
Abstract
Microtubule stabilizers are a mainstay in the treatment of many solid cancers and continue to find utility in combination therapy with molecularly targeted anticancer agents and immunotherapeutics. However, innate and acquired resistance to microtubule stabilizers can limit their clinical efficacy. The taccalonolides are a unique class of microtubule stabilizers isolated from plants of Tacca that circumvent clinically relevant mechanisms of drug resistance. Although initial reports suggested that the microtubule-stabilizing activity of the taccalonolides was independent of direct tubulin binding, additional studies have identified that potent C-22, C-23 epoxidized taccalonolides covalently bind the Aspartate 226 residue of β-tubulin and that this interaction is critical for their microtubule-stabilizing activity. The taccalonolides have distinct properties as compared to other microtubule stabilizers with regard to their biochemical effects on tubulin structure and dynamics that promote distinct cellular phenotypes. Some taccalonolides have demonstrated in vivo antitumor efficacy in drug-resistant tumor models with exquisite potency and long-lasting antitumor efficacy as a result of their irreversible target engagement. The recent identification of a site on the taccalonolide scaffold that is amenable to modification has provided evidence of the specificity of the taccalonolide–tubulin interaction. This also affords an opportunity to further optimize the targeted delivery of the taccalonolides to further improve their anticancer efficacy and potential for clinical development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The taccalonolides are a unique class of microtubule stabilizers that are produced by several Tacca species (Fig. 1). What we now know as the taccalonolides were first identified in the early 1960s by Dr. Paul Scheuer as the “bitter principle” of the tubers of Tacca leontopetaloides, a starchy food source. Scheuer and his colleagues purified a compound they named taccalin in 1963 as an intensely bitter, light yellow powder with a proposed tetracyclic structure [1]. While the actual structure of the taccalonolides was later found to be much larger than originally proposed, it laid the groundwork for the future study of this class of compounds. In 1987, Chen et al. [2] elucidated the structures of taccalonolides A (1) and B (2) isolated from the rhizomes of Tacca plantaginea. Taccalonolide A (1), the most abundant taccalonolide, demonstrated cytotoxic activity against P-388 leukemia in cell culture as well as antimalarial activity against Plasmodium berghei.

Tacca chantrieri
Over the following decades, two dozen additional taccalonolides were purified from Tacca species. Taccalonolides C–M (3–13) and W–Y (23–25) were first isolated from Tacca plantaginea [3,4,5,6,7], taccalonolides O–Q (15–17) from Tacca subflabellata [8], taccalonolides R–V (18–22) from Tacca paxiana [9], and taccalonolides Z (26) and AA (27) from Tacca integrifolia and Tacca chantrieri, respectively (Table 1) [10]. The discovery of the microtubule-stabilizing effects of the taccalonolides has sustained interest in these compounds as potential anticancer agents and will be the primary focus of this contribution. The taccalonolides and extracts from the leaves and tubers of Tacca leontopetaloides have also demonstrated antitrypanosomal activity [11] and the ability to control against snails [12] leading to a patent for their use as anthelmintic and molluscicidal agents [13].
2 Taccalonolides as Microtubule Stabilizers
The initial bioassays performed with purified taccalonolides were crude measures of cancer cell toxicity in vitro or antiparasitic and nematocidal activities that were not attributable to a specific mechanism of action. However, in 2003, the taccalonolides A (1) and E (5) (for structures see Table 1) were isolated as the bioactive components from Tacca chantrieri extracts that led to paclitaxel-like microtubule bundling and mitotic arrest with the formation of multiple spindle asters in cellular assays (Fig. 2) [14]. The antiproliferative potency of the taccalonolides A (1) and E (5) was found to be in the low micromolar range against human ovarian cancer (SK-OV-3, 1A9) cervical cancer (HeLa), and melanoma (MDA-MB-435 [15]) cell lines, approximately 1000 times less potent than paclitaxel. However, the taccalonolides retained potency in the NCI/ADR-RES paclitaxel-resistant model that expresses high levels of the P-glycoprotein drug efflux pump, a major mechanism of taxane resistance in the clinic. The taccalonolides also retained potency in the 1A9 ovarian cancer cell line that contains mutations in paclitaxel (PTX 10 and PTX 22) binding sites in the human M40 β-tubulin isotype [16, 17].

The effect of taccalonolide-enriched Tacca fractions on microtubule structures in HeLa cervical cancer cells expressing GFP-tagged tubulin. The taccalonolides promote bundling of interphase microtubules (top panels) as well as the formation of multiple asters in mitotic cells that are markedly distinct from the microtubule spindle in normal mitotic cells (bottom panels)
The ability to circumvent these taxane resistance mechanisms was the first indication that their mechanism of action could be distinct from this other plant-derived class of microtubule stabilizers. Although the taccalonolides caused cellular microtubule bundling, mitotic arrest with multipolar spindles, and subsequent apoptosis similar to paclitaxel, the taccalonolides were also distinct in that they promoted the formation of spindle poles greater in number than paclitaxel, further suggesting the possibility of a distinct mechanism of action. Together, these findings first demonstrated that the taccalonolides were a structurally novel class of microtubule stabilizers produced from a plant source with micromolar potency that were able to circumvent mechanisms of drug resistance to the taxanes potentially through a distinct mechanism.
