
Abstract
Liver regeneration is a unique process in which the organ returns to its original size, histologic structure and normal metabolic function. The past several decades have identified many of the mechanisms associated with regeneration after hepatic injury. Liver regeneration in acute liver failure, as well as acute on chronic liver injury, has provided crucial information regarding pathways of regeneration beyond what the classical models of partial hepatectomy have shown. There is a growing body of data regarding cellular differentiation and stem cell involvement in the regenerative process of the liver that has been uncovered in these clinically relevant models. In this chapter, we describe the common and unique details of these myriad molecular mechanisms, including immunomodulation, microRNAs, and the gut-liver axis. Key to these pathways are the role of cellular signaling and stem cells as they relate to liver regeneration in both acute and acute on chronic injury.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Acute liver failure
- Acute on chronic liver failure
- Cytokines
- Growth factors
- Homeostasis
- microRNAs
- Partial hepatectomy
- Regeneration
- Stem cells
-
Liver regeneration is a tightly regulated process of coordinating cytokines, growth factors, inflammation, and cell fate.
-
Emerging pathophysiologic mechanisms of this process, or processes include the gut-liver axis, microRNAs, the Hippo-YAP pathway, and stem cell function.
-
Promising therapeutics include immunomodulation, microRNA technology, and stem cell therapy.
Introduction
The study of liver regeneration has evolved dramatically over the past century, and our understanding stems from early experimental models of liver injury by partial hepatectomy [1] and carbon tetrachloride [2] to the modern discovery of liver progenitor cells. Similarly, there has been an evolution in understanding of liver regeneration in liver disease as relates to its temporal course—from chronic liver injury to acute liver failure (ALF), as well as the recently defined unique entity of acute-on-chronic liver failure (ACLF). Our goal in this review is to clearly describe liver regeneration in the acute injury setting (such as ischemic, toxic, and surgical insults), as well as in acute on chronic liver injury (additional insult in those already with chronic hepatic fibrosis and cirrhosis). It is beneficial for the reader to understand modern concepts in liver regeneration in the setting of ALF and the preclinical and animal models developed in its study. Historically, the 2/3 partial hepatectomy (PHx) model has been used as the apical model for liver injury—an acute insult which under certain circumstances can lead to liver failure in its host and does not cause persistent injury in remaining hepatocytes. This is referenced throughout as a means of understanding pathways of liver regeneration.
Background
The liver is the organ in the body whose purpose is to maintain homeostasis of essential functions involving (i) proteins, cholesterol, and hormone metabolism and synthesis; (ii) biotransformation of bilirubin and medications, bile salt synthesis for nutritional utilization, immune regulation via the reticuloendothelial system; and (iii) storage of glycogen, lipids, and essential vitamins and minerals. Given the complexity of function, it follows that the liver has complex and unique mechanisms to maintain normal cell function and repair of injured cells. These unique characteristics include rapid initiation of mitosis from quiescent hepatocytes, the ability to synchronize this process between varying hepatic cell types, and an astonishing ability to regulate an essential hepatic mass.
Early models first suggested the presence of an extra-hepatic “humoral” factor(s) that initiated the regeneration process. Early studies in PHx models demonstrated restoration of liver mass to preoperative weight, and that irreversible necrosis ensued and regeneration failed after a greater degree of hepatic resection [1, 3]. These investigators found a significant increase in mitotic activity and DNA synthesis in rat hepatocytes from the normal partner induced by cross circulation from a partially hepatectomized donor [4]. Later work demonstrated a “wave” of mitoses in the injured liver, progressing from periportal to pericentral regions in a synchronous manner that exhibited a cell-autonomous function [5]. In fact, early xenotransplantation studies using mouse hepatocytes implanted into rat liver were noted to follow the same time course of regeneration as if they were still intrinsically in the mouse liver, and were not significantly impacted by the surrounding cellular milieu [6].
These findings and others led to search for a “master” mitogen that promotes the initiation of synchronized liver regeneration and maintains that process but only until the appropriate liver mass was attained. Ultimately, this expedition has demonstrated an ever-expanding catalog of contributors to the process of hepatic regeneration rather than a single factor. Our hope is to delineate the basis for the modern understanding of hepatic regeneration—first through classical pathways, and then through the modern era of microRNAs and stem cells.
Classical Pathways of Liver Regeneration
The classical description of liver regeneration focuses on signaling cascades, both intra- and extra-hepatic. These cascades occur with rapid precision, affecting the hepatocytes and the surrounding cellular matrix. This process has traditionally been termed “priming and progression,” as hepatic regeneration is first preceded by a signal to hepatocytes “priming” them for mitosis and division, then prompting progression from G0 [7]. This early signal is insufficient to push hepatocytes through the cell cycle and a second signal, a mitogen of extra-hepatic origin, is necessary for cellular progression through G1 and mitosis. These mechanisms are further described later in the chapter.
Intracellular Signaling Pathways
The study of the molecular mechanisms of liver regeneration requires identifying signaling pathways that stimulate a rapid response to hepatocyte injury. Transcription factors, such as STAT3, NF-κB, and β-catenin and their post-translational impact have been studied, and support an expedient mechanism of cell cycle regulation and gene expression. The hedgehog signaling pathway goes beyond liver development, but is upregulated in regeneration after PHx [8]. Further regulators of these processes will be discussed here [9, 10].
Role of Tumor-Necrosis Factor Alpha (TNF-α)
TNF-α is a signaling protein and inflammatory cytokine primarily produced by macrophages/monocytes during acute inflammation and has a diverse range of signaling events within cells. It plays a significant role in liver regeneration both after PHx as well as CCl4 induced injuries [11]. In the priming phase, TNF-α acts on hepatocytes to enter the cell cycle for regeneration [12]. It exerts many of its effects by binding to two types of receptors, namely TNFR-1 and TNFR-2. In CCL4 induced liver injury, TNFR-1 knockout mice had impairment in cellular replication and delay in liver weight recovery of which both processes were reversed with IL-6 treatment. In PHx models, TNFR-1 knockout mice had severely impaired DNA synthesis of transcription factors, which recovered after injection of IL-6 [13]. In wild-type mice, treatment with anti-TNF prior to PHx increased the IL-6 levels whereas untreated mice had no effect [14]. These series of experiments showed that TNF-α initiates a cascade of intracellular signaling via TNFR-1 receptor, eventually leading cells to enter the proliferation phase.
Intracellular signaling pathways initiated by TNF-α have been well studied. In the context of liver regeneration, NF-κB and STAT3 are transcription factors with key roles in the intracellular cascade of signals for proliferation of hepatocytes. NF-κB is essential to maintaining hepatocyte homeostasis, including cell survival and apoptosis [15]; and plays a crucial role during development [16]. It has been well established that NF-κB activation in Kupffer cells is crucial for liver regeneration after PHx [17]; and inactivation of NF-κB in both Kupffer cells and hepatocyte have been shown to impair cellular proliferation after PHx [18].
Role of Interleukin 6 (IL-6)
Interleukin 6 has broad biological functions including pro-inflammatory, mediation of acute phase reactions, regeneration, and carcinogenesis. It is involved in two distinct pathways for signal transduction, both of which are important in liver regeneration—classical and trans-signaling [19]. In the classical pathway, IL-6 binds to membrane protein receptor IL-6-R (also known as glycoprotein GP-80) of effector cells. After binding, IL-6—gp80 complex interacts with gp-130, leading to homodimerization of the complex, autophosphorylation of gp-130 and activation of cytoplasmic tyrosine kinase JAK1. This subsequently activates STAT3, STAT1 and also leads to RAS/Map signal pathway activation [20]. Of note, IL-6 receptors that are expressed at the surface membrane are restricted to only certain types of cells, including hepatocytes, some epithelial cells and leukocytes. However, in the second pathway for signal transduction, known as trans-signaling, soluble IL-6R is cleaved from the cell membrane by metalloproteinase ADAM17 and shed into serum and cytoplasm [21]. The complex, IL-6-sIL-6R can then activate gp130 in a similar fashion to homodimerization in other types of cells and induce the cellular signaling cascade. The signaling cascade will have variable effects depending on the cell type, concentration of gp-130 and serum levels of sIL-6R, making IL-6 a somewhat pleiotropic cytokine.
In liver regeneration, Kupffer cells are the likely source of IL-6, demonstrated in bone marrow transplant and macrophage-specific IL-6 knockout experiments [22]. In rats, after hepatectomy, serum levels of TNF-α and then IL-6 were elevated within a few hours and subsequently associated with significant activation of transcription factors STAT3 and C/EBPβ/nuclear factor-IL-6 resulting in enhanced transcription of these genes. The results were suggestive that these may trigger G0/G1 phase transition in hepatocytes after partial hepatectomy [20]. The integral role of IL-6 for liver regeneration was demonstrated in IL-6 knockout mice, which after PHx had ALF due to lack of DNA synthesis and a G1 phase response [23]. This was also associated with reduced STAT3 activation and decreased expression of various factors involved in cell cycle regulation. Moreover, when these mice were injected with IL-6, hepatocyte proliferation was restored, and liver failure was averted, indicating the fundamental importance of this cytokine in liver regeneration.
