
Abstract
Liver cirrhosis represents the end stage of chronic liver disease, characterized by excessive scar tissue (fibrosis/cirrhosis), intense inflammatory cell infiltration, and liver loss-of-function, leading to multiple organ failures. It is generally accepted that activation of hepatic stellate cells, the main fibrogenic cell type in the liver, is key to fibrosis and cirrhosis development and progression. Recent advances in the fields of liver physiology and immunology unraveled new pathways for hepatic stellate cell activation and highlighted the pivotal role for inflammation- and immune-cell-derived signals in liver fibrogenesis. As such, deciphering the complexity of the integrated signals seems to be the mandatory approach to identify therapeutic targets. Ultimately, the modulation of fibrogenic and immune cell abilities to sense and react to changes in their microenvironment may hold the key to controlling or even reversing liver fibrosis and cirrhosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Points-
Liver cirrhosis represents the end stage of chronic liver disease, characterized by excessive scar tissue (fibrosis), intense inflammatory cell infiltration, and liver loss-of-function, leading to multiple organ failure.
-
Fibrogenic cells in the liver may be of different origins, but activated stellate cells represent the main source of extracellular matrix components.
-
Fibrogenic cells sense changes in their microenvironment especially under inflammatory conditions and respond to a plethora of inflammatory stimuli including cytokines, danger-associated molecular patterns, and pathogen-associated molecular patterns.
-
Both innate and adaptive immune cells are actively participating in the initiation, progression, and resolution of liver fibrosis. Current efforts are made to decipher the complex interplay between the immune system and liver disease.
-
Immunomodulation represents a promising therapeutic approach in the control of liver fibrosis.
The classical development of chronic liver diseases, whatever the etiology, follows a well-established pattern. Chronic or severe acute liver injury leads to the initiation of various inflammatory processes that comprise the activation of local immune cells, as well as the recruitment and activation of circulating cells. The liver also possesses potent regenerative capacities, characterized by the ability for parenchymal or liver-resident progenitor cells to proliferate in response to hepatic function alteration. Additionally, fibrogenic cells of different origins may be activated and deposit scar tissue. When tissue scarring is excessive, this is termed “fibrosis” and serves as the soil for advanced liver disease or cirrhosis, ultimately leading to liver failure.
A considerable amount of data supports the limitless relevance of studying the interactions between liver fibrosis and inflammation, in terms of virtually all immune cell types and phenotypes as well as immunological processes performed by professional or non-professional immune cells. Accordingly, new discoveries are made on a regular basis in the field of liver immunology and fibrosis and will further increase our knowledge of all the finely tuned cellular processes implicated and consequently lead to numerous advances for patients in the near future.
What Is Liver Fibrosis, and How Does It Evolve to Liver Cirrhosis?
Liver cirrhosis is defined as a final stage of chronic liver disease in which excessive parenchymal cell necrosis and scar tissue accumulation impedes blood flow, leading to liver loss-of-function and, consequently, the accumulation of toxics in the blood mainstream. Every year about 5–7% of previously asymptomatic cirrhotic patients exhibit multiple organ failure when decompensation has occurred [1, 2]. Complications include ascites, peritonitis, hepatic encephalopathy, hepatorenal syndrome, hepatopulmonary syndrome, and hypersplenism. Another important risk for cirrhotic patients is acute-on-chronic liver failure (ACLF), described as acute decompensation and a high short-term mortality occurring after an acute insult (e.g., drug-induced liver injury, viral hepatitis, or alcohol consumption) on a compensated cirrhotic liver [2, 3]. Importantly, systemic inflammation is constantly observed in patients with decompensated cirrhosis and ACLF patients, emphasizing the close interaction between immune cells and pathogenesis [2, 4]. Histologically, cirrhosis is characterized by regenerative nodules representing an attempt of the remaining liver cells to regenerate the organ and restore liver functions. These regenerative nodules are surrounded by fibrous septa made of extracellular matrix bridging the portal tracts. The fibrotic tissue is densely composed of fibrogenic cells, innate and adaptive immune cells, and pseudo-ductular structures, in a process termed as ductular reaction (see below) [5]. Hence, liver fibrosis and accompanying inflammation and ductular cell proliferation may be regarded as unbalanced regenerative processes [6]. At later stages, liver transplant may represent the only therapeutic option for cirrhotic patients, which represents a challenge due to organ donor shortage.
Virtually any chronic liver disease can lead to cirrhosis, whether caused by alcohol abuse, viral hepatitis, chemical toxicity, autoimmune liver diseases, non-alcoholic fatty liver disease, or genetic predispositions, among others. Whatever the etiology, the common soil for liver cirrhosis is fibrosis [3]. Thus, a lot of efforts are being put toward limiting or even reversing liver fibrosis. Among the different strategies, fibrogenic cell inhibition and immune system modulation represent the most promising approaches.
Liver fibrosis is defined as an excessive accumulation of extracellular matrix, mainly consisting of type I and III collagens, laminin, and hyaluronic acid. This scar tissue progressively occupies larger areas and replaces functional liver parenchyma. Myofibroblasts derived from activated hepatic stellate cells (HSCs) are considered to be the main source of extracellular matrix in the liver [7, 8]. HSCs are located in the space of Disse in the healthy liver, between the hepatic sinusoids lined by liver sinusoidal endothelial cells, and the basolateral surface of the hepatocytes. One of their main functions in the healthy liver is the storage of retinoids (vitamin A) in perinuclear droplets. Upon activation by various stimuli (detailed below), HSCs progressively lose their vitamin A droplets and adopt a myofibroblast phenotype notably defined by intense extracellular matrix deposition, alpha-smooth muscle actin expression, as well as migratory properties [8]. Myofibroblasts are characterized by their contractility, participating in the increase of portal resistance observed upon liver fibrosis. This contractility is induced by enthothelin-1 and angiotensin-II [9, 10]. The signals that activate or inhibit HSCs have been extensively reviewed previously [11].
Collagen deposition typically occurs around the remains of hepatic parenchyma and fibrotic septa expand from the periportal area in advanced stages of liver fibrosis, a feature referred to as bridging fibrosis. It is thus remarkable that HSCs, considered to be the main producers of extracellular matrix, seem to migrate to the perilobular areas before laying down collagen fibers. These facts raised doubts on the origin of the putative fibrogenic cells in the liver. Thus alternative cellular sources of fibrogenic cells have been identified, namely, portal fibroblasts and bone-marrow-derived circulating fibrogenic cells, or fibrocytes, among other potential candidates [12,13,14,15,16]; even epithelial cells (i.e., hepatocytes and biliary epithelial cells) undergoing epithelial-to-mesenchymal transition have been studied [15, 17, 18]. Although there is still some debate regarding the relative contribution of each cell type, HSC-derived myofibroblasts are still considered as the main collagen-producing cells in chronic liver diseases.