The cellular microtubule-stabilizing activity of taccalonolides A (1) and E (5) was confirmed by Buey et al. [18] who demonstrated that 5 µM taccalonolide A (1) and 10 µM taccalonolide E (5) induced microtubule bundling, multipolar spindles, and multiple micronuclei in A549 adenocarcinomic human alveolar basal epithelial cells. However, these cellular microtubule effects could not be recapitulated in biochemical tubulin binding or polymerization assays. Furthermore, taccalonolides A (1) and E (5) did not promote tubulin assembly at concentrations as high as 66 µM with 60 µM GTP–tubulin when analyzed by either centrifugation or electron microscopy, and there was no evidence of taccalonolide binding to cross-linked or native microtubules. The taccalonolides only weakly displaced the paclitaxel-site probe Flutax-2, and the effect was not concentration dependent or observed in preincubation experiments leading to the conclusion that any observed Flutax-2 displacement was artifactual. Taccalonolide A (1) was also unable to promote microtubule polymerization even in non-denatured cytosolic extracts [19], further suggesting that the cellular microtubule-stabilizing activity of this taccalonolide was not the result of direct binding to microtubules or interactions with other soluble cellular factors that regulate microtubule polymer mass.
In spite of the inability to detect a direct interaction with microtubules and in being less potent than other classes of microtubule stabilizers, there was a continued interest in the taccalonolides due to their potential inability to interact with tubulin in biochemical preparations and their efficacy against taxane-resistant cancer cells. These studies were expanded by the evaluation of taccalonolides A (1), B (2), E (5), and N (14) as compared to other classes of microtubule-targeted drugs in cell lines representing clinical mechanisms of taxane resistance, including overexpression of P-glycoprotein, MRP7, and βIII-tubulin [20]. All four taccalonolides retained in vitro efficacy in taxane-resistant human ovarian cancer cell lines expressing P-glycoprotein, human embryonic kidney cell lines overexpressing MRP7, and HeLa cervical cancer cell lines expressing βIII-tubulin. Taccalonolides A (1) and E (5) were also found to be effective in vivo in a P-glycoprotein-expressing multidrug-resistant syngeneic murine mammary adenocarcinoma model Mam17/ADR that is resistant to both paclitaxel and doxorubicin [20].
Surprisingly, although the taccalonolides were on average over 100-fold less potent than the taxanes in vitro, they demonstrated in vivo efficacy at concentrations comparable to or even lower than those used for paclitaxel. Further studies also demonstrated in vivo efficacy of taccalonolides A (1), E (5), N (14), and B (2) in the mammary 16/c syngeneic tumor model at total doses of 20–90 mg/kg, which were comparable to a total dose of 74 mg/kg paclitaxel [10]. These data not only confirmed that the taccalonolides were a novel class of microtubule stabilizers that can circumvent clinically relevant forms of drug resistance, but also demonstrated in vivo antitumor efficacy in paclitaxel-sensitive and -resistant tumor models at doses much lower than expected from studies based on their in vitro potency.
The lack of biochemical tubulin-polymerizing activity of the taccalonolides prompted additional cellular studies to elucidate the mechanism of cellular microtubule stabilization and how these effects were distinct from those of the taxanes. One intriguing finding was that gross bundling of interphase microtubules occurred at concentrations of taccalonolide A (1) that were equal to or less than those that promoted antiproliferative effects, whereas the concentration of paclitaxel required to observe cellular microtubule bundling was over 30-fold greater than its antiproliferative IC50 value, further suggesting a mechanistic difference between these two microtubule stabilizers [19]. This was particularly significant as it coincided with reports suggesting that the interphase effects of microtubule-targeting agents contribute to their antitumor efficacy in the clinic [21, 22]. It was also found that the cellular effects of the taccalonolides were highly persistent, providing long-term antiproliferative and cytotoxic efficacy even after only short periods of drug exposure and subsequent removal from the culture medium. This cellular persistence was not observed for other classes of microtubule stabilizers, including paclitaxel, further highlighting mechanistic differences between the taccalonolides and the taxanes. A high degree of cellular persistence has been associated with potent in vivo efficacy of the clinically approved microtubule destabilizer eribulin [23], providing a rationale for the unexpected in vivo potency of the taccalonolides.
3 Identification of Epoxidized Taccalonolides
Although early experiments demonstrated that taccalonolides A and E enriched preparations had microtubule stabilizing activity that was distinct from that of the taxanes, there were two issues that confounded a full understanding of their mechanism of action. One is the aforementioned lack of interaction with purified tubulin and the second was the inconsistent potency of different preparations of taccalonolides A (1) and E (5).
The original characterization of the microtubule-stabilizing effects of taccalonolide A (1) in 2004 demonstrated low micromolar potency, but follow-up studies by our same group in 2008 using a newly purified batch of taccalonolide A (1) were approximately 10-fold more potent. This inconsistency in the potency of our taccalonolide A (1) batches from preparation to preparation led to a rigorous evaluation of the chemical and biological properties of each of our HPLC fractions, including those before and after the prominent taccalonolide peak. To our surprise, we found that the microtubule-stabilizing potency did not comigrate perfectly with fractions that contained the highest taccalonolide A (1) levels. A careful chemical interrogation of the most potent HPLC fractions identified a sample containing compounds with the taccalonolide backbone possessing an unanticipated epoxide at positions C-22 and C-23 as opposed to the double bond in taccalonolide A (1). While this was a trace product that was not in sufficient quantity to purify fully from the natural product, the identification of the presence of this product led to its efficient semisynthesis from abundant 22,23-alkene taccalonolides (Scheme 1).