In acute-on-chronic liver injury models, it has been noted that there is a shift from the IL-6/STAT3 pathway. Chronic liver injury attenuates liver generation because the Kupffer cells in these liver models produce reduced levels of IL-6. In cases of acute injury against a backdrop of chronic liver disease, there is a robust innate response with IFN-γ, which then activates the STAT1 pathway [24]. Unlike that of STAT3, the STAT1 pathway is inhibitory and blocks liver regeneration. In liver injury models, there is a balance between the IL-6/STAT3 and IFN-γ/STAT1 pathways that controls liver regeneration [25]. Studies have shown that an imbalance of the two pathways can lead to impairment of liver regeneration. In acute-on-chronic liver injury models, it was shown that IL-22 recombinant dimer enhanced STAT3 pathway over the STAT1 pathway, which then enhanced liver regeneration [26]. IL-22 is a cytokine produced by multiple immune cells, and its key targets include nonhematopoietic epithelial and stromal cells, where it can promote proliferation and play a role in tissue regeneration. This novel approach has been shown experimentally and has therapeutic potential for liver injuries and ACLF.
Immune Regulation in the Regenerating Liver
The liver serves as the initial sensor of all intestinal venous blood draining the gut, with the gut-liver axis being a complex and highly regulated system of immune tolerance in the setting of constant bombardment with toxins and a plethora of microbial antigens [27]. The gut-liver axis also serves as a reservoir for immune regulatory cells, most notably Kupffer cells—the resident macrophages of the liver. These Kupffer cells represent the majority of all tissue macrophages, including cells present in hepatic sinusoids [28]. The interactions between the Kupffer cells and intestinal venous blood promote their cell signaling and makes them an fundamental aspect of hepatic regeneration.
In the setting of hepatocyte injury, macrophage number and division is upregulated, while concurrently promoting recruitment of other inflammatory cells to liver tissue [29]. The decisive role of Kupffer cells and other recruited macrophages in the process of liver regeneration remains in question, as studies assessing both activation and depletion show varying outcomes. Hepatocyte-protective effects with macrophage inactivation are offset by results showing that macrophage depletion delays regeneration and loss of NF-κB activation, as well as recruitment of infiltrating macrophages [29,30,31,32]. Perhaps in part due to macrophage polarization and the M1/M2 phenotype, it is clear that Kupffer cells likely shift between phenotypes through the hepatocyte repair process as well as hepatic fibrosis [33].
Many of the mediators of regeneration discussed in this review are signaling molecules or cytokines that are essential to the normal function of the immune system. Their role in the immune response to regeneration has been elucidated with the importance of each molecule changing over time. Early studies in rodent models bred to be athymic, germ free, and lipopolysaccharide (LPS) resistant implicated the innate immune response in liver regeneration [34]. The Toll like receptor 4 (TLR4) is an essential binding protein for routine immunity; and TLR4 knockout models have demonstrated intact hepatocyte regeneration. Knockouts, however, lacking signaling protein MyD88 (a common adaptor molecule required for signaling mediated by TLR) showed a significant decrease in regeneration [35, 36]. Complement pathways have also been implicated in hepatocyte regeneration, as C3 and C5 knockout models again demonstrated impaired hepatic regeneration [37].
After PHx, macrophage colony stimulating factor (CSF-1) serum levels increased proportionately to the amount of tissue resected and shown to accelerate the regenerative proecess [38, 39]. ALF in humans appears to provide a clinically representative model in which the immune response is altered. As example, toxic overdoses of acetaminophen decrease levels of circulating monocytes, and increase hepatic populations of circulation-derived macrophages as well as Kupffer cells compared to normal controls. These immune changes were seen as a result of elevated serum levels of chemokine ligands 2 and 3, interleukins 6 and 10, and transforming growth factor ß1 [29]. The acetaminophen ALF model has also demonstrated that serum levels of macrophage CSF-1 may predict mortality in this population, as lower levels were associated with a worse prognosis [40].
These translational studies and others support the concept of utilizing immune-modulating therapies in persons with ALF and ACLF to promote hepatocyte regeneration by targeting specific pathways [41]. To this end, numerous studies have evaluated the role of estrogens and androgens, corticosteroids, and exogenous stimulating factors in patients with ALF and ACLF [42,43,44,45,46,47].
Growth Factors
Subsequent to liver injury, and after the G0 to G1 phase transition in hepatocytes, growth factors play an important role as the cell progresses through G1. Two growth factors and their respective receptors that are particularly relevant and critical to liver regeneration are epidermal growth factor (EGF) and hepatocyte growth factor (HGF).
Epidermal Growth Factor and Its Receptor (EGFR)
During liver regeneration, the EGFR on hepatocyte is activated by one of many ligands, which leads to proliferation and survival of the cell. Several ligands are found to be upregulated during liver injury and PHx, including EGF, transforming growth factor (TGF)-α, heparin-binding EGF (HB-EGF), and amphiregulin [48]. These ligands are synthesized from various sources, adding to the redundancy in upregulation during liver regeneration. EGFR knockout mice have multiple developmental defects mostly in the endothelium and neural tissue, and usually they are not viable for longer than 8 days. PHx in mice with a conditional knockout of EGFR has significant liver regeneration delay and death, mostly driven by lack of regeneration from cell cycle arrest and reduced levels and activity of cyclin D1, among other cellular factors [49].
HB-EGF is produced by Kupffer cells and sinusoidal endothelial cells to act in a paracrine manner [48]. In HB-EGF knockout models, the delay in hepatocyte proliferation was only transient, possibly because of upregulation of TGF-α as a compensatory mechanism. In PHx models, HB-EGF levels was directly correlated to the degree of hepatectomy in that 1/3 PHx had undetectable serum levels whereas 2/3 PH had increased levels, which subsequently correlated with DNA replication. Moreover, HB-EGF administered to 1/3 PHx mice resulted in >15-fold increase in DNA replication [50]. TGF-α and amphiregulin are produced by hepatocytes to act in an autocrine manner. PHx in mice lacking TGF-α surprisingly did not show any abnormality in liver regeneration, perhaps in part because of multiple redundant pathways. In contrast, defects in amphiregulin expression showed impaired cellular proliferation [51, 52]. EGF is secreted by salivary gland and Brunner’s gland in the gut to act in endocrine manner. Early studies reported impaired regeneration in its absence, and upregulation when recombinant EGF was administered to PHx mice [53].
Hepatocyte Growth Factor
Hepatocyte growth factor (HGF), also known as scatter factor (SF) is a paracrine growth factor primarily secreted by mesenchymal cells of the liver (primarily Kupffer cells and endothelial cells). HGF expression is upregulated in these cells as well as several other organs in response to liver injury. HGF production is also augmented by distant organs, in response to cytokines produced during liver injury (as discussed above, IL-6 and TNF-α play key roles here), highlighting some “endocrine-like” nature of this growth factor [54]. HGF stimulates epithelial cell proliferation, motility, morphogenesis and angiogenesis. In acute liver injury models, using CCl4 induced hepatitis, rats that were given anti-HGF IgG showed reduced numbers of proliferating hepatocytes [55]. Specifically, HGF acts via tyrosine phosphorylation of c-Met receptor, a transmembrane protein that is activated by binding of HGF, and induces intracellular cascade promoting the wide array of cellular functions. In knockout models for c-Met, the organisms fail embryonic development and have significant liver abnormalities [56]. Also, in mice with the knockout c-Met gene in liver, regeneration after PHx was delayed due to disruption of the cell cycle [57]. Another study showed that deletion of c-Met in liver cells in a non-inducible manner showed severe liver necrosis and jaundice after PHx [58]. Additional studies have shown that c-Met is not only important in cell survival but has a crucial function in liver regeneration and cannot be compensated by other growth factors [59]. It is not surprising that a cellular and functional loss of liver endothelial cells, together with their regenerative angiocrine functions, are associated with decreased hepatocyte proliferation and regeneration in ACLF compared to ALF patients [60].

