
Abstract
The therapeutic benefits of immune modulatory agents in autoimmunity revealed the pathologic relevance of B cells, plasma cells, and autoantibodies to these diseases. They have also rejuvenated interest in how B cells mediate multiple effector functions including antibody production, antigen presentation to T cells, costimulation, and the production of immune modulatory cytokines. Considerable experience in repurposing these drugs from autoimmunity and cancer immunotherapy, as well as new drug development in transplantation, has yielded important advances in the treatment of antibody-mediated rejection episodes and novel drug development aimed at desensitization to human leukocyte antigens (HLA). After years of slow progress, we now stand on an important threshold that promises many advances in the care of allosensitized patients. Critical to this are efforts to encourage basic scientists, clinical investigators, industry, National Institutes of Health (NIH), our academic societies, and the U.S. Food and Drug Administration (FDA) to continue support of these important objectives. Modification of alloimmunity and alloantibodies will also have relevance to xenotransplantation where the xenoantibodies present a formidable obstacle to advancement of this important therapy. Working together, we can advance transplant therapeutics where biologic agents are likely to play novel and important roles. Here we discuss current and emerging drugs in this area.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Mechanisms of Allosensitization
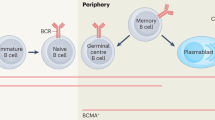
Sensitization to alloantigen (human leukocyte antigen [HLA] class I or class II), molecules derived from the allograft, occurs through a process of antigen presentation to naïve T cells that mature to T follicular helper (Tfh) cells in the regional lymph nodes or spleen (germinal centers) of allograft recipients. In the germinal center, Tfh cell activation is critical for generation of de novo alloantibody production and is driven primarily by the production of interleukin (IL)-6 and IL-21. Naïve B cells subsequently become activated by Tfh cells, and the persistence of IL-6 enhances development of memory B cells and plasmablasts.
IL-6 production in plasmablasts enhances germinal center formation and terminal development of donor-specific antibody (DSA)-producing plasma cells [1, 2]. Alloantibody migrates to the graft and initiates complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC), with resultant antibody-mediated rejection (AMR). DSA production usually results from inadequate immunosuppression or activation of established memory responses to alloantigens in sensitized individuals. Regardless, once established, alloimmmune responses persist for the life of the allograft and beyond, creating a highly HLA-sensitized individual. For a more comprehensive overview of the etiology of allosensitization and approaches to management, several recent papers are recommended [2,3,4].
Antibody-Mediated Rejection (AMR)
AMR is an increasingly recognized, severe form of allograft rejection, characterized by several pathologic variants resistant to treatment with standard immunosuppressive agents. Significant advances have occurred to identify patients at risk for AMR and the pathologic features associated with this diagnosis [2,3,4]. The immunopathology of AMR suggests an important role for antibodies, B cells, and plasma cells. Here, intravenous immune globulin (IVIg), rituximab, and/or plasmapheresis are commonly used for the treatment of acute AMR [5,6,7,8,9].
Despite successes, post-transplant AMR, chronic active AMR (cAMR), and transplant glomerulopathy remain significant problems that are only modestly amenable to these therapies. Data from the Deterioration in Kidney Allograft Function (DeKAF) study show that in the current era of immunosuppression, most graft losses have evidence of cAMR with C4d+ staining [10]. It is estimated that 5000 allografts are lost each year in the USA, primarily from cAMR [11]. The current treatment paradigms rely on reduction of antibody levels to prevent AMR.
AMR is often seen in patients who are noncompliant or receiving inadequate immunosuppressive therapy and those who receive HLA-incompatible transplants. Additionally, transplant glomerulopathy usually results from persistent DSA-positivity which dissipates allograft function, resulting in graft failure and return to dialysis with devastating emotional consequences for patients and their families, and financial consequences for the healthcare system [12,13,14,15,16]. No current therapy is approved by the US Food and Drug Administration (FDA), and patients are often treated with combination therapies making analysis of efficacy difficult. Thus, the scope of antibody-induced injury in the transplant population is significant and increasing [10]. Here, there is a large unmet clinical need.
One critical obstacle in the successful treatment of AMR is addressing the long-lived nature of plasma cells and persistent DSA, despite treatment strategies [17]. An “ideal” treatment option would eliminate circulating DSAs, inhibit CDC and ADCC, and rebound DSA generation.