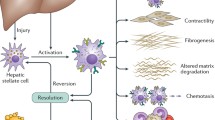
A process called ductular reaction is a hallmark in most chronic liver diseases [5]. It is defined as immune cell accumulation, fibrosis, and ductular cell proliferation in the periportal area. Numerous studies have reported close interactions between these three events. In brief, liver injury leads to the recruitment of immune cells including monocyte-derived macrophages and T lymphocytes, which in turn favor fibrogenic cell activation, as well as ductular cell (liver progenitor cells, or biliary cells) proliferation [19,20,21,22,23,24,25,26,27,28]. Fibrogenesis has been suggested to favor ductular cell proliferation, and reciprocally, ductular cells are known to release fibrogenic factors [5, 29,30,31]. Moreover, fibrogenic cells and ductular cells release pro-inflammatory mediators, thus participating in the maintenance of an inflammatory microenvironment and tissue injury [32,33,34,35]. This vicious circle may hold the key to the control of chronic liver disease progression, and numerous efforts are put toward identifying key therapeutic targets that may impede these processes (Fig. 35.1).

Classical liver disease history. Inflammation plays a critical role not only in inducing liver fibrogenesis during chronic liver injury but also in promoting liver fibrosis resolution and liver regeneration. ECM extracellular matrix
HSCs Sense Changes in Their Microenvironment Under Inflammatory Conditions
HSCs are well located and equipped to sense changes in their microenvironment. Indeed, their cytoplasmic protrusions expand toward the liver sinusoidal cells and hepatocytes [8, 36, 37]. HSCs, and by extension myofibroblasts, possess a complete arsenal of sensing receptors that detect changes in their microenvironment especially inflammatory conditions (please see reference [38]). Toll-like receptors (TLRs) are among these key receptors, and TLR types 1–9 have been proposed to be expressed by HSCs [33, 39, 40]. Most notably, direct TLR 2, 3, 4, and 9 activation on HSCs have been described as some of the mechanisms leading to inflammation and fibrosis progression (as detailed below) [41]. The TLRs and injury-related changes in the liver microenvironment have been described as crucial mediators of numerous inflammatory and fibrogenic processes through immune or parenchymal cell stimulation, but that will not be discussed in this section (reviewed elsewhere [42, 43]).
Danger-Associated Molecular Patterns (DAMPs) (Such as High-Mobility Group Protein 1 [HMG-1], Mitochondrial DNA [mtDNA])
DAMPs are molecules that are released upon cell death or exposed atypically at the cell membrane under stressing conditions. DAMPs are often described as the mediators of sterile inflammation, a process that initiates immune responses and tissue regeneration/fibrosis, independently of pathogens [44]. A variety of DAMPs have been implicated in liver disease and fibrosis, from nucleus or mitochondrial DNA to acute-phase proteins and protein chaperones [42]. For instance, TLR9 stimulation on HSCs, by apoptotic hepatocyte-derived nucleus DNA, led to the immobilization of migrating HSCs at the site of injury and to their activation into a collagen-producing phenotype [45]. Another example has been the proposed mechanism that TLR3 activation would induce the release of exosomes from HSCs, which would then stimulate interleukin (IL)-17A production by γδ T cells – a potent pro-inflammatory and pro-fibrogenic cytokine [46]. High mobility group box 1 (HMGB1) is similarly regarded as a crucial enhancer of liver fibrosis. Indeed, it has been shown that HMGB1 released by damaged hepatocytes activates HSCs toward a pro-fibrogenic phenotype, through TLR4 activation and endoplasmic stress induction [47]. However, liver (i.e., hepatocyte and biliary cell) HMGB1-deficient mice had similar liver inflammation and fibrosis in a hepatocarcinogenesis model [48].
Pathogen-Associated Molecular Patterns (PAMPs) (Such as Bacterial Products)
It has been shown that LPS-mediated TLR4 stimulation on HSCs favors their activation toward collagen-producing cells through reduced TGFβ pseudo-receptor bone morphogenetic protein (BMP), bone morphogenetic protein (BMP), and activin membrane-bound inhibitor homolog (BAMBI) expression, thus rendering them more responsive to transforming growth factor (TGF)β1 stimulation [33]. This study also demonstrated the importance of systemic inflammation and more specifically of intestinal bacterial product leakage in initiating and perpetuating liver fibrosis. Indeed, antibiotic treatment reduced bile duct-ligation-induced liver damage and tissue fibrosis [33]. These effects have been specifically attributed to TLR4 expression on HSCs and not Kupffer cells. Moreover, TLR9 stimulation by bacterial- or mitochondrial-derived DNA is known to promote liver fibrosis [49]. Lastly, it has been demonstrated that lipopolysaccharides (LPS) treatment (i.e., TLR4 activation) on HSCs downregulates miR-29 expression, favoring HSC activation and increased collagen expression [50].
Other Inflammatory Mediators (Such as Apoptotic Bodies, Extracellular Vesicles)
HSCs can be activated when phagocytosing damaged hepatocyte-derived apoptotic bodies [51]. Of note, macrophages that phagocyte apoptotic bodies also adopt an anti-inflammatory phenotype, notably characterized by enhanced TGFβ1 release [52]. This macrophage polarization has been questioned by another study reporting that cell debris phagocyting monocyte-derived macrophages adopt a phenotype favoring fibrosis resolution, through increased matrix-metalloproteinase expression [53]. Extracellular vesicles (EVs) allow for intracellular component sharing among cells, and EVs are increasingly studied in the field of liver diseases and fibrosis [54,55,56]. EVs are implicated in cell-to-cell communication and can also be used to deliver therapeutic agents to targeted cell populations. Indeed, studies using mesenchymal stromal/stem cell-derived EVs have reported promising results in ameliorating liver fibrosis and inflammation in rodent models [57, 58]. More recently, it has been shown that liver stem cell-derived EVs reduced ductular reaction and liver fibrosis in the multidrug resistance 2 knockout (Mdr2−/−) mice, via Lethal-7 microRNA and notably by reducing NF-κB and IL-13 signaling pathways in liver tissue [59]. EV cargos may also prove to be detrimental, since another group demonstrated that HSC-derived platelet-derived growth factor receptor (PDGFR)α-enriched EVs directly promote liver fibrosis in vivo [60]. As stated above, it has been demonstrated that apoptotic body engulfment by HSCs leads to fibrosis progression, thus shedding the light on the need for state-of-the-art EV isolation protocols to appropriately discriminate between EVs and apoptotic bodies [61, 62].
Cytokines Regulate Liver Fibrosis Initiation, Progression, or Resolution
Liver fibrosis is the consequence of a multitude of events, to include chronic tissue injury, inflammation, and fibrogenic cell activation. Here, we mainly discuss several cytokines that play an important role in regulating HSC activation in the liver. Some factors are produced by multiple cell types and have been shown to have redundant functions. Table 35.1 summarizes the main cytokine implications in liver fibrosis.