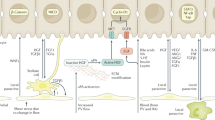
Molecular mechanisms of regeneration after acetaminophen (N-acetyl-para-aminophenol; APAP)-induced liver injury. Liver regeneration after APAP overdose involves a complex time- and dose-dependent interplay of several signaling mediators. Several proliferative signaling pathways that control cell cycle machinery, including growth factor signaling via epidermal growth factor receptor (EGFR) and c-MET [receptor for hepatocyte growth factor (HGF)], cytokine signaling [tumor necrosis factor (TNF)-α/NF-κB and IL-6/STAT-3], Wnt/β-catenin, and bile acid signaling are activated after APAP overdose, potentially contributing to liver regeneration. Some of these proliferative signaling pathways including Wnt/β-catenin and TNF-α/NF-κB signaling are inhibited after severe APAP overdose (others such as EGFR/c-MET and IL-6/STAT-3 signaling remain activated), which is accompanied by unchecked DNA damage and activation of antiproliferative pathways [transforming growth factor (TGF)–β and p53/p21] leading to cell cycle arrest and impaired liver regeneration. Angiogenesis and the restoration of microvasculature during normal liver regeneration involve the activation of vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) signaling, which also indirectly contributes to hepatocyte proliferation via the stimulation of HGF release from endothelial cells. Top, hematoxylin and eosin–stained liver sections that are normal (left) and necrotic (right). Bottom, regenerating liver, shown as proliferating cell nuclear antigen (PCNA)-positive hepatocytes (brown nuclear staining). FXR farnesoid X receptor, Fzld frizzled protein, G0 gap 0 phase, G1 gap 1 phase, G2 gap 2 phase, GSH glutathione, GSK glycogen synthase kinase, ILK integrin-linked protein kinase, M mitosis phase, MAPK mitogen-activated protein kinase, NAPQI N-acetyl-p-benzoquinone imine, S synthesis phase, TNFR TNF receptor. (Figure and Caption source: Bhushan, Bharat et al., Liver Regeneration after Acetaminophen Hepatotoxicity, The American Journal of Pathology, Volume 189, Issue 4, 719–729) [61]
The Role of Metabolism in Liver Regeneration
It is now well recognized that bile acids play a major role in liver regeneration. PHx models with external biliary drainage demonstrated reduced regenerative capacity and those with carbon tetrachloride induced injury showed increased hepatocyte restoration with supplementation of bile acids. This later effect was shown to be related to increased FOXM1 signaling, which is a key transcription factor in cell cycle progression [62, 63]. This finding was confirmed in a human clinical study of patients undergoing PHx, where reduced liver volumes were observed at day 7 with external drainage of bile [64].
Nuclear receptor farsenoid X receptor (FXR) is a key receptor in the mechanisms of bile acid signaling, and is expressed in numerous tissues, including the liver and small bowel. It acts via multiple pathways in regulating bile acid, lipid homeostasis, including other key metabolic pathways in the body [65]. PHx and carbon tetrachloride toxicity in FXR knockout murine models demonstrated reduced early liver regeneration; and supplementation of bile acids did not ameliorate those effects. These studies also demonstrated that FXR binds to a fibroblast growth factor, which interacts with cytochrome P450 as a key pathway in bile acid synthesis. More specifically, decreased FOXM1 expression was associated with impaired bile acid production and liver regeneration [66]. To further delineate the role of FXR in liver regeneration, a study of hepatic and intestine-specific FXR knockout mice showed that hepatic FXR was necessary for induction of FOXM1, while this finding was also observed in intestine-specific FXR knockouts [67].
The role of gut microbiota and bile acid homeostasis is a popular topic with enormously important clinical implications. In the setting of liver injury, or PHx, there is increased bacterial translocation across gut mucosa and exposure to byproducts of the microbiome [68]. The composition of the microbiome is implicated in altering bile acid homeostasis via changes in primary and secondary bile acid synthesis. Reduced microbiome diversity in cirrhotic humans leads to decreased conversion of primary to secondary bile acids in this population. This could, in part, explain one at least one mechanism for the hepatic dysfunction and risk for liver failure in persons with acute on chronic liver failure [69].
Platelets and Platelet-Derived Factors
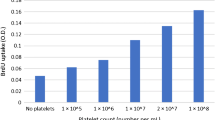
Evidence suggests an essential role for platelets and platelet-derived factors in liver regeneration after PHx. Platelets accumulate in the liver remnant following PHx in human and murine models. While an elevated platelet count stimulates liver regeneration after PHx, regeneration is significantly delayed when platelets are depleted or functionally impaired [70]. Several clinical studies have shown worse outcomes with regard to mortality, liver dysfunction, and reduced volumes of regeneration; and related to the finding that activated platelets secrete growth factors. Fibrinogen is one such factor that has been shown to deposit in the liver after PHx. and inhibition of fibrinogen deposition leads to decreased hepatocyte proliferation [71]. Studies in murine and human models after PHx suggest a unique mechanism in which intrahepatic fibrin(ogen) deposition drives platelet accumulation and ultimately promotes hepatic regeneration after PHx [72].
Paracrine Mediators
Wnt/β-catenin Pathway in Liver Regeneration
The Wnt/β-catenin pathway plays a critical role in liver regeneration, development, and normal physiology. In the absence of Wnt signaling, β-catenin is marked for degradation by a complex involving the tumor suppressor protein APC. When activated, free β-catenin will translocate to the nucleus and mediates target gene transcription via T-cell factor proteins [73]. β-catenin levels are tightly regulated, with a significant proportion typically bound to either the APC complex or E-cadherin at the cell membrane [74]. Following PHx, cytosolic β-catenin levels increase with subsequent translocation to the nucleus. The significance of β-catenin in liver regeneration after injury has been studied in β-catenin knockout models, where there is a delay in hepatocyte proliferation and decreased liver mass during early regeneration [75]. Acetaminophen-induced liver injury also serves as a clinically relevant model for the role of β-catenin in hepatic regeneration (Fig. 4.1). Murine models of acetaminophen overdose demonstrated activation of β-catenin with a subsequent increased expression of glutamine synthase (a β-catenin target), and ultimately increased cyclin-D1, thereby, promoting cellular proliferation [76]. Similarly, liver tissues from biopsies of persons with acetaminophen-induced liver injury have demonstrated correlation between nuclear β-catenin localization and spontaneous liver regeneration.
The Wnt/β-catenin pathway is also involved in the “metabolic zonation” of the liver during organogenesis and regeneration, via APC regulation. Based on varying signaling patterns, hepatocytes express a gradient between respective periportal and pericentral phenotypes and their associated metabolic activities [77, 78]. This pathway also drives architectural development during regeneration in PHx models by increasing levels of β-catenin and E-cadherin, for coordination of cell–cell adhesion [79, 80].
Transforming Growth Factor β
Transforming growth factor beta (TGF-β) is a key factor in termination of liver regeneration. Early studies showed TGF-β to be a strong inhibitor of DNA synthesis in mitogen-stimulated hepatocytes, and this effect decreased in a time-dependent manner in hepatocytes that were isolated from the regenerating liver [81]. A corroborating study showed that TGF-β mRNA expression increased after PHx and peaked after the first round of hepatocyte cellular division has occurred. This increase in levels of TGF-β was countered by a reduction in TGF-β receptor expression after the liver injury [82]. The importance of receptor expression in regeneration was shown in TGF-β receptor knockout models demonstrating an increase in hepatocyte proliferation with corresponding increase in liver mass. This inverse relationship was likely mediated by inhibition of cyclin D1 and arrest in the G1 phase of the cell cycle [83].
Beta-2 spectrin (β2SP) has been shown to be another key receptor in TGF-β signaling. Murine knockout models of β2SP resulted in dysfunctional hepatocyte cell cycle progression and delayed liver regeneration after PHx, in a p53-independent fashion [84]. These data suggested that TGF-β plays a coordinating role in regeneration, rather than simply acting as a terminal signal.
Hippo/YAP Regeneration Pathway
Another pathway critical to regulation of liver mass and progenitor cell determination is the Yap/Hippo pathway. The transcription coactivator Yes-associated protein (YAP1) is the main effector of the pathway, with nuclear localization negatively controlled by Hippo upstream signaling. Hippo activation leads to phosphorylation and activation of mammalian Sterile20-like (MST) 1 and 2, which in turn phosphorylate and activate large tumor suppressor kinases (LATS) 1 and 2. When Hippo is turned off, YAP can translocate to the nucleus and bind to transcription factors, leading to transcription of genes involved in cell survival, growth, and proliferation. LATS phosphorylation of YAP1 prevents its translocation to the nucleus and therefore interactions with transcription factors and the Hippo/Yap pathway [85]. Induction of YAP1 in transgenic models with resultant overexpression created a 4-fold increase in liver size via an increase in cell number, and this effect was reversible with interruption of YAP1 expression [86]. Hepatocyte overexpression of YAP led to rapid growth of progenitor-like populations of hepatocytes, and increased nuclear localization of YAP1 has been associated with hepatocellular carcinoma [87]. Interestingly, while YAP protein levels increased significantly, mRNA levels did not reflect this large increase suggesting post-translational modification or inhibition of degradation during regeneration [88]. With greater understanding of the molecular mechanisms involved in this pathway, the list of regulators has grown significantly, and the pathway is seen as an integral part of the “hepatostat.”