Therapeutic Approaches to Treatment of AMR
The Anti-CD20 Monoclonal Antibodies
Rituximab
Rituximab is a chimeric monoclonal antibody (mAb) aimed at CD20, a cell surface antigen highly expressed on pre-B- and mature B-lymphocytes, but not differentiated plasma cells. In a single center, double-blind, placebo-controlled study by van den Hoogen et al., patients were randomized to receive induction therapy with a single dose of rituximab or placebo, with standard immunotherapy. The primary endpoint was the incidence of biopsy-proven rejection 6 months post-transplant [18]. Induction with rituximab did not result in significant reductions in allograft rejection at 6 months versus placebo. However, in high-risk patients (retransplants, DSA+), a significant reduction in allograft rejection was seen with rituximab.
Our group has published a blinded, placebo-controlled study of IVIg + rituximab versus IVIg + placebo for desensitization which demonstrated an essential role of rituximab in prevention of DSA rebound, AMR/cAMR, transplant glomerulopathy, and improved post-transplant graft survival. The study was unblinded due to severe adverse events in the IVIg + placebo group, including AMR (n = 3) and graft loss (n = 2) (P = 0.06). Although no significant differences were seen in DSAs at transplant, rapid DSA rebound associated with severe AMR occurred within 1 month post-transplant. DSAs trended downward in those who received IVIg + rituximab, and no cases of AMR were observed on for-cause and 6-month protocol biopsies. Additionally, the IVIg + rituximab group showed significant benefits in renal function at 6 and 12 months (P = 0.04) [19].
Zachary et al. so elegantly demonstrated the ability of rituximab to prevent anamnestic responses to alloantigens post-transplant. In 24 patients sensitized to HLA antigens, who did not have HLA antibody before transplantation, no post-transplant antibody to HLA antigens was detected in 10 rituximab-treated patients. However, HLA antibody was detected in 13 of 16 cases without rituximab treatment (P = 0.00006). Thus, elimination of peripheral HLA-specific B cells in patients who are sensitized to HLA antigens, but lacking detectable antibody abrogates an anamnestic response and risk for AMR [20].
Kohei et al. showed the administration of rituximab in ABO-incompatible transplant recipients (n = 57) resulted in long-term prevention of dnDSAs and low incidence of AMR versus a cohort of ABO-compatible living donor transplants not receiving rituximab (n = 83). The 5-year graft survival rates were 98.1% with rituximab versus 90.3% in the ABO-compatible group. At 2 years post-transplant, the incidence of AMR in the rituximab group was 3.5% versus 22.9% in the ABO-compatible patients [21].
Recent data from a randomized, placebo-controlled clinical trial showed that anti-CD20mAb did not add benefit to treatment of AMR with plasmapheresis + IVIg [22]. However, there were many troubling issues in this trial that likely limit the validity of the results. First, rituximab was given on the same day as IVIg. This likely limits the efficacy and half-life of rituximab due to IVIg blocking of the Fc neonatal receptors (FcRn) on endothelium that are responsible for recycling IgG molecules and extending their half-life. The investigators also performed plasma exchange <24 hours after IVIg + rituximab, removing the majority of the drug [23, 24].
Another randomized, placebo-controlled study assessed the efficacy of rituximab for treatment of AMR. This multicenter trial aimed to enroll 25 patients per arm but (due to cost) enrolled 25 in total (12 rituximab, 13 placebo). The authors found no difference in outcomes at 1 year. Despite good intentions, this study was not powered to formulate the conclusions put forward.
From our work in desensitization and that of others, rituximab shows benefits in removing memory B cells and limiting antibody rebound after other treatments (i.e., IVIg, plasmapheresis, IdeS) [19, 20].
Obinutuzumab
Obinutuzumab is a humanized, type II, immunoglobulin-G1 anti-CD20mAb, FDA-approved for chronic lymphocytic leukemia. Obinutuzumab, unlike rituximab is a more powerful depleter of B cells, operating through ADCC and direct apoptosis, with little effect through CDC. This is important since the concomitant use of complement inhibitors (i.e., eculizumab) with rituximab likely diminishes efficacy of rituximab, as its primary mode of B-cell depletion is through CDC.
In vitro obinutuzumab was twofold more efficient in reducing B-cell cytotoxicity. Specifically, obinutuzumab exhibited a more potent activation of natural killer (NK) cells and neutrophils, and a more effective Fcγ receptor interaction [25,26,27].
A Phase Ib, open-label study of single and repeat doses of obinutuzumab to assess pharmacokinetics, pharmacodynamics, and safety in highly HLA-sensitized end-stage renal disease patients awaiting kidney transplantation did not show benefit in reducing calculated panel-reactive antibody (cPRA) values, although ~50% of patients ultimately underwent transplantation [28]. Thus, obinutuzumab, a novel type 2 anti-CD20mAb with superior B-cell depletional activity and efficacy against plasmablasts, could be of benefit in prevention and treatment of AMR.
