1999a) Synthesis and characterization of binding of 5-[76Br]bromo-3-[[2(S)-azetidinyl]methoxy]pyridine, a novel nicotinic acetylcholine receptor ligand, in rat brain. J Neurochem 73:1264–1272
Sihver W, Fasth KJ, Ögren M et al (1998) In vitro evaluation of 11C-labeled (S)-nicotine, (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl)isoxazole, and (R,S)-1-methyl-2-(3-pyridyl)azetidine as nicotinic receptor ligands for positron emission tomography studies. J Neurochem 71:1750–1760
Sihver W, Fasth KJ, Ögren M et al (1999b) In vivo positron emission tomography studies on the novel nicotinic receptor agonist [11C]MPA compared with [11C]ABT-418 and (S)(−) [11C]nicotine in rhesus monkeys. Nucl Med Biol 26:633–640
Sihver W, Langström B, Nordberg A (2000a) Ligands for in vivo imaging of nicotinic receptor subtypes in Alzheimer brain. Acta Neurol Scand Suppl 176:27–33
Sihver W, Nordberg A, Langström B et al (2000b) Development of ligands for in vivo imaging of cerebral nicotinic receptors. Behav Brain Res 113:143–157
Small E, Shah HP, Davenport JJ et al (2010) Tobacco smoke exposure induces nicotine dependence in rats. Psychopharmacology 208:143–158
Smith DF, Jakobsen S (2007) Stereoselective neuroimaging in vivo. Eur Neuropsychopharmacol 17:507–522
Smith ML, Souza FG, Bruce KS et al (2014) Acetylcholine receptors in the retinas of the alpha7 nicotinic acetylcholine receptor knockout mouse. Mol Vis 20:1328–1356
Sobrio F, Quentin T, Dhilly M et al (2008) Radiosynthesis and ex vivo evaluation of [11C]-SIB-1553A as a PET radiotracer for β4 selective subtype nicotinic acetylcholine receptor. Nucl Med Biol 35:377–385
Soloway SB (1976) Naturally occurring insecticides. Environ Health Perspect 14:109–117
Song C, Leonard BE (2005) The olfactory bulbectomised rat as a model of depression. Neurosci Biobehav Rev 29:627–647
Sorokina EG, Vol’pina OM, Semenova Zh B et al (2012) Autoantibodies to the α7 subunit of the neuronal acetylcholine receptor in craniocerebral trauma in children. Neurosci Behav Physiol 42:740–744
Spang JE, Patt JT, Westera G et al (2000) Comparison of N-[11C]methyl-norchloroepibatidine and N-[11C]methyl-2-(2-pyridyl)-7-azabicyclo[2.2.1]heptane with N-[11C]methyl-epibatidine in small animal PET studies. Nucl Med Biol 27:239–247
Spurden DP, Court JA, Lloyd S et al (1997) Nicotinic receptor distribution in the human thalamus: autoradiographical localization of [3H]nicotine and [125I] alpha-bungarotoxin binding. J Chem Neuroanat 13:105–113
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–40
Staley JK, Krishnan-Sarin S, Cosgrove KP et al (2006) Human tobacco smokers in early abstinence have higher levels of β2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci 26:8707–8714
Steinlein OK, Mulley JC, Propping P et al (1995) A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 11:201–203
Stephens SH, Logel J, Barton A et al (2009) Association of the 5′-upstream regulatory region of the α7 nicotinic acetylcholine receptor subunit gene (CHRNA7) with schizophrenia. Schizophr Res 109:102–112
Suhara T, Inoue O, Kobayashi K et al (1994) An acute effect of triazolam on muscarinic cholinergic receptor binding in the human brain measured by positron emission tomography. Psychopharmacology 113:311–317
Sullivan JP, Donnelly-Roberts D, Briggs CA et al (1996) A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro pharmacological properties of a novel, high affinity α4β2 nicotinic acetylcholine receptor ligand. Neuropharmacology 35:725–734
Sultzer DL, Melrose RJ, Riskin-Jones H et al (2017) Cholinergic receptor binding in alzheimer disease and healthy aging: assessment in vivo with positron emission tomography imaging. Am J Geriatr Psychiatry 25:342–353