Idea of Hepatostat
The recently coined term “hepatostat” defines the homeostatic mechanisms ensuring appropriate liver size and architecture following injury or stress [89]. Species-specific regenerative follows a typical time course, with final restoration of liver mass in 5–7 days in rodents and 3–4 months in humans after partial hepatectomy. However, this process of proliferation does not only involve mitosis and cellular division, but rather a still incompletely understood concept of cell fate and replication. It has long been appreciated that hepatocytes divide at differing rates depending on location, with periportal and zone 2 hepatocytes accounting for as much as 80% of all cell division; and that nuclear ploidy affects this geographic difference [90]. While the size of an organ was determined, primarily by the number and size of its cells, this was not confirmed in the liver until relatively recently. Liver regeneration after a 30% hepatectomy was achieved solely through hypertrophy, without cellular division. Meanwhile, cellular hypertrophy preceded proliferation in the 70% hepatectomy model and both hypertrophy and proliferation contributed equally to hepatocyte cell mass [91, 92].
The Role of microRNAs in Liver Regeneration
MicroRNAs (miRNA) are evolutionarily conserved, short non-coding RNAs, which play an integral role in virtually all biological pathways. MiRNAs are transcribed as primary transcripts (pri-miRNA) by RNA polymerase II. They then undergo cleavage by an RNAse III enzyme to release pre-miRNA hairpins that are exported to the cytoplasm where the nascent miRNA undergoes further processing by protein complexes (Dicer, RNAse III enzymes, TRBP) to produce mature miRNAs. These mature non-coding sequences can then bind to complementary sites on target messenger RNA transcripts to induce either translational pause or transcriptional degradation for regulation of these genes [93]. In the past decade, extensive studies have shown critical roles of miRNA in almost all aspects of liver development, including hepatic and biliary specification and differentiation, hepatocyte and HSC development, metabolic functions, liver zonation, as well as liver regeneration [94]. Most recently, it has been reported that specific regeneration-associated miRNAs, are predictive of outcome and patient selection for liver transplantation in both acute and chronic liver disease [95].
MicroRNA-122
MicroRNA-122 (miR-122) is the most abundantly found in liver tissue constituting 70% of the total miRNA pool in the liver; and its concentration is almost undetectable in other tissues. Its role has been described as one of the key factors in normal liver functions as well as pathogenesis of liver diseases [96]. It has been associated with improved prognosis clinically in patients suffering from acute liver failure, which has also been demonstrated in the mouse model [97]. In acetaminophen-induced murine liver injury, there was a dose- and duration-dependent increase in circulating miR-122 levels [98]. It has been shown to promote levels of FoxA1 genes (responsible for liver specific transcripts such as albumin and transthyretin) and HNF4a (Hepatic nuclear factor 4 alpha, responsible for the development of various organs including liver). This miRNA also has been known to alter the balance of the mesenchymal-to-epithelial transition (MET) and vice-versa; suggesting links to carcinogenesis [99].
MicroRNA-21
Another miRNA, miR-21, has been well studied in its function in cell proliferation after cellular injury. miRNA upregulates liver regeneration acting via multiple pathways, including those associated with PTEN (Phosphatase and tensin homolog), a well-documented tumor-suppressor gene that inhibits cell growth and tumor development [100]. This gene is downregulated by increased levels of miR-21 after PHx and the downregulation/loss of PTEN leads to increased activity of AKT and mTOR kinase signaling, cell cycle progression and cellular proliferation [101,102,103]. In vivo studies showing correlation between miR-21 and PTEN requires further investigation. Another pathway which is activated by increased miR-21 pathway is Pellino-1, a mediator of IL-1R/TLR signaling, and inhibition of NF-κB signaling pathway; and together, it is postulated that they form negative feedback loop to regulate NF-κB pathway [104]. Dysregulation of miRNA-21 has been implicated in the pathogenesis of multiple chronic liver diseases including hepatocellular carcinoma, NAFLD, viral liver diseases, and liver fibrosis [105].
Antiapoptotic miRNA, miR-221, has been implicated in acceleration of hepatocyte proliferation which has been demonstrated in experiments with AAV-mediated overexpression of this miRNA in PHx in vivo mouse models [106]. The proposed mechanism is that the overexpression of miR-221 leads to rapid S-phase entry of hepatocytes by targeting p27, p57 and Arnt mRNA, contributing to rapid proliferation. miR-221 has also been shown to protect from Fas induced acute liver failure by p53 upregulated modulation of apoptosis [107].
The Role of Stem Cells
Stem cells, by definition, have the ability to self-renew and differentiate into multiple cell line lineages. During embryonic development, the liver is generated from primarily endodermal-derived cells called hepatoblasts, which then differentiate into either hepatocytes or cholangiocytes, the two types of epithelial cells in the liver. However, the role of stem cells in liver regeneration after hepatectomy or injury is still debated. In PHx models, the remnant liver cells are not widely injured, and regeneration occurs primarily by hypertrophy and proliferation of mature hepatocytes. In rat model bile duct ligation studies, labeled hepatocytes were injected into their livers prior to bile duct ligation, and these rats were treated with diaminodiphenylmethane (DAMP), a biliary toxin, or sham. In both experiments, regenerated cholangiocytes were labeled, indicating a trans-differentiation from hepatocytes, with higher contribution in DAPM treatment group [108]. The trans-differentiation was driven primarily via the NOTCH pathway, and experiments with Cre-induced transgenic models led the induced hepatocytes to express biliary epithelial cell markers. Blockage of this cascade significantly impaired the trans-differentiation as well as repressed YAP levels, suggesting cross talk between the NOTCH pathway and Hippo/YAP [109].
The PHx model, however, does not completely replicate the pathology of most liver diseases, which often are associated with hepatocyte damage/death from inflammatory and fibrogenic responses. In acute liver diseases as well as acute-on-chronic liver failure due to various toxin-induced (e.g. alcohol or drug related), metabolic (fatty liver diseases) and infectious (viral hepatitis), regeneration often requires the activation of a unique cell population called liver progenitor cells (LPC) [110]. While their site of origin is still unclear, but most studies have focused on canal of Hering as the potential source. In literature, they have been given various names, including “ductular hepatocytes”, “atypical ductal cells”, “intermediate hepatobiliary cells” or “hepatic/liver progenitor cells”. The term “oval cells” is primarily used in rat models, which are only present in damaged liver [111]. The most established protocol used to induce oval cells is 2-acetylaminofluorene (2-AAF)/PHx systems, where hepatocyte proliferation is blocked by 2-AAF prior to PHx. Using this method, it was shown that oval cells have the biopotential to differentiate into both hepatocytes and cholangiocytes [112]. 2-AAF/PHx system does not work in mice, so other methods have been used (such as 3,5-diethyoxycarbonyl-1,4-dihidro-collidine (DDC)-containing diet or Choline-deficient ethionine-supplemented diet (CDE)), to induce hepatic injury [113, 114]. These methods serve as models for varying type of liver injury and their potential therapeutic targets. For example, DDC-induced liver injury acts as model for biliary fibrosis, and CDE induces fatty liver, which is used as model for NASH. The resultant oval cells from these various models are not truly the same, and therefore, the use of the term “oval cells” is becoming less common and “LPC” is broadly used.
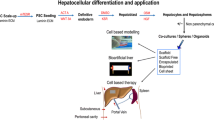
Liver transplantation is the only realistic option when regeneration does not compensate for the loss of metabolic function. While the yearly trend has been in the positive direction for the number of transplants throughout the United States, a significant number of patients die every year while on the liver transplant waitlist. For this reason, regeneration medicine, especially with the use of stem cells has been widely investigated worldwide. In the last decade, several hepatic differentiation protocols for mesenchymal stem cells (MSCs) have been described (Fig. 4.2) [115]. In vitro, co-culture of MSCs with primary liver cells induces differentiation of MSCs into hepatocyte-like cells (HLCs) [116]. In CCl4-induced murine models of liver failure, transplantation of MSC-derived hepatocytes have been shown to restore liver function, and a similar finding has been reported in drug-induced ALF [117, 118]. There are multiple ongoing trials for use of mesenchymal stem cell transfusions in patients with liver diseases. Most recently, it has been reported that mesenchymal stromal cells promote liver regeneration by inhibiting the activation of innate immune cells and activating those of the adaptive immune system including T (Tregs) and B (Bregs) regulatory cells [119].