Costimulation Blockade
CTLA4Ig (Belatacept): An Inhibitor of T and B Cells
Recent studies have highlighted the importance of costimulatory blockade in controlling B-cell-directed immune responses to allografts [29]. Post-transplant generation of dnDSA is recognized as a major cause for allograft failure. To date, the immunosuppressive regimen associated with low dnDSA development is a failure to maintain therapeutic levels of tacrolimus [30]. Recent clinical trials using the novel costimulatory-blocking IgG Fc fusion protein containing CTLA4 (CTLA4-Ig) show that kidney transplant recipients treated with belatacept have better graft survival, graft function, and a lower proportion of dnDSAs versus cyclosporine [31]. Chen et al. showed that CTLA4-Ig treatment of allosensitized mice resulted in significant suppression of B memory cell responses [32].
Our group conducted dosing experiments in a mouse model of allogeneic sensitization to evaluate the efficacy of CTLA4-Ig treatment in DSA suppression. We found that CTLA4-Ig significantly inhibited dnDSA IgM and IgG production in mice sensitized to HLA-A2+ skin grafts. In longitudinal experiments, we found that CTLA4-Ig administered during T-cell priming 90 days after primary skin graft exposure had a long-lasting effect in reducing DSA IgG memory responses to HLA-A2+ skin grafts. The inhibitory effect of CTLA4-Ig in suppressing DSA memory responses was significantly enhanced by the addition of an anti-IL-6R antibody. We also demonstrated that dnDSA suppression by CTLA4-Ig is due to inhibition of T-dependent B-cell activation secondary to Tfh cell inhibition. In in vitro experiments with alloreactive plasma cells, we found that CTLA4-Ig inhibited plasma cell proliferation and Ig production. This suggests that plasma cells may depend on costimulation through CD28/B7 as a mechanism of activation [33].
Leibler et al. recently investigated the mechanisms involved in the control of humoral responses by analyzing the effect of belatacept on different steps of the B-cell-mediated response in humans. In vitro belatacept reduced plasmablast differentiation, Ig production, and the expression of the major transcription factor involved in plasma cell function, Blimp-1, in a T-cell-independent manner. Belatacept reduced the expression of CD86 on antigen-presenting cells (APCs). Additionally, belatacept blocked CD28-mediated activation of Tfh in an autologous Tfh-memory B-cell model.
In kidney transplant recipients treated with belatacept, investigators demonstrated that patients treated with belatacept had reduced effector B cells and activated Tfh cells compared with calcineurin inhibitor-treated patients. They concluded that belatacept modulates B-cell function directly at the level of B cell–Tfh interaction and these interactions are likely responsible for the modulation of humoral immunity seen in belatacept-treated patients [34]. This paper is of great interest since belatacept may emerge as an important agent for prevention and treatment of AMR.
Anti-plasma Cell Therapies
Daratumumab
Daratumumab is an anti-CD38mAb which induces potent CDC and ADCC against CD38+ cells in patients with multiple myeloma. Daratumumab is the first anti-CD38mAb, and received FDA breakthrough status in 2015 [35]. CD38 is a type II transmembrane glycoprotein heavily involved in intracellular signaling via cell adhesion, calcium-dependent signal cascade, activation of NK cells, and IgG1 production from B- and T-cell signal transmission [36]. CD38 is more highly expressed on malignant cells and is present on the surface of short- and long-lived plasma cells. In a dose escalation study for multiple myeloma, daratumumab significantly reduced bone marrow plasma cells. CD38+ T regulatory cells (Tregs) and CD38+ B regulatory cells (Bregs) were decreased by daratumumab administration. This mechanism may provide a potential treatment of plasma cell-induced AMR. Overall, the safety profile of daratumumab is acceptable [37]. However, the impact on depletion of Tregs/Bregs by daratumumab may be a concern for induction of cell-mediated rejection in HLA-sensitized patients.
Proteasome Inhibitors
Bortezomib
Targeting antibody-producing plasma cells was felt to be a superior strategy to use of anti-CD20mAb in treating AMR. Several centers promoted the use of bortezomib, a proteasome inhibitor approved for the treatment of multiple myeloma. Data supporting the use of bortezomib were limited to single centers, and evidence on efficacy and safety from a larger cohort of patients with AMR was lacking. However, two recent papers have addressed this issue.
First, Eskandary et al. reported results of the first prospective, randomized, placebo-controlled trial of bortezomib in patients with late active AMR (BORTEJECT Trial) [38]. Of 744 patients, 44 met study criteria and were randomized to two cycles of bortezomib (n = 21) or placebo (n = 23). In direct contradiction to previous reports, these investigators found that bortezomib had no effect on outcomes over 2 years of follow-up (GFR slope −4.7 versus −5.2 ml/min per 1.73 m2 per year). Proteinuria, DSA and AMR histology, and molecular microscopic analysis did not differ between the two groups. Bortezomib therapy was associated with more drug-related side effects [38].