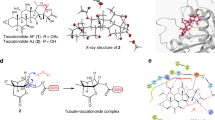
Synthesis of taccalonolides AF (28) and AJ (29) via epoxidation of taccalonolides A (1) and B (2), respectively
Remarkably, the 22,23-epoxidation epoxidation of taccalonolides A (1) and B (2) to generate taccalonolides AF (28) and AJ (29), respectively, resulted in taccalonolide microtubule stabilizers with low nanomolar potency in cells. Additionally, these potent taccalonolides were able to effectively bind and polymerize tubulin in biochemical preparations, a property that was never observed for the non-22,23-epoxidized taccalonolides [24]. With this knowledge in hand, our group has revisited the activity of taccalonolides A (1) and B (2), in particular, and found that the microtubule-stabilizing activity of these compounds can be diminished by additional rounds of purification with highly purified material being completely devoid of any antiproliferative, cytotoxic, or cellular microtubule-stabilizing effects (unpublished observations).
Together, these data demonstrate that the previously reported micromolar potency biological activities of 22,23-alkene taccalonolides, including A (1), E (5), B (2), and N (14) among others, are likely a result of small amounts of material that was oxidized to generate a 22,23-epoxide. These formerly undetected trace amounts of nanomolar potency epoxidized taccalonolide were sufficient to promote cellular efficacy in the micromolar range that was attributed to the more abundant 22,23-alkene taccalonolides. In contrast, the low quantities of epoxidized taccalonolides in these preparations were insufficient to promote biochemical tubulin polymerization, which requires near equimolar concentrations to tubulin, providing a rationale for why cellular but not biochemical microtubule polymerization could be observed in the early evaluations of taccalonolides A (1) and E (5). To provide further evidence for this rationale, Peng et al. demonstrated that the semisynthetic introduction of a 22,23-epoxide to ten additional purified taccalonolides was sufficient to improve their antiproliferative potency, some into the sub-nanomolar range. Furthermore, there has been minimal to no batch-to-batch variation in potency among preparations of the 22,23-epoxidized taccalonolides AF (28) and AJ (29) isolated from different plant sources by different laboratory groups, providing confidence that these are indeed the bioactive component of Tacca species that have been investigated for decades. Importantly, the potent taccalonolides, AF (28) and AJ (29), retain many of the same biological properties that were previously ascribed to taccalonolides A (1) and B (2), including the ability to circumvent clinically relevant drug resistance mechanisms, a high degree of cellular persistence, and in vivo antitumor efficacy [25, 26].
4 Identification of a Direct Interaction Between the Taccalonolides and Tubulin
Equipped with an understanding of the role of the 22,23-epoxide in the microtubule-stabilizing activity of the taccalonolides and semisynthetic strategies to convert the naturally abundant 22,23-alkene into the potent 22,23-epoxy-taccalonolides, there was a renewed approach in understanding the molecular interactions between the taccalonolides and tubulin/microtubules. Unlike the 22,23-alkene taccalonolides A (1) and E (5), the 22,23-epoxy-taccalonolides were indeed sufficient to polymerize purified tubulin in a similar manner to other microtubule-stabilizing agents. However, there was a significant lag time associated with taccalonolide-induced tubulin polymerization in contrast to the almost immediate polymerization induced by the taxanes (Fig. 3) [26].

Comparison of the biochemical tubulin polymerization activities of paclitaxel and taccalonolide AJ. Left: paclitaxel (10 µM) promotes the immediate polymerization of purified tubulin (20 µM) as compared to a vehicle control. Right: in contrast, taccalonolide AJ (10 µM) dependent polymerization of purified tubulin (20 µM) is associated with a lag time of 8–10 min
Additionally, the microtubules formed in the presence of taccalonolide AJ (29) were highly resistant to cold-induced depolymerization as determined both turbidometrically and by electron microscopy [26]. This was markedly distinct from microtubules induced by other stabilizers, including paclitaxel and laulimalide (also named fijianolide B), which were subject to cold-induced depolymerization. Together, these results suggested that the taccalonolides promoted a distinct mechanism of microtubule polymerization from other classes of microtubule stabilizers.
To address whether the taccalonolides bind to the same site as paclitaxel and laulimalide on microtubules, synergism and displacement studies were employed. Synergistic effects were observed between taccalonolide AF (28) and either paclitaxel or laulimalide [26], further indicating that the taccalonolides bind to a site pharmacologically distinct from the two major stabilizer-binding sites on tubulin. Displacement studies using equimolar concentrations of taccalonolide AJ (29) with either laulimalide or paclitaxel with purified tubulin demonstrated some competition between taccalonolide AJ (29) and paclitaxel [26]. However, it was notable that prior addition of taccalonolide AJ (29) before paclitaxel was required to observe decreased paclitaxel binding. This temporal effect on taxane displacement was the first indication that the taccalonolides might be interacting in an irreversible manner with tubulin. Indeed, after interaction with purified tubulin, the taccalonolides could not be extracted from either the supernatant or the microtubule pellet [26].
Mass spectrometric analysis confirmed that the m/z 212–230 peptic fragment of β-tubulin was lost after incubation with taccalonolide AJ (29) and replaced by a peptide that was increased by the molecular weight of 29 [26]. Together, these results demonstrated that the 22,23-epoxy-taccanolides covalently bound β-tubulin within the β212–230 region, which includes the βHis229 residue that is the covalent binding site of the cyclostreptin and zampanolide microtubule stabilizers. Additional hydrogen–deuterium exchange mass spectrometry was employed to determine that taccalonolide AJ-induced microtubule stabilization did not involve profound stabilization of the M-loop of tubulin, which is associated with taxane and zampanolide-induced microtubule stabilization, but instead promoted dramatic inter-protofilament stability as a mechanism of microtubule stabilization [26].