Major Cytokines That Promote Liver Fibrosis
Transforming growth factor beta 1 (TGFβ1) and platelet-derived growth factor (PDGF) are the two major cytokines that promote HSC activation and proliferation, respectively. TGFβ1 is considered the most prominent fibrogenic factor that favors HSC activation and fibrogenesis through the activation of Smads 2 and 3. Many types of cells can release TGFβ1, including HSCs, hepatocytes, T cells, and macrophages [63, 64]. In addition, TGFβ1 possesses potent anti-inflammatory properties that may direct immune cells toward a pro-fibrogenic response.
PDGF is long recognized as a potent mitogen for HSCs by targeting PDFGRα on these cells [65, 66]. Different sources of PDGF have been identified, such as Kupffer cells and proliferating cholangiocytes [67]. PDFGRα expression is highly upregulated after HSC activation and is induced by TGFβ1 [68]. Moreover, it has been shown that targeting PDFGRα in hepatocytes may result in lowering PDGFRα expression on HSCs, thus reducing their activation and liver fibrosis [69]. Therapeutic approaches such as the use of dominant negative soluble PDGFβ receptor or PDGF receptor signaling inhibitor (imatinib) have generated promising results in counteracting liver fibrosis [70, 71].
IL-17A (IL-17)-producing cells are frequently observed in the liver of patients suffering from alcoholic steatohepatitis and a variety of other chronic liver diseases associated with liver fibrosis [19, 72]. IL-17 levels are strongly increased upon liver fibrosis, and IL-17 has also been shown to correlate with disease progression and a poor prognosis in a variety of liver diseases [19, 73, 74]. More specifically, IL-17A-deficient animals exhibited reduced fibrosis and inflammation in the bile duct ligation model [73, 74]. Recombinant IL-17 strongly increased production of pro-inflammatory mediators such as IL-6 and TNFα in macrophages [74]. IL-17-receptor is ubiquitously expressed in the organism and has been shown to directly induce fibrogenic cell activation by favoring HSC to myofibroblast activation and enhancing collagen expression [73]. Furthermore, IL-17 directly induced collagen type I production in myofibroblasts through signal transducer and transcription factor 3 (STAT3) activation. Another study reported that IL-17A sensitizes HSCs to TGFβ1-mediated activation [75]. Interestingly, Th17 cells are also a potent source of IL-22, which exerts anti-fibrotic effects (see below) [73, 76].
Defined as Th2-profile cytokines, IL-4 and IL-13 are often associated and display similar functions [77]. Although both cytokines are crucial in host defense against infection, they also exert potent fibrogenic functions that have been long described. Indeed, IL-4 has been shown to increase TGFβ1 production in fibroblasts, and IL-13 is a potent pro-fibrogenic cytokine that directly acts on myofibroblasts [78,79,80]. Both IL-4 and IL-13 have also been shown to directly stimulate collagen expression and production in cultured fibroblasts [81, 82]. Lastly, IL-4 levels have been correlated with advanced fibrosis development in HCV patients [83].
Major Cytokines That Attenuate Liver Fibrosis
Interferon-gamma (IFN-γ) is considered a major negative regulator of liver fibrosis. IFN-γ directly reduces myofibroblast activation and collagen production in culture [84,85,86,87]. Moreover, IFN-γ is known to induce a cytotoxic NK cell phenotype in the liver, directed against activated HSCs [88]. Additionally, IFN-γ is known to increase liver injury in acute models such as Concanavalin A or lipopolysaccharides [89, 90]. Accordingly, in a model of methionine- and choline-deficient high-fat, or in a model of primary sclerosing cholangitis (Mdr2−/− mice), it has been reported that IFN-γ-deficient mice had reduced liver inflammation and fibrosis as compared to IFN-γ-expressing mice, possibly due to reduced tissue injury [91, 92]. These results highlight the complex roles of IFN-γ in favoring both tissue injury and repair mechanisms and orientating the immune system toward an anti-fibrotic response and inhibiting fibrogenic gene expression on HSCs.
IL-22, mainly produced by Th17 and Th22 cells, opposes the anti-fibrogenic effects of IL-17 by inducing HSC senescence and protecting against hepatocellular damage [76]. Mechanistically, IL-22-induced HSC senescence was prevented when STAT3 signaling was blunted. Accordingly, IL-22 deletion exacerbated liver fibrosis and IL-22 administration prevented bile-duct ligation liver fibrosis [73]. In addition, IL-22 has potent hepatoprotective roles, by favoring hepatocyte survival through STAT3 activation, thereby inhibiting liver fibrosis [93, 94].
Other Cytokines That May Have Dual Roles in the Control of Liver Fibrosis
TNFα, one of the most potent inflammatory cytokines, is upregulated during tissue injury responses and is participating in tissue injury by favoring hepatocyte apoptosis. TNFα administration enhanced liver fibrosis through increasing HSC survival through induction of tissue inhibitor of metalloproteinase 1 (TIMP-1) expression, an effect that has been reported to be mediated by Kupffer cell activation [95]. TNFα and LPS stimulated HSCs harbored reduced BAMBI expression, resulting in increased TGFβ1 signaling [96]. Additionally, HSCs isolated from TNR-receptor 1- and/or TNR-receptor 2-deficient mice showed reduced proliferation in response to PDGF and a reduced expression of collagen [97]. Contrastingly, direct treatment of isolated HSCs by TNFα led to reduced collagen expression but increased cell proliferation in other studies [98,99,100]. Therefore, TNFα seems to have contradictory effects on HSCs, which may be linked to their integration into a more complex microenvironment that exposes fibrogenic cells to a multitude of activating and inhibitory signals. TNFα stimulation induced activation of apoptosis signal-regulating kinase 1 (ASK1) and subsequently activated the p38/JNK signaling pathway, promoting liver fibrosis [101, 102]. Although an early phase II trial reported that selonsertib, an ASK1 inhibitor, showed promising results in reducing liver fibrosis in nonalcoholic steatohepatitis patients [103], selonsertib failed to reduce liver fibrosis in a phase III clinical trial (STELLAR-4, NCT03053063).
IL-6 not only plays an important role in protecting against hepatocellular damage and promoting liver regeneration but also acts as a pro-inflammatory cytokine. However, IL-6 can also promote HSC activation and survival via the activation of STAT3, thereby enhancing liver fibrosis [104,105,106]. Therefore, the effect of IL-6 on liver disease progression depends on the balance between its beneficial and detrimental functions.
Several studies reported that IL-1-receptor or IL-1β deficient mice were resistant to liver fibrosis [107, 108]. It has been questioned, however, whether these effects were due to direct effects of IL-1β or IL-1α on HSCs or indirect by favoring a pro-inflammatory environment. In addition, macrophage-derived IL-1β was demonstrated to induce fibrogenic gene expression in myofibroblasts from the liver [109].