Moreno Gonzales et al. recently reported that 32 doses of bortezomib for desensitization were not well tolerated and had only a modest impact on anti-HLA antibodies [39]. Thus, the role of bortezomib as a future therapeutic agent in treatment and prevention of AMR is questionable, and may be more effective when combined with other therapies.
Carfilzomib
Ensor et al. reported on carfilzomib-based therapy for treatment of AMR in lung transplant recipients. Carfilzomib is a second-generation proteasome inhibitor that irreversibly binds the 26s proteasome and permanently inhibits activity. These investigators found that treatment with carfilzomib resulted in significant reductions in DSAs and improvement in lung allograft function during the treatment period in 10 of 14 patients. However, seven deaths occurred in carfilzomib responders due to allograft failure. The authors suggest that severe AMR may not be amenable to intermittent carfilzomib therapy [40]. It is also likely that rebound DSA responses after cessation of carfilzomib accelerated the decline in allograft function. Carfilzomib + CTLA4-Ig may offer an excellent therapeutic approach for prevention and treatment of AMR [41].
IL-6 and IL-6R Inhibitors
IL-6 was first described as a multifunctional cytokine that directed the development and maturation of B cells to plasma cells and sustained antibody production [1, 42,43,44]. In this regard, the role of IL-6 in induction of Tfh cells is critical for initiation of adaptive immune responses, progression of naive B cells to plasma cells, and production of high-affinity antibodies [1, 44, 45]. Additionally, persistence of IL-6/IL-6R (IL-6 receptor) signaling inhibits Treg cell development, thus enhancing Tfh and Th17 pathogenic antibody and inflammatory functions. Importantly, anti-IL-6/IL-6R therapies are known to have effects in reducing Th17 and Tfh cells, which block autoimmunity and reduce pathogenic antibody production. The ability of anti-IL-6/IL-6R therapy to inhibit Tfh activity and reduce alloreactive B cells, plasmablasts, and DSA production is a significant consideration in the prevention and treatment of alloantibody-induced injury.
IL-6 is a growth factor critical for B cells and plasma cells and is produced by plasmablasts, resulting in new germinal center formation. IL-6 inhibition significantly reduces Th17 and Tfh cells, plasmablasts, and upregulates Treg cells. Clinical trials of anti-IL-6/IL-6R therapies have been completed or are now underway in kidney transplantation for the treatment and prevention of AMR [46,47,48,49,50]. Data on the two most important IL-6 inhibitors are below.
Tocilizumab
Tocilizumab is a first-in-class mAb directed at the IL-6R. Tocilizumab was FDA-approved for treatment of rheumatoid arthritis and juvenile idiopathic arthritis in 2011. Tocilizumab resulted in reductions in peripheral pre- and postswitch memory B cells, IgG+ and IgA+ B cells, and significantly reduced B-cell hyper-reactivity.
Our group conducted a single-center phase I/II open-label study in HLA-sensitized patients with end-stage renal disease who had failed desensitization with IVIg + rituximab ± plasmapheresis. All patients received IVIg 10% 2 g/kg on days 1 and 30, and tocilizumab 8 mg/kg on day 15, then monthly for 6 months. Of the ten patients, five received transplants (two were withdrawn due to noncompliance with protocol pretransplant). Mean time to transplant from first desensitization decreased from 25 ± 10.5 to 8 ± 5.4 months with tocilizumab treatment. Reductions in immunodominant DSAs were seen in all transplanted patients at transplant (p = 0.024) and most significantly at 12 months (p = 0.0003). All patients received six tocilizumab doses post-transplant and 6-month protocol biopsies showed no evidence of rejection. The estimated glomerular filtration rate (eGFR) at 12 months in transplanted patients was 60 ± 25 ml/min [51].
In another single center, open-label study conducted by our group, highly sensitized patients with DSA+ cAMR, who failed IVIg + rituximab ± plasmapheresis therapy, received tocilizumab 8 mg/kg monthly for 6–25 months. A total of 36 patients were assessed (between 2011 and 2016) for allograft loss, patient survival, DSA reduction, and improvement in biopsy results. They were compared with a historical cohort treated with standard-of-care therapy (IVIG ± rituximab ± plasmapheresis) (n = 39). The median follow-up was 3.26 years (maximum 7 years). Four of 36 tocilizumab-treated patients had graft failure (11.1%). Repeat biopsies in tocilizumab-treated patients showed significant reductions in glomerulitis + peritubular capillaritis and C4d+ scores. This contrasted to 21 of 39 graft losses (54%) in patients treated with standard-of-care therapy. At 6 years post-cAMR diagnosis, the tocilizumab-treated patients had a graft and patient survival probability of 80% and 91%, respectively [52].