Extracellular vesicles as paracrine mediators in liver disease and therapeutic potential of mesenchymal stem/stromal cells. After ischemia reperfusion injury (I/R) or hepatectomy, hepatocytes (1) HPCs (2) release EVs with the ability to induce hepatocyte proliferation. (3) HPC-derived EVs stimulate LSEC and macrophage production of proliferative cytokines such as IL25 and IL17B. (4) On the other hand, free fatty acids induce the production of hepatocyte-derived EVs that result in the activation of quiescent HeSCs and pro-inflammatory macrophages (M1). (5) During chronic hepatitis C virus infection, EVs secreted by HCV-infected hepatocytes induce activation of HeSCs. (6) EVs secreted by hepatocytes after alcohol injury (containing CD40L and miRNAs) induce activation of monocytes and HeSCs. It seems to be a balance between EVs derived from active or quiescent HeSCs that promotes or inhibits fibrogenesis. Activated HeSC-derived EVs induce activation of quiescent HeSCs through CCN2 (7), and quiescent HeSCs inhibit activated HeSCs transferring Twist1 or miRNA199a-5p (8). LSEC-derived EVs could also regulate HeSC activation (9). MSC-EVs induce hepatocyte proliferation, reduce oxidative stress and apoptosis, and modulate inflammatory response by carrying GPX1 or SK2 (10). Engineered MSC-EVs transfer miRNA-122, miRNA 181-5p and miRNA-223 as potentially key modulators. The effects of MSC-EVs on HeSCs, hepatic macrophages, LSEC and infiltrated cells populations remain poorly explored. Green arrows: Inactivation of HeSCs; Red arrows: Activation of HeSCs; Blue arrow: Proliferative effect; Color spots represent EVs from different cell origin; NCDase Neutral ceramidase, SK2 Sphingosine kinase 2, S1P Sphingosine-1-phosphate, IL Interleukin, SK1 Sphingosine kinase 1, CCN2 Connective tissue growth factor, Twist1: Basic helix-loop-helix transcription factor; GPX1 Glutathione peroxidase 1, HCV Hepatitis C virus, EVs Extracellular vesicles. (Figure and Caption Source: Fiore EJ, Domínguez LM, Bayo J, García MG, Mazzolini GD. Taking advantage of the potential of mesenchymal stromal cells in liver regeneration: Cells and extracellular vesicles as therapeutic strategies. World J Gastroenterol 2018; 24(23): 2427–2440) [120]
Acute-on-Chronic Liver Failure
Previous discussions in this chapter have focused on regeneration of a previously healthy liver after an acute insult. Once cirrhosis is present the natural progression to decompensated disease is a direct consequence of impaired liver function which ensues from a decrease in functional hepatocyte mass and disruption of hepatic architecture. This results clinically in an increased risk of bleeding, susceptibility to infection, and multisystem organ dysfunction—all of which are associated with a higher incidence of short-term mortality [121]. Rather than the natural progression of those with cirrhosis to develop decompensated disease, acute-on-chronic liver failure (ACLF) is an acute insult in patients with cirrhosis, which leads to rapid clinical deterioration in those individuals with previously compensated cirrhosis [122]. Typical clinical events that can precipitate ACLF include infections, gastrointestinal bleeding, viral hepatitis, drug toxicity or ischemic injury. It is noteworthy that persons with ACLF demonstrate upwards of 5-times the risk of mortality at both 28 days and 90 days [123, 124]. While infection can trigger ACLF, it is well recognized that the innate immune system can initiate an inflammatory response in the absence of infection, termed sterile inflammation. The process occurs via the release of host-derived products, called damage-associated molecular patterns (DAMPs) [125]. These DAMPs, which include interleukins, mitochondrial DNA, and bile acids, interact with immune cells and initiate an inflammatory signal through chemokine and cytokine release, which sustains and amplifies the inflammatory response [126, 127]. Therefore, in ACLF with reduced hepatic reserve and chronic circulatory dysfunction, hepatocyte death causes release of DAMPs and incites inflammation with resultant further liver failure.
In addition to diminished functional capacity, the ACLF population also demonstrates a significantly altered immune milieu, with significant alterations in pro- and anti-inflammatory cytokines such as TNF-α, interleukins, and interferons [128]. Levels of inflammatory markers such as IL-6 are lower in ACLF than in those patients with sepsis. However, the induction of TNF-α production and HLA-DR expression is significantly diminished with resultant dysfunction of regulatory monocytes and macrophages [129]. Kupffer cell populations are depleted in the setting of both ALF and ACLF, and it is hypothesized that the loss of these phagocytes leads to increased levels of circulating microbial antigens and exposure to DAMPs [126].
This alteration of immune function and cytokine milieu in ACLF therefore has a significant effect on hepatic regenerative capacity and serves as the basis behind emerging therapies to enhance recovery and regeneration. Macrophages, as a key driving force of injury in ACLF, as well as upstream cytokines, which stimulate macrophage activity are attractive targets for potential therapies [129]. Although clinical trials involving molecular targeting in ACLF are limited, studies using endogenous stem cells to enhance tissue repair and therapies targeting inflammatory pathways and programmed cell death pathways have shown promise. As mentioned previously, g-CSF therapy mobilizes bone marrow derived stem cells in an effort to enhance hepatic tissue repair [45, 130].
Conclusion
We are hopeful that this review gives the reader a solid introduction and overview to the science and multi-faceted complex nature of liver regeneration. From growth factors, immune modulation, and metabolic changes to microRNA and stem cells, the breadth of influences on hepatic repair in part explains why this continues to be a nascent field of study. Given the significant heterogeneity in both the etiology of liver injury and associated repair mechanisms, the study of hepatocyte regeneration in ALF and ACLF will no doubt continue to evolve. Based on studies to date, it would be realistic to imagine therapeutic interventions after acute liver injury that could include infusion of NF-κB to stimulate Kupffer cells, macrophage colony stimulating factor to promote macrophage infiltrations into injured tissue, or heparin-binding epidermal growth factor to stimulate hepatic DNA replication. Most recently, it was demonstrated that administering a transfusion of readily-available platelets or fibrinogen can independently promote hepatic regeneration [72]. Mechanistically, we are capable of in vivo manipulation of miR-122 or miR-21 to stimulate or inhibit hepatocyte regeneration, depending on the unique clinical scenario. It is highly probably that near future therapeutic approaches to regenerate liver would include delivery of potent and durable hepatic mesenchymal stem cells into patients with ALF or ACLF as a means of promoting hepatocyte (and other liver cell) regeneration, preservation and a return to normal liver function.
We have attempted to highlight known pathways of cellular repair. Most notably, however, we recognize that elucidation of the interplay of these elements with host and microbiome factors is necessary for a more complete understanding of the mechanisms involved in hepatic regeneration.
Questions
-
1.
All of the following mediators promote liver regeneration, except:
-
(a)
Wnt/β-catenin pathway
-
(b)
Transforming Growth Factor β pathway
-
(c)
IL-6/STAT3 pathway
-
(d)
TNF-α/TNFR pathway
-
(e)
HGF
-
(a)
-
2.
Which of the following pathways drives the architectural development?
-
(a)
Wnt/β-catenin pathway
-
(b)
Transforming Growth Factor β pathway
-
(c)
IL-6/STAT3 pathway
-
(d)
TNF-α/TNFR pathway
-
(e)
HGF
-
(a)
-
3.
What is the key difference in IL-6/STAT3 pathway in liver regeneration during hepatectomy compared to acute-on-chronic liver failure?
-
(a)
There is no difference in these two liver regeneration models
-
(b)
IL-6//STAT3 is upregulated in ACLF and downregulated in PHx
-
(c)
IL-6 levels are upregulated in PHx models, however, due to dysfunction of Kupffer cells, IL-6 production is not robust in ACLF
-
(d)
IFN-γ/STAT1 pathway acts in synergistic fashion with IL-6/STAT3 in both models of liver injury to promote liver regeneration
-
(a)
-
4.
Which of the following is true regarding microRNA-122 (miR-122)?
-
(a)
This microRNA is almost never seen in hepatocytes
-
(b)
In mouse models, it’s presence has been shown to have poor outcomes in liver failure
-
(c)
This microRNA suppresses the level of FoxA1 gene to reduce the liver specific transcripts such as albumin
-
(d)
This microRNA promotes HNF4a gene, which is responsible for the development of various organs including liver
-
(a)
-
5.
In acute-on-chronic liver failure, which of the following mechanisms drive further liver failure?
-
(a)
Patients with ACLF have reduced hepatic reserve and chronic circulatory dysfunction
-
(b)
Hepatocyte death causes release of DAMPs (damage-associated molecular patterns) and incites inflammation with resultant further liver failure
-
(c)
ACLF population have a significantly altered immune milieu, with significant alterations in pro- and anti-inflammatory cytokines which can hinder liver regeneration
-
(d)
Kupffer cell populations are depleted in the setting ACLF, and loss of these phagocytes leads to increased levels of circulating microbial antigens and exposure to DAMPs
-
(e)
All of the above are true
-
(a)
Answers
Question 1: Answer: b. Transforming Growth Factor β pathway
Explanation:
Transforming growth factor beta (TGF-β) is a key factor in termination of liver regeneration. TGF-β is a strong inhibitor of DNA synthesis in mitogen-stimulated hepatocytes. TGF-β mRNA expression increases after partial hepatectomy (PHx) and peaks after the first round of hepatocyte cellular division has occurred. This increase in levels of TGF-β is countered by a reduction in TGF-β receptor expression after the liver injury. In TGF-β-receptor knockout models, there is an increase in hepatocyte proliferation with corresponding increase in liver mass. This inverse relationship is likely mediated by inhibition of cyclin D1 and arrest in the G1 phase of the cell cycle.