IL-10 is a potent anti-fibrotic cytokine, and accordingly, IL-10-deficient mice develop a stronger immune response and a more severe fibrosis than wild-type mice following repeated CCl4 injections [110, 111]. Direct effects of IL-10 on HSCs were related to senescence induction and a decrease in HSC viability [112].
IL-33 is generally associated with tissue regeneration. For instance in the liver, it has been shown to promote biliary cell proliferation [113]. Moreover, IL-33 expression is higher in human and mouse fibrotic livers as compared to normal liver samples [114]. Accordingly, IL-33 has been characterized as a pro-fibrogenic factor being produced by activated HSCs and increasing their collagen production, as well as a pro-fibrogenic immune environment [114,115,116,117,118].
Immune Cells Regulate Liver Fibrogenesis
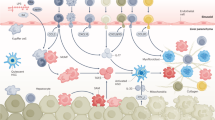
Liver fibrosis is seemingly always associated with liver inflammation (with the apparent exception of hemochromatosis) [119, 120]. Virtually all myeloid and lymphoid immune cells are implicated in liver fibrosis initiation, progression, and/or resolution (Fig. 35.2) [55, 121]. While immune cells respond to tissue injury by clearing DAMPs and PAMPs, an unbalanced response or chronic inflammation can enhance tissue damage and lead to liver fibrosis. Indeed, inflammatory processes are tightly regulated, and a slight imbalance results in either aggravated pathology or recovery. While specific factors produced by immune or parenchymal cells have been discussed (see above), we herein briefly describe the putative and sometimes contradictory roles of immune cell populations in the liver over the course of liver fibrosis.

Immune cell implication in liver fibrosis prevention or promotion. This figure represents the main immune cell types and subpopulations implicated in liver fibrosis progression or resolution. HSC hepatic stellate cell, IL interleukin, IFN interferon, MAIT mucosal-associated invariant T cells, MFB myofibroblast, NK natural killer, TGF transforming growth factor, Th Helper T cell, TNFα tumor necrosis factor alpha
Liver-Resident Macrophages
Kupffer cells are the liver-resident macrophages and are renowned to exert sentinel functions in healthy conditions. Kupffer cells are thus considered to be among the first immune cells to sense changes associated with liver injury [122]. During liver disease initiation, they are notably a potent source of chemokines for other immune cell types. Due to the difficulties in distinguishing Kupffer cells and monocyte-derived macrophages upon chronic liver injury, some reports may inadvertently confound these two cell types, and macrophage-depleting approaches may impact both compartments [123]. Accordingly, macrophage depletion by using clodronate-loaded liposomes at early stages of liver fibrosis prevents excessive scarring, while macrophage depletion at later stages dampens fibrosis resolution in the CCl4 model [124].
Monocyte-Derived Macrophages
Mononuclear cell infiltration (more specifically monocyte accumulation) is a classical feature of liver fibrosis. Monocytes can activate toward a plethora of phenotypes, including pro- or anti-inflammatory and pro- or anti-fibrogenic phenotypes [123, 125]. They are thus regarded as crucial orchestrators of liver disease due to their potent cytokine secretion. C–C motif chemokine receptor 2 (CCR2) is crucial for monocyte recruitment, since CCR2-deficient mice had reduced numbers of liver macrophages after bile duct ligation [126]. Accordingly, the use of a CCR2/CCR5 antagonist (cenicriviroc) has shown promising effects for the treatment of nonalcoholic steatohepatitis with fibrosis [127]. Nonetheless, monocytes are also crucial in liver regeneration, and monocyte/macrophage-depleting methods have been proven to sometimes delay or prevent tissue repair mechanisms [128,129,130,131,132].
Neutrophils
Neutrophils are among the first responders to liver injury and are mainly characterized by their potent roles in aggravating tissue injury through reactive oxygen species release [133, 134]. Consequently, neutrophil recruitment and activation are generally regarded to promote chronic pro-inflammatory and pro-fibrogenic environment. On the other hand, neutrophils are crucial in pathogen clearance that is necessary for inflammation resolution [135].
Natural Killer (NK) and NKT Cells
A clear role of NK cells in liver fibrosis is to control fibrosis progression through killing activated HSCs and producing IFN-γ that induces HSC apoptosis and cell cycle arrest [88, 136, 137]. In contrast, CD1d-dependent invariant NKT cells play dual roles in regulating liver fibrogenesis; for example, NKT cells not only can promote fibrosis progression, through IL-4 and IL-13 production [138], but may also attenuate liver fibrosis by killing HSCs and producing IFN-γ [139, 140].
T Lymphocytes
CD4+ T lymphocytes, also termed T-helper cells, are regarded as immune response orchestrators due to their very distinct and intense immune system-mediating cytokine release. Th1, Th2, and Th17 are the most studied and well-characterized activation phenotypes in liver disease. While Th1 cells are classically regarded as anti-fibrogenic through the promotion of anti-fibrogenic responses and IFN-γ-mediated fibrogenic cell death, Th2 cells are considered to be pro-fibrogenic through IL-4 and IL-13 production [141,142,143]. Th17 cells, on the other hand, have been described as having contradictory roles in liver disease. Th17 cells are mainly characterized by IL-17A and IL-22 production, which exert opposing functions in liver disease, IL-17A being pro-inflammatory and pro-fibrogenic and IL-22 favoring tissue regeneration and inducing HSC senescence (discussed above) [74, 76]. Cytotoxic CD8+ T lymphocytes play detrimental roles in alcoholic liver disease, notably by directly killing parenchymal cells [144]. Furthermore, autoreactive CD8+ T cells are considered to be the main drivers of biliary cell damage in primary biliary cholangitis, by targeting biliary epithelial cells [145].
B Lymphocytes
B lymphocytes represent a major lymphocyte population in the liver [146]. Despite identical initial injury, B-cell-deficient (JH−/−) mice showed reduced fibrosis 16 weeks after CCl4-induced liver fibrosis [146]. Furthermore, B cells accumulate in the liver from Mdr2−/− mice, and B-cell ablation by intravenous injections of anti-mouse CD20 monoclonal antibody promoted HSC senescence-mediated fibrosis resolution and was also associated with reduced TNFα and NF-κB activation [147].
Mucosal-Associated Invariant T (MAIT) Cells
MAIT cells have recently gained a lot of interest due to their antibacterial activity and are especially enriched in the human liver [148]. They are innate-like T cells mostly characterized as CD161+CD8+ T-cells and by the invariant TCR-chain, Vα7.2-Jα33 [149, 150]. MAIT cells have notably been shown to accumulate at the portal tracts around biliary ducts in human cholangiopathies and have thus been suggested to play a role in bile duct diseases [151]. Moreover, IL-7 is produced by hepatocytes under inflammatory conditions, and IL-7-stimulated MAIT cells dramatically upregulated their IL-17A production [148]. Similarly, repetitive IL-12 stimulation upregulated IL-17A production by MAIT cells [152]. In this same study, the authors demonstrated that although MAIT cells are less frequent in fibrotic than in the healthy liver, the remaining MAIT cells have adopted a pro-fibrogenic phenotype that further accentuates liver fibrosis, notably through IL-17A [152].