Clazakizumab
Clazakizumab is an immunoglobulin G1 (IgG1) mAb aimed at the IL-6 ligand. Clazakizumab has been evaluated extensively in patients with rheumatoid arthritis, but is not FDA-approved for any condition [53]. Our center has recently initiated two phase I/II, open-label, single-arm exploratory studies.
First, an AMR study (n = 10) will examine the safety and tolerability of clazakizumab 25 mg subcutaneously every 4 weeks in DSA+ patients with cAMR transplant glomerulopathy on biopsy [NCT03380377] [48]. Second, a desensitization study will assess 10 highly sensitized patients with cPRA >50% awaiting either a living donor or deceased donor kidney transplant. Eligible patients will receive plasmapheresis + IVIg followed by 6 months of clazakizumab 25 mg subcutaneously until transplantation. If transplanted, patients will receive six additional doses [NCT03444103] [47]. An additional placebo-controlled study assessing the utility of clazakizumab for treatment of cABMR is being conducted in Vienna and Berlin [46]. A large blinded, placebo-controlled multicenter study in cAMR is set to start in 2019.
IgG-Degrading Enzyme of Streptococcus pyogenes (IdeS, imlifidase)
Imlifidase is a novel drug that is being developed for desensitization and treatment of AMR in kidney transplant patients. Imlifidase is an immunomodulating enzyme that cleaves all four IgG antibody subclasses into F(ab’)2 and Fc fragments at the lower hinge region with high specificity. Other immunoglobulins, including IgA, IgM, IgE, and IgD, are not affected by the administration of imlifidase [54]. A critical observation in early assessments of IdeS in vitro is the inability of F(ab)’2 fragments to mediate CDC and ADCC [55].
In 2015, a single-arm, single-center, Phase II dose-finding study was completed in Sweden using Imlifidase in HLA-sensitized patients awaiting kidney transplantation to evaluate safety, tolerability, pharmacokinetics, and efficacy of imlifidase. The results demonstrated that imlifidase treatment eliminated all HLA antibodies detected by Luminex single antigen bead assays 6 hours after infusion and, more importantly, eliminated all complement-activating (C1q+) HLA antibodies 1 hour postinfusion [56].
We have also shown that IdeS is a potent inhibitor of ADCC [limiting NK-cell γ-IFN (interferon) release induced by anti-HLA antibodies binding to target endothelial cells] [57]. These initial observations led to the development of an open-label, Phase I/II study of imlifidase for desensitization in 25 highly sensitized patients with DSAs undergoing living and deceased donor kidney transplantation in Sweden and USA. All patients received imlifidase infusion prior to transplantation. The objective of the study was to assess the ability of imlifidase to eliminate DSAs in patients who were DSA+, with a positive crossmatch at time of transplantation. The patient group at Cedars-Sinai received desensitization therapy with IVIg 2 g/kg + rituximab, while the patients in Uppsala did not receive desensitization.
Of the 25 patients who received IdeS, 24 were successfully transplanted. AMR occurred at a mean of 2 weeks post-transplant in three patients in the Swedish arm due to rebound DSA, with C4d-positivity on for-cause biopsies. At Cedars-Sinai, AMR occurred in two patients at 2 and 5 months post-transplant, which correlated with an increase in DSA intensity and resolved with treatment. The differences in rebound times likely reflect the post-transplant use of IVIg + rituximab in the US patients. Long-term outcomes for these patients have been good [58]. A trial of imlifidase for treating AMR is now underway. In summary, imlifidase may represent an important breakthrough in prevention and treatment of AMR.
Important Considerations When Administering Biologic Agents for AMR
IdeS (imlifidase)
Imlifidase cleaves human and rabbit IgG at the hinge region creating F(ab)’2 and Fc fragments of all IgG molecules in the body in 4–6 hours after administration. The half-life of IdeS is approximately 8–12 hours, but IgG cleaving capacity may last for up to 4 days. This poses a problem for induction therapy post-transplant. Prior to performing the first clinical desensitization trials, we found that alemtuzumab was rapidly cleaved by imlifidase-treated sera up to 4 days post-transplant [58]. We altered our induction protocol to use high-dose steroids on days 1–4 and alemtuzumab on day 4. This gave similar T-cell depletion as seen in non-imlifidase-treated patients. Since thymoglobulin is also cleaved by IdeS, one should use a similar approach or consider horse anti-thymocyte globulin. Additionally, the post-transplant administration of IVIg + rituximab should be delayed for 4–5 days after IdeS administration.