These biochemical studies were confirmed by Wang et al., who reported the first crystal structure of tubulin complexed with taccalonolide AJ (29) (Fig. 4), demonstrating that the 22,23-epoxide of 29 binds covalently to the Asp226 residue on β-tubulin [27]. Their data suggested that this covalent interaction promotes a conformation shift in the M-loop of tubulin that favors the binding of GTP in the E-site of tubulin. While the authors suggested in the supplemental data that the previously assigned stereochemistry of the 22,23-epoxide may need to be revisited, they did not ultimately promote this adjustment in configuration. However, additional crystallographic data of taccalonolide AJ (29) in the absence of tubulin and elucidation of the reaction mechanism (Scheme 2) confirmed that the stereochemistry of the 22,23-epoxide originally described by Li et al. needed to be revised [28, 29].

Taccalonolide AJ binding covalently to Asp226 on β-tubulin as determined by X-ray crystallography of the T2R-TTL-taccalonolide AJ complex

Reaction mechanism of the covalent binding of taccalonolide AJ to β-tubulin Asp226
Based on these data, Sanchez-Murcia et al. undertook extensive in silico modeling and molecular dynamics simulations to elucidate further the unique interaction between the 22,23-epoxy-tacconolides and β-tubulin [30]. They proposed that the nucleophilic attack on C-22 by the OD1/OD2 carboxylate of βAsp226 and opening of the 22,23-epoxide is facilitated by the long-lived hydrogen bond interaction of the carboxylate with the side-chain hydroxy of Thr223 and enhanced stabilization mediated via water-bridged hydrogen bonds. The C-22 carbon was suggested to undergo initial addition, indicating that the epoxide is non-protonated prior to nucleophilic attack. They further emphasize that the 22,23-epoxide is essential for covalent bond formation between the taccalonolide and tubulin. Their detailed molecular analysis of the putative interactions provides an important framework for additional biochemical and molecular biological studies exploring their functional importance.
The discrepancy between the lack of taccalonolide-induced M-loop stabilization detected by hydrogen–deuterium exchange mass spectrometry and the observed conformational shift of the M-loop in the taccalonolide AJ-tubulin crystal structure [27] was clarified by Balaguer et al. [25]. This elegant study compared the binding, allosteric effects, and tubulin polymerization dynamics of the three known covalent microtubule stabilizers: zampanolide, cyclostreptin, and the potent 22,23-epoxy-taccanolides.
The crystal structure of cyclostreptin bound to β-tubulin corrected previous literature suggesting covalent interactions with βThr220 and βAsn228 and instead confirmed that cyclostreptin and zampanolide both bind covalently to βHis229 [25]. Furthermore, they demonstrated that cyclostreptin-dependent tubulin polymerization was associated with a similar lag period that had been observed with taccalonolide AJ (29). In contrast, zampanolide rapidly induced tubulin polymerization in a manner similar to taxane microtubule stabilizers. Superimposition of the crystal structures of each of these compounds with tubulin demonstrated that extensive M-loop interactions and helical stabilization were correlated with stabilizers that promoted a strong initial rate of assembly [25]. In contrast, stabilizers such as taccalonolide AJ (29) that only promote partial M-loop structuring without inducing a helical confirmation were associated with a significant lag time prior to the initiation of tubulin polymerization. Together, these data demonstrate that while the taccalonolides do promote some structuring of the M-loop of β-tubulin, these interactions are not as significant as those promoted by the taxanes, which result in a delay in the initiation of microtubule stabilization by the taccalonolides.
5 Cellular Effects of Taccalonolide-Induced Microtubule Stabilization
Given the distinct biochemical interaction of the taccalonolides with tubulin, studies were undertaken to evaluate the effects of the 22,23-epoxy-tacconolides as compared to taxanes on microtubule dynamics in biochemical preparations and in live cells [31]. While paclitaxel and taccalonolide AJ (29) had similar overall effects on the microtubule dynamics of purified tubulin that promoted overall stabilization, taccalonolide AJ (29) demonstrated a greater suppression of catastrophe frequency likely as a result of its irreversible binding. In contrast, paclitaxel had a greater effect on microtubule rescue frequency than taccalonolide AJ (29). Similar effects were observed when microtubule dynamicity was evaluated in live cells with 29 having a greater impact on microtubule catastrophe and paclitaxel affecting rescue frequency to a larger extent. These differences in cellular microtubule dynamics were found to underlie the distinct microtubule aster morphology observed in cells treated with the taccalonolides as compared to the taxanes [14, 32].
Real-time spindle formation was evaluated in live cells expressing GFP-tagged tubulin upon treatment with the taccalonolides or paclitaxel compared to vehicle controls. While cells entered mitosis at similar rates with similar effects on aster formation, differences were noted in the consolidation of these asters by paclitaxel but not the taccalonolides during extended mitotic arrest. This aster consolidation in paclitaxel-treated cells led to the previously described phenotype of 2–3 asters per cell in contrast to the taccalonolides that result in an average over five asters per cell (Fig. 5). The finding that the taccalonolides suppress microtubule catastrophe and inhibit aster consolidation to a greater extent than paclitaxel demonstrates that these distinct effects on microtubule dynamicity between the test compounds can lead to the formation of different cellular microtubule structures that could contribute toward distinct biological readouts.