Roles of Immune Cells in Fibrosis Resolution
Despite the crucial roles of inflammation in initiating liver fibrosis, there is considerable amount of data enlightening the role of immune cells in fibrosis resolution. Accordingly, it has been demonstrated that macrophage depletion at early stages prevents CCl4-induced liver fibrosis, while macrophage depletion during liver fibrosis resolution stage, on the other hand, leads to fibrosis perpetuation [124]. The role of macrophage-mediated fibrosis resolution could be explained by the fact that bone-marrow-derived macrophages are required for natural killer (NK) cell recruitment. NK cells will then release TNF-related apoptosis-inducing ligand (TRAIL) and IFN-γ and subsequently induce fibrogenic cell apoptosis [88, 153]. Another major function of NK cells is the production of IFN-γ, which is known to oppose TGFβ1 fibrogenic signaling and inhibit HSC fibrogenicity through STAT1 activation [154].
Potential Anti-fibrotic Therapeutic Approaches by Targeting Immune Components
Withdrawal of the causative agents of liver injury has been shown to effectively prevent disease worsening and even allow for fibrosis or cirrhosis regression in hepatitis B and C infected patients, autoimmune diseases, non-alcoholic steatohepatitis, and more disputably in alcoholic patients [155,156,157,158]. As discussed above, many immune components have been implicated in the pathogenesis of liver fibrogenesis. Some of these components could be used as therapeutic targets for the treatment of liver fibrosis. For example, one potential approach to modulate HSC activation is to alter the TGFβ1 and unfolded protein response (UPR) [159]. More specifically, upon extracellular matrix protein assembly, fibrogenic cells experience increased ER stress, leading to the UPR and allowing for a proper protein folding and trafficking out of the cell while favoring cell survival. TGFβ1 is known to increase ECM protein production and to induce ER stress as well as UPR [159]. Procollagen I export blockade through transport and Golgi organization protein 1 (TANGO1) impairment led to HSC death in basal conditions due to enhanced ER stress, which was further increased upon concomitant TGFβ1 stimulation [159]. In addition, targeting the UPR through pharmacological inhibition of C/EBPβ-p300 may result in limiting fibrogenic cell activation and liver fibrosis [160].
IFN-γ is one of the most potent anti-fibrotic cytokines and was examined in clinical trials for the treatment of liver fibrosis with some beneficial effects, but long-term benefits were not observed [161, 162]. These disappointing results were likely due to low efficacy and adverse effects from IFN-γ therapy because IFN-γ strongly inhibits liver regeneration by targeting hepatocytes and induces inflammation by targeting immune cells. Researchers have been trying to develop fibroblast-targeted IFN-γ via the fusion of PDGF-β receptor recognizing peptide and IFN-γ, which had increased anti-fibrotic potency and improved safety profile in experimental models of liver fibrosis in vivo [163].
IL-22 has many beneficial functions in the liver, including hepatoprotective, proliferative, anti-oxidative, and anti-fibrotic functions [76]. More importantly, IL-22 therapy may generate limited side effects because IL-22 mainly targets epithelial cells as well as HSCs without affecting immune cells. Thus, IL-22 is a promising drug for the treatment of liver failure and may also have therapeutic potential for the treatment of liver fibrosis [164]. Indeed, a clinical trial shows promising results regarding IL-22Fc treatment for severe alcoholic hepatitis [165].
In summary, many immunological factors play important roles in controlling liver fibrogenesis. Further understanding of their functions may help identify novel therapeutic targets and effective strategies for the treatment of liver fibrosis in the future.
Abbreviations
- ACLF:
-
Acute-on-chronic liver failure
- ASK1:
-
Apoptosis signal − regulating kinase 1
- BAMBI:
-
Bone morphogenetic protein (BMP) and activin membrane-bound inhibitor homolog
- CCR2:
-
C–C motif chemokine receptor 2
- DAMP:
-
Danger-associated molecular pattern
- ER:
-
Endoplasmic reticulum
- EV:
-
Extracellular vesicle
- HMGB − 1:
-
High − mobility group box 1
- HSC:
-
Hepatic stellate cell
- IFN:
-
Interferon
- IL:
-
Interleukin
- MAIT:
-
Mucosal-associated invariant T cells
- Mdr2:
-
Multidrug resistance gene 2
- mtDNA:
-
Mitochondrial DNA
- NK:
-
Natural killer
- PAMP:
-
Pathogen-associated molecular pattern
- PDGFR:
-
Platelet-derived growth factor receptor
- STAT3:
-
Signal transducer and transcription factor 3
- TGF:
-
Transforming growth factor
- TIMP − 1:
-
Tissue inhibitor of metalloproteinases-1
- TLR:
-
Toll-like receptor
- TNFα:
-
Tumor necrosis factor alpha
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- UPR:
-
Unfolded protein response
References
Bernardi M, Moreau R, Angeli P, Schnabl B, Arroyo V. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol. 2015;63:1272–84.
Arroyo V, Moreau R, Kamath PS, Jalan R, Ginès P, Nevens F, et al. Acute-on-chronic liver failure in cirrhosis. Nat Rev Dis Primers. 2016;2:16041.
Lackner C, Tiniakos D. Fibrosis and alcohol-related liver disease. J Hepatol. 2019;70:294–304.
Claria J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al. Systemic inflammation in decompensated cirrhosis: characterization and role in acute-on-chronic liver failure. Hepatology. 2016;64:1249–64.
Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology. 2019;69:420–30.
Cordero-Espinoza L, Huch M. The balancing act of the liver: tissue regeneration versus fibrosis. J Clin Invest. 2018;128:85–96.
Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42.
Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72.
Rockey DC. Vascular mediators in the injured liver. Hepatology. 2003;37:4–12.
Shao R, Yan W, Rockey DC. Regulation of endothelin-1 synthesis by endothelin-converting enzyme-1 during wound healing. J Biol Chem. 1999;274:3228–34.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411.
Lemoinne S, Cadoret A, Rautou PE, El Mourabit H, Ratziu V, Corpechot C, et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology. 2015;61:1041–55.
Kisseleva T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology. 2017;65:1039–43.
Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111:E3297–305.
Wells RG, Schwabe RF. Origin and function of myofibroblasts in the liver. Semin Liver Dis. 2015;35:97–106.
Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823.
Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol Asp Med. 2019;65:2–15.
Xu J, Kisseleva T. Bone marrow-derived fibrocytes contribute to liver fibrosis. Exp Biol Med (Maywood). 2015;240:691–700.
Guillot A, Gasmi I, Brouillet A, Ait-Ahmed Y, Calderaro J, Ruiz I, et al. Interleukins-17 and 27 promote liver regeneration by sequentially inducing progenitor cell expansion and differentiation. Hepatol Commun. 2018;2:329–43.