The other factors (a, c, d, e) all promote hepatocyte regeneration via various mechanisms.
Question 2: Answer: a. Wnt/β-catenin pathway
Explanation:
This pathway promotes architectural development during regeneration in PHx models by increasing levels of β-catenin and E-cadherin, for coordination of cell-cell adhesion. This pathway is also involved in the “metabolic zonation” of the liver during organogenesis and regeneration.
Question 3: Answer: c. IL-6 levels are upregulated in PHx models…
Explanation:
Due to the dysfunction of Kupffer cells, IL-6 production is not robust in ACLF. This, in part, underscores the importance of non-parenchymal cells in liver regeneration from whatever cause…be it surgical removal of a portion of the liver, or toxin-induced injury.
Question 4: Answer: d. This microRNA promotes HNF4a gene, which is responsible for the development of various organs including liver.
Explanation:
The other answers are incorrect because miR-122 is almost exclusively seen in hepatocytes, and its presence has been shown to be associated with improved prognosis in liver failure. Also, miR-122 promotes the level of FoxA1 gene expression, which increases liver specific transcripts.
Question 5: Answer: e. All of the above are true.
Explanation:
These are all salient points about patients with ACLF which puts them at a higher risk of further liver injury.
References
Higgins G. Experimental pathology of the liver: restoration of the liver in the white rate following partial surgical removal. Arch Pathol. 1931;12:186–202.
Rabinovici N, Wiener E. Liver regeneration after partial hepatectomy in carbon tetrachloride-induced cirrhosis in the rat. Gastroenterology. 1961;40(3):416–22. https://doi.org/10.1016/S0016-5085(61)80075-9.
Panis Y, McMullan DM, Emond JC. Progressive necrosis after hepatectomy and the pathophysiology of liver failure after massive resection. Surgery. 1997;121(2):142–9.
Moolten FL, Bucher NL. Regeneration of rat liver: transfer of humoral agent by cross circulation. Science. 1967;158(3798):272–4.
Rabes HM. Kinetics of hepatocellular proliferation as a function of the microvascular structure and functional state of the liver. Ciba Foundation Symposium 55 - hepatotrophic factors. 1978.
Weglarz TC, Sandgren EP. Timing of hepatocyte entry into DNA synthesis after partial hepatectomy is cell autonomous. Proc Natl Acad Sci U S A. 2000;97(23):12595–600. https://doi.org/10.1073/pnas.220430497.
Fausto N, Laird AD, Webber EM. Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. FASEB J. 1995;9(15):1527–36.
Sadri A-R, Jeschke MG, Amini-Nik S. Cellular and molecular cascades during liver regeneration. Surg Res Open J. 2015;2(2):53–61. https://doi.org/10.17140/SROJ-2-110.
Cressman DE, Diamond RH, Taub R. Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology. 1995;21(5):1443–9.
Cressman DE, Greenbaum LE, Haber BA, Taub R. Rapid activation of post-hepatectomy factor/nuclear factor κB in hepatocytes, a primary response in the regenerating liver. J Biol Chem. 1994;269(48):30429–35.
Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech. 2000;50(3):184–95. https://doi.org/10.1002/1097-0029(20000801)50:3<184::Aid-jemt2>3.0.Co;2-h.
Kang LI, Mars WM, Michalopoulos GK. Signals and cells involved in regulating liver regeneration. Cell. 2012;1(4):1261–92. https://doi.org/10.3390/cells1041261.
Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94(4):1441–6.
Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, Diehl AM. Antibodies to tumor necrosis factor-α inhibit liver regeneration after partial hepatectomy. Am J Phys. 1992;263(4 Pt 1):G579–85. https://doi.org/10.1152/ajpgi.1992.263.4.G579.
Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–7. https://doi.org/10.1038/ni0302-221.
Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376(6536):167–70. https://doi.org/10.1038/376167a0.
Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43(2 Suppl 1):S45–553. https://doi.org/10.1002/hep.20969.
Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–90. https://doi.org/10.1016/j.cell.2005.04.014.
Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64(6):1403–15. https://doi.org/10.1016/j.jhep.2016.02.004.
Streetz KL, Luedde T, Manns MP, Trautwein C. Interleukin 6 and liver regeneration. Gut. 2000;47(2):309–12.
Mackiewicz A, Schooltink H, Heinrich PC, Rose-John S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J Immunol. 1992;149(6):2021–7.
Aldeguer X, Debonera F, Shaked A, Krasinkas AM, Gelman AE, Que X, et al. Interleukin-6 from intrahepatic cells of bone marrow origin is required for normal murine liver regeneration. Hepatology. 2002;35(1):40–8. https://doi.org/10.1053/jhep.2002.30081.
Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274(5291):1379–83.
Sun R, Gao B. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-γ). Gastroenterology. 2004;127(5):1525–39. https://doi.org/10.1053/j.gastro.2004.08.055.
Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, et al. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110(10):1503–13. https://doi.org/10.1172/JCI15841.
Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E, et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J Hepatol. 2020;72:736–45. https://doi.org/10.1016/j.jhep.2019.11.013.
Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. 2014;6(237):237–66. https://doi.org/10.1126/scitranslmed.3008618.
Bouwens L, Baekeland M, De Zanger R, Wisse E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology. 1986;6(4):718–22.
Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. 2012;56(2):735–46. https://doi.org/10.1002/hep.25657.
Abshagen K, Eipel C, Kalff JC, Menger MD, Vollmar B. Loss of NF-κB activation in Kupffer cell-depleted mice impairs liver regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1570–7. https://doi.org/10.1152/ajpgi.00399.2006.
Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, Pohl LR. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002;15(12):1504–13.
You Q, Holt M, Yin H, Li G, Hu CJ, Ju C. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem Pharmacol. 2013;86(6):836–43. https://doi.org/10.1016/j.bcp.2013.07.006.
Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59(5):2034–42. https://doi.org/10.1002/hep.26754.
Cornell RP, Liljequist BL, Bartizal KF. Depressed liver regeneration after partial hepatectomy of germ-free, athymic and lipopolysaccharide-resistant mice. Hepatology. 1990;11(6):916–22.
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–8.
Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, et al. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology. 2005;41(3):443–50. https://doi.org/10.1002/hep.20603.
Markiewski MM, DeAngelis RA, Strey CW, Foukas PG, Gerard C, Gerard N, et al. The regulation of liver cell survival by complement. J Immunol. 2009;182(9):5412–8. https://doi.org/10.4049/jimmunol.0804179.
Matsumoto K, Miyake Y, Umeda Y, Matsushita H, Matsuda H, Takaki A, et al. Serial changes of serum growth factor levels and liver regeneration after partial hepatectomy in healthy humans. Int J Mol Sci. 2013;14(10):20877–89. https://doi.org/10.3390/ijms141020877.
Sauter KA, Waddell LA, Lisowski ZM, Young R, Lefevre L, Davis GM, et al. Macrophage colony-stimulating factor (CSF1) controls monocyte production and maturation and the steady-state size of the liver in pigs. Am J Physiol Gastrointest Liver Physiol. 2016;311(3):G533–47. https://doi.org/10.1152/ajpgi.00116.2016.
Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology. 2015;149(7):1896–909. https://doi.org/10.1053/j.gastro.2015.08.053.
Possamai LA, Thursz MR, Wendon JA, Antoniades CG. Modulation of monocyte/macrophage function: a therapeutic strategy in the treatment of acute liver failure. J Hepatol. 2014;61(2):439–45. https://doi.org/10.1016/j.jhep.2014.03.031.
Aldrighetti L, Pulitano C, Arru M, Finazzi R, Catena M, Soldini L, et al. Impact of preoperative steroids administration on ischemia-reperfusion injury and systemic responses in liver surgery: a prospective randomized study. Liver Transpl. 2006;12(6):941–9. https://doi.org/10.1002/lt.20745.
Eagon PK, Porter LE, Francavilla A, DiLeo A, Van Thiel DH. Estrogen and androgen receptors in liver: their role in liver disease and regeneration. Semin Liver Dis. 1985;5(1):59–69. https://doi.org/10.1055/s-2008-1041758.
Francavilla A, Polimeno L, DiLeo A, Barone M, Ove P, Coetzee M, et al. The effect of estrogen and tamoxifen on hepatocyte proliferation in vivo and in vitro. Hepatology. 1989;9(4):614–20.