Distinct mitotic spindle structures in normal cells (left), paclitaxel-treated cells (middle), and taccalonolide-treated cells (right)
Rohena et al. [32] investigated the microtubule-associated mitotic effects initiated by three structurally and functionally diverse microtubule-stabilizing agents: taccalonolide AJ (29), laulilamide, and paclitaxel. Each microtubule stabilizer initiated distinct mitotic defects and differentially dysregulated the expression of key mitotic kinases. Taccalonolide AJ (29) produced the most profound defects in centrosome maturation, separation, and disjunction as observed by indirect immunofluorescence of the centrosomal-associated proteins rootletin, Nek2, and γ-tubulin [32]. However, taccalonolide AJ-treated cells also contained the more peripheral centrosomal protein pericentrin at every spindle aster, suggesting these structures facilitated the maintenance and stability of the multiple, highly focused asters observed in taccalonolide-treated cells as compared to the other two microtubule stabilizers, which only contained two pericentrin foci [32]. Not surprisingly, these defects in centrosomal structures were accompanied by mitotic signaling defects, including enhanced Eg5 phosphorylation by taccalonolide AJ-treated cells as compared to those treated with the other stabilizers [32].
6 In Vivo Antitumor Efficacy of 22,23-Epoxy-tacconolides
The antitumor efficacy of the epoxy-taccalonolides AF (28) and AJ (29) was evaluated initially in a MDA-MB-231 flank triple-negative breast cancer xenograft murine model. Taccalonolide AF (28) exhibited antitumor efficacy at a total dose of 5 mg/kg that produced a greater degree of tumor regression than 40 mg/kg paclitaxel [26]. Additional antitumor studies with the potent 22,23-epoxidation products of taccalonolides T (20) and AI (30) also demonstrated antitumor efficacy in a MDA-MB-231 xenograft model [33]. However, taccalonolide AJ (29) did not demonstrate antitumor effects even at the LD40 dose of 2 mg/kg [26]. These results suggested that taccalonolides AF and AJ, with similar biochemical and cellular microtubule-stabilizing activities, may have distinct pharmacokinetic properties.
Initial efforts to characterize differences in the chemical stability of taccalonolides AF (28) and AJ (29) demonstrated that the C-15 acetoxy group of taccalonolide AF (28) was hydrolyzed in aqueous solutions to generate AJ (29) [26]. In vivo pharmacokinetic properties were evaluated for both taccalonolides AJ (29) and AF (29) in the same strain of nude mice that were utilized for xenograft studies. AJ (29) was demonstrated to have an elimination half-life of 8.1 min, when administered systemically, while the half time of AF (28) was 44.1 min [34]. AJ (29) had excellent and persistent antitumor efficacy when administered directly into the tumor, suggesting that the lack of antitumor efficacy demonstrated with systemic administration of AJ (29) was likely due to its short half-life in vivo [34].
Given the fact that the C-15 acetyl group on taccalonolide AF (28), which demonstrated in vivo efficacy, was effectively hydrolyzed in aqueous solution to generate taccalonolide AJ (29), which does not have a therapeutic window for systemic administration in vivo, we hypothesized that semisynthesis of taccalonolides with C-15 substitutions could provide increased stability of an active antitumor drug to provide an increased therapeutic window. The in vitro biological activities of 28 novel taccalonolides with mono substitutions at C-7 or C-15 or disubstitutions at C-7 and C-25 ranged in antiproliferative potency from 2.4 nM to >20 μM [29]. However, no improved stability or therapeutic window was observed with isovalerate, cyclopropyl, isobutyrate, or formate substituents at C-7 or C-15. Additionally, substitutions at C-25 completely abrogated in vitro activity, likely due to interference with the covalent binding of the 22,23-epoxide to β226 of tubulin. The two most potent taccalonolides in vitro, isovalerate modifications at C-7 or C-15, were evaluated for in vivo antitumor efficacy by intratumoral injection in the drug-resistant human NCI/ADR-RES xenograft murine model. Similarly to taccalonolide AJ (29), the isovalerate-modified taccalonolides demonstrated potent in vivo efficacy when directly administered to the tumor and notably caused long-term antineoplastic efficacy for over a month after the final dose was administered [29]. These results demonstrate that targeted delivery of the taccalonolides provides for long-term efficacy in drug-resistant tumor models and led to studies to identify a handle on the taccalonolides that could be used for tumor targeting strategies.
7 Taccalonolide Conjugates Provide Evidence of Specificity and a Handle for the Generation of Targeted Agents
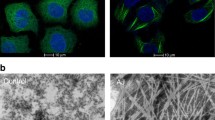
Data from previous semisynthetic efforts as well as an interrogation of the taccalonolide AJ-tubulin crystal structure led to the identification of C-6 as a possible handle amenable to functionalization. Indeed, modification of this site provided a stable fluorescein-tagged taccalonolide that retained microtubule-stabilizing activity and could be visualized colocalizing with microtubules (Fig. 6) [35].