Knight B, Matthews VB, Akhurst B, Croager EJ, Klinken E, Abraham LJ, et al. Liver inflammation and cytokine production, but not acute phase protein synthesis, accompany the adult liver progenitor (oval) cell response to chronic liver injury. Immunol Cell Biol. 2005;83:364–74.
Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59:1393–405.
Van Hul N, Lanthier N, Espanol Suner R, Abarca Quinones J, van Rooijen N, Leclercq I. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am J Pathol. 2011;179:1839–50.
Strick-Marchand H, Masse GX, Weiss MC, Di Santo JP. Lymphocytes support oval cell-dependent liver regeneration. J Immunol. 2008;181:2764–71.
Hines IN, Kremer M, Isayama F, Perry AW, Milton RJ, Black AL, et al. Impaired liver regeneration and increased oval cell numbers following T cell-mediated hepatitis. Hepatology. 2007;46:229–41.
Knight B, Lim R, Yeoh GC, Olynyk JK. Interferon-gamma exacerbates liver damage, the hepatic progenitor cell response and fibrosis in a mouse model of chronic liver injury. J Hepatol. 2007;47:826–33.
Tirnitz-Parker JE, Viebahn CS, Jakubowski A, Klopcic BR, Olynyk JK, Yeoh GC, et al. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology. 2010;52:291–302.
Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. 2012;143:188–98. e7
Jiang A, Okabe H, Popovic B, Preziosi ME, Pradhan-Sundd T, Poddar M, et al. Loss of Wnt secretion by macrophages promotes hepatobiliary injury after administration of 3,5-diethoxycarbonyl-1,4-dihydrocollidine diet. Am J Pathol. 2019;189:590–603.
He Y, Wu GD, Sadahiro T, et al. Interaction of CD44 and hyaluronic acid enhances biliary epithelial proliferation in cholestatic livers. Am J Physiol Gastrointest Liver Physiol. 2008;295:G305–12.
Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology. 2016;63:217–32.
Wu N, Meng F, Invernizzi P, Bernuzzi F, Venter J, Standeford H, et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-beta1 biliary secretion in mice. Hepatology. 2016;64:865–79.
Syal G, Fausther M, Dranoff JA. Advances in cholangiocyte immunobiology. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1077–86.
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32.
Kruglov EA, Nathanson RA, Nguyen T, Dranoff JA. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2006;290:G765–71.
Puche JE, Lee YA, Jiao J, Aloman C, Fiel MI, Muñoz U, et al. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology. 2013;57:339–50.
Friedman SL, Roll FJ, Boyles J, Arenson DM, Bissell DM. Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J Biol Chem. 1989;264:10756–62.
Melton AC, Yee HF. Hepatic stellate cell protrusions couple platelet-derived growth factor-BB to chemotaxis. Hepatology. 2007;45:1446–53.
Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–79.
Wang B, Trippler M, Pei R, Lu M, Broering R, Gerken G, et al. Toll-like receptor activated human and murine hepatic stellate cells are potent regulators of hepatitis C virus replication. J Hepatol. 2009;51:1037–45.
Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–55.
Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–35.
Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–94.
Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol. 2012;3:138.
Wree A, Mehal WZ, Feldstein AE. Targeting cell death and sterile inflammation loop for the treatment of nonalcoholic steatohepatitis. Semin Liver Dis. 2016;36:27–36.
Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46:1509–18.
Seo W, Eun HS, Kim SY, Yi HS, Lee YS, Park SH, et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology. 2016;64:616–31.
He Q, Fu Y, Ding X, Li D, Wang Z, Tian D, et al. High-mobility group box 1 induces endoplasmic reticulum stress and activates hepatic stellate cells. Lab Investig. 2018;98:1200–10.
Hernandez C, Huebener P, Pradere JP, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest. 2018;128:2436–51.
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34. e7
Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–18.
Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Investig. 2003;83:655–63.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8.
Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:E3186–95.
Livshits MA, Khomyakova E, Evtushenko EG, Lazarev VN, Kulemin NA, Semina SE, et al. Isolation of exosomes by differential centrifugation: theoretical analysis of a commonly used protocol. Sci Rep. 2015;5:17319.
Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70:249–59.
Szabo G, Momen-Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2017;14:455–66.
Ohara M, Ohnishi S, Hosono H, Yamamoto K, Yuyama K, Nakamura H, et al. Extracellular vesicles from amnion-derived mesenchymal stem cells ameliorate hepatic inflammation and fibrosis in rats. Stem Cells Int. 2018;2018:3212643.
Mardpour S, Hassani SN, Mardpour S, Sayahpour F, Vosough M, Ai J, et al. Extracellular vesicles derived from human embryonic stem cell-MSCs ameliorate cirrhosis in thioacetamide-induced chronic liver injury. J Cell Physiol. 2018;233:9330–44.
McDaniel K, Wu N, Zhou T, Huang L, Sato K, Venter J, et al. Amelioration of ductular reaction by stem cell derived extracellular vesicles in MDR2 knockout mice via Lethal-7 microRNA. Hepatology. 2019;69:2562–78.
Kostallari E, Hirsova P, Prasnicka A, Verma VK, Yaqoob U, Wongjarupong N, et al. Hepatic stellate cell-derived platelet-derived growth factor receptor-alpha-enriched extracellular vesicles promote liver fibrosis in mice through SHP2. Hepatology. 2018;68:333–48.
Jiang JX, Mikami K, Shah VH, Torok NJ. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a Rho guanosine triphosphatase-dependent mechanism. Hepatology. 2008;48:1497–505.
Jiang JX, Mikami K, Venugopal S, Li Y, Torok NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51:139–48.
Takehara T, Tatsumi T, Suzuki T, Rucker EB 3rd, Hennighausen L, Jinushi M, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–97.
Kitani A, Fuss I, Nakamura K, Kumaki F, Usui T, Strober W. Transforming growth factor (TGF)-beta1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-beta1-mediated fibrosis. J Exp Med. 2003;198:1179–88.
Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;148:785–800.
Pinzani M, Gesualdo L, Sabbah GM, Abboud HE. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J Clin Invest. 1989;84:1786–93.
Grappone C, Pinzani M, Parola M, Pellegrini G, Caligiuri A, DeFranco R, et al. Expression of platelet-derived growth factor in newly formed cholangiocytes during experimental biliary fibrosis in rats. J Hepatol. 1999;31:100–9.
Pinzani M, Gentilini A, Caligiuri A, De Franco R, Pellegrini G, Milani S, et al. Transforming growth factor-beta 1 regulates platelet-derived growth factor receptor beta subunit in human liver fat-storing cells. Hepatology. 1995;21:232–9.
Lim BJ, Lee WK, Lee HW, Lee KS, Kim JK, Chang HY, et al. Selective deletion of hepatocyte platelet-derived growth factor receptor alpha and development of liver fibrosis in mice. Cell Commun Signal. 2018;16:93.