Garg V, Garg H, Khan A, Trehanpati N, Kumar A, Sharma BC, et al. Granulocyte colony-stimulating factor mobilizes CD34+ cells and improves survival of patients with acute-on-chronic liver failure. Gastroenterology. 2012;142(3):505–12. https://doi.org/10.1053/j.gastro.2011.11.027.
Saha BK, Mahtab MA, Akbar SMF, Noor EASM, Mamun AA, Hossain SMS, et al. Therapeutic implications of granulocyte colony stimulating factor in patients with acute-on-chronic liver failure: increased survival and containment of liver damage. Hepatol Int. 2017;11(6):540–6. https://doi.org/10.1007/s12072-017-9814-1.
Yamashita Y, Shimada M, Hamatsu T, Rikimaru T, Tanaka S, Shirabe K, Sugimachi K. Effects of preoperative steroid administration on surgical stress in hepatic resection: prospective randomized trial. Arch Surg. 2001;136(3):328–33.
Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213(2):286–300. https://doi.org/10.1002/jcp.21172.
Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A. 2007;104(43):17081–6. https://doi.org/10.1073/pnas.0704126104.
Mitchell C, Nivison M, Jackson LF, Fox R, Lee DC, Campbell JS, Fausto N. Heparin-binding epidermal growth factor-like growth factor links hepatocyte priming with cell cycle progression during liver regeneration. J Biol Chem. 2005;280(4):2562–8. https://doi.org/10.1074/jbc.M412372200.
Berasain C, Garcia-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J, Avila MA. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology. 2005;128(2):424–32.
Russell WE, Kaufmann WK, Sitaric S, Luetteke NC, Lee DC. Liver regeneration and hepatocarcinogenesis in transforming growth factor-α-targeted mice. Mol Carcinog. 1996;15(3):183–9. https://doi.org/10.1002/(sici)1098-2744(199603)15:3<183::Aid-mc4>3.0.Co;2-j.
Noguchi S, Ohba Y, Oka T. Influence of epidermal growth factor on liver regeneration after partial hepatectomy in mice. J Endocrinol. 1991;128(3):425–31.
Kinoshita T, Hirao S, Matsumoto K, Nakamura T. Possible endocrine control by hepatocyte growth factor of liver regeneration after partial hepatectomy. Biochem Biophys Res Commun. 1991;177(1):330–5.
Burr AW, Toole K, Chapman C, Hines JE, Burt AD. Anti-hepatocyte growth factor antibody inhibits hepatocyte proliferation during liver regeneration. J Pathol. 1998;185(3):298–302. https://doi.org/10.1002/(sici)1096-9896(199807)185:3<298::Aid-path88>3.0.Co;2-b.
Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699–702. https://doi.org/10.1038/373699a0.
Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A. 2004;101(29):10608–13. https://doi.org/10.1073/pnas.0403412101.
Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101(13):4477–82. https://doi.org/10.1073/pnas.0306068101.
Paranjpe S, Bowen WC, Bell AW, Nejak-Bowen K, Luo JH, Michalopoulos GK. Cell cycle effects resulting from inhibition of hepatocyte growth factor and its receptor c-Met in regenerating rat livers by RNA interference. Hepatology. 2007;45(6):1471–7. https://doi.org/10.1002/hep.21570.
Shubham S, Kumar D, Rooge S, Maras JS, Maheshwari D, Nautiyal N, et al. Cellular and functional loss of liver endothelial cells correlates with poor hepatocyte regeneration in acute-on-chronic liver failure. Hepatol Int. 2019;13:777–87. https://doi.org/10.1007/s12072-019-09983-y.
Bhushan B, Apte U. Liver regeneration after acetaminophen hepatotoxicity: mechanisms and therapeutic opportunities. Am J Pathol. 2019;189(4):719–29. https://doi.org/10.1016/j.ajpath.2018.12.006.
Naugler WE. Bile acid flux is necessary for normal liver regeneration. PLoS One. 2014;9(5):e97426. https://doi.org/10.1371/journal.pone.0097426.
Suzuki H, Iyomasa S, Nimura Y, Yoshida S. Internal biliary drainage, unlike external drainage, does not suppress the regeneration of cholestatic rat liver after partial hepatectomy. Hepatology. 1994;20(5):1318–22.
Otao R, Beppu T, Isiko T, Mima K, Okabe H, Hayashi H, et al. External biliary drainage and liver regeneration after major hepatectomy. Br J Surg. 2012;99(11):1569–74. https://doi.org/10.1002/bjs.8906.
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–44.
Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312(5771):233–6. https://doi.org/10.1126/science.1121435.
Zhang L, Wang YD, Chen WD, Wang X, Lou G, Liu N, et al. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology. 2012;56(6):2336–43. https://doi.org/10.1002/hep.25905.
Wang XD, Soltesz V, Andersson R, Bengmark S. Bacterial translocation in acute liver failure induced by 90% hepatectomy in the rat. Br J Surg. 1993;80(1):66–71.
Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58(5):949–55. https://doi.org/10.1016/j.jhep.2013.01.003.
Murata S, Ohkohchi N, Matsuo R, Ikeda O, Myronovych A, Hoshi R. Platelets promote liver regeneration in early period after hepatectomy in mice. World J Surg. 2007;31(4):808–16. https://doi.org/10.1007/s00268-006-0772-3.
Beier JI, Guo L, Ritzenthaler JD, Joshi-Barve S, Roman J, Arteel GE. Fibrin-mediated integrin signaling plays a critical role in hepatic regeneration after partial hepatectomy in mice. Ann Hepatol. 2016;15(5):762–72. https://doi.org/10.5604/16652681.1212587.
Groeneveld D, Pereyra D, Veldhuis Z, Adelmeijer J, Ottens P, Kopec AK, et al. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood. 2019;133(11):1245–56. https://doi.org/10.1182/blood-2018-08-869057.
Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol. 2012;4(11):a007906. https://doi.org/10.1101/cshperspect.a007906.
Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, et al. Hepatocyte growth factor induces Wnt-independent nuclear translocation of β-catenin after Met-β-catenin dissociation in hepatocytes. Cancer Res. 2002;62(7):2064–71.
Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of β-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131(5):1561–72. https://doi.org/10.1053/j.gastro.2006.08.042.
Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, Monga SP. β-Catenin activation promotes liver regeneration after acetaminophen-induced injury. Am J Pathol. 2009;175(3):1056–65. https://doi.org/10.2353/ajpath.2009.080976.
Gougelet A, Torre C, Veber P, Sartor C, Bachelot L, Denechaud PD, et al. T-cell factor 4 and β-catenin chromatin occupancies pattern zonal liver metabolism in mice. Hepatology. 2014;59(6):2344–57. https://doi.org/10.1002/hep.26924.
Leibing T, Geraud C, Augustin I, Boutros M, Augustin HG, Okun JG, et al. Angiocrine Wnt signaling controls liver growth and metabolic maturation in mice. Hepatology. 2018;68(2):707–22. https://doi.org/10.1002/hep.29613.
Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Changes in WNT/β-catenin pathway during regulated growth in rat liver regeneration. Hepatology. 2001;33(5):1098–109. https://doi.org/10.1053/jhep.2001.23786.
Nelson WJ, Nusse R. Convergence of Wnt, β-catenin, and cadherin pathways. Science. 2004;303(5663):1483–7. https://doi.org/10.1126/science.1094291.
Nakamura T, Tomita Y, Hirai R, Yamaoka K, Kaji K, Ichihara A. Inhibitory effect of transforming growth factor-β on DNA synthesis of adult rat hepatocytes in primary culture. Biochem Biophys Res Commun. 1985;133(3):1042–50.
Chari RS, Price DT, Sue SR, Meyers WC, Jirtle RL. Down-regulation of transforming growth factor beta receptor type I, II, and III during liver regeneration. Am J Surg. 1995;169(1):126–31.
Ko TC, Yu W, Sakai T, Sheng H, Shao J, Beauchamp RD, Thompson EA. TGF-β1 effects on proliferation of rat intestinal epithelial cells are due to inhibition of cyclin D1 expression. Oncogene. 1998;16(26):3445–54. https://doi.org/10.1038/sj.onc.1201902.
Thenappan A, Shukla V, Abdul Khalek FJ, Li Y, Shetty K, Liu P, et al. Loss of transforming growth factor β adaptor protein β-2 spectrin leads to delayed liver regeneration in mice. Hepatology. 2011;53(5):1641–50. https://doi.org/10.1002/hep.24111.
Patel SH, Camargo FD, Yimlamai D. Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology. 2017;152(3):533–45. https://doi.org/10.1053/j.gastro.2016.10.047.
Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17(23):2054–60. https://doi.org/10.1016/j.cub.2007.10.039.
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61. https://doi.org/10.1101/gad.1602907.
Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157(6):1324–38. https://doi.org/10.1016/j.cell.2014.03.060.