Fluorescein-tagged taccalonolide (green) colocalized with β-tubulin immunofluorescence (orange) in fixed HCC1937 triple-negative breast cancer cells after 24 h treatment
These efforts were expanded to eventually generate a C-6-fluorescein taccalonolide conjugate that retained in vitro potency in the low nanomolar range and provided the stability to perform detailed imaging and cellular binding studies [28]. Optimization of the taccalonolide–fluorescein probe included the addition of pivaloyl-protected groups on fluorescein to quench fluorescence of the probe prior to cellular import, which provided the ability to monitor uptake and binding in live cells with no need to remove excess probe from the surrounding medium (Fig. 7).

Flu-tacca-7 is a cell-permeable fluorescent taccalonolide containing pivalate protective groups on the fluorophore that prevent fluorescence as well as target engagement prior to intracellular esterase cleavage. Upon cellular entry and pivalate deprotection to generate flu-tacca-8, the probe can directly bind tubulin and fluorescently label intracellular microtubules. The quenching provided by the pivalate groups permits live cellular imaging without the need to remove excess probe from the medium, providing a no-wash, irreversible fluorogenic labeling system for cellular microtubules
Serendipitously, the C-6 fluorescein modification actually improved the biochemical tubulin-polymerizing activity of the taccalonolides by making additional contacts with tubulin [28]. However, the pivalate protective groups prevented a direct interaction with tubulin in biochemical assays, demonstrating that the pivalate modification simultaneously prevented fluorescence and target engagement prior to cellular hydrolysis. The taccalonolide–fluorescein probe was found to be superior to commercially available taxane probes with regard to its microtubule staining without the addition of carrier molecules or removing excess probe from the medium [28]. It also provided microtubule staining under conditions that are not amenable to visualization with reversible taxane-based probes, including chilled conditions where microtubules are sensitive to depolymerization or in cells with high expression of drug efflux transporters [28].
The fluorescent taccalonolide probe strikingly colocalized with β-tubulin by immunofluorescence in human cancer cells, and the interaction was retained throughout immunoblotting to demonstrate a specific interaction between tubulin and the labeled taccalonolide [28]. A taccalonolide probe lacking the 22,23-epoxide completely abrogated this colocalization and binding, providing the first direct evidence of the exquisite specificity of the covalent interaction between the 22,23-epoxide of the taccalonolides and tubulin. This provided an unprecedented opportunity to use mutational analysis of an ectopically expressed form of tubulin to systematically evaluate the relative contribution of β-tubulin residues to taccalonolide binding with a focus on those that would be predicted to facilitate this interaction based on the crystallographic and modeling data [27, 30]. Consistent with these data, βAsp226 was critical for the covalent interaction between the taccalonolides and tubulin [28]. Additionally, βLys19 and βLeu219 were also critical for taccalonolide binding, βHis229 and βThr223A had a moderate effect on binding, and βArg278 did not influence binding [28]. These data provide critical insight into the taccalonolide pharmacophore that will be highly valuable in strategies to optimize target binding and, potentially, to facilitate the synthesis of new classes of taccalonolide-like small molecules. Overall, this study provided insight into the target specificity and detailed drug–target interactions of the taccalonolides and strategies to further develop targeted taccalonolides.
8 Other Bioactive Compounds Isolated from Tacca Species
In addition to the taccalonolides, other bioactive compounds have been isolated from Tacca species. Most intriguingly, a microtubule destabilizer, taccabulin A (31) (Table 2), was isolated from the roots and rhizomes of Tacca species [36], which was the first study reporting the isolation of both a microtubule stabilizer and microtubule destabilizer from the same natural product source. Taccabulin A (31) effectively displaced colchicine binding to tubulin, suggesting that it binds within the colchicine pharmacophore, and demonstrated synergistic effects when combined with the taccalonolides [36]. Similar to the taccalonolides and other colchicine site-binding agents, taccabulin A (31) retained efficacy in drug-resistant models, including those that express elevated levels of the P-glycoprotein drug efflux pump or the β-III isotype of tubulin [36]. Six additional retro-chalcones, taccabulins B–E (32–35) and evelynins A (36) and B (37), were also isolated from Tacca extracts. Evelynin A (36) and B (37), as well as taccabulin D (34) demonstrated some cytotoxic activity toward cancer cells in vitro but with no evidence of microtubule stabilizing or destabilizing activities [37, 38]. Other classes of compounds isolated from Tacca species include withanolides, glucosides, steroidal glycosides, diarylheptanoids, and diarylheptanoid glycosides [39,40,41,42,43,44,45,46,47,48,49].
9 Conclusion
In the 60 years since the taccalonolides were first identified as the bitter principle of Tacca tubers, they have continually provided interesting and often unanticipated discoveries. These include the finding in 1987 that the structure of the taccalonolides was more complicated than initially proposed, the elucidation of their mechanism of action as microtubule stabilizers in 2003, and the critical nature of the 22,23-epoxide for direct tubulin binding in 2013. This last finding is somewhat of a cautionary tale in natural products research that describes how a potent minor constituent, in this case 22,23-epoxy-tacconolides, could be responsible for the activity originally ascribed to more naturally abundant compounds. We now know that taccalonolides without a 22,23-epoxide lack the ability to bind and polymerize tubulin and have no detectable antiproliferative activity against cancer cell lines in culture. In contrast, potent 22,23-epoxy-taccanolides, including AJ (29) and AF (28), have the ability to covalently and irreversibly bind the Asp226 residue of β-tubulin to promote a distinct profile of microtubule stability as compared to other classes of clinically approved microtubule stabilizers. Most notably, some of these taccalonolides have demonstrated in vivo antitumor efficacy in drug-resistant breast and ovarian cancer models that persists for extended periods after drug treatment due to their covalent binding. Continued efforts to improve the therapeutic window for systemic administration and/or promote localized drug delivery based on the recent identification of a drug handle on the taccalonolide backbone may provide for their development as novel anticancer agents for the treatment of drug-resistant disease.