Borkham-Kamphorst E, Herrmann J, Stoll D, Treptau J, Gressner AM, Weiskirchen R. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell activation and attenuates liver fibrosis. Lab Investig. 2004;84:766–77.
Neef M, Ledermann M, Saegesser H, Schneider V, Widmer N, Decosterd LA, et al. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J Hepatol. 2006;44:167–75.
Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–57.
Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–76.e3.
Guillot A, Hamdaoui N, Bizy A, Zoltani K, Souktani R, Zafrani ES, et al. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology. 2014;59:296–306.
Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-beta receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol. 2014;193:3925–33.
Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–9.
Guo L, Hu-Li J, Zhu J, Pannetier C, Watson C, McKenzie GJ, et al. Disrupting Il13 impairs production of IL-4 specified by the linked allele. Nat Immunol. 2001;2:461–6.
Kodera T, McGaha TL, Phelps R, Paul WE, Bona CA. Disrupting the IL-4 gene rescues mice homozygous for the tight-skin mutation from embryonic death and diminishes TGF-beta production by fibroblasts. Proc Natl Acad Sci U S A. 2002;99:3800–5.
Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–56.
Gieseck RL 3rd, Ramalingam TR, Hart KM, Vannella KM, Cantu DA, Lu WY, et al. Interleukin-13 activates distinct cellular pathways leading to ductular reaction, steatosis, and fibrosis. Immunity. 2016;45:145–58.
Aoudjehane L, Pissaia A Jr, Scatton O, Podevin P, Massault PP, Chouzenoux S, et al. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab Investig. 2008;88:973–85.
Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–85.
Batsaikhan B, Lu MY, Yeh ML, Huang CI, Huang CF, Lin ZY, et al. Elevated interleukin-4 levels predicted advanced fibrosis in chronic hepatitis C. J Chin Med Assoc. 2019;82:277–81.
Mallat A, Preaux AM, Blazejewski S, Rosenbaum J, Dhumeaux D, Mavier P. Interferon alfa and gamma inhibit proliferation and collagen synthesis of human ito cells in culture. Hepatology. 1995;21:1003–10.
Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, et al. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology. 1996;23:1189–99.
Rockey DC, Maher JJ, Jarnagin WR, Gabbiani G, Friedman SL. Inhibition of rat hepatic lipocyte activation in culture by interferon-gamma. Hepatology. 1992;16:776–84.
Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–3.
Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–52.
Tsuji H, Mukaida N, Harada A, Kaneko S, Matsushita E, Nakanuma Y, et al. Alleviation of lipopolysaccharide-induced acute liver injury in Propionibacterium acnes-primed IFN-gamma-deficient mice by a concomitant reduction of TNF-alpha, IL-12, and IL-18 production. J Immunol. 1999;162:1049–55.
Cao Q, Batey R, Pang G, Russell A, Clancy R. IL-6, IFN-gamma and TNF-alpha production by liver-associated T cells and acute liver injury in rats administered concanavalin A. Immunol Cell Biol. 1998;76:542–9.
Luo XY, Takahara T, Kawai K, Fujino M, Sugiyama T, Tsuneyama K, et al. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol. 2013;305:G891–9.
Ravichandran G, Neumann K, Berkhout LK, Weidemann S, Langeneckert AE, Schwinge D, et al. Interferon-gamma-dependent immune responses contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol. 2019;71(4):773–82.
Kong X, Feng D, Mathews S, Gao B. Hepatoprotective and anti-fibrotic functions of interleukin-22: therapeutic potential for the treatment of alcoholic liver disease. J Gastroenterol Hepatol. 2013;28(Suppl 1):56–60.
Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–42.
Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. 2006;55:415–24.
Liu C, Chen X, Yang L, Kisseleva T, Brenner DA, Seki E. Transcriptional repression of the transforming growth factor beta (TGF-beta) Pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by nuclear factor kappaB (NF-kappaB) p50 enhances TGF-beta signaling in hepatic stellate cells. J Biol Chem. 2014;289:7082–91.
Tarrats N, Moles A, Morales A, Garcia-Ruiz C, Fernandez-Checa JC, Mari M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology. 2011;54:319–27.
Hernandez-Munoz I, de la Torre P, Sanchez-Alcazar JA, García I, Santiago E, Muñoz-Yagüe MT, et al. Tumor necrosis factor alpha inhibits collagen alpha 1(I) gene expression in rat hepatic stellate cells through a G protein. Gastroenterology. 1997;113:625–40.
Osawa Y, Hoshi M, Yasuda I, Saibara T, Moriwaki H, Kozawa O. Tumor necrosis factor-alpha promotes cholestasis-induced liver fibrosis in the mouse through tissue inhibitor of metalloproteinase-1 production in hepatic stellate cells. PLoS One. 2013;8:e65251.
Houglum K, Buck M, Kim DJ, Chojkier M. TNF-alpha inhibits liver collagen-alpha 1(I) gene expression through a tissue-specific regulatory region. Am J Phys. 1998;274:G840–7.
Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. 2019;10:100.
Xiang M, Wang PX, Wang AB, Zhang XJ, Zhang Y, Zhang P, et al. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J Hepatol. 2016;64:1365–77.
Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. 2018;67(2):549–59.
Xiang DM, Sun W, Ning BF, Zhou TF, Li XF, Zhong W, et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut. 2018;67:1704–15.
Kagan P, Sultan M, Tachlytski I, Safran M, Ben-Ari Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS One. 2017;12:e0176173.
Kwon HJ, Won YS, Park O, Chang B, Duryee MJ, Thiele GE, et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology. 2014;60:146–57.
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6.
Gieling RG, Wallace K, Han YP. Interleukin-1 participates in the progression from liver injury to fibrosis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1324–31.
Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280–92.
Louis H, Van Laethem JL, Wu W, Quertinmont E, Degraef C, Van den Berg K, et al. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology. 1998;28:1607–15.
Thompson K, Maltby J, Fallowfield J, McAulay M, Millward-Sadler H, Sheron N. Interleukin-10 expression and function in experimental murine liver inflammation and fibrosis. Hepatology. 1998;28:1597–606.
Huang YH, Chen MH, Guo QL, Chen YX, Zhang LJ, Chen ZX, et al. Interleukin10 promotes primary rat hepatic stellate cell senescence by upregulating the expression levels of p53 and p21. Mol Med Rep. 2018;17:5700–7.
Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124:3241–51.
Marvie P, Lisbonne M, L’Helgoualc’h A, Rauch M, Turlin B, Preisser L, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. 2010;14:1726–39.
Kotsiou OS, Gourgoulianis KI, Zarogiannis SG. IL-33/ST2 axis in organ fibrosis. Front Immunol. 2018;9:2432.
Gao Y, Liu Y, Yang M, Guo X, Zhang M, Li H, et al. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget. 2016;7:33649–61.
Weiskirchen R, Tacke F. Interleukin-33 in the pathogenesis of liver fibrosis: alarming ILC2 and hepatic stellate cells. Cell Mol Immunol. 2017;14:143–5.