Suzuki T, Hide I, Matsubara A et al (2006) Microglial α7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 83:1461–1470
Taly A, Corringer PJ, Guedin D et al (2009) Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 8:733–750
Tanibuchi Y, Wu J, Toyohara J et al (2010) Characterization of [3H]CHIBA-1001 binding to α7 nicotinic acetylcholine receptors in the brain from rat, monkey, and human. Brain Res 1348:200–208
Taylor P, Radic Z, Kreienkamp HJ et al (1994) Expression and ligand specificity of acetylcholinesterase and the nicotinic receptor: a tale of two cholinergic sites. Biochem Soc Trans 22:740–745
Teaktong T, Graham AJ, Johnson M et al (2004) Selective changes in nicotinic acetylcholine receptor subtypes related to tobacco smoking: an immunohistochemical study. Neuropathol Appl Neurobiol 30:243–254
Teodoro R, Scheunemann M, Deuther-Conrad W et al (2015) A promising PET tracer for imaging of α7 nicotinic acetylcholine receptors in the brain: design, synthesis, and in vivo evaluation of a dibenzothiophene-based radioligand. Molecules 20:18387–18421
Teodoro R, Scheunemann M, Wenzel B et al (2018) Synthesis and radiofluorination of novel fluoren-9-one based derivatives for the imaging of α7 nicotinic acetylcholine receptor with PET. Bioorg Med Chem Lett 28:1471–1475
Thal DR, Braak H (2005) Post-mortem diagnosis of Alzheimer’s disease (in German). Pathologe 26:201–213
Thiessen JD, Shams E, Stortz G et al (2016) MR-compatibility of a high-resolution small animal PET insert operating inside a 7 T MRI. Phys Med Biol 61:7934–7956
Thompson HJ, Lifshitz J, Marklund N et al (2005) Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma 22:42–75
Tiepolt S, Becker GA, Wilke S et al (2018) (+)-[18F]flubatine ein neuer α4β2* nikotinischer acetylcholin-rezeptor (nAChR) PET radioligand - ergebnisse der first-In-Human Studie bei Patienten mit Alzheimer Demenz (AD) und gesunden Probanden (HC). Nuklearmedizin 57:A37
Tomizawa M, Cowan A, Casida JE (2001) Analgesic and toxic effects of neonicotinoid insecticides in mice. Toxicol Appl Pharmacol 177:77–83
Toyohara J, Ishiwata K, Sakata M et al (2010) In vivo evaluation of α7 nicotinic acetylcholine receptor agonists [11C]A-582941 and [11C]A-844606 in mice and conscious monkeys. PLoS One 5:e8961
Toyohara J, Sakata M, Wu J et al (2009) Preclinical and the first clinical studies on [11C]CHIBA-1001 for mapping α7 nicotinic receptors by positron emission tomography. Ann Nucl Med 23:301–309
Tregellas JR, Tanabe J, Rojas DC et al (2011) Effects of an alpha 7-nicotinic agonist on default network activity in schizophrenia. Biol Psychiatry 69:7–11
Tsukada H, Miyasato K, Kakiuchi T et al (2002) Comparative effects of methamphetamine and nicotine on the striatal [11C]raclopride binding in unanesthetized monkeys. Synapse 45:207–212
Unwin N (2005) Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346:967–989
Vafaee MS, Gjedde A, Imamirad N et al (2015) Smoking normalizes cerebral blood flow and oxygen consumption after 12-hour abstention. J Cereb Blood Flow Metab 35:699–705
Valette H, Bottlaender M, Dollé F et al (2005) Acute effects of physostigmine and galantamine on the binding of [18F]fluoro-A-85380: A PET study in monkeys. Synapse 56:217–221
Valette H, Bottlaender M, Dollé F et al (1997) An attempt to visualize baboon brain nicotinic receptors with N-[11C]ABT-418 and N-[11C]methyl-cytisine. Nucl Med Commun 18:164–168
Valette H, Bottlaender M, Dollé F et al (1999) Imaging central nicotinic acetylcholine receptors in baboons with [18F]fluoro-A-85380. J Nucl Med 40:1374–1380
Valette H, Dollé F, Saba W et al (2007) [18F]FPhEP and [18F]F2PhEP, two new epibatidine-based radioligands: evaluation for imaging nicotinic acetylcholine receptors in baboon brain. Synapse 61:764–770