Michalopoulos GK. Hepatostat: liver regeneration and normal liver tissue maintenance. Hepatology. 2017;65(4):1384–92. https://doi.org/10.1002/hep.28988.
Grisham JW. A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer Res. 1962;22:842–9.
Abu Rmilah A, Zhou W, Nelson E, Lin L, Amiot B, Nyberg SL. Understanding the marvels behind liver regeneration. Wiley Interdiscip Rev Dev Biol. 2019;8(3):e340. https://doi.org/10.1002/wdev.340.
Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22(13):1166–75. https://doi.org/10.1016/j.cub.2012.05.016.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–24. https://doi.org/10.1038/nrm3838.
Chen Y, Verfaillie CM. MicroRNAs: the fine modulators of liver development and function. Liver Int. 2014;34(7):976–90. https://doi.org/10.1111/liv.12496.
Salehi S, Tavabie OD, Verma S, McPhail MJW, Farzaneh F, Bernal W, et al. Serum miRNA signatures in recovery from acute and chronic liver injury and selection for liver transplantation. Liver Transpl. 2020;26:811–22. https://doi.org/10.1002/lt.25781.
Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122–a key factor and therapeutic target in liver disease. J Hepatol. 2015;62(2):448–57. https://doi.org/10.1016/j.jhep.2014.10.004.
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12(9):735–9.
Wang K, Zhang S, Marzolf B, Troisch P, Brightman A, Hu Z, et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci U S A. 2009;106(11):4402–7. https://doi.org/10.1073/pnas.0813371106.
Deng XG, Qiu RL, Wu YH, Li ZX, Xie P, Zhang J, et al. Overexpression of miR-122 promotes the hepatic differentiation and maturation of mouse ESCs through a miR-122/FoxA1/HNF4a-positive feedback loop. Liver Int. 2014;34(2):281–95. https://doi.org/10.1111/liv.12239.
Gil A, Rodriguez-Escudero I, Stumpf M, Molina M, Cid VJ, Pulido R. A functional dissection of PTEN N-terminus: implications in PTEN subcellular targeting and tumor suppressor activity. PLoS One. 2015;10(4):e0119287. https://doi.org/10.1371/journal.pone.0119287.
Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2(3):135–64. https://doi.org/10.18632/oncotarget.240.
Chen X, Song M, Chen W, Dimitrova-Shumkovska J, Zhao Y, Cao Y, et al. MicroRNA-21 contributes to liver regeneration by targeting PTEN. Med Sci Monit. 2016;22:83–91. https://doi.org/10.12659/msm.896157.
Yan-nan B, Zhao-yan Y, Li-xi L, Jiang Y, Qing-jie X, Yong Z. MicroRNA-21 accelerates hepatocyte proliferation in vitro via PI3K/Akt signaling by targeting PTEN. Biochem Biophys Res Commun. 2014;443(3):802–7. https://doi.org/10.1016/j.bbrc.2013.12.047.
Marquez RT, Wendlandt E, Galle CS, Keck K, McCaffrey AP. MicroRNA-21 is upregulated during the proliferative phase of liver regeneration, targets Pellino-1, and inhibits NF-κB signaling. Am J Physiol Gastrointest Liver Physiol. 2010;298(4):G535–41. https://doi.org/10.1152/ajpgi.00338.2009.
Zhang T, Yang Z, Kusumanchi P, Han S, Liangpunsakul S. Critical role of microRNA-21 in the pathogenesis of liver diseases. Front Med. 2020;7:7. https://doi.org/10.3389/fmed.2020.00007.
Yuan Q, Loya K, Rani B, Mobus S, Balakrishnan A, Lamle J, et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57(1):299–310. https://doi.org/10.1002/hep.25984.
Sharma AD, Narain N, Handel EM, Iken M, Singhal N, Cathomen T, et al. MicroRNA-221 regulates FAS-induced fulminant liver failure. Hepatology. 2011;53(5):1651–61. https://doi.org/10.1002/hep.24243.
Michalopoulos GK, Barua L, Bowen WC. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology. 2005;41(3):535–44. https://doi.org/10.1002/hep.20600.
Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27(7):719–24. https://doi.org/10.1101/gad.207803.112.
Van Haele M, Snoeck J, Roskams T. Human liver regeneration: an etiology dependent process. Int J Mol Sci. 2019;20(9):2332. https://doi.org/10.3390/ijms20092332.
Dolle L, Best J, Mei J, Al Battah F, Reynaert H, van Grunsven LA, Geerts A. The quest for liver progenitor cells: a practical point of view. J Hepatol. 2010;52(1):117–29. https://doi.org/10.1016/j.jhep.2009.10.009.
Evarts RP, Nagy P, Nakatsukasa H, Marsden E, Thorgeirsson SS. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 1989;49(6):1541–7.
Akhurst B, Croager EJ, Farley-Roche CA, Ong JK, Dumble ML, Knight B, Yeoh GC. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology. 2001;34(3):519–22. https://doi.org/10.1053/jhep.2001.26751.
Preisegger KH, Factor VM, Fuchsbichler A, Stumptner C, Denk H, Thorgeirsson SS. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Investig. 1999;79(2):103–9.
Lee CW, Chen YF, Wu HH, Lee OK. Historical perspectives and advances in mesenchymal stem cell research for the treatment of liver diseases. Gastroenterology. 2018;154(1):46–56. https://doi.org/10.1053/j.gastro.2017.09.049.
Qihao Z, Xigu C, Guanghui C, Weiwei Z. Spheroid formation and differentiation into hepatocyte-like cells of rat mesenchymal stem cell induced by co-culture with liver cells. DNA Cell Biol. 2007;26(7):497–503. https://doi.org/10.1089/dna.2006.0562.
Kuo TK, Hung SP, Chuang CH, Chen CT, Shih YR, Fang SC, et al. Stem cell therapy for liver disease: parameters governing the success of using bone marrow mesenchymal stem cells. Gastroenterology. 2008;134(7):2111–21. https://doi.org/10.1053/j.gastro.2008.03.015.
Tan CY, Lai RC, Wong W, Dan YY, Lim SK, Ho HK. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res Ther. 2014;5(3):76. https://doi.org/10.1186/scrt465.
Hu C, Wu Z, Li L. Mesenchymal stromal cells promote liver regeneration through regulation of immune cells. Int J Biol Sci. 2020;16(5):893–903. https://doi.org/10.7150/ijbs.39725.
Fiore EJ, Dominguez LM, Bayo J, Garcia MG, Mazzolini GD. Taking advantage of the potential of mesenchymal stromal cells in liver regeneration: cells and extracellular vesicles as therapeutic strategies. World J Gastroenterol. 2018;24(23):2427–40. https://doi.org/10.3748/wjg.v24.i23.2427.
Olson JC, Kamath PS. Acute-on-chronic liver failure: concept, natural history, and prognosis. Curr Opin Crit Care. 2011;17(2):165–9. https://doi.org/10.1097/MCC.0b013e328344b42d.
Forbes SJ, Newsome PN. Liver regeneration - mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol. 2016;13(8):473–85. https://doi.org/10.1038/nrgastro.2016.97.
Fernandez J, Acevedo J, Wiest R, Gustot T, Amoros A, Deulofeu C, et al. Bacterial and fungal infections in acute-on-chronic liver failure: prevalence, characteristics and impact on prognosis. Gut. 2018;67(10):1870–80. https://doi.org/10.1136/gutjnl-2017-314240.
Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144(7):1426–37. https://doi.org/10.1053/j.gastro.2013.02.042.
Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13(2):88–110. https://doi.org/10.1038/nrgastro.2015.200.
Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The role of monocytes and macrophages in acute and acute-on-chronic liver failure. Front Immunol. 2018;9:2948. https://doi.org/10.3389/fimmu.2018.02948.
Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Transl Res. 2017;3(Suppl 1):170–88. https://doi.org/10.18053/jctres.03.2017S1.003.
Jalan R, Gines P, Olson JC, Mookerjee RP, Moreau R, Garcia-Tsao G, et al. Acute-on chronic liver failure. J Hepatol. 2012;57(6):1336–48. https://doi.org/10.1016/j.jhep.2012.06.026.
Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology. 2015;148(3):603–15. https://doi.org/10.1053/j.gastro.2014.11.045.
Jalan R. Novel approaches and therapeutics in acute-on-chronic liver failure. Liver Transpl. 2016;22(S1):14–9. https://doi.org/10.1002/lt.24621.
Disclosure Statement
The authors declare no competing nor commercial and/or financial conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Leventhal, T.M., KC, M., Steer, C.J. (2020). Liver Regeneration in Acute and Acute-on-Chronic Liver Failure. In: Pyrsopoulos, N. (eds) Liver Failure. Springer, Cham. https://doi.org/10.1007/978-3-030-50983-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-50983-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50982-8
Online ISBN: 978-3-030-50983-5
eBook Packages: MedicineMedicine (R0)