Tan Z, Liu Q, Jiang R, Lv L, Shoto SS, Maillet I, et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell Mol Immunol. 2018;15:388–98.
Bridle KR, Crawford DH, Ramm GA. Identification and characterization of the hepatic stellate cell transferrin receptor. Am J Pathol. 2003;162:1661–7.
Ryan E, Byrnes V, Coughlan B, Flanagan AM, Barrett S, O’Keane JC, et al. Underdiagnosis of hereditary haemochromatosis: lack of presentation or penetration? Gut. 2002;51:108–12.
Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127:55–64.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.
Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. 2019;3:730–43.
Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65.
Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident Kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193:344–53.
Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185–97.
Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754–67.
Snyder RJ, Lantis J, Kirsner RS, Shah V, Molyneaux M, Carter MJ. Macrophages: a review of their role in wound healing and their therapeutic use. Wound Repair Regen. 2016;24:613–29.
Chazaud B. Macrophages: supportive cells for tissue repair and regeneration. Immunobiology. 2014;219:172–8.
Abshagen K, Eipel C, Kalff JC, Menger MD, Vollmar B. Loss of NF-kappaB activation in Kupffer cell-depleted mice impairs liver regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1570–7.
Gehring S, Dickson EM, San Martin ME, van Rooijen N, Papa EF, Harty MW, et al. Kupffer cells abrogate cholestatic liver injury in mice. Gastroenterology. 2006;130:810–22.
Elsegood CL, Chan CW, Degli-Esposti MA, Wikstrom ME, Domenichini A, Lazarus K, et al. Kupffer cell-monocyte communication is essential for initiating murine liver progenitor cell-mediated liver regeneration. Hepatology. 2015;62:1272–84.
Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–23.
Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47(phox)-oxidative stress pathway in neutrophils. Gut. 2017;66:705–15.
Rajkovic IA, Williams R. Abnormalities of neutrophil phagocytosis, intracellular killing and metabolic activity in alcoholic cirrhosis and hepatitis. Hepatology. 1986;6:252–62.
Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71.
Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–28.
de Lalla C, Galli G, Aldrighetti L, Romeo R, Mariani M, Monno A, et al. Production of profibrotic cytokines by invariant NKT cells characterizes cirrhosis progression in chronic viral hepatitis. J Immunol. 2004;173:1417–25.
Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta. 1832;2013:1061–9.
Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, et al. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology. 2009;49:1683–94.
Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–94.
Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–58.
Madala SK, Pesce JT, Ramalingam TR, Wilson MS, Minnicozzi S, Cheever AW, et al. Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J Immunol. 2010;184:3955–63.
Chedid A, Mendenhall CL, Moritz TE, French SW, Chen TS, Morgan TR, et al. Cell-mediated hepatic injury in alcoholic liver disease. Veterans Affairs Cooperative Study Group 275. Gastroenterology. 1993;105:254–66.
Tanakaa A, Leung PS, Young HA, Gershwin ME. Toward solving the etiological mystery of primary biliary cholangitis. Hepatol Commun. 2017;1:275–87.
Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115:3072–82.
Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, et al. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology. 2018;67:1970–85.
Tang XZ, Jo J, Tan AT, Sandalova E, Chia A, Tan KC, et al. IL-7 licenses activation of human liver intrasinusoidal mucosal-associated invariant T cells. J Immunol. 2013;190:3142–52.
Walker LJ, Kang YH, Smith MO, Tharmalingham H, Ramamurthy N, Fleming VM, et al. Human MAIT and CD8alphaalpha cells develop from a pool of type-17 precommitted CD8+ T cells. Blood. 2012;119:422–33.
Ussher JE, Willberg CB, Klenerman P. MAIT cells and viruses. Immunol Cell Biol. 2018;96:630–41.
Jeffery HC, van Wilgenburg B, Kurioka A, Parekh K, Stirling K, Roberts S, et al. Biliary epithelium and liver B cells exposed to bacteria activate intrahepatic MAIT cells through MR1. J Hepatol. 2016;64:1118–27.
Bottcher K, Rombouts K, Saffioti F, Roccarina D, Rosselli M, Hall A, et al. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic stellate cell activation. Hepatology. 2018;68:172–86.
Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ, Zhao Y, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. 2017;67:770–9.
Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–51.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367–78.e5; quiz e14–5.
Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381:468–75.
Verrill C, Markham H, Templeton A, Carr NJ, Sheron N. Alcohol-related cirrhosis – early abstinence is a key factor in prognosis, even in the most severe cases. Addiction. 2009;104:768–74.
Serpaggi J, Carnot F, Nalpas B, Canioni D, Guéchot J, Lebray P, et al. Direct and indirect evidence for the reversibility of cirrhosis. Hum Pathol. 2006;37:1519–26.
Maiers JL, Kostallari E, Mushref M, deAssuncao TM, Li H, Jalan-Sakrikar N, et al. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology. 2017;65:983–98.
Liu Z, Li C, Kang N, Malhi H, Shah VH, Maiers JL. Transforming growth factor beta (TGFbeta) cross-talk with the unfolded protein response is critical for hepatic stellate cell activation. J Biol Chem. 2019;294:3137–51.
Wu YJ, Cai WM, Li Q, Liu Y, Shen H, Mertens PR, et al. Long-term antifibrotic action of interferon-gamma treatment in patients with chronic hepatitis B virus infection. Hepatobiliary Pancreat Dis Int. 2011;10:151–7.
Pockros PJ, Jeffers L, Afdhal N, Goodman ZD, Nelson D, Gish RG, et al. Final results of a double-blind, placebo-controlled trial of the antifibrotic efficacy of interferon-gamma1b in chronic hepatitis C patients with advanced fibrosis or cirrhosis. Hepatology. 2007;45:569–78.
van Dijk F, Olinga P, Poelstra K, Beljaars L. Targeted therapies in liver fibrosis: combining the best parts of platelet-derived growth factor BB and interferon gamma. Front Med (Lausanne). 2015;2:72.
Gao B, Xiang X. Interleukin-22 from bench to bedside: a promising drug for epithelial repair. Cell Mol Immunol. 2019;16:666–7.
Arab JP, Sehrawat T, Simonetto DA, Verma VK, Feng D, Tang T, et al. An open label, cohort dose escalation study to assess the safety and efficacy of IL-22 agonist F-652 in patients with alcoholic hepatitis. Hepatology. 2018;68:1454A.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Guillot, A., Gao, B. (2020). Immunopathogenesis of Liver Cirrhosis. In: Gershwin, M.E., M. Vierling, J., Tanaka, A., P. Manns, M. (eds) Liver Immunology . Springer, Cham. https://doi.org/10.1007/978-3-030-51709-0_35
Download citation
DOI: https://doi.org/10.1007/978-3-030-51709-0_35
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-51708-3
Online ISBN: 978-3-030-51709-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)