Valette H, Xiao Y, Peyronneau MA et al (2009) 18F-ZW-104: a new radioligand for imaging neuronal nicotinic acetylcholine receptors—In vitro binding properties and PET studies in baboons. J Nucl Med 50:1349–1355
Valiyaveettil M, Alamneh YA, Miller SA et al (2013) Modulation of cholinergic pathways and inflammatory mediators in blast-induced traumatic brain injury. Chem Biol Interact 203:371–375
Vaupel DB, Stein EA, Mukhin AG (2007) Quantification of α4β2* nicotinic receptors in the rat brain with microPET(R) and 2-[18F]F-A-85380. NeuroImage 34:1352–1362
Vaupel DB, Tella SR, Huso DL et al (2005) Pharmacological and toxicological evaluation of 2-fluoro-3-(2(S)-azetidinylmethoxy)pyridine (2-F-A-85380), a ligand for imaging cerebral nicotinic acetylcholine receptors with positron emission tomography. J Pharmacol Exp Ther 312:355–365
Verbois SL, Hopkins DM, Scheff SW et al (2003a) Chronic intermittent nicotine administration attenuates traumatic brain injury-induced cognitive dysfunction. Neuroscience 119:1199–1208
Verbois SL, Scheff SW, Pauly JR (2002) Time-dependent changes in rat brain cholinergic receptor expression after experimental brain injury. J Neurotrauma 19:1569–1585
Verbois SL, Scheff SW, Pauly JR (2003b) Chronic nicotine treatment attenuates alpha 7 nicotinic receptor deficits following traumatic brain injury. Neuropharmacology 44:224–233
Verbois SL, Sullivan PG, Scheff SW et al (2000) Traumatic brain injury reduces hippocampal alpha7 nicotinic cholinergic receptor binding. J Neurotrauma 17:1001–1011
Villablanca AC (1998) Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. J Appl Physiol 84:2089–2098
Villemagne VL, Horti A, Scheffel U et al (1997) Imaging nicotinic acetylcholine receptors with fluorine-18-FPH, an epibatidine analog. J Nucl Med 38:1737–1741
Visanji NP, O’Neill MJ, Duty S (2006) Nicotine, but neither the alpha4beta2 ligand RJR2403 nor an alpha7 nAChR subtype selective agonist, protects against a partial 6-hydroxydopamine lesion of the rat median forebrain bundle. Neuropharmacology 51:506–516
von Horsten S, Schmitt I, Nguyen HP et al (2003) Transgenic rat model of Huntington’s disease. Hum Mol Genet 12:617–624
Wallace TL, Bertrand D (2013) Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem Pharmacol 85:1713–1720
Wang HY, Lee DH, D’Andrea MR et al (2000) β-Amyloid(1-42) binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem 275:5626–5632
Wang J, Lindstrom J (2018) Orthosteric and allosteric potentiation of heteromeric neuronal nicotinic acetylcholine receptors. Br J Pharmacol 175:1805–1821
Wang MH, Yoshiki H, Anisuzzaman AS et al (2011) Re-evaluation of nicotinic acetylcholine receptors in rat brain by a tissue-segment binding assay. Front Pharmacol 2:65
Wang S, Fang Y, Wang H et al (2018) Design, synthesis and biological evaluation of 1,4-diazobicylco[3.2.2]nonane derivatives as α7-nicotinic acetylcholine receptor PET/CT imaging agents and agonists for Alzheimer’s disease. Eur J Med Chem 159:255–266
Waterhouse RN (2003) Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol 5:376–389
Wecker L, Pollock VV, Pacheco MA et al (2010) Nicotine-induced up regulation of alpha4beta2 neuronal nicotinic receptors is mediated by the protein kinase C-dependent phosphorylation of alpha4 subunits. Neuroscience 171:12–22
Wehner J, Weissler B, Dueppenbecker PM et al (2015) MR-compatibility assessment of the first preclinical PET-MRI insert equipped with digital silicon photomultipliers. Phys Med Biol 60:2231–2255
Weiss S, Nosten-Bertrand M, McIntosh JM et al (2007a) Nicotine improves cognitive deficits of dopamine transporter knockout mice without long-term tolerance. Neuropsychopharmacology 32:2465–2478