References
Scheuer PJ, Swanholm CE, Madamba LA, Hudgins WR (1962) Constituents of Tacca leontopetaloides. Lloydia 26:133
Chen Z-L, Wang B-D, Chen M-Q (1987) Steroidal bitter principles from Tacca plantaginea; structures of taccalonolide A and B. Tetrahedron Lett 28:1673
Chen Z, Wang B, Shen J (1988) Taccalonolide C and D, two pentacyclic steroids of Tacca plantaginea. Phytochemistry 27:2999
Shen J, Chen Z, Gao Y (1996) The pentacyclic steroidal constituents of Tacca plantaginea: taccalonolide E and F. Chin J Chem 9:92
Chen ZL, Shen JH, Gao YS, Wicht M (1997) Five taccalonolides from Tacca plantaginea. Planta Med 63:40
Shen J, Chen Z, Gao Y (1996) Taccalonolides from Tacca plantaginea. Phytochemistry 42:891
Yang J-Y, Zhao R-H, Chen C-X, Ni W, Teng F, Hao X-J, Liu H-Y (2008) Taccalonolides W-Y, three new pentacyclic steroids from Tacca plantaginea. Helv Chim Acta 91:1077
Huang Y, Liu JK, Muhlbauer A, Henkel T (2002) Three novel taccalonolides from the tropical plant Tacca subflabellata. Helv Chim Acta 85:2553
Muehlbauer A, Seip S, Nowak A, Tran VS (2003) Five novel taccalonolides from the roots of the Vietnamese plant Tacca paxiana. Helv Chim Acta 86:2065
Peng J, Risinger AL, Fest GA, Jackson EM, Helms G, Polin LA, Mooberry SL (2011) Identification and biological activities of new taccalonolide microtubule stabilizers. J Med Chem 54:6117
Dik VT, Vihiior B, Bosha JA, Yin TM, Ebiloma GU, de Koning HP, Igoli JO, Gray AI (2016) Antitrypanosomal activity of a novel taccalonolide from the tubers of Tacca leontopetaloides. Phytochem Anal 27:217
Abdel-Aziz A, Brain K, Bashir AK (1990) Screening of Sudanese plants for mollusicicidal activity and identification of leaves of Tacca leontopetaloides (L.) O. Kuntze (Taccaceae) as a potential new exploitable resource. Phytother Res 4:62
Muehlbauer A, Gehling M, Velten R, Andersch W, Erdelen C, Harder A, Marczok P, Nauen R, Turberg A, Tran VS, Adam G, Liu J (2001) Isolation and preparation of taccalonolides for controlling animal pests. Kunming Institute of Botany, Chinese Academy of Sciences, Bayer AG, Germany, p 113
Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC Jr, Hemscheidt TK, Mooberry SL (2003) Taccalonolides E and A: plant-derived steroids with microtubule-stabilizing activity. Cancer Res 63:3211
Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD (2007) MDA-MB-435 cells are derived from M14 melanoma cells — a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat 104:13
Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS (1997) Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 272:17118
Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, Fojo T (2000) A common pharmacophore for epothilone and taxanes: molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc Natl Acad Sci USA 97:2904
Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Diaz JF (2005) Microtubule interactions with chemically diverse stabilizing agents: thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem Biol 12:1269
Risinger AL, Mooberry SL (2011) Cellular studies reveal mechanistic differences between taccalonolide A and paclitaxel. Cell Cycle 10:2162
Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL (2008) The taccalonolides: microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res 68:8881
Sackett DL, Fojo T (2011) Taccalonolides: a microtubule stabilizer poses a new puzzle with old pieces. Cell Cycle 10:3233
Komlodi-Pasztor E, Sackett DL, Fojo AT (2012) Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 18:51
Towle MJ, Salvato KA, Wels BF, Aalfs KK, Zheng W, Seletsky BM, Zhu X, Lewis BM, Kishi Y, Yu MJ, Littlefield BA (2011) Eribulin induces irreversible mitotic blockade: implications of cell-based pharmacodynamics for in vivo efficacy under intermittent dosing conditions. Cancer Res 71:496
Li J, Risinger AL, Peng J, Chen Z, Hu L, Mooberry SL (2011) Potent taccalonolides, AF and AJ, inform significant structure-activity relationships and tubulin as the binding site of these microtubule stabilizers. J Am Chem Soc 133:19064
Balaguer FA, Muhlethaler T, Estevez-Gallego J, Calvo E, Gimenez-Abian JF, Risinger AL, Sorensen EJ, Vanderwal CD, Altmann KH, Mooberry SL, Steinmetz MO, Oliva MA, Prota AE, Diaz JF (2019) Crystal structure of the cyclostreptin-tubulin adduct: implications for tubulin activation by taxane-site ligands. Int J Mol Sci 20:1392
Risinger AL, Li J, Bennett MJ, Rohena CC, Peng J, Schriemer DC, Mooberry SL (2013) Taccalonolide binding to tubulin imparts microtubule stability and potent in vivo activity. Cancer Res 73:6780