Neonatal Fc Receptors and Half-Life of IgG
A common characteristic of IgG molecules is their long serum half-life of 3–4 weeks. This is related to IgG’s interaction with the FcRn. The function of FcRn is twofold. First, FcRn binds to serum IgG that has been endocytosed into lysosomes by endothelial cells or myeloid cells under low pH conditions. Here, the IgG is recycled back to the cell surface and, under neutral pH conditions, released back into the serum. Second, FcRn are expressed on the villi of placentas that are exposed to maternal blood and, after the 28th week of gestation, are responsible for endocytosing and transporting IgG molecules at a high rate from mother to neonate [23, 24]. In animal models of FcRn-knockouts, there is a dramatic reduction in IgG half-life from weeks to a few days.
It is also known that IVIg can occupy FcRn and thus accelerate the turnover of pathogenic antibodies. This may explain why large amounts of IVIg are needed for therapeutic activity in autoimmune and alloimmune disorders. Here, the antibodies in IVIg preparations compete with pathological autoantibodies and alloantibodies for FcRn binding. This explanation is supported by the observations that IVIg resulted in a reduction of approximately 50% in autoantibody half-life in a rat model of immune thrombocytopenic purpura (ITP) and in a neonatal mouse model of bullous pemphigoid [24].
Although FcRn saturation by IVIg is likely important in accelerating the clearance of pathogenic antibodies, it could also result in the rapid clearance of therapeutic antibodies. Little information is available, although there is one report of accelerated clearance of eculizumab when given with high-dose IVIg for treatment of multiple sclerosis [59]. In this regard, it is critical to avoid dosing therapeutic mAbs in temporal proximity to high-dose IVIg. This is likely to rapidly diminish the therapeutic efficacy of the monoclonal antibodies. This is important since reports assessing the efficacy of rituximab in the treatment of AMR in a placebo-controlled trial concluded that rituximab added no benefit to IVIg and plasmapheresis. Here, rituximab was given immediately after IVIg, which likely diminished the half-life and efficacy of rituximab [25]. In our practice, we wait ~7 days after high-dose IVIg administration to proceed with any chimeric or humanized mAbs. We have recently completed a trial to more thoroughly assess the impact of concomitant IVIg treatment on half-life of humanized mAb (anti-C5) therapy [NCT02878616] [60].
Summary
Modification of alloimmunity and alloantibodies will have relevance to all solid organ allotransplantation and to xenotransplantation, where xenoantibodies present a formidable obstacle. The ease of administration of biologic agents will likely change our views of immunosuppression to one of immune modulation that will ultimately result in better, more effective, and less toxic allograft-sustaining therapies, and also increase patient compliance. It is critical to advance transplant therapeutics to the next level where biologic agents are likely to play important roles in addressing the persisting barriers to successful transplantation created by alloimmunity.
Abbreviations
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- AMR:
-
Antibody-mediated rejection
- cAMR:
-
Chronic active antibody-mediated rejection
- CDC:
-
Complement-dependent cytotoxicity
- dnDSA:
-
De novo donor-specific antibody
- DSA:
-
Donor-specific antibody
- HLA:
-
Human leukocyte antigen
- IVIg:
-
Intravenous immunoglobulin
- Tfh:
-
T follicular helper
References
Chavele KM, Merry E, Ehrenstein MR. Cutting edge: circulating plasmablasts induce the differentiation of human T follicular helper cells via IL-6 production. J Immunol. 2015;194:2482–5.
Jordan SC, Ammerman N, Choi J, et al. Novel therapeutic approaches to allosensitization and antibody-mediated rejection. Transplantation. 2019;103:262–72.
Loupy A, Lefaucheur C. Antibody-mediated rejection of solid-organ allografts. N Engl J Med. 2018;379:1150–60.
Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest. 2017;127:2492–504.
Jordan SC, Tyan D, Stablein D, et al. Evaluation of intravenous immunoglobulin as an agent to lower allosensitization and improve transplantation in highly sensitized adult patients with end-stage renal disease: report of the NIH IGO2 trial. J Am Soc Nephrol. 2004;15:3256–62.
Marfo K, Lu A, Ling M, Akalin E. Desensitization protocols and their outcome. Clin J Am Soc Nephrol. 2011;6:922–36.
Montgomery RA, Lonze BE, King KE, et al. Desensitization in HLA-incompatible kidney recipients and survival. N Engl J Med. 2011;365:318–26.
Vo AA, Lukovsky M, Toyoda M, et al. Rituximab and intravenous immune globulin for desensitization during renal transplantation. N Engl J Med. 2008;359:242–51.