Weiss S, Tzavara ET, Davis RJ et al (2007b) Functional alterations of nicotinic neurotransmission in dopamine transporter knock-out mice. Neuropharmacology 52:1496–1508
Wheaton P, Mathias JL, Vink R (2011) Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol 31:745–757
Whiteaker P, Davies AR, Marks MJ et al (1999) An autoradiographic study of the distribution of binding sites for the novel α7-selective nicotinic radioligand [3H]-methyllycaconitine in the mouse brain. Eur J Neurosci 11:2689–2696
Whitehouse PJ, Kellar KJ (1987) Nicotinic and muscarinic cholinergic receptors in Alzheimer’s disease and related disorders. J Neural Transm Suppl 24:175–182
Wildeboer KM, Stevens KE (2008) Stimulation of the alpha4beta2 nicotinic receptor by 5-I A-85380 improves auditory gating in DBA/2 mice. Brain Res 1224:29–36
Wong DF, Kuwabara H, Horti AG et al (2018) Brain PET imaging of α7-nAChR with [18F]ASEM: reproducibility, occupancy, receptor density, and changes in schizophrenia. Int J Neuropsychopharmacol 21:656–667
Wu J, Liu Q, Tang P et al (2016) Heteromeric α7β2 nicotinic acetylcholine receptors in the brain. Trends Pharmacol Sci 37:562–574
Xiao Y, Kellar KJ (2004) The comparative pharmacology and up-regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J Pharmacol Exp Ther 310:98–107
Xiong Y, Mahmood A, Chopp M (2013) Animal models of traumatic brain injury. Nat Rev 14:128–142
Yang Y, Bec J, Zhou J et al (2016) A prototype high resolution small-animal PET scanner dedicated to mouse brain imaging. J Nucl Med 57:1130–1135
Yong T, Zheng MQ, Linthicum DS (1997) Nicotine induces leukocyte rolling and adhesion in the cerebral microcirculation of the mouse. J Neuroimmunol 80:158–164
Yoshida K, Engel J, Liljequist S (1982) The effect of chronic ethanol administration of high affinity 3H-nicotinic binding in rat brain. Naunyn Schmiedeberg’s Arch Pharmacol 321:74–76
Zafonte R, Friedewald WT, Lee SM et al (2009) The citicoline brain injury treatment (COBRIT) trial: design and methods. J Neurotrauma 26:2207–2216
Zanardi A, Leo G, Biagini G et al (2002) Nicotine and neurodegeneration in ageing. Toxicol Lett 127:207–215
Zhang JH, Akula MR, Kabalka GW (2001) 3-((2,4-dimethyl-5-[123I]iodo)benzylidene)-anabaseine: a potent SPECT agent for imaging lung cancer. J Label Compd Radiopharm 44:S359–S361
Zhang Q, Lu Y, Bian H et al (2017) Activation of the alpha7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am J Transl Res 9:971–985
Zhang Y, Pavlova OA, Chefer SI et al (2004) 5-substituted derivatives of 6-halogeno-3-((2-(S)-azetidinyl)methoxy)pyridine and 6-halogeno-3-((2-(S)-pyrrolidinyl)methoxy)pyridine with low picomolar affinity for α4β2 nicotinic acetylcholine receptor and wide range of lipophilicity: potential probes for imaging with positron emission tomography. J Med Chem 47:2453–2465
Zoli M, Pistillo F, Gotti C (2015) Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96:302–311
Zwart R, Bodkin M, Broad LM et al (2004) Common structural and pharmacological properties of 5-HT3 receptors and α7 nicotinic acetylcholine receptors. In: Silman I, Soreq H, Anglister L, Michaelson D, Fisher A (eds) Cholinergic mechanisms: function and dysfunction. Tylor & Francis, London
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Brust, P. et al. (2021). Preclinical and Clinical Aspects of Nicotinic Acetylcholine Receptor Imaging. In: Dierckx, R.A., Otte, A., de Vries, E.F., van Waarde, A., Lammertsma, A.A. (eds) PET and SPECT of Neurobiological Systems. Springer, Cham. https://doi.org/10.1007/978-3-030-53176-8_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-53176-8_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53175-1
Online ISBN: 978-3-030-53176-8
eBook Packages: MedicineMedicine (R0)