Wang Y, Yu Y, Li GB, Li SA, Wu C, Gigant B, Qin W, Chen H, Wu Y, Chen Q, Yang J (2017) Mechanism of microtubule stabilization by taccalonolide AJ. Nature Commun 8:15787
Du L, Yee SS, Ramachandran K, Risinger AL (2020) Elucidating target specificity of the taccalonolide covalent microtubule stabilizers employing a combinatorial chemical approach. Nature Commun 11:654
Ola ARB, Risinger AL, Du L, Zammiello CL, Peng J, Cichewicz RH, Mooberry SL (2018) Taccalonolide microtubule stabilizers generated using semisynthesis define the effects of mono acyloxy moieties at C-7 or C-15 and disubstitutions at C-7 and C-25. J Nat Prod 81:579
Sanchez-Murcia PA, Mills A, Cortes-Cabrera A, Gago F (2019) Unravelling the covalent binding of zampanolide and taccalonolide AJ to a minimalist representation of a human microtubule. J Comput Aided Mol Des 33:627
Risinger AL, Riffle SM, Lopus M, Jordan MA, Wilson L, Mooberry SL (2014) The taccalonolides and paclitaxel cause distinct effects on microtubule dynamics and aster formation. Mol Cancer 13:41
Rohena CC, Peng J, Johnson TA, Crews P, Mooberry SL (2013) Chemically diverse microtubule stabilizing agents initiate distinct mitotic defects and dysregulated expression of key mitotic kinases. Biochem Pharmacol 85:1104
Peng J, Risinger AL, Li J, Mooberry SL (2014) Synthetic reactions with rare taccalonolides reveal the value of C-22,23 epoxidation for microtubule stabilizing potency. J Med Chem 57:6141
Risinger AL, Li J, Du L, Benavides R, Robles AJ, Cichewicz RH, Kuhn JG, Mooberry SL (2017) Pharmacokinetic analysis and in vivo antitumor efficacy of taccalonolides AF and AJ. J Nat Prod 80:409
Du L, Risinger AL, Yee SS, Ola ARB, Zammiello CL, Cichewicz RH, Mooberry SL (2019) Identification of C-6 as a new site for linker conjugation to the taccalonolide microtubule stabilizers. J Nat Prod 82:583
Risinger AL, Peng J, Rohena CC, Aguilar HR, Frantz DE, Mooberry SL (2013) The bat flower: a source of microtubule-destabilizing and -stabilizing compounds with synergistic antiproliferative actions. J Nat Prod 76:1923
Peng J, Risinger AL, Da C, Fest GA, Kellogg GE, Mooberry SL (2013) Structure-activity relationships of retro-dihydrochalcones isolated from Tacca sp. J Nat Prod 76:2189
Peng J, Jackson EM, Babinski DJ, Risinger AL, Helms G, Frantz DE, Mooberry SL (2010) Evelynin, a cytotoxic benzoquinone-type retro-dihydrochalcone from Tacca chantrieri. J Nat Prod 73:1590
Liu HY, Ni W, Xie BB, Zhou LY, Hao XJ, Wang X, Chen CX (2006) Five new withanolides from Tacca plantaginea. Chem Pharm Bull 54:992
Yokosuka A, Mimaki Y, Sashida Y (2003) Chantriolides A and B, two new withanolide glucosides from the rhizomes of Tacca chantrieri. J Nat Prod 66:876
Yokosuka A, Mimaki Y, Sashida Y (2004) Taccasterosides A–C, novel C28-sterol oligoglucosides from the rhizomes of Tacca chantrieri. Chem Pharm Bull 52:1396
Li L, Ni W, Li XR, Hua Y, Fang PL, Kong LM, Pan LL, Chen Li Y, CX, Liu HY, (2011) Taccasubosides A-D, four new steroidal glycosides from Tacca subflabellata. Steroids 76:037
Shwe HH, Aye M, Sein MM, Htay KT, Kreitmeier P, Gertsch J, Reiser O, Heilmann J (2010) Cytotoxic steroidal saponins from the rhizomes of Tacca integrifolia. Chem Biodivers 7:610
Misico RI, Nicotra VE, Oberti JC, Barboza G, Gil RR, Burton G (2011) Withanolides and related steroids. Prog Chem Org Nat Prod 94:127
Yokosuka A, Mimaki Y (2007) New glycosides from the rhizomes of Tacca chantrieri. Chem Pharm Bull 55:273
Yokosuka A, Mimaki Y, Sakuma C, Sashida Y (2005) New glycosides of the campesterol derivative from the rhizomes of Tacca chantrieri. Steroids 70:257
Yokosuka A, Mimaki Y, Sashida Y (2002) Spirostanol saponins from the rhizomes of Tacca chantrierii and their cytotoxic activity. Phytochemistry 6:731
Yokosuka A, Mimaki Y, Sashida Y (2002) Steroidal and pregnane glycosides from the rhizomes of Tacca chantrieri. J Nat Prod 65:1293
Yokosuka A, Mimaki Y, Sakagami H, Sashida Y (2002) New diarylheptanoids and diarylheptanoid glucosides from the rhizomes of Tacca chantrieri and their cytotoxic activity. J Nat Prod 65:283
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yee, S.S., Du, L., Risinger, A.L. (2020). Taccalonolide Microtubule Stabilizers. In: Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Asakawa, Y., Liu, JK. (eds) Progress in the Chemistry of Organic Natural Products 112. Progress in the Chemistry of Organic Natural Products, vol 112. Springer, Cham. https://doi.org/10.1007/978-3-030-52966-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-52966-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-52965-9
Online ISBN: 978-3-030-52966-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)