Vo AA, Petrozzino J, Yeung K, et al. Efficacy, outcomes, and cost-effectiveness of desensitization using IVIG and rituximab. Transplantation. 2013;95:852–8.
Sellarés J, DeFreitas DG, Mengel M, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12:388–99.
Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol. 2012;8:348–57.
Solez K, Colvin RB, Racusen LC, et al. Banff ‘05 meeting report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’). Am J Transplant. 2007;7:518–26.
Gloor J, Cosio F, Lager DJ, et al. The spectrum of antibody-mediated renal allograft injury: implications for treatment. Am J Transplant. 2008;8:1367–73.
Lefaucheur C, Loupy A, Vernerey D, et al. Antibody-mediated vascular rejection of kidney allografts: a population-based study. Lancet. 2013;381:313–9.
Jordan SC, Reinsmoen N, Peng A, et al. Advances in diagnosing and managing antibody-mediated rejection. Pediatr Nephrol. 2010;25:2035–45.
Haas M, Sis B, Racusen LC, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant. 2014;14:272–83.
Schrezenmeier E, Jayne D, Dorner T. Targeting B cells and plasma cells in glomerular diseases: translational perspectives. J Am Soc Nephrol. 2018;29:741–58.
van den Hoogen MW, Kamburova EG, Baas MC, et al. Rituximab as induction therapy after renal transplantation: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Transplant. 2015;15:407–16.
Vo AA, Choi J, Cisneros K, et al. Benefits of rituximab combined with intravenous immunoglobulin for desensitization in kidney transplant recipients. Transplantation. 2001;98:312–9.
Zachary AA, Lucas DP, Montgomery RA, et al. Rituximab prevents an anamnestic response in patients with cryptic sensitization to HLA. Transplantation. 2013;95:701–4.
Kohei N, Hirai T, Omoto K, et al. Chronic antibody-mediated rejection is reduced by targeting B-cell immunity during an introductory period. Am J Transplant. 2012;12:469–76.
Moreso F, Crespo M, Ruiz JC, et al. Treatment of chronic antibody mediated rejection with intravenous immunoglobulins and rituximab: a multicenter, prospective, randomized, double-blind clinical trial. Am J Transplant. 2018;18:927–35.
Roopenian DC, Christianson GJ, Sproule TJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170:3528–33.
Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–25.
Reddy V, Klein C, Isenberg DA, et al. Obinutuzumab induces superior B-cell cytotoxicity to rituximab in rheumatoid arthritis and systemic lupus erythematosus patient samples. Rheumatology. 2017;56:1227–37.
Obinutuzumab (Gazyva) [package insert]. Genentech, Inc., San Francisco; 2017.
Jain P, Kanagal-Shamanna R, Wierda W, et al. Membranoproliferative glomerulonephritis and acute renal failure in a patient with chronic lymphocytic leukemia: response to obinutuzumab. Hematol Oncol Stem Cell Ther. 2017;10:151–4.
A study of obinutuzumab to evaluate safety and tolerability in hypersensitized adult participants with end stage renal disease awaiting transplantation. Available from: https://ClinicalTrials.gov/show/NCT02586051.
Leibler C, Thiolat A, Elsner RA, El Karoui K, Samson C, Grimbert P. Costimulatory blockade molecules and B-cell-mediated immune response: current knowledge and perspectives. Kidney Int. 2019;95:774–86.
Davis S, Gralla J, Klem P, et al. Lower tacrolimus exposure and time in therapeutic range increase the risk of de novo donor-specific antibodies in the first year of kidney transplantation. Am J Transplant. 2018;18:907–15.
Vincenti F. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med. 2016;374:2600–1.
Chen J, Wang Q, Yin D, et al. Cutting edge: CTLA-4Ig inhibits memory B cell responses and promotes allograft survival in sensitized recipients. J Immunol. 2015;195:4069–73.
Kim I, Wu G, Chai NN, et al. Immunological characterization of de novo and recall alloantibody suppression by CTLA4Ig in a mouse model of allosensitization. Transpl Immunol. 2016;38:84–92.
Leibler C, Thiolat A, Hénique C, et al. Control of humoral response in renal transplantation by belatacept depends on a direct effect on B cells and impaired T follicular helper-B cell crosstalk. J Am Soc Nephrol. 2018;29:1049–62.
Dolgin E. Cancer’s true breakthroughs. Nat Med. 2013;19:660–3.
de Weers M, Tai YT, Van Der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–8.
Daratumumab (Darzalex) [package insert]. Janssen Biotech, Inc, Horsham; 2017.
Eskandary F, Regele H, Baumann L, et al. A randomized trial of bortezomib in late antibody-mediated kidney transplant rejection. J Am Soc Nephrol. 2018;29:591–605.
Gonzales MAM, Gandhi MJ, Schinstock CA, et al. 32 doses of bortezomib for desensitization is not well tolerated and is associated with only modest reductions in anti-HLA antibody. Transplantation. 2017;101:1222–7.
Ensor CR, Yousem SA, Marrari M, et al. Proteasome inhibitor carfilzomib-based therapy for antibody-mediated rejection of the pulmonary allograft: use and short-term findings. Am J Transplant. 2017;17:1380–8.
Kwun J, Burghuber C, Manook M, et al. Successful desensitization with proteasome inhibition and costimulation blockade in sensitized nonhuman primates. Blood Adv. 2017;1:2115–9.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16:448–57.
Liu X, Jones GW, Choy EH, et al. The biology behind interleukin-6 targeted interventions. Curr Opin Rheumatol. 2016;2:152–60.
Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res. 2014;2:288–94.
Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol. 2012;12:417–30.
A pilot trial of clazakizumab in late ABMR. Available from: https://ClinicalTrials.gov/show/NCT03444103.
Clazakizumab in highly-HLA sensitized patients awaiting renal transplant. Available from: https://ClinicalTrials.gov/show/NCT03380962.
Clazakizumab for chronic and active Antibody mediated rejection post-kidney transplant. Available from: https://ClinicalTrials.gov/show/NCT03380377.
A safety study of tocilizumab to Improve transplant rates in highly sensitized patients awaiting kidney transplantation. Available from: https://ClinicalTrials.gov/show/NCT01594424.
Cedars-Sinai Medical C. A safety study of tocilizumab to improve transplant rates in highly sensitized patients awaiting kidney transplantation. 2014 November. Available from: https://ClinicalTrials.gov/show/NCT01594424.
Vo AA, Choi J, Kim I, et al. A phase I/II trial of the interleukin-6 receptor-specific humanized monoclonal (tocilizumab) + intravenous immunoglobulin in difficult to desensitize patients. Transplantation. 2015;99:2356–63.
Choi J, Aubert O, Vo AA, et al. Assessment of tocilizumab (anti-interleukin-6 receptor monoclonal) as a potential treatment for chronic antibody-mediated rejection and transplant glomerulopathy in HLA-sensitized renal allograft recipients. Am J Transplant. 2017;17:2381–9.
Weinblatt ME, Mease P, Mysler E, et al. The efficacy and safety of subcutaneous clazakizumab in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate: results from a multinational, phase IIb, randomized, double-blind, placebo/active-controlled, dose-ranging study. Arthritis Rheumatol. 2015;67:2591–600.
von Pawel-Rammingen U, Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin. EMBO J. 2002;21:1607–15.
Tradtrantip L, Asavapanumas N, Verkman AS. Therapeutic cleavage of anti-aquaporin-4 autoantibody in neuromyelitis optica by an IgG-selective proteinase. Mol Pharmacol. 2013;83:1268–75.
Phase II Study, evaluation of safety and efficacy of IdeS in chronic kidney disease. Available from: https://ClinicalTrials.gov/show/NCT02224820.
Toyoda M, Shin BH, Ge S, et al. Impact of desensitization on antiviral immunity in HLA-sensitized kidney transplant recipients. J Immunol Res. 2017;2017:5672523.
Jordan SC, Lorant T, Choi J, et al. IgG endopeptidase in highly sensitized patients undergoing transplantation. N Engl J Med. 2017;377:442–53.
Fitzpatrick AM, Mann CA, Barry S, et al. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J Peripher Nerv Syst. 2011;16:84–91.
Study in end-stage renal disease patients awaiting kidney transplant to investigate the potential effect of IVIG treatment on the pharmacokinetics and pharmacodynamics of LFG316. Available from: https://ClinicalTrials.gov/show/NCT02878616.
Acknowledgments
The authors acknowledge the team members of the Kidney Transplant Program, Transplant Immunology Laboratory, and HLA Laboratory at Cedars-Sinai Medical Center for their commitment to improving lives through transplantation. The authors also want to acknowledge the patients who kindly participated in clinical trials to evaluate these potentially important drugs.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jordan, S.C., Ammerman, N., Vo, A. (2020). Evolving Approaches to Treatment of Allosensitization and Antibody-Mediated Rejection. In: Cooper, D.K.C., Byrne, G. (eds) Clinical Xenotransplantation. Springer, Cham. https://doi.org/10.1007/978-3-030-49127-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-49127-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-49126-0
Online ISBN: 978-3-030-49127-7
eBook Packages: MedicineMedicine (R0)